

Background: Eradication of primary human leukemia cells represents a major challenge. Therapies have not substantially changed in over 30 years.
Results: Using normal versus leukemia specimens enriched for primitive cells, we document aberrant regulation of glutathione metabolism.
Conclusion: Aberrant glutathione metabolism is an intrinsic property of human leukemia cells.
Significance: Interventions based on modulation of glutathione metabolism represent a powerful means to improve therapy.
Keywords: Anticancer Drug, Cancer Stem Cells, Leukemia, Redox Regulation, Tumor Metabolism, CD34+, Glutathione, Human, Parthenolide
Abstract
The development of strategies to eradicate primary human acute myelogenous leukemia (AML) cells is a major challenge to the leukemia research field. In particular, primitive leukemia cells, often termed leukemia stem cells, are typically refractory to many forms of therapy. To investigate improved strategies for targeting of human AML cells we compared the molecular mechanisms regulating oxidative state in primitive (CD34+) leukemic versus normal specimens. Our data indicate that CD34+ AML cells have elevated expression of multiple glutathione pathway regulatory proteins, presumably as a mechanism to compensate for increased oxidative stress in leukemic cells. Consistent with this observation, CD34+ AML cells have lower levels of reduced glutathione and increased levels of oxidized glutathione compared with normal CD34+ cells. These findings led us to hypothesize that AML cells will be hypersensitive to inhibition of glutathione metabolism. To test this premise, we identified compounds such as parthenolide (PTL) or piperlongumine that induce almost complete glutathione depletion and severe cell death in CD34+ AML cells. Importantly, these compounds only induce limited and transient glutathione depletion as well as significantly less toxicity in normal CD34+ cells. We further determined that PTL perturbs glutathione homeostasis by a multifactorial mechanism, which includes inhibiting key glutathione metabolic enzymes (GCLC and GPX1), as well as direct depletion of glutathione. These findings demonstrate that primitive leukemia cells are uniquely sensitive to agents that target aberrant glutathione metabolism, an intrinsic property of primary human AML cells.
Introduction
A major challenge in developing effective anti-cancer agents is to identify tumor-specific properties that are sufficiently selective to achieve eradication of malignant cells with minimal toxicity to normal tissues. Although numerous “targeted” strategies have been proposed in recent years, many of these approaches fail to show sufficient activity against the broad range of cell types commonly encountered in primary human tumors (1). Although the reasons behind these observations are complex, it appears likely that genetic, epigenetic, and cellular heterogeneity is a significant factor underlying the limitations of targeted agents (2). Consequently, therapies focused on fundamental physiological properties, e.g. mitochondrial characteristics, which may be shared irrespective of intra-tumoral heterogeneity may be of great value in achieving optimal therapeutic results (3). Such common properties must also be sufficiently tumor-specific to permit an adequate therapeutic index.
Of the cellular properties known to differ in cancer versus normal cells, redox state is perhaps the most prevalent. An altered redox balance has been reported for many forms of cancer, and presumably reflects a plausible therapeutic target (4). Notably, most conventional forms of chemotherapy employ drugs that induce cellular oxidative stress (5), suggesting that tumor cells may be preferentially sensitive to at least some conditions in which response to oxidative insult is required. However, the relative contribution of redox perturbation to tumor cell death and the mechanisms by which such agents may function in a tumor-specific fashion are not well understood. Thus, to better understand the underlying biology of oxidative state, and the properties that make cells susceptible to redox perturbation, we evaluated the characteristics of primary human hematopoietic cells derived from patients with acute myelogenous leukemia (AML)3 in comparison to normal controls.
The major mechanisms controlling cellular oxidative balance involve the glutathione system, thioredoxin proteins (TXNs), catalase (CAT), and superoxide dismutases (SODs). As comprehensively reviewed elsewhere (6, 7), and illustrated in Fig. 1A, glutathione is created by a process that begins with ligation of glutamate to cysteine to make γ-glutamylcysteine. This rate-limiting step of glutathione biosynthesis is catalyzed by the glutamate-cysteine ligase holoenzyme, which is composed of a catalytic (GCLC) and modulator subunit (GCLM). γ-Glutamylcysteine is subsequently ligated to glycine by glutathione synthetase (GSS) to produce the reduced form of glutathione (GSH). GSH then functions in three major roles: 1) to reduce H2O2 to H2O via the action of glutathione peroxidase (GPXs); 2) to detoxify harmful intrinsic and/or extrinsic electrophiles such as 4-hydroxynonenal, a process that is catalyzed by glutathione S-transferases (GSTs); and 3) to participate in protein S-glutathionylation process, a redox-mediated protein regulation mechanism (6). TXN regulates protein functions via a cysteine thiol-disulfide exchange mechanism mediated by its redox-active CXXC motif (8). CAT reduces H2O2 to H2O and O2 (9). Finally, SOD1 and SOD2 are important for destroying superoxide radicals (O2⨪) in cytoplasm and mitochondria, respectively (10).
FIGURE 1.

Primitive primary human AML cells differentially express genes required for control of redox state. A, schematic diagram showing major antioxidant machineries required for control of the redox state. B, expression of major antioxidant proteins in freshly isolated primary human CD34+ NBM (n = 4) and CD34+ AML (n = 9) specimens. Lysates from an equal number of cells (100,000) were loaded in each lane. The total amount of protein was quantified and presented as micrograms of protein per lane. C, relative mRNA expression of major antioxidant genes in freshly isolated primary human CD34+ NBM (n = 4) and CD34+ AML (n = 9) specimens. Mean expression of HPRT1, GUSB, and TBP was used as reference to internally normalize the expression of each gene within each specimen. Average expression of each gene in CD34+ NBM (n = 4) cells was set to 1, and the relative expression of each gene in each specimen was calculated accordingly and presented as dot plot. Mean ± S.E. of each group is presented. * indicates a significant difference.
Our studies have focused on the relative status of these mechanisms in primitive (CD34+) primary leukemia and normal cell types. Our findings indicate the intrinsic balance of glutathione, TXN, CAT, and SOD is aberrant in human leukemia populations. Leukemic cells show major changes in the relative abundance of enzymes that are required for glutathione biosynthesis and homeostasis, as well as abnormal levels of reduced and oxidized glutathione species. To explore the consequences of this unique biological condition with regard to therapeutic challenge, we examined how various agents modulate glutathione homeostasis in malignant and normal tissue. Our findings indicate agents such as parthenolide (PTL) and piperlongumine (PLM) have a dramatic inhibitory effect on the leukemic glutathione system, whereas only a limited and transient perturbation in normal cells. This preferential effect is strongly linked to their selective toxicity toward leukemia and other cancer cell types. Importantly, we have previously shown that PTL effectively eradicates AML stem and progenitor populations (11), cells that are typically resistant/refractory to conventional chemotherapy (12, 13). Thus, we propose that therapeutic targeting of glutathione metabolism represents a potentially powerful strategy to induce selective toxicity toward a broad range of primary leukemia cells, including malignant stem and progenitor populations.
EXPERIMENTAL PROCEDURES
Human Specimens
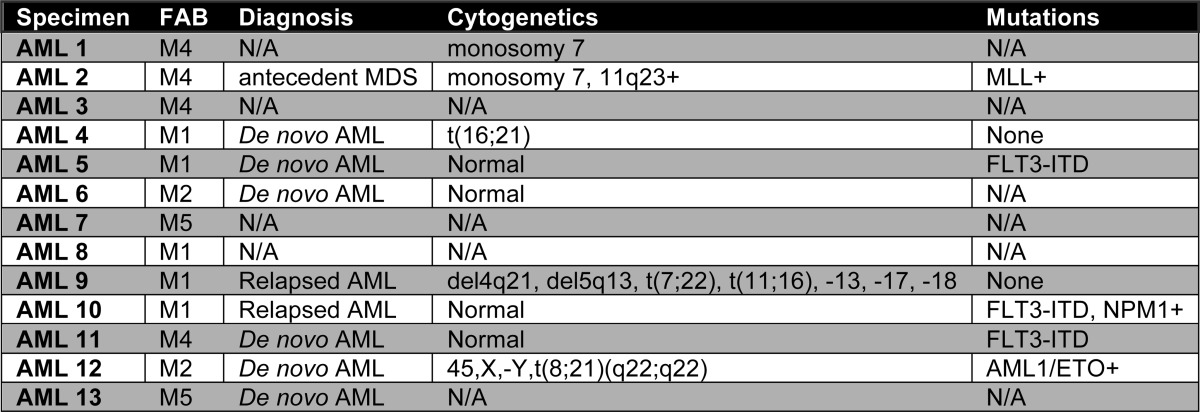
Normal bone marrow was obtained from volunteer donors who gave informed consent on a Research Subjects Review Board approved protocol at the University of Rochester Medical Center. AML specimens were obtained from apheresis product, peripheral blood, or bone marrow of patients who gave informed consent for sample procurement on the University of Rochester tissue procurement protocol. Clinical information of AML specimens is detailed in Table 1. FAB subtype information was determined by flow-based analysis. Total bone marrow mononuclear cells were isolated by standard Ficoll procedures (GE Healthcare), cryo-preserved in freezing medium Cryostor CS10 (BioLife Solutions), and stored in liquid nitrogen. Total mononuclear cells were further enriched for CD34 positive cells using the MACS CD34 enrichment kit (Miltenyi Biotec, Auburn, CA) and standard procedure. Enrichment purity was measured by flow cytometry-based quantification of cells stained positive for CD34 antigen.
TABLE 1.
Clinical information of primary human AML specimens
N/A, information not available.
Cell Lysates and Western Blot
NBM or AML cells were counted and lysed at 20 million/ml in RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% deoxycholic acid, 1% Triton X-100, 0.25 mm EDTA, 5 mm NaF) with freshly added protease inhibitors (1 mm PMSF, 1× protease inhibitor cocktail (Roche Applied Science), 0.1 mm Na3O4). Protein concentration was determined by the Bradford assay (Bio-Rad). Cell lysates were probed with primary antibodies against SOD1 (Santa Cruz), SOD2 (Upstate), CAT (Calbiochem), GCLC (Calbiochem), GCLM (Abcam), GSS (Abcam), GPX1 (Calbiochem), GSR (Calbiochem), GSTP1 (Abcam), TXN (Santa Cruz), heme oxygenase 1 (Stressgene), CASPASE-3 (Cell Signaling), poly(ADP-ribose) polymerase (Cell Signaling), ACTIN (Santa Cruz), IKKB (Cell Signaling), SLC7A11 (Novus), and GAPDH (Santa Cruz), followed by HRP-conjugated secondary antibodies (Santa Cruz). Chemiluminescence was detected using the automated Gel Doc XR+ system and Image Lab software (Bio-Rad) or x-ray films (Thermo).
Quantitative Real-time PCR
Total mRNA was isolated with the RNeasy plus mini-kit (Qiagen) according to the manufacturer's instructions. mRNA purity and quantity were determined with NanoDrop (Thermo). mRNA samples were reverse transcribed into cDNA using the iScript One-Step RT-PCR Kit (Bio-Rad). Quantitative Real-time PCR was performed with LightCycler480 real-time PCR (Roche) using LightCycler 480 SYBR Green I Master Mix reagent (Roche). To determine relative mRNA expression in CD34+ AML versus CD34+ NBM specimens, expression of each gene was first internally normalized to the mean expression of HPRT1, GUSB, and TBP. Average expression of each gene in CD34+ NBM (n = 4) cells was set to 1, and the relative expression of each gene in each specimen was calculated accordingly. To determine shRNA mediated knock-down efficiency, expression of each gene was first internally normalized to GAPDH and then used for comparison. Primer sequences for Q-RT-PCR were: SOD1-F, 5′-AGGCCCCTTAACTCATCT-3′, SOD1-R, 5′-CTACAGGTACTTTAAAGCAACTCT-3′; SOD2-F, 5′-AAGGGAGATGTTACAGCCCAGATA-3′, SOD2-R, 5′-TCCAGAAAATGCTATGATTGATATGAC-3′; CAT-F, 5′-TTTCCCAGGAAGATCCTGAC-3′, CAT-R, 5′-ACCTTGGTGAGATCGAATGG-3′; GCLC-F, 5′-TTGAGGCCAACATGCGAAA-3′, GCLC-R, 5′-AGGACAGCCTAATCTGGGAAATG-3′; GCLM-F, 5′-GGCACAGGTAAAACCAAATAGTAAC-3′, GCLM-R, 5′-CAAATTGTTTAGCAAATGCAGTCA-3′; GSS-F, 5′-AGCGTGCCATAGAGAATGAG-3′, GSS-R, 5′-ATCCCGGAAGTAAACCACAG-3′; GPX1-F, 5′-CCCTCTGAGGCACCACGGT-3′, GPX1-R, 5′-TAAGCGCGGTGGCGTCGT-3′; GSR-F, 5′-CAGTGGGACTCACGGAAGAT-3′, GSR-R, 5′-TTCACTGCAACAGCAAAACC-3′; GSTP1-F, 5′-CTGGTGGACATGGTGAATGAC-3′, GSTP1-R, 5′-CGCCTCATAGTTGGTGTAGATGA-3′; TXN-F, 5′-GTATTCCAACGTGATATTCCTTGAAG-3′, TXN-R, 5′-GCTTTTCCTTATTGGCTCCAG-3′; GAPDH-F, 5′-TGCACCACCAACTGCTTAGC-3′, GAPDH-R, 5′-GGCATGGACTGTGGTCATGAG-3′; TBP-F, 5′-GAGCTGTGATGTGAAGTTTCC-3′, TBP-R, 5′-TCTGGGTTTGATCATTCTGTAG-3′; GUSB-F, 5′-GAAAATATGTGGTTGGAGAGCTCATT-3′, GUSB-R, 5′-CCGAGTGAAGATCCCCTTTTTA-3′; HPRT1-F, 5′-TGAGGATTTGGAAAGGGTGT-3′, HPRT1-R, 5′-GAGCACACAGAGGGCTACAA-3′.
Glutathione Measurements
Glutathione quantification is based on the principle that reduced glutathione (GSH) and 5,5′-dithiobis(2-nitrobenzoic) acid can react and produce free TNB molecules that emit at 412 nm. Therefore, the amount of GSH is directly proportional to the rate of TNB production over time. The original method is detailed here (14). Briefly, glutathione samples were prepared by lysing NBM and AML cells in freshly made glutathione assay lysis buffer (16 mm KH2PO4, 82 mm K2HPO4, 5 mm EDTA, 0.1% Triton X-100, 6 mg/ml of sulfosalicylic acid). Supernatants were collected and used for three measurements: 1) amount of total glutathione (GSH + GSSG) by measuring the rate of TNB production over time; 2) amount of oxidized glutathione (GSSG) by first derivatizing GSH with 2-vinylpyridine (Sigma) and then measuring the rate of TNB production over time; 3) quantification of total protein with Bradford assay (Bio-Rad). After completing these three measurements, the GSH level is determined using following formula: [GSH] = [Total glutathione]-2X[GSSG]. The glutathione level is expressed as nanomole/mg of total protein.
Glutathione Turnover Rate
NBM and AML cells were cultured in DMEM-based serum-free media supplemented with [6-13C,2-15N]l-cystine (Cambridge Isotope, CNLM-4244) to a final concentration of 100 μm. Over time, cells were collected, quickly washed with ice-cold PBS, and pellets were snap frozen and stored in −80 °C freezer. For glutathione extraction, cell pellets were extracted three consecutive times with a −20 °C 50:50 mixture of 100% methanol and TBA solution (10 mm tribuytlamine, 15 mm acetic acid, 97:3 (water:methanol)). The resulting supernatants were combined in a single tube, transferred to HPLC sample vials, and analyzed with reverse-phase liquid chromatography (LC) coupled to a triple-quadrupole mass spectrometer running in negative mode (Thermo Quantum Ultra). Specific chromatography conditions and mass spectrometry parameters were as described previously (15). Glutathione was measured utilizing an MRM scan in negative mode specific for a 306 to 143 transition with a collision energy of 17 eV. Labeled glutathione was quantified using additional MRM scans specific for the extent of 13C and 15N labeling. The dominant labeled glutathione isoform that accumulated had a +4 m/z ratio, i.e. an MRM scan of 310 to 147. The resulting metabolite signal intensities were analyzed by the Xcalibar software (Thermo Electron Corp.).
Cell Culture and Drug Treatments
Isolated CD34+ normal bone marrow, CD34+, or bulk primary human AML cells were cultured and/or treated at 1 million/ml in Iscove's modified Dulbecco's medium-based serum-free media. For drug treatments, cells were preincubated in the media for 1 h before the addition of drugs, and the treatment time was 24 h. PTL (Biomol) and PLM (Tocris) were dissolved in 100% DMSO to make a 200 mm stock solution and stored in aliquots at −20 °C. For treatment, a 200 mm stock solution as first diluted with 100% DMSO to 10 mm, and then diluted with sterile PBS to 1 mm before treating the cells at the indicated concentrations. Ara-C (cytarabine, Sigma) was diluted in PBS to 1 mm concentration before use. IDA (idarubicin, Pfizer) was diluted in PBS to 100 μm before use.
Cell Viability
After treatment, PTL-, PLM-, Ara-C-, or IDA-treated cells were washed with ice-cold FACS buffer (PBS with 0.4% FBS) and stained at 4 °C in Annexin-V binding buffer (10 mm HEPES/NaOH, pH 7.4, 140 mm NaCl, 2.5 mm CaCl2) containing Annexin-V-FITC and DAPI. Stained cells were analyzed immediately by flow cytometry. Viable cells were scored as Annexin-V-negative and DAPI-negative. For drug synergy studies, treated cells were first stained at 4 °C for 30 min with anti-CD34-PE (BD Biosciences) antibody before staining with apoptotic markers. A high throughput sampler (HTS, BD Biosciences) was used to allow automated sampling processes.
Competitive Binding Assay and Targets Identification
Methods were slightly modified from our previous study (16). Briefly, isolated CD34+ AML cells were resuspended in Iscove's modified Dulbecco's medium-based serum-free media, and split into 3 groups. Group A is treated with DMSO control for the first hour and 20 μm biotin for the second hour; group B is treated with DMSO control for the first hour and 20 μm melampomagnolide B (MMB)-biotin for the second hour; group C is treated with 20 μm PTL for the first hour and 20 μm MMB-biotin for the second hour. After the treatments were finished, cells were washed with PBS, and lysed in Buffer F (10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 30 mm sodium pyrophosphate, 50 mm NaF, 5 μm ZnCl2, 1% Triton X-100) with freshly added proteinase inhibitors (1 mm PMSF, 1× protease inhibitor cocktail, 0.1 mm Na3OV4). About 5% of lysates were saved as input control, and the remaining lysates were incubated with streptavidin (SA) beads (Thermo) at 4 °C for 2 h to pull down binding targets of PTL. After incubation, beads were sequentially washed once with PBS, three times with high salt wash buffer (500 mm NaCl in 0.1 m NaOAc, pH 5.0), three times with low pH wash buffer (0.1 m glycine-HCl, pH 2.8), and one last time in 1× PBS. After the wash steps, the SA beads were boiled for 10 min in 2× SDS-PAGE sample buffer to elute down all pull-down products. For LC/MS-based identification of PTL targets, isolated CD34+ AML cells were treated with 20 μm biotin or 20 μm MMB-biotin for 2 h, and then lysed and incubated with SA beads to pull down PTL binding targets as described above. On-bead trypsin digestion was performed to release peptide fragments of PTL-binding targets. The digestion products were then analyzed by nanospray LC-MS/MS, using C13 reverse-phase liquid chromatography resin (Michrom), custom packed 5 cm × 75-μm fused silica column, which was coupled in-line to an ion trap mass spectrometer (Finnigan LTQ, Thermo Scientific). MS/MS-acquired data were searched against human amino acid sequences within the NCBI protein database, using MASCOT software (Matrix Science).
Modeling of PTL Targets Binding Events
The molecular model of GCLC and GPX1 were built with Modeler 9v7 (17) based on template PDB codes 3IG5 and 2F8A, respectively, whereas the GCLM model combines the aligned portions from two templates, PDB codes 1MZR (18) and 1R38 (19). Several loop regions in the models without any template were built and refined with the ab initio loop module of Modeler. A heterodimer model of GCLC-GCLM complex was constructed with the protein-protein docking program ClusPro (20), with a restraint to position Cys194 of GCLC and Cys553 of GCLM adjacent to each other. The covalent docking of PTL onto the targeted cysteines was performed with Prime software (version 9.3, Schrodinger, LLC, New York).
Knock Down and Overexpression Studies
The coding sequences of GCLC (NM_001498.2) and GPX1 (NM_000581.2) are targeted by following shRNA sequences: sh-GCLC #A: 5′-TGAAAGTGCTTCAAGGGTAAT-3′; sh-GCLC #B: 5′-GTCATCAATGTACCAATATTT-3′; sh-GPX1 #A: 5′-CCGCTTCCAGACCATTGACAT-3′; sh-GPX1 #B: 5′-GGTTCGAGCCCAACTTCATGC-3′. These shRNA sequences were constructed into pLKO.1-GFP vector, which contains a human U6 promoter to drive shRNA expression and an IRES-GFP to label shRNA-expressing cells. The GCLC overexpression plasmid was constructed by cloning GCLC cDNA into pLVX-EF1a-IRES-mCherry vector (Clontech). pLVX vector contains an EF1a promoter to drive GCLC expression and IRES-mCherry to label overexpressing cells. The detailed methods for generating lentiviral particles, and infecting AML cells are described here (21). Briefly, each knock-down or overexpression construct was co-transfected with pPax2 (provides packaging proteins) and pMD2.G (provides vesicular stomatitis virus-g envelope protein) plasmids into 293TN (System Bioscience) cells to produce lentiviral particles that were used to infect AML cell line M9-ENL cells. 3–5 days post-infection, if needed, GFP-positive or mCherry-positive M9-ENL cells were sorted on FACS Aria cell sorter (BD Bioscience) for subsequent experiments.
In Vivo Competitive Engraftment Assay
M9-ENL cells were infected with lentiviruses containing sh-GCLC, sh-GPX1, or sh-scramble sequences. At 72–96 h post-infection, these cells are used to inject immune-deficient NSG mice. Immediately before injection, we determined the GFP+ % at day 0. At 11 days post-injection, we sacrificed all groups of mice, harvested their bone marrow cells, and quantified the percentage of GFP+ cells within cells that were stained positive for human CD45 surface antigen. A ratio of GFP+ cells at day 11 relative to day 0 was calculated for each group. This ratio indicates the relative in vivo engraftment potential of M9-ENL cells expressing each shRNA.
Identification of Genes Correlated to PTL Resistance
High-throughput screening of PTL sensitivity in cell lines was carried out according to the following procedures. A total of 307 human cancer cell lines were obtained from the American Type Culture Collection (ATCC), the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ), the Japanese Collection of Research Bioresources (JCRB), or the European Collection of Cell Cultures (ECACC). Cells were grown in RPMI 1640 or DMEM/F-12 growth medium (Invitrogen) supplemented with 5% FBS and maintained at 37 °C in a humidified atmosphere at 5% CO2. Cells were seeded at ≈15% confluence in 96-well microplates (BD Biosciences) in medium supplemented with 5% FBS. After overnight incubation, cells were treated with three concentrations of PTL (100 nm, 1 μm, and 10 μm) by using a Caliper Sciclone ALH3000 multichannel liquid-handling work station (Caliper Life Sciences). After 72 h, cells were fixed in 4% formaldehyde in PBS and stained in a 1:5,000 solution of the fluorescent nucleic acid stain Syto60 (Molecular Probes). Quantitation of fluorescence was carried out at excitation and emission wavelengths of 630 and 695 nm, respectively, using the SpectraMax M5 plate reader (Molecular Devices). The mean of triplicate values for each drug concentration was compared with untreated wells, and a ratio was calculated (viability ratio). 32 cell lines with a viability ratio (treated/untreated cell number) of less than 0.75 after treatment with 1 μm PTL for 3 days were designated as a sensitive group, whereas 26 cells lines with a viability ratio of more than 0.75 after treatment with 10 μm PTL were designated as a resistant group. Comparative whole-genome expression profiling was performed on untreated cells using Affymetrix U133 X3P Microarrays. Expression data were normalized using GCRMA (22). The PAM algorithm was used to generate a gene expression signature to differentiate PTL-sensitive from PTL-resistant cell lines (23). Of the 58 cell lines (32 sensitive and 26 resistant) corresponding to these criteria half were randomly selected (13 insensitive, 16 sensitive) to be used as a training set and the other half as a test set for the PAM analysis.
Combination Index Calculation
Combination index (CI) was calculated by Calcusyn software (Biosoft). Briefly, viability of cells treated 24 h with each drug alone or dual drug combinations at various dose combinations were determined by flow cytometry. The results were input into Calcusyn software, which applies a Chou-Talalay method to calculate the CI value for each specific dose combo: CI <1, =1, and >1 indicates synergism, additive effect, and antagonism, respectively (24).
Statistics
Unless otherwise indicated, statistical analyses were performed using two-tailed (non-directional), type three (unequal variance) Student's t test. For statistical comparisons between glutathione levels and glutathione turnover rates in NBM versus AML cells (Fig. 2, A–D and G), the two-sided Mann-Whitney U test was used.
FIGURE 2.

Primary human AML cells have aberrant glutathione metabolism. Amount of reduced (GSH) (A), oxidized (GSSG) (B), total glutathione (sum of GSH + GSSG) (C), and GSH to GSSG ratio (D) in each specimen. Measurement of glutathione turnover rate (E). Time-dependent increase of newly synthesized glutathione ([13C,15N]glutathione) as percentage of total glutathione in CD34+ AML cells (n = 5) (F) and CD34+ NBM cells (n = 3) (G). H, glutathione turnover at 8 h post-culturing CD34+ NBM (n = 3) or CD34+ AML (n = 5) cells in media with [13C,15N]cystine. In A–D and H, each dot represents the value of each specimen. Median ± IQR (Inter-Quartile Range) of each group is presented as error bar. * indicates a significant difference.
RESULTS
Primitive Primary Human AML Cells Differentially Express Genes Required for Control of Redox State
Extensive previous studies have demonstrated that more primitive hematopoietic stem and progenitor cells reside within a small subset of bone marrow cells expressing the CD34 surface antigen (25). In addition, it has been demonstrated that primitive AML cells also generally express CD34 and are more resistant to conventional chemotherapy (12, 13). Thus, to focus our studies specifically on the most critical subpopulations, we isolated CD34+ cells from AML patients (Table 1) and normal bone marrow (NBM) donors for studies presented in this article (please note: we did not attempt to isolate more purified populations of stem cells, due to recent reports indicating substantial inter-patient variability in the cell surface phenotype of AML stem cells) (26, 27).
To investigate differences in the antioxidant machinery of primary AML versus normal hematopoietic cells, we compared protein expression of all major antioxidant genes. Because recent studies have demonstrated many conventionally used “housekeeping” genes are differentially expressed in cancer versus normal tissues (28, 29), and we consistently found that the GAPDH protein level differs significantly in CD34+ AML versus CD34+ NBM cells (data not shown), we chose to compare protein expression of antioxidant genes from the same number of cells or the same amount of protein lysates. As shown in Fig. 1B, probing the lysates from equal numbers of CD34+ AML (n = 9) and NBM cells (n = 4) revealed comparable levels of SOD1 protein expression. However, SOD2 and CAT protein expression appeared to be down-regulated in about half of the AML specimens examined, suggesting SOD and/or CAT functions are compromised in some AML specimens. In contrast, we found that the majority of AML specimens had up-regulated protein expression of glutathione pathway components including GCLC, GCLM, and GSS, which are required for glutathione biosynthesis, and GPX1 and GSR, which are involved in glutathione homeostasis. In addition, the GSTP1 protein, which is known to be overexpressed in several hematopoietic malignances including childhood acute lymphoblastic leukemia (30) and chronic lymphoblastic leukemia (31), was also up-regulated in our cohort of primary AML specimens. Last, TXN protein was up-regulated in a majority of AML patients as well. To exclude the possibility that the differences described above were due to a higher protein yield in leukemic versus normal specimens, we also compared the expression of these antioxidant genes within equal amounts of total protein from each specimen. The results consistently demonstrated a global up-regulation of glutathione pathway components in CD34+ AML specimens (data not shown).
To gain further insight on the differential expression of redox genes, we compared their mRNA expression as well. We used mean expression of HPRT1, GUSB, and TBP as reference to internally normalize the expression of each gene within each specimen. Average expression of each gene in CD34+ NBM (n = 4) cells was set to 1, and the relative expression of each gene in each specimen was calculated accordingly. Shown in Fig. 1C, in 9 of 10 genes examined (except for GCLM), mRNA levels between AML and NBM cells mirrored the direction of differences we observed at the protein level. For example, CD34+ AML cells have significantly down-regulated expression of SOD2 (0.16-fold, p = 0.046), and up-regulated expression of GSS (1.22-fold, p = 0.041), GPX1 (3.42-fold, p = 0.016), and TXN (1.27-fold, p = 0.041) mRNA (Fig. 1C), consistent with the differences we observed in our Western blot analysis (Fig. 1B). For many genes the degree of difference at the protein level surpasses the differences in mRNA expression, suggesting redox genes may be regulated at both the transcriptional and translational levels. Importantly, of the 6 genes directly involved in glutathione biosynthesis and homeostasis (GCLC, GCLM, GSS, GPX1, GSR, and GSTP1), 5 had elevated mRNA and protein expression in the AML specimens, indicating a global up-regulation of the glutathione pathway in CD34+ primary AML cells.
Primary Human AML Cells Have Aberrant Glutathione Metabolism
To directly compare glutathione pathway activity in primary human AML and normal cells, we quantified the amount of reduced (GSH) and oxidized (GSSG) glutathione in isolated CD34+ NBM (n = 4) and CD34+ AML (n = 11) cells. Our results show that, compared with CD34+ NBM cells, the GSH level is significantly lower and the GSSG level is generally higher in CD34+ AML cells (Fig. 2, A and B). As a result, CD34+ AML cells have significantly less total glutathione (sum of GSH and GSSG) as well as a significantly decreased GSH to GSSG ratio (Fig. 2, C and D). These data indicate an aberrant glutathione homeostasis in CD34+ primary AML cells.
We next compared the glutathione turnover rate in CD34+ AML and CD34+ NBM cells. CD34+ AML and NBM cells were isolated and cultured in media with isotope-labeled cystine ([13C,15N]cystine). We measured newly synthesized glutathione ([13C,15N]glutathione) as the percentage of total glutathione over time in each cell type (Fig. 2E). Over half of the AML specimens tested had a greater glutathione turnover at all time points compared with CD34+ NBM specimens (Fig. 2, F and G). In particular, after 8 h, the percentage of newly synthesized glutathione is significantly higher in CD34+ AML cells compared to CD34+ NBM cells (Fig. 2H). These data indicate the glutathione turnover rate is higher in AML specimens, suggesting glutathione synthesis and consumption are elevated in AML cells.
Primitive Human AML Cells Are Differentially Sensitive to Agents That Deplete Glutathione
The aberrant glutathione metabolism documented in Figs. 1 and 2 suggest that AML cells might be “addicted” to glutathione metabolism and therefore more vulnerable to glutathione pathway inhibition. Consequently, we investigated the activity of several experimental agents that are predicted to modulate glutathione metabolism. We first studied PTL, which contains an active α,β-unsaturated-γ-lactone group (Fig. 3B, red circular area) that should readily react with the free thiol group of glutathione. Indeed, PTL induced a dose-dependent decrease of cellular glutathione within 2 h of treatment in primary AML cells (n = 3, Fig. 3A). We then compared CD34+ cells from AML patients (n = 5) and NBM donors (n = 5) treated with 7.5 μm PTL to determine the change of total glutathione as a function of time. All CD34+ AML specimens experienced >95% of the maximal glutathione depletion after 4 h of PTL treatment (Fig. 3B, red lines). However, normal CD34+ cells treated with the same dose of PTL responded with only 60–80% maximal glutathione depletion at 4 h, followed by a robust rebound of cellular glutathione (up to 100% of untreated state) between 4 and 10 h (Fig. 3B, black lines). These data indicate glutathione homeostasis in AML cells is more vulnerable to PTL treatment. Importantly, this vulnerability correlates directly with cytotoxicity. Consistent with our previous findings (11), 7.5 μm PTL treatment induced significantly more cell death in CD34+ AML cells than CD34+ NBM cells (Fig. 3C). Taken together, these results suggest aberrant glutathione metabolism in AML cells might render AML cells to be hypersensitive to further inhibition of glutathione pathway activities.
FIGURE 3.

Primitive human AML cells are differentially sensitive to agents that deplete glutathione. A, PTL-induced dose-dependent glutathione depletion in primary human AML cells (n = 3). B, structure of PTL and PTL-induced cellular glutathione level change as a function of time. Red lines indicate changes in CD34+ AML cells (n = 5) and AML cell line M9-ENL cells. Black lines indicate changes in CD34+ NBM cells (n = 5). C, percentage of viable cells after being cultured with 7.5 μm PTL for 24 h. D, Western blot showing 7.5 μm PTL induced protein expression change of oxidative stress and apoptosis markers. E, structure of PLM and PLM-induced cellular glutathione level change as a function of time. Red lines indicate changes in CD34+ AML cells (n = 3) and AML cell line M9-ENL cells. Black lines indicate changes in CD34+ NBM cells (n = 2). F, percentage of viable cells after being cultured with 10 μm PLM for 24 h. In C and F, mean ± S.D. of each group is presented as bar graph. ***, p < 0.0005; *, p < 0.05.
Severe and sustained depletion of the cellular glutathione pool is known to induce oxidative stress and stimulate apoptosis (32, 33). We observed the induction of antioxidant defenses such as heme oxygenase 1 at 4 h following PTL treatment (Fig. 3D), the point at which glutathione depletion reaches maximal (Fig. 3B). Onset of apoptosis pathway activity as indicated by cleavage of Caspase-3 and poly(ADP-ribose) polymerase began at 4 h, and maximized at 6 h (Fig. 3D). Importantly, PTL-induced reduction of glutathione clearly occurs before the onset of apoptosis (Fig. 3B), indicating that glutathione loss precedes apoptosis and is not a nonspecific downstream consequence of apoptotic mechanisms.
To expand our studies beyond PTL, we also investigated PLM, a compound with a potentially similar mechanism-of-action. PLM was selected for two reasons. 1) It contains two functional alkene (olefin) groups that are adjacent to each of its ketone groups, indicating that PLM should be a potent electrophile, with chemical activities similar to PTL (34) (Fig. 3E, red circular areas). 2) A recent study reported PLM displays selective toxicity toward many types of cancer cells, an activity associated with induction of reactive oxygen species, although its mechanism of selectivity is not well understood (35). Notably, the toxicity of PLM to hematopoietic malignancies has not been reported up to date.
We first confirmed the ability of PLM to deplete cellular glutathione in a dose-dependent manner. At a 10 μm dose, PLM achieved a very similar degree of glutathione depletion as PTL at 7.5 μm (data not shown). Consequently, we treated CD34+ AML (n = 3) and CD34+ NBM cells (n = 2) with 10 μm PLM and monitored the change of cellular glutathione over time. Similar to the activity of PTL, we observed that a 4-h treatment with PLM induced a >90% maximal glutathione depletion in leukemic cells, but only ∼60% maximal glutathione depletion in CD34+ NBM cells (Fig. 3E). Importantly, consistent with its selective toxicity to many other cancer types (35), 24 h treatment with 10 μm PLM strongly induced cell death in CD34+ AML cells, but only limited toxicity in CD34+ NBM cells (Fig. 3F). Last, whereas PLM treatment in CD34+ AML cells induces severe glutathione depletion as soon as 2 h, poly(ADP-ribose) polymerase and caspase-3 cleavage were only evident at around 6 h post-PLM treatment (data not shown), again indicating that glutathione depletion precedes the onset of cell death.
Overall, these data demonstrate that CD34+ primary AML cells are more sensitive to agents depleting glutathione. In addition, the differential response of CD34+ AML versus NBM cells further indicate an intrinsic difference between mechanisms regulating the glutathione pathway in leukemic and normal cells.
Parthenolide Directly Binds to and Interferes with Multiple Glutathione Pathway Components
To further investigate how PTL affects the cellular glutathione metabolism, we employed more detailed analyses using biochemical and genetic approaches. Reactive cysteine sites are known to be important for enzymatic function of many glutathione pathway components including GCLC, GCLM, and GPX1 (7, 36–40). Given the fact that agents like PTL contain active moieties that readily react with accessible cysteines containing free thiol groups, we reasoned that this class of agents should further inhibit glutathione pathway activities by directly binding to and interfering with enzymes that regulate this pathway. To test this premise, we employed a biotinylated analog of PTL for biochemical studies. As we have previously described, PTL can be converted to the stereoisomer MMB, as a first step in the chemical process of adding a biotin moiety (Fig. 4A). Importantly, MMB-biotin retains the biological activity of PTL, albeit at a slightly reduced potency due to steric hindrance of the biotin moiety. Using MMB-biotin, we previously performed pull-down experiments and identified known binding partners of PTL, such as IKK-β and NF-κB p65 (16). Here we employed an analogous strategy to investigate interactions of PTL with regulators of the glutathione pathway. CD34+ primary human AML cells were treated with MMB-biotin, lysed, and then incubated with streptavidin beads to purify all protein targets of PTL. Subsequent studies using liquid phase-mass spectrometry (LC-MS/MS) identified GCLC and GPX1 as direct binding targets of PTL along with the antioxidant protein TXN (Fig. 4C, chart).
FIGURE 4.

Parthenolide directly binds to and interferes with multiple glutathione pathway components. A, chemical structure of MMB-biotin. Dashed box outlines intact structure of MMB, a stereoisomer of PTL. B, competitive binding assay. C, antioxidant proteins identified as PTL binding targets via competitive binding assay. Input and pull-down products from AML cells treated with biotin (lanes 1 and 4), MMB-biotin (lanes 2 and 5), or PTL followed by MMB-biotin (lanes 3 and 6). Pull-down products from MMB-biotin-treated AML cells were also subjected to LC-MS/MS analysis, and the results were presented in the chart for comparison. + and / signs indicate positive and negative results, respectively. Numbers of peptides recognized by LC-MS/MS were listed as well. D and E, covalent docking results of PTL to GCLC/GCLM holoenzyme complex (D) and GPX1 (E). The l-glutamate binding site of GCLC is enlarged in green box, and the heterodimer interface of GCLC-GCLM complex is in red box. The cysteines covalently attached with PTL are shown as surface representations, whereas the PTL structure is shown as thick sticks. F, Western blots showing time-dependent GCLC and GPX1 protein expression in primary human AML cells (n = 3) and M9-ENL cells treated with 7.5 μm PTL.
To test the specificity of binding events between PTL and its protein targets, we carried out a competitive binding assay. As outlined in Fig. 4B, CD34+ primary AML cells were pretreated with or without PTL followed by MMB-biotin treatment. Lysates were made from each treatment group and then passed over a streptavidin column to enrich for proteins that are directly bound by MMB-biotin. This methodology allowed us to identify specific binding targets of PTL. Using this approach, we successfully pulled down GCLC, GCLM, GPX1, and TXN proteins from cells that were treated with MMB-biotin (Fig. 4C, lane 5). Importantly, a pretreatment with PTL competed the binding of MMB-biotin to GCLC, GCLM, GPX1, and TXN, indicating that these interactions were specific (Fig. 4C, lane 6). IKK-β, a known binding target of PTL was used as a positive control for our assay (41).
To gain insights into these binding events at the molecular level and to predict their functional consequences, we built structures of the human GCLC-GCLM complex and GPX1 protein based on available crystal structures of their homologous proteins, and subsequently modeled docking events of PTL to accessible cysteines (Fig. 4, D and E). Our results show there are multiple cysteines in GCLC, GCLM, and GPX1 proteins that are available for PTL binding. In general, these binding events could result in protein instability or specific enzyme activity loss. For example, Cys249 of GCLC is evolutionarily conserved and resides near the glutamate-binding site (37). Therefore, this binding event would potentially interfere with the enzymatic function of GCLC (Fig. 4D, green box). In addition, PTL readily forms covalent bonds with Cys553 of GCLC and Cys193–Cys194 of GCLM (Fig. 4D, red box), which have been proposed to be involved in inter-subunit disulfide bond formation between GCLC and GCLM (38, 39). Formation of the GCLC and GCLM heterodimer is important for the holoenzyme activity of GCL (7), suggesting such binding events could disrupt GCL holoenzyme formation and consequently diminish its activity. In the case of GPX1, selenocysteine (Se-Cys49) is known as the active center of glutathione peroxidase (42). Therefore, binding between PTL and Se-Cys49 of GPX1 could potentially impair GPX1 enzymatic function as well (Fig. 4E).
To directly study the functional consequence of binding events between PTL and its targets, we measured the expression of proteins following PTL treatment. We found that the GCLC protein level decreased in all primary AML specimens (n = 3) as well as in the leukemic cell line M9-ENL cells (43) (Fig. 4F). The GPX1 protein level also decreased in most cases as well (Fig. 4F). Together, these data demonstrate that PTL targets the glutathione pathway by directly depleting glutathione (Fig. 3A) and interfering with critical glutathione pathway components GCLC and GPX1 (Fig. 4, C–F).
Targeting Glutathione Pathway Is Important for the Anti-leukemia Activity of PTL
PTL is known as an anti-cancer compound with multiple mechanisms of action (44–46), therefore we sought to determine whether targeting the glutathione pathway is important in the context of its overall anti-leukemia activity. We first tested whether genetic approaches that inhibit glutathione metabolism could impair leukemic cell growth and survival. To this end, we employed shRNA-based targeting of GCLC and GPX1 in the AML cell line M9-ENL cells. Sequences targeting each gene were constructed into a lentiviral vector that expresses GFP to mark shRNA-expressing cells (GFP+). At 72–96 h post-lentiviral infection, GFP+ M9-ENL cells were sorted at ∼95% purity for subsequent analyses. Shown in Fig. 5, A and B, for both GCLC and GPX1, two independent shRNA sequences induced efficient reduction of both mRNA and protein. As a result, the viability of M9-ENL cells lacking the expression of GCLC or GPX1 were both significantly lower compared with scramble control cells after 48 h of in vitro culture (Fig. 5C). We also counted the number of cells at 24 and 48 h in culture, and found that all sh-GCLC and sh-GPX1 clones grew much slower compared with control cells (Fig. 5D). These data demonstrate that glutathione pathway inhibition reduces the viability and growth of leukemic cells in vitro.
FIGURE 5.

Targeting glutathione pathway is important for the anti-leukemia activity of PTL. Knockdown of GCLC (A) and GPX1 (B) in M9-ENL cells. C, percentage of viable cells after 48 h culture in vitro. D, number of cells at 24 and 48 h culture in vitro. E, procedure and results of in vivo competitive engraftment assay. F, percentage of viable cells treated with the indicated doses of PTL for 24 h. G, overexpression of GCLC in M9-ENL cells. H, percentage of viable cells treated with indicated doses of PTL for 24 h. Error bars represent mean ± S.D. of triplicates. C indicates control used for statistical comparison, *, p < 0.05; ***, p < 0.0005.
We next wanted to measure the impact of glutathione pathway inhibition to leukemic cell growth in vivo using a competitive engraftment assay. As outlined in the diagram of Fig. 5E, we infected M9-ENL cells with lentiviruses containing sh-GCLC, sh-GPX1, or sh-scramble sequences. At 72–96 h post-infection, these cells were used to inject immune-deficient NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) mice. Immediately before injection, we determined the percentage of GFP+ cells (GFP+ %) at day 0 and found all groups were between 45 and 65% except for sh-GPX1 #B (less than 10%, was not used for subsequent experiments). After 11 days of in vivo engraftment, we sacrificed all groups of mice, harvested their bone marrow cells, and quantified the percentage of GFP+ cells at day 11. Because the cells used for initial injection contain a mixture of shRNA-expressing (GFP+) and wild type (GFP−) cells that will compete with each other to engraft in identical in vivo environment, a ratio of GFP+ % at day 11 relative to day 0 could be calculated to indicate the relative in vivo engraftment potential of M9-ENL cells expressing each shRNA. As shown in Fig. 5E, this ratio is significantly smaller for all sh-GCLC and sh-GPX1 clones compared with sh-SCR control (Fig. 5E). These data indicate that glutathione pathway activity is directly relevant for leukemic cell growth in vivo.
Together, these knock-down studies demonstrate glutathione pathway inhibition alone is harmful to leukemic cells both in vitro and in vivo. In addition, loss-of-function of GCLC or GPX1 can impair leukemic cell survival and growth. Because PTL shows activities that mirror these knock-down studies, PTL inhibits multiple glutathione pathway components including GCLC and GPX1 (Fig. 4), we propose that PTL-induced glutathione pathway inhibition is an important component of its anti-leukemia activity.
Cellular Glutathione Pathway Activity Determines Parthenolide Sensitivity
To further test if targeting the glutathione pathway is important for the anti-leukemia activity of PTL, we measured the sensitivity of leukemic cells with decreased or increased glutathione pathway functions to PTL treatment. Shown in Fig. 5F, we observed all sh-GCLC and sh-GPX1 clones demonstrated significantly increased sensitivity to PTL treatment in vitro. Moreover, when we overexpressed the rate-limiting step enzyme GCLC in M9-ENL cells (Fig. 5G), we dramatically increased the resistance of leukemic cells to PTL treatment (Fig. 5H). Similar data were observed upon treatment with PLM as well (data not shown).
To extend this analysis beyond leukemic cell types, we correlated the relative PTL sensitivity with the global gene expression profile of 307 solid tumor cell lines (multiple histologies) to determine gene functions that are associated with PTL resistance (supplemental Table S1). This study identified GPX2, a gastrointestinal glutathione peroxidase (presumably the solid tumor equivalent of GPX1), as the top ranked gene associated with PTL resistance. In addition, GCLC and SLC7A11 (a subunit of the cystine transporter upstream of glutathione biosynthesis) were correlated with PTL resistance as well. These findings suggest solid tumor cell lines with elevated glutathione pathway activity will be more resistant to PTL. This is consistent with the results of our genetic studies in leukemic cells (Fig. 5, F and H). Together, they indicate glutathione pathway activity is a critical determinant of PTL toxicity in AML cells, as well as other solid tumor types, a conclusion that strongly supports the hypothesis that targeting the glutathione pathway is central to the anti-leukemia activity of PTL.
Parthenolide and Piperlongumine Represent a Novel Class of Anti-leukemic Agents
Our findings of aberrant glutathione metabolism in CD34+ primary AML cells as well the abilities of PTL-like agents in targeting the glutathione pathway raised the possibility that PTL-like agents may represent a fundamentally different type of drug relative to the agents typically employed for leukemia therapy. To investigate this issue in more detail, we first performed a side-by-side efficacy test of PTL and PLM in comparison to the front line AML drug cytarabine (Ara-C) and the anthracycline IDA. Shown in Fig. 6, A and B, when 9 primary human AML specimens were treated with Ara-C or IDA, CD34+ populations in more than half of the specimens displayed a significant level of resistance (Fig. 6, A and B). This is presumably due to the fact that CD34+ AML cells are enriched for more quiescent stem and progenitor cells, and are relatively more resistant to chemotherapy (12, 13). In contrast, 7.5 μm PTL, which is well tolerated by CD34+ NBM cells, showed 75–95% toxicity to 7 of 9 specimens tested (Fig. 6C). We observed similar results when this group of AML cells was treated with PLM, where 10 μm PLM induced 60–95% toxicity to 7 of 9 specimens (Fig. 6D). These findings are noteworthy in that conventional agents, like PTL/PLM, are also known to induce oxidative stress (5), however, as clearly shown in Fig. 6, they are markedly less active toward CD34+ AML cells. This observation prompted us to investigate the mechanisms controlling oxidative stress induction in more detail and to ask whether the differential toxicity of PTL/PLM is correlated to their ability to modulate glutathione pathway activities. To this end, we treated AML specimens with each drug and monitored the change of glutathione following treatment. As shown in Fig. 6E, in striking contrast to PTL and PLM, treatment with Ara-C and IDA did not significantly change total glutathione level in CD34+ AML cells. This finding was also verified in the M9-ENL cell line. When M9-ENL cells were treated with each drug at doses that resulted in comparable cell death (Fig. 6F), PTL and PLM induced dramatic glutathione depletion, but Ara-C and IDA had little to no effect on cellular glutathione contents (Fig. 6G). Together, these data clearly indicate PTL and PLM possess a unique ability to inhibit the cellular glutathione system and are effective against CD34+ primary human AML cells, thereby representing a class of anti-leukemic agents that function by a distinctly different mechanism of action in comparison to standard chemotherapy agents.
FIGURE 6.

Parthenolide and piperlongumine represent a novel class of anti-leukemic agents. Viability of CD34+ AML cells (n = 9) treated 24 h with increasing doses of Ara-C (A), IDA (B), PTL (C), or PLM (D). E, representative graph showing the cellular glutathione level change over time in CD34+ AML cells treated with Ara-C, IDA, PTL, or PLM. F, viability of M9-ENL cells treated 24 h with Ara-C, IDA, PTL, or PLM at the indicated doses. G, cellular glutathione level change over time in M9-ENL cells treated with Ara-C, IDA, PTL, or PLM. In A–D and F, mean ± S.D. of triplicates is presented for each data point. In F, ***, p < 0.0005.
Toxicity of PTL in Combination with Conventional Anti-leukemic Agents Cytarabine and Idarubicin
Our findings of aberrant glutathione metabolism in CD34+ primary AML cells as well the abilities of PTL-like agents in targeting the glutathione pathway raised the possibility that combining PTL-like agents with conventional drugs might be a beneficial strategy. Thus, we tested whether combinations of such agents could act to synergistically induce AML cell death. Three primary AML specimens were treated with either Ara-C or IDA alone or in combination with PTL. The toxicity of each drug combination is presented in Fig. 7, A (PTL + Ara-C) and B (PTL + IDA). In all AML specimens tested, addition of PTL was beneficial in terms of overall cytotoxic activity. Notably, a modest dose of PTL (5 μm) combined with a suboptimal dose of Ara-C (5 μm) or IDA (60 nm) resulted in 82–93% cell death in all specimens tested (Fig. 7, A and B, red bars). To better quantify the effect of PTL with Ara-C or IDA, we calculated the potential synergy of each drug combination, where the CI score indicates synergism (0.4 < CI < 0.6), moderate synergism (0.6 < CI < 0.8), slight synergism (0.8 < CI < 0.9), additivity (CI > 0.9), or antagonism (CI > 1.1). The results of this analysis were converted into a heat map format to better illustrate the data (Fig. 7C). We found that the PTL and Ara-C combination displayed strong to slight synergism in all three AML specimens tested, although a different dose combination was required to achieve maximum synergy for each AML. In the case of the PTL and IDA, the combination showed synergy in one specimen (CD34+ AML2), with additive effects in two additional specimens (CD34+ AML 7 and 8). Together these results suggest agents like PTL can be combined with conventional chemotherapy to target more resistant CD34+ primary AML cells.
FIGURE 7.

Toxicity of PTL in combination with conventional anti-leukemic agents. Viability of CD34+ AML cells treated with PTL + Ara-C (A) or PTL + IDA (B) at various dose combinations. Mean of triplicates is presented. Numbers indicate mean viability. In A, viability of cells treated with 5 μm PTL alone, 5 μm Ara-C alone, and 5 μm PTL + 5 μm Ara-C in combination are highlighted as red bars. In B, viability of cells treated with 5 μm PTL alone, 60 nm IDA alone, and 5 μm PTL + 60 nm IDA in combination are highlighted as red bars as well. C, heat map showing the degree of synergy for each drug combination at fixed dose combos in each AML specimen. Synergism is determined by the CI calculated using Calcusyn software. Color key for each category is presented at the top of graph: synergism (0.4 < CI < 0.6), moderate synergism (0.6 < CI < 0.8), slight synergism (0.8 < CI < 0.9), additive or none (CI > 0.9).
DISCUSSION
AML is a highly heterogeneous disease with multiple types of oncogenic mutations in each individual patient. The role of each mutation as driver or passenger mutation, as well the interplay between various mutations remain poorly understood (2). These issues have made the search for targeted therapies for leukemia a particularly challenging endeavor. As an alternative, analysis of more broadly conserved physiological properties may represent an attractive strategy for developing novel anti-leukemia agents. Here, by comparing CD34+ NBM and AML cells, we identified aberrant glutathione metabolism as a unique and potentially useful property for targeting of primitive (CD34+) primary AML cells.
Our identification of the increased expression of glutathione metabolic enzymes in AML cells reflects several important intrinsic biological differences between primary AML and NBM cells. 1) A recent study found an increased basal level of nuclear Nrf2 protein in many primary AML specimens (47). This is a transcription factor activating the expression of many antioxidant enzymes including the critical components of glutathione pathway such as GCLC, GCLM, GSS, GPX1, GSR, as well as TXN and GSTs. Consistently, our study found up-regulated expression of almost all of these enzymes in CD34+ AML cells (Fig. 1, B and C), suggesting that increased Nrf2 activity in AML cells is potentially responsible for the elevated expression of these genes. 2) We previously demonstrated that primitive AML cells display constitutively active NF-κB signaling (13), which is known to regulate the expression of several antioxidant genes including GCLC (48) and GSTP1 (49). This observation suggests a link between up-regulated expression of GCLC and GSTP1 and elevated NF-κB signaling in primary AML cells.
Our study also found decreased levels of reduced glutathione GSH and increased levels of oxidized glutathione GSSG in CD34+ AML cells. Interestingly, freshly isolated primary chronic lymphoblastic leukemia cells also show a decreased GSH pool compared with normal tissue counterparts (50), suggesting decreased GSH content might be a common property of primary hematopoietic malignant tissues. We propose that the decreased GSH level is due to higher consumption of GSH in several processes required for cancer cell survival including: 1) reduction of reactive oxygen species such as H2O2, 2) proper S-glutathionylation of the proteome in response to oxidative stress, and 3) detoxification of increased production of lipid peroxides. Notably, GSH is particularly important in reducing hydrogen peroxide generated in mitochondria (51), and recent studies identified AML cells as having increased mitochondria mass and oxygen consumption rates (3), suggesting a link between GSH deficiency and abnormal mitochondria functions in leukemia. In addition, a significantly decreased GSH to GSSG ratio further indicates aberrant glutathione homeostasis in AML cells, because the GSH to GSSG ratio is a critical determinant of intracellular redox potential (52). Consistent with this premise, enzymes such as GPX1, GSR, and GSTP1, which are required for the function of GSH in these processes are all up-regulated in CD34+ AML cells, presumably as a survival mechanism (Fig. 1, B and C).
Oxidative stress has been characterized as a common property of AML (53). Moreover, studies show that increased oxidative stress appears to be a key event during leukemic transformation induced by FLT3-ITD, one of the most frequent oncogenic driver mutations in AML (54). In the current study, we identified an aberrant steady state pool of glutathione and global up-regulation of glutathione pathway proteins in AML. These data suggest that CD34+ primary AML cells have increased its glutathione metabolic enzyme expression to compensate for the loss of glutathione, presumably as a mechanism to manage increased oxidative stress induced by oncogenic transformation events. We propose that this unique property of CD34+ primary AML cells can be exploited by a class of agents such as PTL and PLM to achieve their preferential toxicity toward leukemic cell glutathione homeostasis and cell survival.
We observed, within 6 h of PTL or PLM treatment, that the glutathione levels drop faster and to a lower degree in CD34+ AML cells compared with CD34+ NBM cells (Fig. 3). This effect is likely due to a smaller steady state pool of glutathione in AML cells (Fig. 2), which make them more susceptible to PTL/PLM-mediated direct glutathione depletion. Between 6 and 10 h, glutathione levels robustly rebound in CD34+ NBM cells, presumably a cyto-protective antioxidant response of normal cells. However, glutathione levels in CD34+ AML cells generally stayed low, suggesting these cells are less able to efficiently up-regulate antioxidant functions (Fig. 3). We think this is likely because PTL also targets glutathione metabolism enzymes GCLC and GPX1 and induces their degradation (Fig. 4), therefore inhibiting glutathione recovery. Importantly, GCLC and GPX1 are both significantly up-regulated in AML cells, suggesting high protein levels are required for their function in AML cell glutathione homeostasis. Thus, PTL-induced GCLC and GPX1 degradation events are potentially more stressful to AML cells. Consistent with this premise, PTL or PLM treatment induced a significantly higher degree of cell death in AML compared with normal cells (Fig. 3).
Our studies indicate PTL and PLM represent a unique class of anti-leukemia agents that act to perturb multiple glutathione pathway components. However, it is important to note that glutathione inhibition is not the sole activity of PTL/PLM. Another well known activity of PTL is inhibition of NF-κB signaling, which is constitutively active in primitive primary AML cells (13). In addition, toxicity of PTL to other cancer tissues might be attributed to inhibition of epigenetic regulators such as HDAC1 and DNMT1 (55, 56) and suppression of STAT-3 activity (57), which are frequently elevated in cancer cells. Nonetheless, our genetic studies showed knock-down of GCLC or GPX1 as individual perturbation impaired the growth of leukemic cells in vitro (Fig. 5, C and D) and in vivo (Fig. 5E). These data clearly demonstrate that glutathione pathway inhibition alone strongly affects the health of leukemic cells, and therefore highlights the importance of targeting glutathione metabolism as a central component of the anti-leukemia activity of PTL/PLM. Notably, loss-of-function of either GCLC or GPX1 alone did not completely recapitulate the dramatic toxicity of PTL/PLM. We postulate that the drugs are more effective because they can simultaneously target both GCLC and GPX1, and also directly deplete glutathione, thereby providing a much more effective overall inhibition of glutathione metabolism. Furthermore, genetic manipulations require a gene transfer and selection process that is kinetically much slower than drug treatments. This slower process may allow cells to switch to an alternative mechanism for survival. For example, recent studies have shown that in certain cancer types, other antioxidant systems such as the TXN system can be employed by cells as a compensatory mechanism to combat the stress from glutathione pathway inhibition (58). Interestingly, TXN is also a direct binding target of PTL (Fig. 4C), a factor that may contribute to the potency of PTL. The importance of glutathione pathway inhibition in the anti-leukemia activity of PTL/PLM is further supported by the fact that knock-down of GCLC or GPX1 sensitized AML cells to PTL/PLM treatment, and overexpressing the rate-limiting enzyme GCLC vastly de-sensitized AML cells to PTL/PLM treatment (Fig. 5, F–H).
PTL and PLM are clearly distinct from standard AML chemotherapy agents like cytarabine and anthracyclines, which do not affect cellular glutathione and have limited toxicity toward the CD34+ population of AML cells (Fig. 6). CD34+ AML cells are enriched for leukemia stem and progenitor cells, and are regarded as a critical subpopulation to target for improved therapy. Hence, simultaneous eradication of both AML stem cells, as well as bulk disease, are regarded as important objectives toward achieving better clinical outcomes. To this end, we tested the combined effect of PTL with Ara-C (clinically known as cytarabine) or idarubicin (IDA), and found that PTL can substantially increase the activity of both agents toward CD34+ AML cells (Fig. 7). This observation is important because it indicates that transient suppression of glutathione is sufficient to augment the activities of conventional drugs, indicating that PTL and related agents may be useful as adjuvants to current chemotherapy.
Last, despite the fact that PTL and PLM seem to be quite different in their chemical backbone, as noted above, they both share similar active moieties that make them potent electrophiles. Interestingly, using a gene expression profile from PTL-treated AML cells, we previously identified other electrophiles such as 4-hydroxynonenal and 15-Δ-prostaglandin J2 as potent anti-leukemia agents (59). In addition, a recent screen of 303,282 compounds also identified an electrophile as its lead compound that selectively eradicates BJ-ELR (HRASG12V transformed) cells but spares untransformed parental BJ fibroblasts (60). Both PTL and PLM were also reported to have strong preferential toxicity toward BJ-ELR cells, with only limited toxicity to control cells (34). These data suggest certain types of electrophiles might be employing a similar mechanism to achieve their selective toxicity to cancer cells. We reason this mechanism is linked to an aberrant glutathione metabolism acquired during oncogenic transformation. In addition, PLM has been shown to have selective toxicity toward many different types of cancer cell lines (35), suggesting the mechanism of PLM-derived selectivity in primary AML cells described in the current study might be also relevant to many other types of cancer.
Taken together, our findings along with recent data from others indicate that primitive (CD34+) primary AML cells have acquired aberrant glutathione metabolism and are selectively eradicated by agents that target the glutathione pathway. Moreover, aberrant glutathione metabolism might be common to many cancer types, and specific classes of electrophilic compounds represent powerful means by which to target unique physiological properties of cancer cells and augment therapeutic regimens.
Acknowledgments
We gratefully acknowledge critical comments and feedback from Drs. Monica Guzman and Clayton Smith.
This work was supported, in whole or in part, by National Institutes of Health Grant R01AI081773 (to J. M.), Leukemia and Lymphoma Society Grant 6230-11 (to C. T. J.), Department of Defense Grant W81XWH-07-1-0601 (to C. T. J.), New York State Stem Cell Foundation Grant C024964 (to C. T. J.) and R01-CA158275 (to P. A. C.).

This article contains supplemental Table S1.
- AML
- acute myelogenous leukemia
- TXN
- thioredoxin proteins
- CAT
- catalase
- SOD
- superoxide dismutases
- GPX
- glutathione peroxidase
- PTL
- parthenolide
- PLM
- piperlongumine
- IDA
- idarubicin
- CI
- combination index
- NBM
- normal bone marrow
- MMB
- melampomagnolide B
- DMSO
- dimethyl sulfoxide
- LC
- liquid chromatography
- TNB
- 5-thio-2-nitrobenzoic acid.
REFERENCES
- 1. Couzin J. (2002) Cancer drugs. Smart weapons prove tough to design. Science 298, 522–525 [DOI] [PubMed] [Google Scholar]
- 2. Patel J. P., Gönen M., Figueroa M. E., Fernandez H., Sun Z., Racevskis J., Van Vlierberghe P., Dolgalev I., Thomas S., Aminova O., Huberman K., Cheng J., Viale A., Socci N. D., Heguy A., Cherry A., Vance G., Higgins R. R., Ketterling R. P., Gallagher R. E., Litzow M., van den Brink M. R., Lazarus H. M., Rowe J. M., Luger S., Ferrando A., Paietta E., Tallman M. S., Melnick A., Abdel-Wahab O., Levine R. L. (2012) Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366, 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skrtić M., Sriskanthadevan S., Jhas B., Gebbia M., Wang X., Wang Z., Hurren R., Jitkova Y., Gronda M., Maclean N., Lai C. K., Eberhard Y., Bartoszko J., Spagnuolo P., Rutledge A. C., Datti A., Ketela T., Moffat J., Robinson B. H., Cameron J. H., Wrana J., Eaves C. J., Minden M. D., Wang J. C., Dick J. E., Humphries K., Nislow C., Giaever G., Schimmer A. D. (2011) Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20, 674–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trachootham D., Alexandre J., Huang P. (2009) Targeting cancer cells by ROS-mediated mechanisms. A radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591 [DOI] [PubMed] [Google Scholar]
- 5. Conklin K. A. (2004) Chemotherapy-associated oxidative stress. Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 3, 294–300 [DOI] [PubMed] [Google Scholar]
- 6. Lu S. C. (2013) Glutathione synthesis. Biochim. Biophys. Acta 1830, 3143–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Franklin C. C., Backos D. S., Mohar I., White C. C., Forman H. J., Kavanagh T. J. (2009) Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Aspects Med. 30, 86–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arnér E. S., Holmgren A. (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109 [DOI] [PubMed] [Google Scholar]
- 9. Chelikani P., Fita I., Loewen P. C. (2004) Diversity of structures and properties among catalases. Cell Mol. Life Sci. 61, 192–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tainer J. A., Getzoff E. D., Richardson J. S., Richardson D. C. (1983) Structure and mechanism of copper, zinc superoxide dismutase. Nature 306, 284–287 [DOI] [PubMed] [Google Scholar]
- 11. Guzman M. L., Rossi R. M., Karnischky L., Li X., Peterson D. R., Howard D. S., Jordan C. T. (2005) The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105, 4163–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Costello R. T., Mallet F., Gaugler B., Sainty D., Arnoulet C., Gastaut J. A., Olive D. (2000) Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 60, 4403–4411 [PubMed] [Google Scholar]
- 13. Guzman M. L., Neering S. J., Upchurch D., Grimes B., Howard D. S., Rizzieri D. A., Luger S. M., Jordan C. T. (2001) Nuclear factor-κB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98, 2301–2307 [DOI] [PubMed] [Google Scholar]
- 14. Akerboom T. P., Sies H. (1981) Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Methods Enzymol. 77, 373–382 [DOI] [PubMed] [Google Scholar]
- 15. Munger J., Bennett B. D., Parikh A., Feng X. J., McArdle J., Rabitz H. A., Shenk T., Rabinowitz J. D. (2008) Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26, 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nasim S., Pei S., Hagen F. K., Jordan C. T., Crooks P. A. (2011) Melampomagnolide B. A new antileukemic sesquiterpene. Bioorg. Med. Chem. 19, 1515–1519 [DOI] [PubMed] [Google Scholar]
- 17. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 18. Jeudy S., Monchois V., Maza C., Claverie J. M., Abergel C. (2006) Crystal structure of Escherichia coli DkgA, a broad-specificity aldo-keto reductase. Proteins 62, 302–307 [DOI] [PubMed] [Google Scholar]
- 19. Kratzer R., Kavanagh K. L., Wilson D. K., Nidetzky B. (2004) Studies of the enzymic mechanism of Candida tenuis xylose reductase (AKR 2B5). X-ray structure and catalytic reaction profile for the H113A mutant. Biochemistry 43, 4944–4954 [DOI] [PubMed] [Google Scholar]
- 20. Kozakov D., Hall D. R., Beglov D., Brenke R., Comeau S. R., Shen Y., Li K., Zheng J., Vakili P., Paschalidis I. Ch., Vajda S. (2010) Achieving reliability and high accuracy in automated protein docking. ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins 78, 3124–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ashton J. M., Balys M., Neering S. J., Hassane D. C., Cowley G., Root D. E., Miller P. G., Ebert B. L., McMurray H. R., Land H., Jordan C. T. (2012) Gene sets identified with oncogene cooperativity analysis regulate in vivo growth and survival of leukemia stem cells. Cell Stem Cell 11, 359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bolstad B. M., Irizarry R. A., Astrand M., Speed T. P. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 [DOI] [PubMed] [Google Scholar]
- 23. Tibshirani R., Hastie T., Narasimhan B., Chu G. (2002) Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. U.S.A. 99, 6567–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chou T.-C., Talalay P. (1983) Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol. Sci. 4, 450–454 [Google Scholar]
- 25. Krause D. S., Fackler M. J., Civin C. I., May W. S. (1996) CD34. Structure, biology, and clinical utility. Blood 87, 1–13 [PubMed] [Google Scholar]
- 26. Eppert K., Takenaka K., Lechman E. R., Waldron L., Nilsson B., van Galen P., Metzeler K. H., Poeppl A., Ling V., Beyene J., Canty A. J., Danska J. S., Bohlander S. K., Buske C., Minden M. D., Golub T. R., Jurisica I., Ebert B. L., Dick J. E. (2011) Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 17, 1086–1093 [DOI] [PubMed] [Google Scholar]
- 27. Sarry J. E., Murphy K., Perry R., Sanchez P. V., Secreto A., Keefer C., Swider C. R., Strzelecki A. C., Cavelier C., Récher C., Mansat-De Mas V., Delabesse E., Danet-Desnoyers G., Carroll M. (2011) Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J. Clin. Invest. 121, 384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valente V., Teixeira S. A., Neder L., Okamoto O. K., Oba-Shinjo S. M., Marie S. K., Scrideli C. A., Paçó-Larson M. L., Carlotti C. G., Jr. (2009) Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. BMC Mol. Biol. 10, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Kok J. B., Roelofs R. W., Giesendorf B. A., Pennings J. L., Waas E. T., Feuth T., Swinkels D. W., Span P. N. (2005) Normalization of gene expression measurements in tumor tissues. Comparison of 13 endogenous control genes. Lab. Invest. 85, 154–159 [DOI] [PubMed] [Google Scholar]
- 30. Sauerbrey A., Zintl F., Volm M. (1994) P-glycoprotein and glutathione S-transferase π in childhood acute lymphoblastic leukaemia. Br. J. Cancer 70, 1144–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schisselbauer J. C., Silber R., Papadopoulos E., Abrams K., LaCreta F. P., Tew K. D. (1990) Characterization of glutathione S-transferase expression in lymphocytes from chronic lymphocytic leukemia patients. Cancer Res. 50, 3562–3568 [PubMed] [Google Scholar]
- 32. Franco R., Cidlowski J. A. (2009) Apoptosis and glutathione. Beyond an antioxidant. Cell Death Differ. 16, 1303–1314 [DOI] [PubMed] [Google Scholar]
- 33. Armstrong J. S., Steinauer K. K., Hornung B., Irish J. M., Lecane P., Birrell G. W., Peehl D. M., Knox S. J. (2002) Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 9, 252–263 [DOI] [PubMed] [Google Scholar]
- 34. Adams D. J., Dai M., Pellegrino G., Wagner B. K., Stern A. M., Shamji A. F., Schreiber S. L. (2012) Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc. Natl. Acad. Sci. U.S.A. 109, 15115–15120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raj L., Ide T., Gurkar A. U., Foley M., Schenone M., Li X., Tolliday N. J., Golub T. R., Carr S. A., Shamji A. F., Stern A. M., Mandinova A., Schreiber S. L., Lee S. W. (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475, 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Mullenbach G. T., Tabrizi A., Irvine B. D., Bell G. I., Hallewell R. A. (1987) Sequence of a cDNA coding for human glutathione peroxidase confirms TGA encodes active site selenocysteine. Nucleic Acids Res. 15, 5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hamilton D., Wu J. H., Alaoui-Jamali M., Batist G. (2003) A novel missense mutation in the γ-glutamylcysteine synthetase catalytic subunit gene causes both decreased enzymatic activity and glutathione production. Blood 102, 725–730 [DOI] [PubMed] [Google Scholar]
- 38. Tu Z., Anders M. W. (1998) Identification of an important cysteine residue in human glutamate-cysteine ligase catalytic subunit by site-directed mutagenesis. Biochem. J. 336, 675–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fraser J. A., Kansagra P., Kotecki C., Saunders R. D., McLellan L. I. (2003) The modifier subunit of Drosophila glutamate-cysteine ligase regulates catalytic activity by covalent and noncovalent interactions and influences glutathione homeostasis in vivo. J. Biol. Chem. 278, 46369–46377 [DOI] [PubMed] [Google Scholar]
- 40. Sheehan D., Meade G., Foley V. M., Dowd C. A. (2001) Structure, function and evolution of glutathione transferases. Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 360, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kwok B. H., Koh B., Ndubuisi M. I., Elofsson M., Crews C. M. (2001) The anti-inflammatory natural product parthenolide from the medicinal herb Feverfew directly binds to and inhibits IκB kinase. Chem. Biol. 8, 759–766 [DOI] [PubMed] [Google Scholar]
- 42. Asahi M., Fujii J., Takao T., Kuzuya T., Hori M., Shimonishi Y., Taniguchi N. (1997) The oxidation of selenocysteine is involved in the inactivation of glutathione peroxidase by nitric oxide donor. J. Biol. Chem. 272, 19152–19157 [DOI] [PubMed] [Google Scholar]
- 43. Barabé F., Kennedy J. A., Hope K. J., Dick J. E. (2007) Modeling the initiation and progression of human acute leukemia in mice. Science 316, 600–604 [DOI] [PubMed] [Google Scholar]
- 44. Mathema V. B., Koh Y. S., Thakuri B. C., Sillanpää M. (2012) Parthenolide, a sesquiterpene lactone, expresses multiple anti-cancer and anti-inflammatory activities. Inflammation 35, 560–565 [DOI] [PubMed] [Google Scholar]
- 45. Pajak B., Gajkowska B., Orzechowski A. (2008) Molecular basis of parthenolide-dependent proapoptotic activity in cancer cells. Folia Histochem. Cytobiol. 46, 129–135 [DOI] [PubMed] [Google Scholar]
- 46. Kreuger M. R., Grootjans S., Biavatti M. W., Vandenabeele P., D'Herde K. (2012) Sesquiterpene lactones as drugs with multiple targets in cancer treatment. Focus on parthenolide. Anticancer Drugs 23, 883–896 [DOI] [PubMed] [Google Scholar]
- 47. Rushworth S. A., Bowles K. M., MacEwan D. J. (2011) High basal nuclear levels of Nrf2 in acute myeloid leukemia reduces sensitivity to proteasome inhibitors. Cancer Res. 71, 1999–2009 [DOI] [PubMed] [Google Scholar]
- 48. Yang H., Magilnick N., Lee C., Kalmaz D., Ou X., Chan J. Y., Lu S. C. (2005) Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-κB and AP-1. Mol. Cell Biol. 25, 5933–5946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xia C., Hu J., Ketterer B., Taylor J. B. (1996) The organization of the human GSTP1–1 gene promoter and its response to retinoic acid and cellular redox status. Biochem. J. 313, 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trachootham D., Zhang H., Zhang W., Feng L., Du M., Zhou Y., Chen Z., Pelicano H., Plunkett W., Wierda W. G., Keating M. J., Huang P. (2008) Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood 112, 1912–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fernández-Checa J. C., Kaplowitz N., García-Ruiz C., Colell A., Miranda M., Marí M., Ardite E., Morales A. (1997) GSH transport in mitochondria. Defense against TNF-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 273, G7–17 [DOI] [PubMed] [Google Scholar]
- 52. Forman H. J., Zhang H., Rinna A. (2009) Glutathione. Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 30, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Er T. K., Tsai S. M., Wu S. H., Chiang W., Lin H. C., Lin S. F., Wu S. H., Tsai L. Y., Liu T. Z. (2007) Antioxidant status and superoxide anion radical generation in acute myeloid leukemia. Clin. Biochem. 40, 1015–1019 [DOI] [PubMed] [Google Scholar]
- 54. Sallmyr A., Fan J., Datta K., Kim K. T., Grosu D., Shapiro P., Small D., Rassool F. (2008) Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair. Implications for poor prognosis in AML. Blood 111, 3173–3182 [DOI] [PubMed] [Google Scholar]
- 55. Gopal Y. N., Arora T. S., Van Dyke M. W. (2007) Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem. Biol. 14, 813–823 [DOI] [PubMed] [Google Scholar]
- 56. Liu Z., Liu S., Xie Z., Pavlovicz R. E., Wu J., Chen P., Aimiuwu J., Pang J., Bhasin D., Neviani P., Fuchs J. R., Plass C., Li P. K., Li C., Huang T. H., Wu L. C., Rush L., Wang H., Perrotti D., Marcucci G., Chan K. K. (2009) Modulation of DNA methylation by a sesquiterpene lactone parthenolide. J. Pharmacol. Exp. Ther. 329, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sobota R., Szwed M., Kasza A., Bugno M., Kordula T. (2000) Parthenolide inhibits activation of signal transducers and activators of transcription (STATs) induced by cytokines of the IL-6 family. Biochem. Biophys. Res. Commun. 267, 329–333 [DOI] [PubMed] [Google Scholar]
- 58. Sobhakumari A., Love-Homan L., Fletcher E. V., Martin S. M., Parsons A. D., Spitz D. R., Knudson C. M., Simons A. L. (2012) Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One 7, e48175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hassane D. C., Guzman M. L., Corbett C., Li X., Abboud R., Young F., Liesveld J. L., Carroll M., Jordan C. T. (2008) Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood 111, 5654–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weïwer M., Bittker J. A., Lewis T. A., Shimada K., Yang W. S., MacPherson L., Dandapani S., Palmer M., Stockwell B. R., Schreiber S. L., Munoz B. (2012) Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg. Med. Chem. Lett. 22, 1822–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]