

Background: Studies of humans have identified missense mutations in the PHD2 gene associated with erythrocytosis.
Results: Mice bearing a heterozygous disease-associated Phd2 mutation display erythrocytosis equivalent in degree to that observed in Phd2+/− mice.
Conclusion: The human PHD2-associated erythrocytosis is due to a haploinsufficiency, rather than a dominant negative, mechanism.
Significance: The data formally prove that a PHD2 missense mutation can cause erythrocytosis.
Keywords: Erythropoeisis, Erythropoietin, Hypoxia, Hypoxia-inducible Factor, Hypoxia-inducible Factor (HIF), Prolyl Hydroxylase Domain Protein
Abstract
The central pathway for controlling red cell mass is the PHD (prolyl hydroxylase domain protein):hypoxia-inducible factor (HIF) pathway. HIF, which is negatively regulated by PHD, activates numerous genes, including ones involved in erythropoiesis, such as the ERYTHROPOIETIN (EPO) gene. Recent studies have implicated PHD2 as the key PHD isoform regulating red cell mass. Studies of humans have identified erythrocytosis-associated, heterozygous point mutations in the PHD2 gene. A key question concerns the mechanism by which human mutations lead to phenotypes. In the present report, we generated and characterized a mouse line in which a P294R knock-in mutation has been introduced into the mouse Phd2 locus to model the first reported human PHD2 mutation (P317R). Phd2P294R/+ mice display a degree of erythrocytosis equivalent to that seen in Phd2+/− mice. The Phd2P294R/+-associated erythrocytosis is reversed in a Hif2a+/−, but not a Hif1a+/− background. Additional studies using various conditional knock-outs of Phd2 reveal that erythrocytosis can be induced by homozygous and heterozygous knock-out of Phd2 in renal cortical interstitial cells using a Pax3-Cre transgene or by homozygous knock-out of Phd2 in hematopoietic progenitors driven by a Vav1-Cre transgene. These studies formally prove that a missense mutation in PHD2 is the cause of the erythrocytosis, show that this occurs through haploinsufficiency, and point to multifactorial control of red cell mass by PHD2.
Introduction
The oxygen-dependent, differential hydroxylation of specific proline residues within the α subunits of the transcription factor HIF (HIF-α)2 is a critical molecular switch that allows changes in oxygen tension to produce changes in gene transcription (1–3). This hydroxylation is catalyzed by PHD (prolyl hydroxylase domain) proteins (also known as egg laying defective nine homologues). Under normoxia, recognition of prolyl hydroxylated HIF-α by VHL (von Hippel-Lindau tumor suppressor protein), an E3 ubiquitin ligase, allows for rapid degradation of the former (4–6). Conversely, under hypoxia, nonhydroxylated HIF-α escapes degradation, allowing it to dimerize with HIF-β (also known as the aryl hydrocarbon nuclear translocator) and initiate transcriptional responses to this stress. This response to hypoxia has been shown to be a key factor during development, when fetal cells outgrow their nascent vascular system triggering HIF-dependent angiogenesis (7, 8). Furthermore, solid tumor development is also affected by hypoxic responses in areas of the tumor in which vasculature lags behind cell division (9, 10).
Importantly, the PHD:HIF:VHL axis plays a central role in the physiological increase in red cell mass in response to lowered oxygen tension via the production of the EPO hormone (11). EPO is generally present in low amounts in healthy adults, but it can be dramatically up-regulated in response to reduced atmospheric oxygen or in disease states leading to reduced oxygen uptake or delivery. Its primary effect is to induce the expansion and maturation of red blood cell progenitors in the bone marrow by binding to the EPO receptor (EPOR). Binding to EPOR induces a signaling cascade through the JAK-STAT pathway in red cell progenitors, leading to increased red cell mass. During mammalian fetal development, the liver is the main site of EPO synthesis; in adults, EPO production occurs primarily in the kidney (12). Following EPO stimulation of erythroid progenitors, the increased circulating erythrocytes lead to increased oxygen delivery, which is then followed by PHD-mediated hydroxylation of HIF-α and subsequent down-regulation of EPO mRNA as a homeostatic mechanism.
In mammals, the HIF-α and PHD protein families each contains three isoforms. Among the three HIF-α isoforms, HIF-1α and HIF-2α are the best characterized. HIF-1α is expressed ubiquitously, and its stabilization leads to diverse downstream effects including changes in glucose metabolism, apoptosis, and angiogenesis. HIF-2α (also known as endothelial PAS domain-containing protein 1) expression is more restricted. Studies of humans and genetically modified mice have indicated that HIF-2α plays the predominant role in control of erythropoiesis in adult mammals (13–16).
Among the PHD isoforms, PHD2 (also known as egg laying defective nine homologue 1) has emerged as particularly critical. In mice, knock-out of Phd2, but not of Phd1 or Phd3, leads to embryonic lethality (17). Acute global deletion of Phd2 in the mouse leads to elevated serum Epo levels and marked erythrocytosis (18, 19). Importantly, a number of human patients presenting with erythrocytosis have been shown to carry missense mutations in the PHD2 gene (20–22). Two notable features are as follows. First, in all instances, the mutation is heterozygous, which raises the question of whether the mechanism of erythrocytosis is through haploinsufficiency or through a dominant negative effect. Second, these patients typically display serum EPO levels that are within the normal range. This, in turn, raises the question of whether the EPO level is simply inappropriately normal with respect to the elevated red cell mass, whether there may be other EPO-independent effects of the PHD2 mutation, or both.
To gain insight into these issues, here we have focused on modeling the index PHD2 mutation, P317R, which affects an amino acid in the vicinity of the active site of PHD2 and markedly diminishes its activity (21, 23). The resulting knock-in mouse, which harbors a Phd2 P294R mutation, not only formally demonstrates that this mutation is the cause of the erythrocytosis but also shows that it occurs through haploinsufficiency. We also present results from studies of a number of conditional Phd2 knock-outs that employ a conditional Phd2 allele, including some in which Cre recombinase is driven by Pax3, Vav1, and VE-cadh promoters, as well as one bearing a globally expressed, tamoxifen-inducible Cre. Notably, erythrocytosis is observed in a conditional knock-out of Phd2 in hematopoietic precursors using a Vav1-Cre transgene, providing evidence that loss of Phd2 has Epo-independent effects on red cell mass. Taken together, these findings reinforce the central role that PHD2 plays in red cell control, highlight the sensitivity of this response to PHD2 gene dosage, and point to a wider, EPO-independent, role of PHD2 in this process.
EXPERIMENTAL PROCEDURES
Mouse Lines
The construct for generating a P294R mutation in the mouse Phd2 gene was prepared by recombineering (24). In brief, a minitargeting vector was constructed in the vector pL452 (25). This minitargeting vector contained genomic DNA encompassing exon 2 of the mouse Phd2 gene with a nucleotide change encoding for the P294R mutation. The vector contained a neomycin selection cassette flanked by loxP sites (floxed) and additional sequences downstream of exon 2. A retrieval plasmid was constructed in the vector pMC1-DTA (26). This retrieval plasmid contained sequences that flank 11 kb of genomic DNA sequence at the mouse Phd2 locus, as well as a diptheria toxin A negative selection cassette. This retrieval plasmid was used to capture, by recombineering, 11 kb of mouse Phd2 genomic DNA containing exons 2–4 from C57BL/6 bacterial artificial chromosome clone RP23–356I16 (Invitrogen). The resulting product was then used, in the second recombineering step with the minitargeting vector, to generate the final targeting vector. This targeting vector contains a 6.9-kb 5′ arm containing exon 2 with the P294R knock-in mutation, a neomycin selection cassette flanked by loxP sites and a 4.1-kb 3′ arm (Fig. 1A). The presence of the knock-in mutation and the integrity of exons 2, 3, and 4 were confirmed by DNA sequencing.
FIGURE 1.

Generation of Phd2P294R/+ knock-in mouse line. A, targeting strategy. Boxes denote exons, with numbers indicating exon number and black indicating coding sequence. The P294R mutation is marked by an asterisk. neo, neomycin selection cassette; DTA, diptheria toxin A negative selection cassette. The sites of Phd2mouseF (a) and Phd2mouseR (b) primers used for PCR genotyping are shown. Shaded boxes labeled 5′ and 3′ indicate the locations of 5′ and 3′ Southern blot probes, respectively. B, Southern blot screening of ES cell or mouse tail DNA. DNA was digested with EcoRI (3′ Southern) or ScaI (5′ Southern). Phd2 genotypes are shown. Neo selection cassette is indicated by neo. C, dye termination sequencing chromatogram of ES cell DNA showing heterozygous CCA to CGA (P294R) mutation. D, PCR genotyping of wild type and P294R knock-in Phd2 mice. The 0.48-kb band indicates the presence of the P294R Phd2 allele, whereas the 0.39-kb band indicates the presence of the WT allele.
C57BL/6 ES cells were electroporated with the targeting vector and selected using G418 by Caliper Life Sciences. Screening was performed by Southern blotting. Of 192 clones, 5 potential targeted clones were identified. Sequencing of these clones revealed that four had the P294R heterozygous mutation. Two of these clones were injected into C57BL/6 blastocysts to produce chimeras. Chimeric male mice were then mated with C57BL/6 female mice, and germ line transmission (as assessed by PCR and Southern blotting) was obtained with chimeras derived from one of the two targeted ES clones. Mice with germ line transmission of the knock-in allele were then mated with C57BL/6-Gt(ROSA)26Sortm16(Cre)Arte mice (Taconic) to delete the neomycin cassette, followed by further crossing with C57BL/6 mice to segregate the Cre allele, thereby creating Phd2P294R/+ mice. These mice were maintained in a C57BL/6 background.
Mice with a floxed Phd2 exon 2 allele in C57BL/6 background were generated as follows. We prepared a targeting construct, pFRT.loxP.Phd2, with PCR products obtained using C57BL/6 bacterial artificial chromosome clone pRP23–356I16 as a template. The construct contains a 5.6-kb 5′ arm, a floxed Phd2 exon 2, a neomycin positive selection cassette flanked by FRT sites, a 2.5-kb 3′ arm, and a thymidine kinase negative selection cassette (Fig. 2A). Integrity of exons 2 and 3 was confirmed by DNA sequencing. Bruce4 C57BL/6 ES cells were electroporated with the targeting vector and selected using G418 at the University of Pennsylvania School of Medicine Gene Targeting Facility. Of 1,600 clones, 10 clones were identified with correct 5′ and 3′ recombination events by Southern blotting. Southern blotting of the ES cell clone that was employed in these studies using a 3′ probe revealed the expected 6.0-kb band upon hybridization with BglII-digested DNA in addition to the wild type 4.4-kb band (Fig. 2B, left panel). Southern blotting with a 5′ probe revealed the expected 10.5-kb band upon hybridization with ScaI-digested DNA in addition to the wild type 14-kb band (Fig. 2B, right panel).
FIGURE 2.

Generation of Phd2f/f and Phd2+/− mouse lines. A, targeting strategy. Boxes denote exons, with numbers indicating exon number and black indicating coding sequence. neo, neomycin selection cassette; TK, thymidine kinase negative selection cassette. The sites of the PHD2rec55 (a), Pex2–3 3′ (b), Pint1 Sal 5′ (c), and Pint2–3 3′ (d) primers for PCR genotyping are as shown. Shaded boxes labeled 5′ and 3′ indicate the locations of 5′ and 3′ Southern blot probes, respectively. B and C, Southern blots of DNA from ES cells (B) or mouse tail (C). DNA was digested with ScaI (5′ Southern) or Bgl II (3′ Southern). D, PCR genotyping of mice with the indicated Phd2 genotypes. In the left panel, the WT allele produces a 0.73-kb band, whereas the floxed allele produces a 0.84-kb band. In the right panel, the 0.56-kb band indicates the presence of the knock-out allele.
This ES cell clone was injected into C57BL/6J-Tyrc-2J blastocysts to generate chimeras at the University of Pennsylvania School of Medicine Transgenic Core Facility. These chimeras yielded germ line transmission, as assessed by a combination of coat color, PCR, and Southern blotting (Fig. 2C). Mice with germ line transmission of the targeted allele were then mated with C57BL/6-Tg(ACTB-Flpe)2Arte mice (Taconic) to delete the neomycin cassette, followed by further crossing with C57BL/6 mice to segregate the Flp allele, thereby creating mice (Phd2f/+) harboring an allele in which exon 2 has been floxed. Phd2f/f mice were obtained by crossing Phd2f/+ mice (Fig. 2D, left panel).
Phd2+/− mice were generated by mating these Phd2f/f mice with C57BL/6-Gt(ROSA)26Sortm16(Cre)Arte mice to delete exon 2 (Fig. 2D, right panel), followed by further crossing with C57BL/6 mice to segregate the Cre allele. These mice were maintained in a C57BL/6 background.
Pax3-Cre; Phd2f/+, Pax3-Cre; Phd2f/f, Vav1-Cre; Phd2f/f, and VE-cadherin-Cre (VE-cadh-Cre); Phd2f/f mice were generated by crossing the aforementioned Phd2f/f or Phd2f/+ mice with B6;129-Pax3tm1(cre)Joe/J (27, 28), B6.Cg-Tg(Vav1-cre)2Kio/J (29), or B6.Cg-Tg(Cdh5-cre)7Mlia/J (30) mice, as appropriate (all from the Jackson Laboratory). In all cases, the Cre transgene was maintained in hemizygous state. The Pax3-Cre derived mice were maintained in a mixed background. The others were maintained in a C57BL/6 background.
Mice with a floxed Hif2a exon 2 allele have been described (15) and were backcrossed to C57BL/6 mice 10 times. Hif2a+/− mice were generated by mating Hif2af/f mice with C57BL/6-Gt(ROSA)26Sortm16(Cre)Arte mice to delete exon 2, followed by further crossing with C57BL/6 mice to segregate the Cre allele. These mice were then crossed with Phd2P294R/+ mice and were subsequently maintained in a C57BL/6 background. Mice with a floxed Hif1a exon 2 allele (31) in a C57BL/6 background were obtained from the Jackson Laboratory (stock number 007561). Hif1a+/− mice were generated by mating Hif1af/f mice with C57BL/6-Gt(ROSA)26Sortm16(Cre)Arte mice to delete exon 2, followed by further crossing with C57BL/6 mice to segregate the Cre allele. These mice were then crossed with Phd2P294R/+ mice, and these mice were maintained in a C57BL/6 background.
Mice with a tamoxifen-inducible Cre recombinase (fused to the estrogen receptor) and floxed Phd2 alleles were obtained by crossing a mouse line with a floxed Phd2 exon 2 allele (17, 19) with C57BL/6-Gt(ROSA)26Sortm9(cre/Esr1)Arte mice (Taconic) and have been described (32). These mice, hereafter denoted Rosa26-CreER; Phd2f/f, were maintained in a mixed background, as were mice that were derived from them by crossing with the Hif1af/f or Hif2af/f mice. All animal procedures were approved by the Institutional Animal Care and Use Committees at the University of Pennsylvania in compliance with Animal Welfare Assurance.
Southern Blotting
Digoxigenin-labeled probes were generated by PCR using a PCR DIG probe synthesis kit (Roche Applied Science). For the 5′ probe (0.37 kb), the primers were 5′-TTA CAG ATG AGG AAA TGC CTC GGC-3′ and 5′-GCC AAG GTC ATT GGA ACA GAC ATG-3′. For the 3′ probe (0.25 kb), the primers were 5′-GTG AGA AAG GTG TGA GG-3′ and 5′-ACA GAT CTG GAG CTG AC-3′. For both probes, bacterial artificial chromosome clone bacterial artificial chromosome RP23–356I16 was employed as the template. Southern blotting was performed using DIG Easy Hyb, DIG wash and block buffer set, anti-digoxigenin-AP conjugates, and CDP-Star substrate (all from Roche Applied Science).
PCR Genotyping
DNA was isolated from mouse tails (33). The following primers were employed for genotyping the P294R knock-in mutation: Phd2mouseF (5′-CAA ATG GAG ATG GAA GAT GC-3′) and Phd2mouseR (5′-TCA ACT CGA GCT GGA AAC C-3′). The wild type allele produces a PCR product of 0.39 kb, whereas the knock-in mutant allele produces a PCR product of 0.48 kb. The following primers were employed for genotyping the Phd2 alleles that were generated in a C57BL/6 background. For the Phd2 floxed allele (PCR product = 0.73 kb for wild type allele and 0.84 kb for floxed allele), the Phd2Rec55 primer was 5′-AGG GCT TCT GGC ATT AGT TGA CC-3′, and the Pex2–2 3′ primer was 5′-ACA TGT CAC GCA TCT TCC ATC TCC-3′. For the Phd2 knock-out allele (PCR product = 0.56 kb), the Pint1 Sal 5′ primer was 5′-AAT GGC TTG GGA CAA CTC-3′, and the Pint2–3 3′ primer was 5′-GGA CAA CGT TTG GGA GTT GGT AAG-3′. Primer sequences for genotyping of the floxed Hif1a and Hif2a alleles, as well as those for the Phd2 alleles that were maintained in a mixed strain background, have been described (15, 17, 19, 31).
Tamoxifen Administration
A solution of tamoxifen (MP Biomedicals) in corn oil at a final concentration of 10 mg/ml was prepared as described (32). At 6–10 weeks of age, experimental and control mice received a 2-mg dose daily for 5 days by oral gavage.
Hematology Analyses
Blood samples were collected through retro-orbital eye bleeds and placed in heparin-treated collection tubes (Sarstedt). Hemoglobin and blood counts were obtained using a Hemavet FS950 instrument (Drew Scientific). Hematocrit was measured with a CritSpin Microhematocrit centrifuge (StatSpin).
Serum Epo Measurements
Serum was obtained by incubating heparin-free retro-orbital blood samples overnight at 4 °C. Clotted blood was centrifuged at 4,000 × g for 20 min at 4 °C. The supernatant (serum) was used to determine Epo concentration via a commercially available rodent Quantikine Epo Immunoassay Kit (R&D Systems). Plate measurements were performed on a Sunrise microplate reader (Tecan), and absorbance values were compared with a genuine Epo standard curve.
Erythroid Burst-forming Unit (BFU-E) Assay
Bone marrow cells were obtained by flushing of bone marrow from the femurs of 6-week-old mice. Erythrocytes were lysed in ammonium chloride lysis buffer (Stem Cell Technologies), and the remaining cells were washed and resuspended in Iscove's modified Dulbecco's medium with 2% FBS to a concentration of 1 × 106 cells/ml. 1.5 × 105 cells were plated in methylcellulose medium containing varying concentrations of EPO (prepared by mixing appropriate amounts of EPO-containing Methocult M3236 with EPO-deficient M3436; Stem Cell Technologies). BFU-E colonies were counted at 9 days after bone marrow harvest.
Real Time PCR Measurements
Approximately 0.5 cm2 of kidney tissue was isolated from experimental and control mice and snap frozen in liquid nitrogen prior to mRNA extraction. RNA isolation was performed using TRIzol reagent (Invitrogen) following the manufacturer's instructions. Briefly, frozen tissue was homogenized in TRIzol reagent followed by addition of 0.2 volumes of chloroform. The tissue suspension was centrifuged, and the aqueous layer was taken and mixed with isopropanol to precipitate RNA. RNA was washed and resuspended in diethylpyrocarbonate treated water. RNA was quantified by absorbance at 260 nm. cDNA was prepared using the high capacity cDNA reverse transcription kit (Applied Biosystems) and following the manufacturer's instructions. Briefly, 0.8 μg of total RNA was reverse transcribed using random hexamer primers and Multiscribe reverse transcriptase for 60 min at 37 °C. The products of cDNA synthesis were used directly in real time PCRs.
Real time PCRs were prepared using an Applied Biosystems 2× SYBR green PCR mix. For 18 S rRNA measurements, we used the following primers: SG-m18S 5′ primer (5′-TCG GAA CTG AGG CCA TGA TT-3′) and SG-m18S 3′ primer (5′-TAG CGG CGC AAT ACG AAT G-3′. The primers for Phd2 and Epo measurements have previously been described (32). PCRs were performed and measured on an Applied Biosystems 7300 real time PCR system. Threshold CT values were determined automatically by the system and exported for analysis. Fold change values relative to WT samples were computed using the ΔΔCT method (34).
Histologic Analyses
Liver, spleen, heart, pancreas, and kidney tissues were fixed in 10% buffered formalin and embedded in paraffin. Sections were cut and stained with hemotoxylin and eosin following standard procedures. Images of sections were obtained using an BX60 light microscope (Olympus) and a SPOT RT Color camera (Diagnostic Instrument, Inc.), or alternatively, a DM2500 light microscope (Leica) and a DFC420 camera (Leica).
Plasmids
An expression vector for mouse Phd2, pcDNA3-FLAG-mPhd2, was constructed as follows. First, pcDNA3-HA-mPhd2 (1–35) was constructed by subcloning a synthetic oligonucleotide duplex encoding the indicated amino acids into the EcoRI/NotI site of pcDNA3-HA. Then pcDNA3-mPhd2 (36–400) was constructed by subcloning the NotI/StuI fragment of IMAGE clone 3375423 into the NotI/XhoI (blunt) site of pcDNA3. pcDNA3-HA-mPhd2 was constructed by subcloning the 1.6-kb NotI/XbaI fragment of pcDNA3-mPhd2 (36–400) into the NotI/XbaI site of pcDNA3-HA-mPhd2 (36–400). pcDNA3-FLAG-mPhd2 was constructed by subcloning the 1.6-kb EcoRI fragment of pcDNA3-HA-mPhd2 into the EcoRI site of pcDNA3-FLAG.
An expression vector for mPhd2 P294R was constructed by subcloning a mutagenized PCR product into the BsgI/XbaI site of pcDNA3-FLAG-mPhd2. The insert PCR product containing the P294R mutation and a silent KpnI site (used for screening) was produced by overlap extension PCR (35). The 5′ first round PCR product was produced using pcDNA3-FLAG-mPhd2 as a template and the following primers: mPHD2 BsgI forward (5′-CAG ATC GCC GAT GAG GTG CG-3′) and mPR 2 (5′-GCA TCT TCC ATC TCC ATT TCG GTT ATC AAC GTG ACG GAC ATA GCC GGT ACC GTT GCC TGG GTA ACA AGC-3′). The 3′ first round PCR product was produced similarly using the following primers: mPHD2 XbaI reverse (5′-GGC CTC TAG ATG CAT GCT CGA-3′) and mPR3 (5′-GCT TGT TAC CCA GGC AAC GGT ACC GGC TAT GTC CGT CAC GTT GAT AAC CGA AAT GGA GAT GGA AGA TGC-3′). Both PCR products were gel-purified and combined to serve as a template for a second round PCR using the mPHD2 BsgI forward and mPHD2 XbaI reverse primers, the product of which was then digested with BsgI and XbaI to serve as the final insert.
pSV-Sport-HA-HIF-2α has been described (21). pcDNA3-mHif-1α and pcDNA3-mHif-2α have been described (36). pGEX-mHif-1α (631–731) was constructed by amplifying the coding sequence of the indicated amino acids by PCR using pcDNA3-mHif-1α as a template, digesting the 0.3-kb product with BamHI/XhoI, and then subcloning into the BamHI/XhoI site of pGEX-5X-1. pMAL-mHif-1α (631–731) was constructed by subcloning the 0.3-kb BamHI/XhoI fragment of pGEX-mHif-1α (631–731) into the BamHI/XhoI site of pMAL-5X-1. pGEX-mHif-2α (576–693) and pMAL-mHif-2α (576–693) were constructed by subcloning the 0.35-kb SmaI/Bsp120I fragment of pcDNA3-mHif-2α into the SmaI/NotI sites of pGEX-5X-1 or pMAL-5X-1, respectively. Authenticity of plasmids was obtained by dideoxynucleotide sequencing.
Western Blotting
Total protein extracts were prepared from kidney by sonication and dissolution in a modified radioimmune precipitation assay lysis buffer including 1% SDS, 0.5% deoxycholate, 0.5% Nonidet P-40, and protease inhibitors (Sigma). Western blotting was then performed on equal protein amounts (determined by DC protein assay; Bio-Rad) essentially as described (37).
Antibodies
Rabbit polyclonal antibodies to mouse Hif-1α (631–731) were prepared as follows. First, GST-mHif-1α (631–731) and MBP-mHif-1α (631–731) were purified from Escherichia coli transformed with pGEX-mHif-1α (631–731) and pMAL-mHif-1α (631–731), respectively, using affinity chromatography on GSH-Sepharose and amylose-agarose, respectively. Then polyclonal antibodies to MBP-mHif-1α (631–731) were then raised in rabbits and affinity purified on GST-mHif-1α (631–731) coupled to agarose by Covance Research Products. These antibodies did not cross-react with mHif-2α.
Rabbit polyclonal antibodies to mouse Hif-2α (576–693) were prepared as follows. First, GST-mHif-2α (576–693) and MBP-mHif-2α (576–693) were purified from E. coli transformed with pGEX-mHif-2α (576–693) and pMAL-mHif-2α (576–693), respectively, using affinity chromatography on GSH-Sepharose and amylose-agarose, respectively. Then polyclonal antibodies to MBP-mHif-2α (576–693) were then raised in rabbits and affinity purified on GST-mHif-2α (576–693) coupled to agarose by Invitrogen. These antibodies did not cross-react with mHif-1α.
Rabbit polyclonal antibodies to PHD2 (1–63) have been described (37). Anti-FLAG M2 antibody conjugated to alkaline phosphatase was from Sigma. The E7 monoclonal antibody against β-tubulin developed by Dr. Michael Klymkowsky was obtained from the Developmental Studies Hybridoma Bank developed under the asupices of the NICHD and maintained by the University of Iowa Department of Biology (Iowa City, IA).
Luciferase Assays
HEK293 FT cells were cultured in DMEM with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Transfections were performed using Lipofectamine 2000 (Invitrogen) with cells at 70% confluency and at a 2:1 (μl/μg) ratio of Lipofectamine:DNA. The (eHRE)3-Luciferase plasmid containing three copies of the EPO hypoxia response element was used as a reporter and normalized to the activity of pRL-TK as previously described (38). Luciferase activities were measured using the dual luciferase reporter assay (Promega). Hypoxic exposure (1% O2, 5% CO2, 94% N2) was performed using a modular incubator (Billups-Rothenberg). Luciferase activities were measured using a Wallac LB9507 luminometer either 18 h post-transfection or following 18 h of exposure to hypoxia.
Whole Body Plethysmography
Respiration rate and tidal volume were measured via whole body plethysmography. Briefly, a 150-ml holding chamber was connected to a pneumotachometer. Data from this instrument were recorded continuously using a Powerlab instrument interface and analyzed using LabChart 6 software (ADInstruments). Gas was supplied using a Pegas-400 MF mixer (Columbus Instruments). The mice were allowed to equilibrate in a darkened chamber until body movement and breath rate became steady before measurements were taken. The mice were challenged with hypoxia (12% O2) for a period of not more than 10 min and allowed to equilibrate for a minimum of 5 min to the new atmosphere before measurements were taken. Tidal volume and respiratory rate were measured directly and normalized to body mass.
Right Ventricular Pressure Measurements and Echocardiography
Measurement of right ventricular pressure was performed on isoflurane-anesthetized mice. Following a midline cervical incision, a 1.4 French miniaturized pressure catheter (Millar Instruments) was inserted through the right jugular vein to the right ventricle using microsurgical techniques. Measurements of right ventricular pressure and heart rate were recorded using LabChart 6 software. Echocardiograms were recorded using a Vevo770 ultrahigh frequency small animal ultrasound system (Visual Sonics) with a 30-MHz probe and an Integrated Rail System III for image recording.
Statistical Analysis
Hematologic, real time PCR, luciferase assays, and histological measurements were analyzed by ANOVA followed by Tukey HSD post hoc tests, unless otherwise noted. Pulmonary function tests were analyzed by unpaired Student's t tests. Survival curve analysis was done by Log-Rank test comparing each to control Cre negative mice (39). All error bars are presented as ± one standard error of the mean. Analysis was conducted using the R Software Environment for Statistical Computing (40). p < 0.05 was considered significant.
RESULTS
Phd2 P294R Displays Decreased Activity in Vitro
Pro-317 of human PHD2 corresponds to Pro-294 of mouse Phd2 (Fig. 3A) and shows strong conservation across metazoan species (21). In vitro, the P317R mutation in human PHD2 diminishes activity toward HIF (21). Using a luciferase reporter gene containing three copies of a hypoxia response element (HRE), we sought to examine whether the P294R Phd2 substitution homologous to the observed P317R PHD2 mutation from human patients also results in decreased activity. We find that in comparison to wild type Phd2, P294R Phd2 has diminished ability to suppress HRE reporter gene activity under hypoxic or normoxic conditions (Fig. 3B) or upon coexpression with HIF-2α (Fig. 3D). These results support the functional equivalence between the human PHD2 P317R and mouse Phd2 P294R mutations.
FIGURE 3.
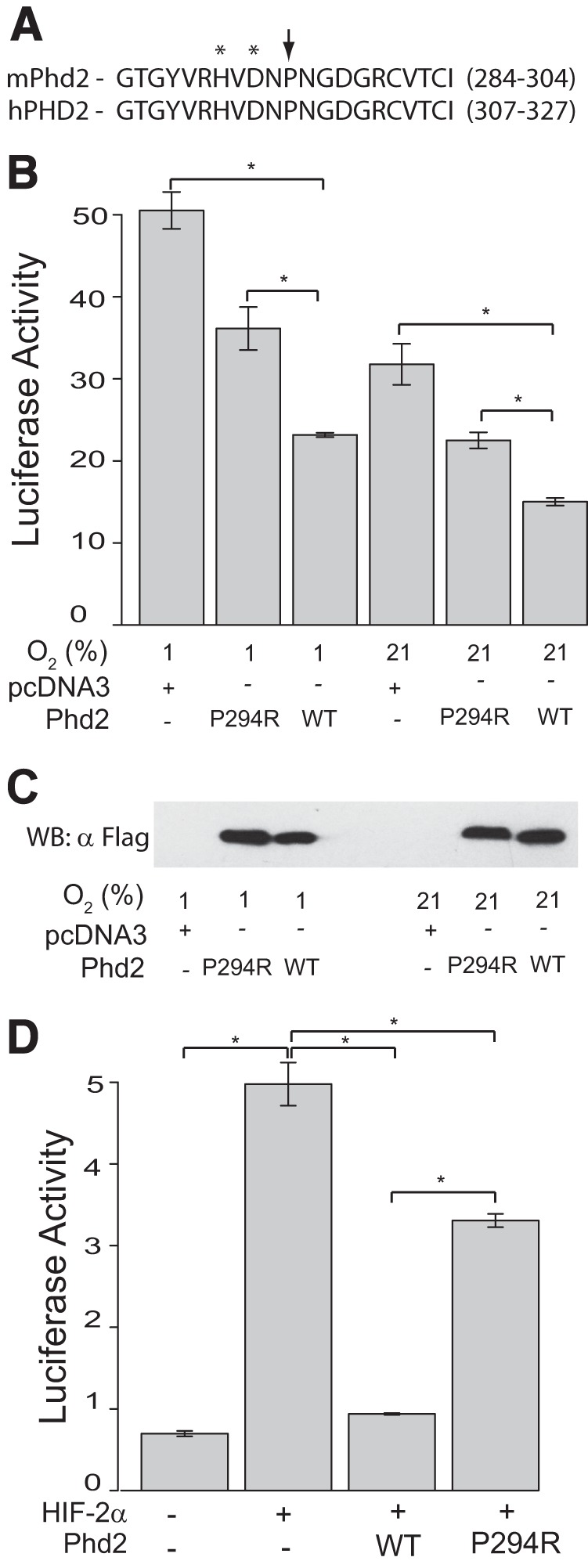
Murine Phd2 P294R has reduced activity toward HIF in vitro. A, sequence alignment for mouse PHD2 (mPhd2) and human PHD2 (hPHD2) for the indicated amino acids. The arrow indicates a mutation site, and asterisks indicate iron chelating residues. B, HEK293 FT cells were transfected with 50 ng of (eHRE)3-Luc, 100 ng of pRL-TK, and 60 ng of either pcDNA3, pcDNA3-FLAG-mPhd2, or pcDNA3-FLAG-mPhd2 P294R. Cells were exposed to the indicated oxygen concentrations, and luciferase activities were then measured (n = 3/group). *, p < 0.05, by ANOVA with pairwise Tukey HSD. C, anti-FLAG Western blot (WB) to assess expression of mPhd2 WT and P294R. D, HEK293 FT cells were transfected with 100 ng of (eHRE)3-Luc, 100 ng of pRL-TK, 300 ng of pSV-Sport-HA-HIF-2α, and 4 ng of pcDNA3-FLAG-mPhd2 or pcDNA3-FLAG-mPhd2 P294R, as indicated. DNA dose was normalized with pcDNA3. Luciferase activities were measured (n = 3/group). *, p < 0.05, by ANOVA with pairwise Tukey HSD.
We generated a knock-in mouse model that harbors the P294R mutation in the Phd2 gene. The targeting strategy is shown in Fig. 1A. Following electroporation into C57BL/6 ES cells and selection using G418, four clones with the correct recombination event and mutation were identified. Southern blotting with a 3′ probe revealed the expected 7.0-kb band upon hybridization with EcoRI-digested DNA in addition to the wild type 6.5-kb band (Fig. 1B, left panel). Southern blotting with a 5′ probe revealed the expected 15.9-kb band upon hybridization with ScaI-digested DNA in addition to the wild type 14-kb band (Fig. 1B, middle panel, lane 2). Sequencing confirmed the presence of a heterozygous P294R mutation in the Phd2 gene (Fig. 1C). Chimeras derived from injection of one of the recombinant ES clones into blastocysts yielded germ line transmission, as assessed by Southern blotting (Fig. 1B, middle panel, lane 3). The neomycin cassette was deleted, and deletion was confirmed by Southern blotting (Fig. 1B, second lane of right panel and lane 5 of middle panel). We subsequently obtained Phd2P294R/+ mice, and a PCR-based genotyping strategy was developed (Fig. 1D). Mating Phd2P294R/+ mice failed to produce any Phd2P294R/P294R mice (Table 1). Crosses of Phd2+/− mice have similarly failed to yield any Phd2−/− mice (17). The Phd2P294R/+ mice were outwardly normal and did not display any significant changes in body weight or survival (data not shown).
TABLE 1.
Phd2 genotypes of litters from Phd2P294R/+ × Phd2P294R/+ cross
The numbers in parentheses indicate the actual numbers observed.
| Genotype | +/+ | P294R/+ | P294R/P294R |
|---|---|---|---|
| % | % | % | |
| Observed | 44 (36) | 56 (45) | 0 (0) |
| Expected | 25 | 50 | 25 |
Phd2P294R/+ Mice Develop Erythrocytosis
We wished to evaluate whether Phd2P294R/+ mice display erythrocytosis and, if so, whether this occurs by haploinsufficiency or by a dominant negative mechanism. For this purpose, we compared these mice to Phd2+/− mice, which we generated in the same C57BL/6 background (Fig. 2). We reasoned that if the mechanism was via haploinsufficiency of Phd2 activity, then the degree of erythrocytosis observed in the two different genotypes would be the same. If, on the other hand, the mechanism was by a dominant negative mechanism (such as inhibition of the wild type protein by the mutant protein), then we would predict that the degree of erythrocytosis observed with the Phd2P294R/+ mice would be greater than that with the Phd2+/− mice. We found that the Phd2P294R/+ mice do indeed display erythrocytosis, as reflected by hematocrit, hemoglobin, or red blood cell counts, and importantly, this erythrocytosis was indistinguishable from that observed with Phd2+/− mice (Fig. 4, A–C). In addition, neither genotype displayed differences in the number of either white blood cell or platelets compared with wild type mice (Fig. 4D and data not shown). These findings formally demonstrate that the P294R missense mutation in Phd2 gene is by itself sufficient to cause erythrocytosis and are consistent with a haploinsufficiency, rather than dominant negative, mechanism.
FIGURE 4.
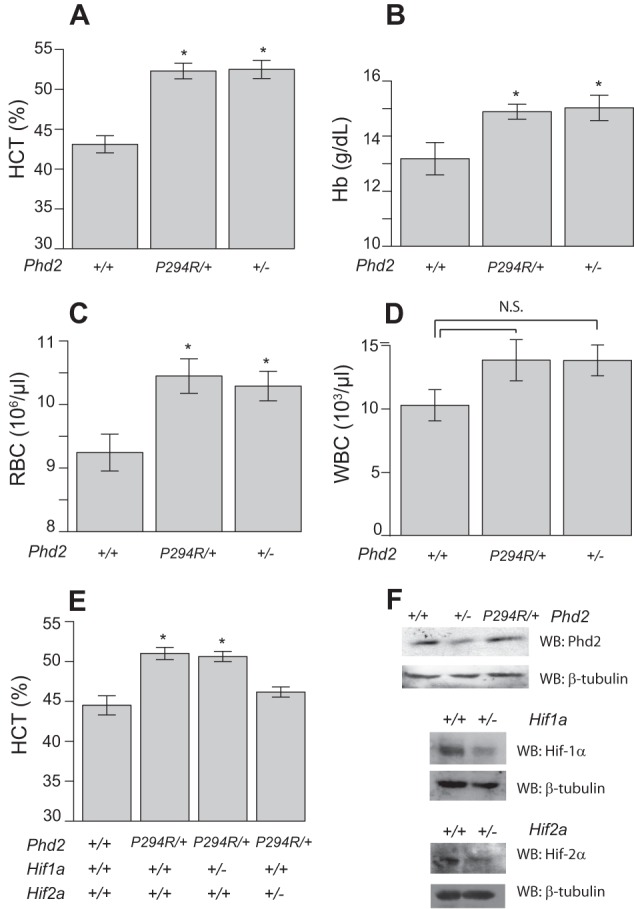
Phd2P294R/+ mice display erythrocytosis. A–D, hematocrit (A, HCT), hemoglobin (B, Hb), red blood cell counts (C, RBC), and white blood cell counts (D, WBC) were measured in mice bearing the indicated Phd2 alleles (age = 12–13 months; n = 8/group). *, p < 0.05 relative to WT controls by ANOVA with pairwise Tukey HSD. E, hematocrit measurements from mice bearing the indicated Phd2, Hif1a, and Hif2a alleles (n = 5/group). *, p < 0.05 relative to WT controls by ANOVA with pairwise Tukey HSD. F, Western blots (WB) examining levels of the indicated proteins in kidney extracts from wild type, Phd2P294R/+, Phd2+/−, Hif1a+/−, and Hif2a+/− animals.
We also examined the effects of concurrent heterozygous loss of Hif1a or Hif2a. We found that the erythrocytosis is Hif2a-dependent (Fig. 4E, compare second and fourth columns). Heterozygous loss of Hif1a, in contrast, was without effect (Fig. 4E, third column). The results are consistent with other studies that implicate HIF-2α as the central HIF-α isoform controlling erythropoiesis (13, 15, 16). We confirmed that Phd2+/−, Hif1a+/−, and Hif2a+/− mice have decreased expression of the relevant proteins by Western blot (Fig. 4F). We also measured Epo levels in Hif2a+/− mice and found that they do not have levels significantly different from wild type controls (608 ± 163 versus 446 ± 33 pg/ml, respectively, n = 3, p > 0.05).
Phd2P294R/+ Mice Exhibit Increased Minute Ventilation under Normoxia
Dysregulation of the HIF pathway can have systemic effects beyond erythrocytosis. For example, humans and mice with the hypomorphic Chuvash (R200W) mutation in the VHL gene display increased minute ventilation under normoxia, hypoxia, or both (41, 42). We therefore measured tidal volume, respiratory frequency, and minute ventilation by whole body plethysmography in Phd2P294R/+ and wild type littermate controls. Under normoxia, Phd2P294R/+ mice exhibited increased minute ventilation (VE) and increased tidal volume (VT) (Fig. 5, A and B). Following exposure to a hypoxic challenge of 12% O2, mice in both groups of mice exhibited hyperventilation, as expected (Fig. 5, D–F). Although there was a trend toward increased minute ventilation in the Phd2P294R/+ mice as compared with controls, it was not statistically significant.
FIGURE 5.
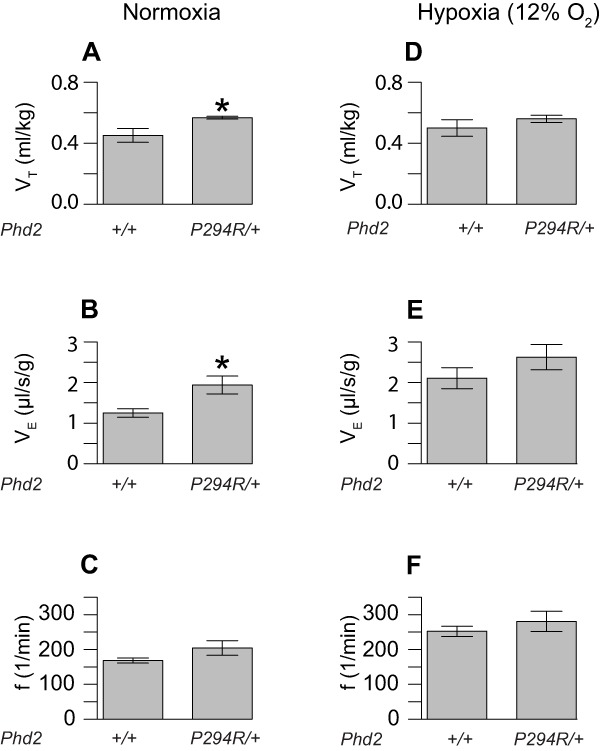
Changes in pulmonary function in Phd2P294R/+ mice. The tidal volume (A and D, VT), minute ventilation (B and E, VE), and frequency (C and F, f) of mice with the indicated Phd2 genotypes (age = 12–13 months) were measured under normoxic (A–C) and hypoxic (12% O2) conditions (D–F) (n = 5/group). *, p < 0.05 relative to WT controls under the same conditions by Student's t test.
Another systemic effect reported with dysregulation of the HIF pathway is pulmonary hypertension. Humans and mice with the Chuvash VHL mutation display pulmonary hypertension (41–43). Moreover, humans and mice with gain of function mutations in the HIF2A gene also display pulmonary hypertension (44–46). Accordingly, we measured right ventricular systolic pressure and heart rate by right ventricular catheterization of Phd2P294R/+ and wild type controls, as well as ventricular wall thickness by echocardiography. We did not observe differences in either right ventricular systolic pressure (31.21 ± 2.03 mm Hg for Phd2+/+ versus 33.50 ± 0.87 mm Hg for Phd2P294R/+, n = 4), heart rate (548.6 ± 13.5 bpm for Phd2+/+ versus 565.0 ± 26.8 bpm for Phd2P294R/+, n = 4) or right ventricular wall thickness (0.254 ± 0.033 mm for Phd2+/+ versus 0.245 ± 0.028 mm for Phd2P294R/+, n = 4, diastolic thickness). We are not aware of any published reports of pulmonary hypertension in human patients with PHD2 mutations.
Phd2P294R/+ Mice Display Inappropriately Normal Serum Epo Levels
Most patients with PHD2-associated erythrocytosis, including those with the original P317R mutation, display normal serum EPO levels despite the elevated hematocrit (14, 21). We measured serum Epo in Phd2P294R/+ mice and found the levels to be similar to those seen in wild type controls or in Phd2+/− mice (Fig. 6A). We also did not observe any increase in renal Epo mRNA from Phd2P294R/+ mice; if anything, there was a trend toward decreased Epo mRNA in either Phd2P294R/+ or Phd2+/− mice (Fig. 6B), although neither was statistically significant. Previous work has established that homozygous loss of all three PHD isoforms is required for hepatic Epo production in the adult mouse (47), and levels of Epo message in the liver in the present studies were at least 25-fold lower than that seen in the kidney (data not shown); thus, the liver is unlikely to be a significant source of Epo in these mice. We did observe a change in spleen weight from Phd2P294R/+ relative to wild type mice; it was not significantly different from that of Phd2+/− mice (Fig. 6C). We observed no significant changes in either heart or liver weights from either Phd2P294R/+ or Phd2+/− mice, relative to wild type (Fig. 6D and data not shown).
FIGURE 6.
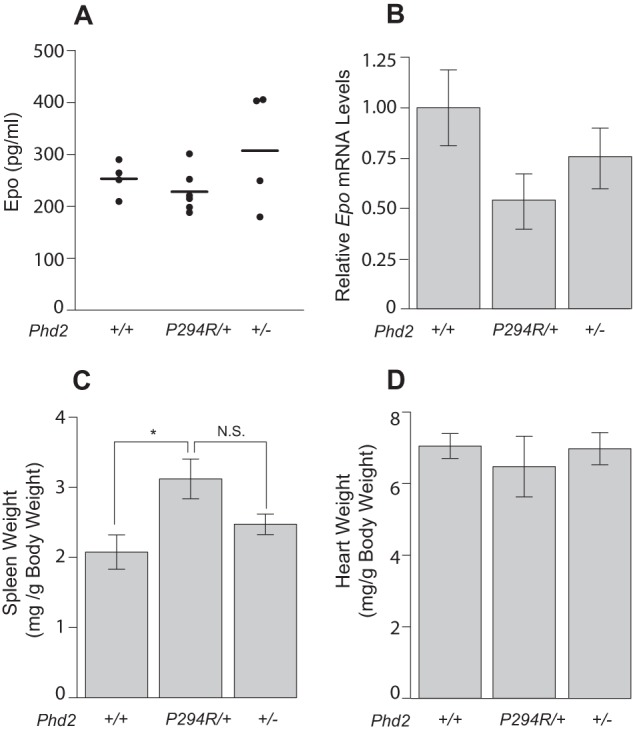
Phd2P294R/+ mice display normal Epo. A, serum Epo concentrations were measured by ELISA assay (n = 4–6/genotype). B, Epo message levels were measured by real time PCR (n = 5/group). The difference between Phd2P294R/+ and WT was not significant (p = 0.075). C and D, spleen (C) and heart (D) weights of mice (age = 12–13 months) with the indicated Phd2 genotypes. (n = 5/group). *, p < 0.05 by Student's t test. N.S., not significant.
Global Conditional Phd2 Knock-out Mice Exhibit Hif2a-dependent Erythrocytosis
The normal serum Epo observed in the Phd2P294R/+ mice contrasts with the dramatic elevation observed upon acute global deletion of Phd2 (18, 19, 32). We confirmed the latter using mice bearing a tamoxifen-inducible, Cre-estrogen receptor fusion protein and floxed Phd2 alleles (Rosa26-CreER; Phd2f/f). Upon tamoxifen treatment, which efficiently deletes Phd2 (data not shown), we observed marked erythrocytosis with hematocrits approaching 80% (Fig. 7A, second column). In agreement with the results obtained with the Phd2P294R/+ mice and other studies (48), we found that this phenotype is Hif-2α-dependent as evidenced by the rescue of the erythrocytosis phenotype by a concurrent deletion of a floxed Hif2a allele (Fig. 7A, fourth column). We also observed that although Rosa26-CreER; Phd2f/f mice suffer from early mortality, survival is comparable to control levels with concurrent loss of Hif2a (Fig. 7D). In contrast, concurrent deletion of Hif1a did not restore hematocrit to normal levels (Fig. 7A, third column). Interestingly, it did improve survival of these mice relative to Phd2 loss alone, although not to levels seen with control mice (Fig. 7D).
FIGURE 7.

Erythrocytosis following acute global Phd2 ablation is Hif2a dependent. A–C, hematocrit (A, HCT), serum Epo (B), and spleen weights (C) were measured following tamoxifen treatment of mice (age = 4 months) with the indicated Phd2, Hif1, and Hif2 genotypes. CreER indicates Rosa26-CreER transgene (n = 5–6/genotype). *, p < 0.05 relative to CreER negative controls by ANOVA with pairwise Tukey HSD. D, survival analysis following tamoxifen administration to mice with the indicated genotypes. Control group consists of Phd2f/f, Phd2f/f; Hif1af/f, and Phd2f/f; Hif2af/f mice. Initial cohort sizes were as follows: control, 22; CreER; Phd2f/f, 9. CreER; Phd2f/f; Hif1af/f = 10. CreER; Phd2f/f; Hif2af/f = 9. E–H, photomicrographs of spleens from Phd2f/f (E), Rosa26-CreER; Phd2f/f (F), Rosa26-CreER; Phd2f/f; Hif1af/f (G), and Rosa26-CreER; Phd2f/f; Hif2af/f (H) mice. Extramedullary hematopoiesis is present in F and G. Magnification, 100×, hematoxylin- and eosin-stained.
This increase in red cell mass in Rosa26-CreER; Phd2f/f mice compared with controls is seen in association with increases in serum Epo production (Fig. 7B, second column) and renal Epo mRNA transcript (data not shown). Both increases are Hif2a-dependent (Fig. 7B, fourth column, and data not shown). Concurrent Hif1a deletion had no significant effect either on serum Epo protein level or on Epo transcript levels relative to Phd2 deletion alone, supporting the primacy of Hif-2α in the induction of erythropoiesis following Phd2 loss.
The acute global deletion of Phd2 also leads to increases in spleen weight (Fig. 7C, second column) and dramatic changes in spleen architecture. Examination of histologic sections from the spleens of tamoxifen treated Rosa26-CreER; Phd2f/f mice revealed extramedullary hematopoiesis (compare Fig. 7, E and F). Concurrent deletion of Hif2a rescued the splenic enlargement and restored splenic architecture to WT relative to Phd2 deletion alone (Fig. 7, C and H). Rescue of either the splenic enlargement or the histological changes was not seen with concurrent deletion of Hif1a (Fig. 7, C and G).
Knock-out of Phd2 in Epo-producing Renal Cells Leads to Dramatic Erythrocytosis
The primary cellular source of EPO in adults are specialized interstitial cells in the renal cortex (49). These cells can be targeted using Cre driven by a Pax3 promoter (13), which targets neural crest-derived cells and somite derivatives. With homozygous Phd2 deletion in this compartment, we observed hematocrits near 90%, greater even than that observed with global conditional knock-outs, consistent with Phd2 playing a pivotal role in controlling hematopoiesis through its action in these interstitial cells (Fig. 8A, third column). Notably, heterozygous deletion in this model also yields increases in hematocrit (Fig. 8A, second column), which were modest and more comparable to those observed in both Phd2P294R/+ and Phd2+/− mice. The findings suggest that heterozygous Phd2 loss in the renal interstitial compartment alone can account for a significant portion of the erythrocytosis in the Phd2P294R/+ and Phd2+/− global heterozygotes. In conjunction with the results from the global tamoxifen-induced conditional Phd2 knock-out discussed above, these studies support Phd2 and Hif-2α as the primary gatekeepers of the Epo response. It also suggests that Phd1 and Phd3 do not substitute for Phd2 in regulating Hif-2α degradation in the Epo-producing kidney interstitial cells.
FIGURE 8.

Pax3-Cre Phd2f/+ and Pax3-Cre Phd2f/f display erythrocytosis. A–C, hematocrit (A, HCT), serum Epo (B), and spleen weights (C) were measured in mice (age = 12 months) with the indicated genotypes (n = 4–7/group). *, p < 0.05; ***, p < 0.001 relative to Phd2f/f controls by ANOVA with pairwise Tukey HSD. D, photomicrograph showing evidence of extramedullary hematopoiesis in spleen from a Pax3-Cre; Phd2f/f mouse. Magnification, 100×, hematoxylin- and eosin-stained. E and F, photomicrographs of Phd2f/f (E) and Pax3-Cre Phd2f/f (F) livers showing increased vascularity in the latter. Magnification, 50×, hematoxylin- and eosin-stained.
Similar to Phd2+/− mice, the Pax3-Cre; Phd2f/+ mice showed no significant increase in serum Epo, despite elevated red blood cell counts relative to wild type littermate control mice (Fig. 8B, middle panel). This, therefore, is another example of a haploinsufficiency state of Phd2 leading to erythrocytosis with normal Epo levels. Similarly, we saw little transcriptional up-regulation of other Hif targets including Pgk1 and Vegfa in kidney and liver of these mice by Real Time PCR (data not shown). Elevated red cell mass independent of increased EPO is a phenotype most commonly associated with primary polycythemias in which the EPOR/JAK-STAT pathway is constitutively activated. However, it should be noted that such conditions typically show depressed production of EPO as a compensatory response to the elevated red cell volume (50). Along these lines, it is noteworthy that both the Phd2P294R/+ and the Phd2+/− mice have levels of circulating Epo that are predominantly in a normal range despite their elevated hematocrits. Thus, this change in the Phd2-Hif-2α-Vhl axis yields normal levels of Epo and a phenotype distinct from both pure primary (bone marrow/EPOR driven) and pure secondary (EPO driven) erythrocytoses.
The splenomegaly phenotype of global conditional Phd2 knock-out was recapitulated in Pax3-Cre; Phd2f/f mice (Fig. 8, C and D), in line with the hypothesis that increased Epo is responsible for extramedullary hematopoiesis. These mice also showed increased cardiac mass (8.34 ± 0.29 mg/g of body weight for Pax3-Cre; Phd2f/f versus 5.05 ± 0.37 mg/g of body weight for Phd2f/f; age = 12 months, n = 5–7/group, p < 0.05), which may be a result of increased blood viscosity associated with their highly elevated hematocrits (>85%). In addition, these mice displayed slightly increased liver weights (65.18 ± 1.18 mg/g of body weight for Pax3-Cre; Phd2f/f versus 55.49 ± 2.29 mg/g of body weight for Phd2f/f; age = 12 months, n = 5–7/group, p < 0.05). Global conditional loss of Phd2 has been reported previously to result in increased liver vascularity (19), and we observed this same phenotype in the more restricted Pax3-Cre deletion mouse line (Fig. 8, E and F). We note both increased number (4.46 ± 0.56 vessels/mm2 for Pax3-Cre; Phd2f/f versus 2.05 ± 0.42 vessels/mm2 for Phd2f/f, n = 5, p < 0.05) and size of vessels (1,486 ± 78 μm2 for Pax3-Cre; Phd2f/f versus 998 ± 178 μm2 for Phd2f/f, n = 5, p < 0.05) in the livers of these mice.
Knock-out of Phd2 in Hematopoietic Precursors Leads to Erythrocytosis
The normal Epo levels in the Phd2P294R/+ mice might raise the question of Epo-independent effects of Phd2 loss of function. One possibility is that there may be alterations in endothelial cells. We therefore examined Phd2 loss in endothelial cell lineages through the use of a VE-cadh-Cre; Phd2f/f mouse line. We did not observe any changes in hematocrit (Fig. 9A). Serum Epo levels were not significantly different from controls (Fig. 9B), and liver, kidney, and spleens appeared normal upon histological examination (data not shown). However, these mice had dramatically increased mortality with less than half surviving to 6 months of age (Fig. 9C). Indeed, it was more comparable to that observed in the global conditional knock-out mice following tamoxifen treatment (Fig. 7D). Interestingly, VE-cadh-Cre; Phd2f/f mice also showed increased heart mass relative to negative controls (Fig. 9D). The increased heart mass is similar to that seen in Pax3-Cre; Phd2f/f mice, even though the latter displayed substantially higher hematocrits (compare Fig. 8A with Fig. 9A).
FIGURE 9.
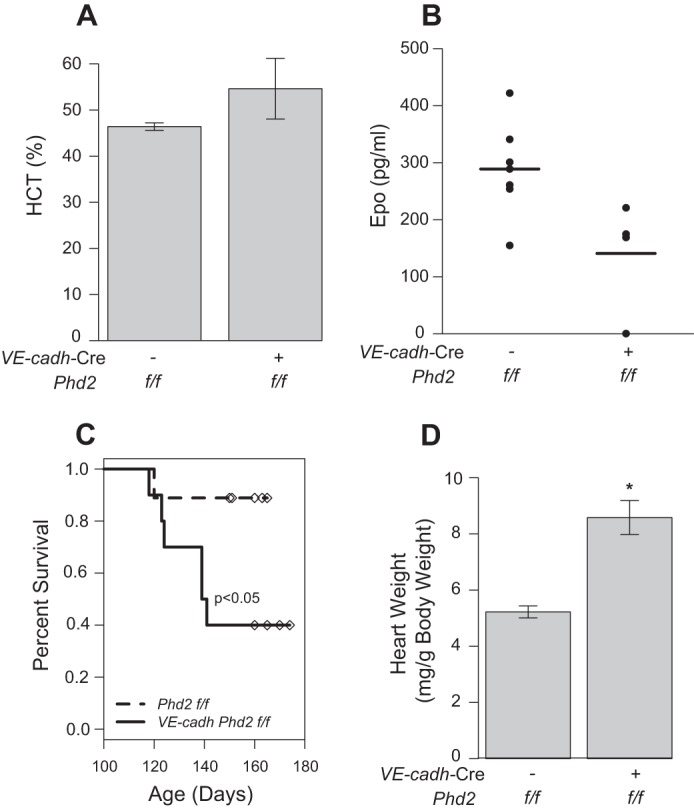
A and B, hematocrit (A, HCT) and serum Epo (B) were measured in VE-cadh-Cre; Phd2f/f or Phd2f/f control mice (age = 5–6 months). No significant differences were found between the two groups by Student's t test (n = 4–7/group). C, survival analysis of VE-cadh-Cre; Phd2f/f or Phd2f/f control mice. Individuals were sacrificed for further analysis at 5–6 months of age and are indicated with overlaid symbols. D, heart weight of VE-cadh-Cre; Phd2f/for Phd2f/f control mice (age = 5–6 months). *, p < 0.05 relative to Phd2f/f controls by Student's t test.
We next considered the possibility that there may be alterations in erythroid precursors upon Phd2 deletion. Using a Vav1-Cre expressing line crossed with our previously discussed homozygous floxed Phd2 mice, we deleted Phd2 in the hematopoietic compartment. The resulting mice developed an erythrocytosis compared with controls (Fig. 10, A–C). Serum Epo levels were not increased (Fig. 10D). We did not observe significant increases in white blood cell or platelet numbers (Fig. 10E and data not shown). The erythrocytosis phenotype required a homozygous deletion in this compartment, because heterozygous conditional knock-out mice showed no change from wild type controls (data not shown). The increase in hematocrit observed with homozygous deletion was relatively mild compared with global conditional Phd2 knock-out or Pax3-Cre; Phd2f/f deletion (hematocrit of ∼60% versus ∼80–90%, compare Fig. 10A versus Figs. 7A and 8A), suggesting that renal-derived Epo is still the predominant mechanism driving global Phd2 inactivation-dependent increases in red cell mass. We next isolated bone marrow cells from Vav1-Cre; Phd2f/f and control Phd2f/f mice and performed BFU-E colony forming assays. Bone marrow cells from Vav1-Cre Phd2f/f mice yielded increased numbers of BFU-E colonies compared with Phd2f/f controls (Fig. 10F). This increased proliferation was apparent at both high (3.0 unit/ml) and low (0.3 unit/ml) concentrations of EPO (Fig. 10F). The data provide evidence for EPO hypersensitivity of erythroid precursors from Vav1-Cre; Phd2f/f mice.
FIGURE 10.
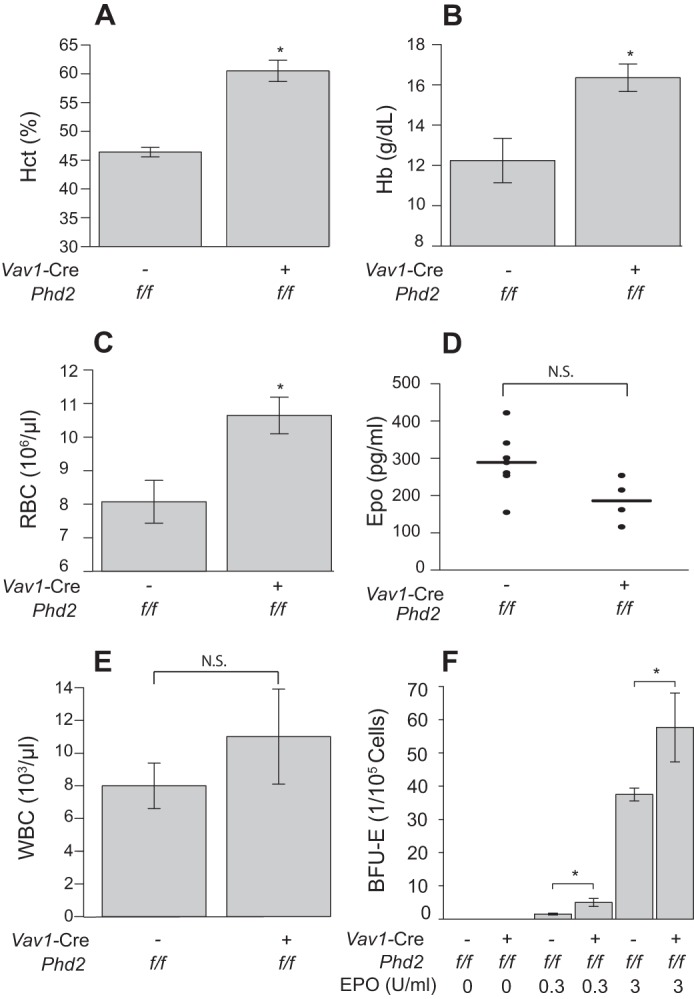
Vav1-Cre; Phd2f/f mice display erythrocytosis. A–E, hematocrit (A, HCT), hemoglobin (B, Hb), red blood cell counts (C, RBC), and white blood cell counts (E, WBC) were measured in mice (age = 12 months) with the indicated genotypes (n = 5/group). Serum Epo (D) was also measured. N.S., not significant. F, BFU-E assays were performed on bone marrow cells obtained from mice with the indicated genotypes (age = 6 weeks). Colonies were counted following 9 days of exposure to medium with the indicated levels of EPO (n = 4/group). *, p < 0.05 relative to Phd2f/f controls by Student's ttest.
DISCUSSION
The Phd2P294R/+ mouse model that we present here accurately recapitulates a number of the key features seen in human patients bearing the homologous mutation (P317R), namely, erythrocytosis and normal Epo levels (Figs. 4 and 6). Indeed, the erythrocytosis is comparable to that seen in Phd2+/− mice, which also display normal Epo levels. These findings are consistent with haploinsufficiency as the mechanism for the human PHD2 mutation-induced erythrocytosis. This, in turn, underscores the sensitivity of the EPO response to PHD2 gene dosage, because a single normal PHD2 allele in both humans and mice is not sufficient to maintain normal red cell mass. It is consistent with the notion of PHD2 as the key “oxygen sensor” of the EPO pathway, because modest changes in activity can translate into measurable changes in red cell mass. Indeed, Pax3-Cre-driven deletion of a single floxed Phd2 allele is also sufficient to induce erythrocytosis (Fig. 8A).
Our data also underscore the notion that PHD2 is playing a major, nonredundant role in the Epo-producing interstitial cells of the kidney. In these cells, which are the primary drivers of the erythropoiesis, alterations in the abundance or activity of PHD2 can lead to significant downstream consequences. Conversely, it appears that in other cell types, such as hepatocytes, loss of PHD2 may be tolerated through the actions of PHD1 and PHD3 (47). As such, a key factor in the influence of any one of these factors is the cell-dependent, relative expression of the other family members. This model is also supported by the observation that heterozygous loss of Hif2a is sufficient to rescue the Phd2P294R/+ associated-erythrocytosis phenotype (Fig. 4E), suggesting that a proper balance of these two proteins is necessary for the normal regulation of erythropoiesis.
The erythrocytosis seen in the Phd2P294R/+ mice as well as in an inducible global deletion model of Phd2 is Hif2a-dependent, consistent with an expanding body of data supporting HIF-2α as the critical HIF isoform regulating erythropoiesis. It is relevant to note that although the hypomorphic P317R mutation in PHD2 that forms the basis of this study shows defects in activity toward both HIF-1α and HIF-2α, the defect toward HIF-2α is more marked (23). This is reminiscent of the hypomorphic Chuvash mutation in VHL, which shows a more marked defect toward Hif-2α as compared with Hif-1α (51).
The results presented here with global inducible knock-out of Phd2 can be compared with a recent report examining conditional Phd2 knock-out using a CD68 promoter-driven Cre, which targets interstitial cells in the kidney, astrocytes, neurons, and hematopoietic cells (48). As in that study, we observe marked erythrocytosis. Indeed, we also observed marked erythrocytosis with Pax3-Cre conditional knock-out, underscoring the importance of the renal interstitial cells. Also, similar to that study, we find that the global knock-out of Phd2 is reversed by concurrent Hif2a, but not Hif1a, deletion. However, in contrast to the recent report, we observe that although loss of Phd2 leads to increased mortality, the concurrent Hif1a deletion improves, rather than worsens, survival.
The conditional knock-outs have allowed us to make additional observations. Deletion in neural crest cells results in increased splenic mass and increased vascularity in the liver. The observation of the latter phenotype bears a notable resemblance to that observed previously in global Phd2 deletion (19). In our Pax3-Cre; Phd2f/f model, hepatic Phd2 recombination was undetectable by PCR (data not shown). Hence, this supports a cell-extrinisic effect. Whether this reflects a response to increased circulating Epo or some other effect will require additional investigation.
Finally, we have identified a role for PHD2 in the hematopoietic compartment in regulating red cell development. The function of PHD2 in this context has not been previously explored, and it may play a role in the fine-tuning of erythropoiesis in response to hypoxia. Erythroid precursors from mice bearing a conditional deletion of Phd2 in this compartment (Vav1-Cre; Phd2f/f) display hypersensitivity to EPO, and indeed these mice display erythrocytosis (Fig. 10). This result is consistent with hypersensitivity to EPO in erythroid precursors obtained from mice with acute global deletion of Phd2 (32), and the effect of the prolyl hydroxylase inhibitor dimethyloxalylglycine on red cell precursors (52). It might also be noted that erythroid precursors from humans and mice harboring the Chuvash VHL mutation also display hypersensitivity to EPO (41, 53). That being said, the Vav1-Cre; Phd2f/f phenotype presented here appears insufficient to completely account for the erythrocytosis in patients or mice with heterozygous PHD2 loss, because Vav1-Cre; Phd2f/+ mice do not display erythrocytosis. Although we did not observe differences in white blood cell or platelet counts in the Vav1-Cre; Phd2f/f mice, it is conceivable that hematopoietic stem cell/progenitor cells may be altered upon Phd2 loss. Evidence for this has recently been presented (54).
The Vav1-Cre; Phd2f/f mice display normal Epo levels. By way of contrast, serum EPO was depressed in a mouse model of polycythemia vera in which animals are markedly hypersensitive to Epo (55). However, these mice develop dramatically increased hematocrits (>75%), compared with those that we report here for the Vav1-Cre; Phd2f/f mice (we also note that the Vav1-Cre should not be active in the Epo producing cells of the kidney). We believe that the minimally perturbed Epo levels are a reflection of the relatively modest erythrocytosis observed in the Vav1-Cre; Phd2f/f animals that is insufficient to robustly down-regulate Hif-2α-mediated Epo gene transcription. In a similar manner, we believe that the modest erythrocytosis observed in the Pax3-Cre; Phd2f/+ mice is not sufficient to down-regulate Epo levels. These mice presumably have haploinsufficiency of Phd2 in the neural crest lineage (from which the renal Epo-producing cells are derived), and we would propose that their Epo level is inappropriately normal, much in the same way that the Phd2P294R/+ or Phd2+/− mice display a modest erythrocytosis (caused by Phd2 haploinsufficiency) and normal Epo levels.
Acknowledgments
We thank Adrian Flores, Paul Furlow, and Dr. Quan Zhao for assistance in the generation of targeting vectors. We thank Dr. Tobias Raabe of the University of Pennsylvania Gene Targeting facility for performing the ES cell electroporation of the pFRT.loxP.Phd2 targeting vector, and Dr. Jean Richa of the University of Pennsylvania Transgenic core facility for performing injections of correctly targeted ES cells into blastocysts. We thank Dr. Daisheng Song for assistance with genotyping. We thank Dr. Li Chen and Dr. Liu Xi of the Penn Cardiovascular Institute Mouse Cardiovascular Physiology and Microsurgery Core for performing the right ventricular pressure and echocardiography measurements, respectively; Amy Ziober for providing the hematoxylin/eosin-stained slides; and Dr. Emidio Pistilli and Dr. Predrag Krajacic for guidance with the plethysmography. We thank the NCI for providing the plasmid pL452 and E. coli SW102 for recombineering and Dr. Guo-Hua Fong (University of Connecticut Health Center) for providing the Phd2 floxed mouse line that was employed in the acute global deletion experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-CA153347 (to F. S. L.).
This work is dedicated to the memory of Dr. Yulan Sun.
- HIF
- hypoxia-inducible factor
- BFU-E
- erythroid burst-forming unit
- EPO
- erythropoietin
- EPOR
- erythropoietin receptor
- floxed
- flanked by loxP sites
- ANOVA
- analysis of variance
- HRE
- hypoxia response element
- HSD
- honestly significant differences
- PHD
- prolyl hydroxylase domain protein
- VHL
- von Hippel Lindau protein.
REFERENCES
- 1. Kaelin W. G., Jr., Ratcliffe P. J. (2008) Oxygen sensing by metazoans. The central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 [DOI] [PubMed] [Google Scholar]
- 2. Majmundar A. J., Wong W. J., Simon M. C. (2010) Hypoxia inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Semenza G. L. (2007) Life with oxygen. Science 318, 62–64 [DOI] [PubMed] [Google Scholar]
- 4. Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J. M., Lane W. S., Kaelin W. G. (2001) HIFα targeted for VHL-mediated destruction by proline hydroxylation. Implications for O2 sensing. Science 292, 464–468 [DOI] [PubMed] [Google Scholar]
- 5. Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]
- 6. Yu F., White S. B., Zhao Q., Lee F. S. (2001) HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. U.S.A. 98, 9630–9635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adelman D. M., Maltepe E., Simon M. C. (1999) Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev. 13, 2478–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kotch L. E., Iyer N. V., Laughner E., Semenza G. L. (1999) Defective vascularization of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev. Biol. 209, 254–267 [DOI] [PubMed] [Google Scholar]
- 9. Mazzone M., Dettori D., Leite de Oliveira R., Loges S., Schmidt T., Jonckx B., Tian Y.-M., Lanahan A. A., Pollard P., Ruiz de Almodovar C., De Smet F., Vinckier S., Aragonés J., Debackere K., Luttun A., Wyns S., Jordan B., Pisacane A., Gallez B., Lampugnani M. G., Dejana E., Simons M., Ratcliffe P., Maxwell P., Carmeliet P. (2009) Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136, 839–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skuli N., Majmundar A. J., Krock B. L., Mesquita R. C., Mathew L. K., Quinn Z. L., Runge A., Liu L., Kim M. N., Liang J., Schenkel S., Yodh A. G., Keith B., Simon M. C. (2012) Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Invest. 122, 1427–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jelkmann W. (2011) Regulation of erythropoietin production. J. Physiol. 589, 1251–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haase V. H. (2013) Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 27, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kapitsinou P. P., Liu Q., Unger T. L., Rha J., Davidoff O., Keith B., Epstein J. A., Moores S. L., Erickson-Miller C. L., Haase V. H. (2010) Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 116, 3039–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee F. S., Percy M. J. (2011) The HIF pathway and erythrocytosis. Annu. Rev. Pathol. 6, 165–192 [DOI] [PubMed] [Google Scholar]
- 15. Gruber M., Hu C. J., Johnson R. S., Brown E. J., Keith B., Simon M. C. (2007) Acute postnatal ablation of Hif-2α results in anemia. Proc. Natl. Acad. Sci. U.S.A. 104, 2301–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Scortegagna M., Ding K., Zhang Q., Oktay Y., Bennett M. J., Bennett M., Shelton J. M., Richardson J. A., Moe O., Garcia J. A. (2005) HIF-2α regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood 105, 3133–3140 [DOI] [PubMed] [Google Scholar]
- 17. Takeda K., Ho V. C., Takeda H., Duan L.-J., Nagy A., Fong G.-H. (2006) Placental but not heart defects are associated with elevated hypoxia-inducible factor α levels in mice lacking prolyl hydroxylase domain protein 2. Mol. Cell. Biol. 26, 8336–8346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Minamishima Y. A., Moslehi J., Bardeesy N., Cullen D., Bronson R. T., Kaelin W. G., Jr. (2008) Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 111, 3236–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takeda K., Aguila H. L., Parikh N. S., Li X., Lamothe K., Duan L.-J., Takeda H., Lee F. S., Fong G.-H. (2008) Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 111, 3229–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ladroue C., Carcenac R., Leporrier M., Gad S., Le Hello C., Galateau-Salle F., Feunteun J., Pouysségur J., Richard S., Gardie B. (2008) PHD2 mutation and congenital erythrocytosis with paraganglioma. New Engl. J. Med. 359, 2685–2692 [DOI] [PubMed] [Google Scholar]
- 21. Percy M. J., Zhao Q., Flores A., Harrison C., Lappin T. R., Maxwell P. H., McMullin M. F., Lee F. S. (2006) A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc. Natl. Acad. Sci. U.S.A. 103, 654–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Percy M. J., Furlow P. W., Beer P. A., Lappin T. R., McMullin M. F., Lee F. S. (2007) A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 110, 2193–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pappalardi M. B., Martin J. D., Jiang Y., Burns M. C., Zhao H., Ho T., Sweitzer S., Lor L., Schwartz B., Duffy K., Gontarek R., Tummino P. J., Copeland R. A., Luo L. (2008) Biochemical characterization of human prolyl hydroxylase domain protein 2 variants associated with erythrocytosis. Biochemistry 47, 11165–11167 [DOI] [PubMed] [Google Scholar]
- 24. Copeland N. G., Jenkins N. A., Court D. L. (2001) Recombineering. A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2, 769–779 [DOI] [PubMed] [Google Scholar]
- 25. Liu P., Jenkins N. A., Copeland N. G. (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13, 476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yagi T., Ikawa Y., Yoshida K., Shigetani Y., Takeda N., Mabuchi I., Yamamoto T., Aizawa S. (1990) Homologous recombination at c-fyn locus of mouse embryonic stem cells with use of diphtheria toxin A-fragment gene in negative selection. Proc. Natl. Acad. Sci. U.S.A. 87, 9918–9922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lang D., Lu M. M., Huang L., Engleka K. A., Zhang M., Chu E. Y., Lipner S., Skoultchi A., Millar S. E., Epstein J. A. (2005) Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature 433, 884–887 [DOI] [PubMed] [Google Scholar]
- 28. Degenhardt K. R., Milewski R. C., Padmanabhan A., Miller M., Singh M. K., Lang D., Engleka K. A., Wu M., Li J., Zhou D., Antonucci N., Li L., Epstein J. A. (2010) Distinct enhancers at the Pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev. Biol. 339, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Boer J., Williams A., Skavdis G., Harker N., Coles M., Tolaini M., Norton T., Williams K., Roderick K., Potocnik A. J., Kioussis D. (2003) Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol. 33, 314–325 [DOI] [PubMed] [Google Scholar]
- 30. Alva J. A., Zovein A. C., Monvoisin A., Murphy T., Salazar A., Harvey N. L., Carmeliet P., Iruela-Arispe M. L. (2006) VE-Cadherin-Cre-recombinase transgenic mouse. A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759–767 [DOI] [PubMed] [Google Scholar]
- 31. Ryan H. E., Poloni M., McNulty W., Elson D., Gassmann M., Arbeit J. M., Johnson R. S. (2000) Hypoxia-inducible factor-1α is a positive factor in solid tumor growth. Cancer Res. 60, 4010–4015 [PubMed] [Google Scholar]
- 32. Li X., Sutherland S., Takeda K., Fong G.-H., Lee F. S. (2010) Integrity of the prolyl hydroxylase domain protein 2:erythropoietin pathway in aging mice. Blood Cells Mol. Dis. 45, 9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laird P. W., Zijderveld A., Linders K., Rudnicki M. A., Jaenisch R., Berns A. (1991) Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 19, 4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 35. Higuchi R., Krummel B., Saiki R. K. (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments. Study of protein and DNA interactions. Nucleic Acids Res. 16, 7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu C.-J., Wang L.-Y., Chodosh L. A., Keith B., Simon M. C. (2003) Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol. Cell. Biol. 23, 9361–9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song D., Li L.-S., Heaton-Johnson K. J., Arsenault P. R., Master S. R., Lee F. S. (2013) Prolyl hydroxylase domain protein 2 (PHD2) binds a Pro-Xaa-Leu-Glu motif, linking it to the heat shock protein 90 pathway. J. Biol. Chem. 288, 9662–9674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu F., White S. B., Zhao Q., Lee F. S. (2001) Dynamic, site-specific interaction of hypoxia-inducible factor-1α with the von Hippel-Lindau tumor suppressor protein. Cancer Res. 61, 4136–4142 [PubMed] [Google Scholar]
- 39. Bewick V., Cheek L., Ball J. (2004) Statistics review 12. Survival analysis. Crit Care 8, 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. R Development Core Team (2010) R Environment for Statistical Computing, R Foundation for Statistical Computing, Vienne Austria [Google Scholar]
- 41. Hickey M. M., Richardson T., Wang T., Mosqueira M., Arguiri E., Yu H., Yu Q.-C., Solomides C. C., Morrisey E. E., Khurana T. S., Christofidou-Solomidou M., Simon M. C. (2010) The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J. Clin. Invest. 120, 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith T. G., Brooks J. T., Balanos G. M., Lappin T. R., Layton D. M., Leedham D. L., Liu C., Maxwell P. H., McMullin M. F., McNamara C. J., Percy M. J., Pugh C. W., Ratcliffe P. J., Talbot N. P., Treacy M., Robbins P. A. (2006) Mutation of von Hippel-Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med. 3, e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bushuev V. I., Miasnikova G. Y., Sergueeva A. I., Polyakova L. A., Okhotin D., Gaskin P. R., Debebe Z., Nekhai S., Castro O. L., Prchal J. T., Gordeuk V. R. (2006) Endothelin-1, vascular endothelial growth factor and systolic pulmonary artery pressure in patients with Chuvash polycythemia. Haematologica 91, 744–749 [PubMed] [Google Scholar]
- 44. Formenti F., Beer P. A., Croft Q. P., Dorrington K. L., Gale D. P., Lappin T. R., Lucas G. S., Maher E. R., Maxwell P. H., McMullin M. F., O'Connor D. F., Percy M. J., Pugh C. W., Ratcliffe P. J., Smith T. G., Talbot N. P., Robbins P. A. (2011) Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway. von Hippel-Lindau disease and HIF-2α gain-of-function mutation. FASEB J. 25, 2001–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gale D. P., Harten S. K., Reid C. D., Tuddenham E. G., Maxwell P. H. (2008) Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2α mutation. Blood 112, 919–921 [DOI] [PubMed] [Google Scholar]
- 46. Tan Q., Kerestes H., Percy M. J., Pietrofesa R., Chen L., Khurana T. S., Christofidou-Solomidou M., Lappin T. R., Lee F. S. (2013) Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain-of-function mutation. J. Biol. Chem. 288, 17134–17144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Minamishima Y. A., Kaelin W. G., Jr (2010) Reactivation of hepatic EPO synthesis in mice after PHD loss. Science 329, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Franke K., Kalucka J., Mamlouk S., Singh R. P., Muschter A., Weidemann A., Iyengar V., Jahn S., Wieczorek K., Geiger K., Muders M., Sykes A. M., Poitz D. M., Ripich T., Otto T., Bergmann S., Breier G., Baretton G., Fong G.-H., Greaves D. R., Bornstein S., Chavakis T., Fandrey J., Gassmann M., Wielockx B. (2013) HIF-1α is a protective factor in conditional PHD2-deficient mice suffering from severe HIF-2α-induced excessive erythropoiesis. Blood 121, 1436–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Obara N., Suzuki N., Kim K., Nagasawa T., Imagawa S., Yamamoto M. (2008) Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood 111, 5223–5232 [DOI] [PubMed] [Google Scholar]
- 50. Mossuz P., Girodon F., Donnard M., Latger-Cannard V., Dobo I., Boiret N., Lecron J. C., Binquet C., Barro C., Hermouet S., Praloran V. (2004) Diagnostic value of serum erythropoietin level in patients with absolute erythrocytosis. Haematologica 89, 1194–1198 [PubMed] [Google Scholar]
- 51. Hickey M. M., Lam J. C., Bezman N. A., Rathmell W. K., Simon M. C. (2007) von Hippel-Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2α signaling and splenic erythropoiesis. J. Clin. Invest. 117, 3879–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flygare J., Rayon Estrada V., Shin C., Gupta S., Lodish H. F. (2011) HIF1α synergizes with glucocorticoids to promote BFU-E progenitor self-renewal. Blood 117, 3435–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ang S. O., Chen H., Hirota K., Gordeuk V. R., Jelinek J., Guan Y., Liu E., Sergueeva A. I., Miasnikova G. Y., Mole D., Maxwell P. H., Stockton D. W., Semenza G. L., Prchal J. T. (2002) Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 32, 614–621 [DOI] [PubMed] [Google Scholar]
- 54. Singh R. P., Franke K., Kalucka J., Mamlouk S., Muschter A., Gembarska A., Grinenko T., Willam C., Naumann R., Anastassiadis K., Stewart A. F., Bornstein S., Chavakis T., Breier G., Waskow C., Wielockx B. (2013) HIF prolyl hydroxylase 2 (PHD2) is a critical regulator of hematopoietic stem cell maintenance during steady-state and stress. Blood 121, 5158–5166 [DOI] [PubMed] [Google Scholar]
- 55. Akada H., Yan D., Zou H., Fiering S., Hutchison R. E., Mohi M. G. (2010) Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood 115, 3589–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]