

Background: Calcium promotes gastric wound repair, but the in vivo mechanisms are unknown.
Results: Signaling pathways sequentially mobilize intracellular and extracellular calcium as a requirement to repair gastric lesions. PMCA1 mediates extracellular calcium increases.
Conclusion: Endogenous calcium is an essential second and third messenger driving gastric repair.
Significance: Signaling and ion flux pathways are newly identified targets for enhancing gastric repair.
Keywords: Calcium, Calcium Imaging, Calcium Signaling, Cell Migration, Fluorescence Resonance Energy Transfer (FRET), Fura Red, Yellow Cameleon, Photodamage, Two-photon Microscopy
Abstract
We report that a localized intracellular and extracellular Ca2+ mobilization occurs at the site of microscopic epithelial damage in vivo and is required to mediate tissue repair. Intravital confocal/two-photon microscopy continuously imaged the surgically exposed stomach mucosa of anesthetized mice while photodamage of gastric epithelial surface cells created a microscopic lesion that healed within 15 min. Transgenic mice with an intracellular Ca2+-sensitive protein (yellow cameleon 3.0) report that intracellular Ca2+ selectively increases in restituting gastric epithelial cells adjacent to the damaged cells. Pretreatment with U-73122, indomethacin, 2-aminoethoxydiphenylborane, or verapamil inhibits repair of the damage and also inhibits the intracellular Ca2+ increase. Confocal imaging of Fura-Red dye in luminal superfusate shows a localized extracellular Ca2+ increase at the gastric surface adjacent to the damage that temporally follows intracellular Ca2+ mobilization. Indomethacin and verapamil also inhibit the luminal Ca2+ increase. Intracellular Ca2+ chelation (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid/acetoxymethyl ester, BAPTA/AM) fully inhibits intracellular and luminal Ca2+ increases, whereas luminal calcium chelation (N-(2-hydroxyetheyl)-ethylendiamin-N,N,N′-triacetic acid trisodium, HEDTA) blocks the increase of luminal Ca2+ and unevenly inhibits late-phase intracellular Ca2+ mobilization. Both modes of Ca2+ chelation slow gastric repair. In plasma membrane Ca-ATPase 1+/− mice, but not plasma membrane Ca-ATPase 4−/− mice, there is slowed epithelial repair and a diminished gastric surface Ca2+ increase. We conclude that endogenous Ca2+, mobilized by signaling pathways and transmembrane Ca2+ transport, causes increased Ca2+ levels at the epithelial damage site that are essential to gastric epithelial cell restitution in vivo.
Introduction
Intracellular Ca2+ is a ubiquitous second messenger that influences numerous cellular processes, but, recently, extracellular Ca2+ has also been ascribed a role as a third messenger that can integrate and coordinate signals among diverse cells (1, 2). Recognizing the possibility of more dispersed roles for Ca2+, in many biological systems there is now less certainty about the sites at which Ca2+ drives biological responses.
The greatest uncertainty is in the understanding of in vivo responses in mammals because of the technical hurdles of measuring Ca2+ in such environments (3). There is long-standing evidence that Ca2+ plays an important role in regulating protective mechanisms of the gastric epithelium, including bicarbonate secretion and mucus secretion (4–6). However, discrimination of Ca2+ roles is limited to amphibian and/or in vitro models. Despite this, some of the most elegant support for the third messenger hypothesis comes from the study of amphibian gastric glands. In healthy tissue, carbachol increases both intracellular and luminal Ca2+ in amphibian gastric glands, and this Ca2+ extrusion into the gastric gland lumen is mediated by the plasma membrane Ca-ATPase (PMCA)2 (7). Furthermore, luminal Ca2+ is sufficient to elicit gastric secretions independently of intracellular Ca2+ changes (7, 8). Thus, luminal Ca2+ contributes to the regulatory control of normal physiologic functions in the amphibian stomach. In mammalian systems, reports have only defined the presence of agonist-stimulated intracellular Ca2+ mobilization in isolated gastric glands or isolated surface cells (7, 9, 10).
In contrast, even in the gastric system, the roles of Ca2+ during the tissue response to injury are only roughly defined. Critchlow et al. (4) first demonstrated that an adequate extracellular Ca2+ level is required for the restitution of isolated frog gastric mucosa after hyperosmotic injury, but the variable Ca2+ found in human diets raises questions about the physiologic significance of the finding. Conversely, in response to extensive gastric damage caused by application of taurocholate, 1 m NaCl, or 50% ethanol into rat stomach, investigators observed that gastric luminal Ca2+ increases in the collected gastric effluent (11–13). Because the luminal Ca2+ could be due to a nonspecific Ca2+ release from dying cells, it remains unclear whether these early observations could be extrapolated to the more modest and punctuate damage observed in response to physiologically relevant stressors (e.g. non-steroidal anti-inflammatory drugs or Helicobacter pylori) (14–16). For the measurement of intracellular Ca2+ during gastric injury, only primary cultured rabbit gastric epithelial cells have been used to demonstrate intracellular Ca2+ mobilization during wound repair (17). Thus, there is limited understanding of the roles of Ca2+ as either a second or third messenger during tissue injury of the stomach.
In this study, intravital confocal and two-photon microscopy is used in vivo to measure intracellular Ca2+ in epithelial cells and extracellular Ca2+ in the space adjacent to the mouse gastric surface epithelium. At the site of microscopic lesions, we observed highly localized intracellular and extracellular Ca2+ mobilizations that are both the result of cellular signaling pathways and are required to promote repair of the epithelium.
EXPERIMENTAL PROCEDURES
Animal Husbandry and Surgery
Experiments used C57Bl/6 mice, YC3.0 transgenic mice on a 129J × C57Bl/6J background (18, 19), PMCA1+/− mice, PMCA4−/− mice, and PMCA1+/− PMCA4−/− mice. For experiments examining knockout genotypes, wild-type controls were +/+ genotypes from the PMCA1 or PMCA4 colony. All animals were used for experiments at 3–6 months of age, were fed a standard rodent chow diet, and had free access to water. All animal procedures were approved by the University of Cincinnati Institutional Animal Care and Use Committee.
The surgical preparation of animals has been described previously (3, 20, 21). Briefly, mice were anesthetized with inactin (10 mg/kg intraperitoneally, Sigma) and ketamine (50 mg/kg intraperitoneally), and then the exposed gastric mucosa protruded into a perfusion chamber on the stage of an inverted confocal/two-photon microscope (Zeiss LSM 510 NLO), with the microscope stage enclosed and heated to keep the body temperature of the animal at ∼37 °C. The mucosal surface was exposed to pH 5 solution (150 mm NaCl and 4 mm homopipes; Research Organics). In some experiments, solutions also contained Fura-Red (100 μm, Invitrogen) and/or HEDTA (10 mm, Fluka), BAPTA/AM (250 μm, Calbiochem), or 2-APB (100 μm, Tocris Bioscience) (22). In some experiments, 10 mm NaCl was replaced with 10 mm CaCl2. U-73122 (30 mg/kg, intraperitoneally, Tocris Bioscience) (23–25), indomethacin (5 mg/kg, subcutaneously, Sigma) (20), or verapamil (3 mg/kg, subcutaneously, Sigma) (26) were administered 1 h before damage induction.
Live Tissue Quantitative Imaging
The surface epithelium of a YC mouse gastric corpus was excited at 840 nm with a femtosecond-pulsed titanium sapphire laser for two-photon fluorescence excitation using 90–100 milliwatt light power into the scanhead. At each time point, images were collected simultaneously for directly excited CFP fluorescence (435–485 nm), FRET fluorescence from YFP (535–590 nm), and confocal reflectance (reflected 840-nm light to show cell/tissue structure). In experiments using tissue from other mouse strains, two-photon excitation at 730 nm was used to simultaneously monitor tissue autofluorescence (435–485 nm) and confocal reflectance. Fura-Red fluorescence was imaged (> 560 nm) in response to alternating excitation at 458 or 488 nm. Imaging regions of the gastric surface were selected in which perfusion solutions could rapidly access the epithelial layer.
Induction of Laser-induced Microlesions in Superficial Gastric Epithelium
The method for inducing photodamage (PD) in the gastric surface epithelium with a two-photon laser has been described previously (3, 20, 21). Briefly, after collecting a set of control images using minimal Ti-Sa (840 or 730 nm) laser power, a small rectangle region (≈200 μm2) of gastric mucosa was scanned repetitively at high laser power (550 or 350 milliwatt average laser power, respectively), for 150 iterations (requiring 5–10 s). The damage-repair cycle was measured independently several times per animal in different locations of the corpus.
Image Analysis
All image analyses were performed with Metamorph software (v. 6.3, Molecular Devices). In the time course series of images collected from each experiment, we measured the maximal damage area. As described previously, the changing size of the damaged area was used to estimate the rates of epithelial restitution by fitting a single exponential decay curve to the 10 min of data subsequent to the time when maximal damage was observed. Results report the exponential rate constant in units of s−1 (20).
Intracellular free Ca2+ was measured semiquantitatively using the YC mouse. The FRET/CFP ratio was obtained by averaging 3–5 circular regions (diameter = 16 μm) from the epithelial cells adjacent to the damaged area. The background-corrected FRET/CFP ratio image was calculated and normalized to a value of 1 in the pre-damage base line to compensate for daily instrument settings.
To semiquantitatively measure extracellular luminal Ca2+ values adjacent to the site of damage (< 20 μm of distance from the gastric surface), background-corrected 458/488 fluorescence ratio images were calculated, and values were obtained by averaging 3–5 non-overlapping circular regions (diameter = 16 μm) from every ratio image. Regions with dye binding to luminal mucus were excluded from the analysis. Ratio values were normalized for daily instrument settings by comparison to Fura-Red dye in perfusate solution prior to tissue exposure.
Isolation of mRNA using Laser Capture Microdissection
Frozen sections (5 μm thick) were cut in a −20 °C cryostat. Slides were dehydrated through an ethanol series, then xylene, and air-dried. Using a laser capture microdissection microscope (Veritas model, Arcturus), four non-overlapping gastric cell populations were collected separately: surface epithelium (< 10 μm from the gastric surface), mid-gland region, basal gland region (final 50 μm at the base of gastric glands), and muscle cells. Tissue regions were collected onto HS LCM Caps (Arcturus), and total RNA in the collected samples was extracted and isolated using the PicoPure RNA isolation kit (Arcturus).
Detection of mRNA Using Reverse Transcriptase PCR and Real-time PCR
Single-stranded cDNA was synthesized by iScript cDNA synthesis kit (Bio-Rad). Conventional reverse transcriptase polymerase chain reaction was performed using the Advantage 2 polymerase mixture (Clontech) on a thermal cycler (MJ Research). Real-time PCR was used to compare the level of gene expression by amplification with an ABI 7500 (Applied Biosystems) using SYBR Green I dye (Applied Biosystems) for the detection of PCR products. To analyze entire gastric mucosa separate from muscle layers, the PMCA knockout mouse corpus was blunt-dissected under a stereo microscope, and RNA was extracted by TRI reagent (Molecular Research Center). The PMCA1 gene was amplified by QuantiTect primer (Mm_Atp2b1_1_SG, Qiagen). Other PCR primers are described in supplemental Fig. S1.
Immunofluorescence
Cryosections (10 μm) were prepared. Antigen retrieval was performed for each antibody using antigen unmasking solution (Vector) for 10 min. To remove nonspecific binding, sections were first incubated with goat serum (20%) for 30 min, followed by Fab fragment goat anti-mouse IgG (0.13 mg/ml, Jackson ImmunoResearch Laboratories) for 30 min. Sections were then incubated with the primary antibodies for 30 min at room temperature. The primary antibodies to PMCA (5F10, mouse monoclonal anti-mouse PMCA, Thermo Scientific) or PMCA4 (JA9, mouse monoclonal anti-mouse PMCA4) were used at a 1:100 dilution. NHE1 antibody (S1A, polyclonal rabbit antiserum) was coincubated at a 1:100 dilution (27). Secondary antiserum was used at a 1:100 dilution (Alexa Fluor 555-labeled goat anti-mouse IgG2a and Alexa Fluor 488-labeled goat anti-rabbit IgG, Invitrogen) and incubated for 30 min at room temperature. Nuclear DNA staining was performed by incubation with Hoechst 33342 (Invitrogen) at 1 μg/ml for 1 min.
Western Blot Analysis
The mouse corpus was blunt-dissected under a stereo microscope to separate the gastric mucosa from the muscle layer. Then, 700 μl radioimmune precipitation assay buffer (Sigma) with protease inhibitor (Roche) was added to isolated mucosa. Protein (30 μg) was separated on a 4–15% gradient gel and transferred to a PVDF membrane (Immobilon-P, Millipore). Membranes were incubated for 1 h with Odyssey blocking buffer (Li-Cor Biosciences), incubated 1 h at room temperature with 5F10 (1:1000 dilution), and then incubated for 1 h in goat anti-mouse secondary antibody (IRDye 800, 1:1000 dilution, Li-Cor Biosciences). The antibodies were stripped (25 mm glycine-HCl (pH 2) with 1% SDS, 30 min), incubated for 1 h at room temperature with mouse monoclonal anti-mouse GAPDH (1:10,000 dilution, Sigma), and then incubated for 1 h in goat anti-mouse secondary antibody (IRDye 800, 1:10,000 dilution, Li-Cor Biosciences). Blots were quantified using an Odyssey infrared imaging system (Li-Cor Biosciences).
Statistical Analysis
All values are reported from representative experiments as the mean ± S.E. from multiple experiments. Statistical significance was determined using unpaired Student's t test or one-way analysis of variance with post hoc Dunnett's multiple comparison test. p < 0.05 was considered significant.
RESULTS
In preliminary studies, conventional Ca2+-sensitive dyes (indo-1, fura-2, or fluo-4) could not be loaded into gastric tissue in vivo. Therefore, we measured free Ca2+ using YC transgenic mice. Two-photon excitation at 840 nm minimized tissue autofluorescence in wild-type C57BL/6 mouse stomach, efficiently excited CFP, and minimally excited YFP directly (data not shown). We used 840-nm excitation to estimate free Ca2+ from YC transgenic tissue as the ratio of FRET-stimulated YFP fluorescence divided by directly stimulated CFP fluorescence.
The gastric surface epithelial cells of YC transgenic mice uniformly express YC fluorescence (data not shown). When a single gastric surface epithelial cell in the YC mouse was photodamaged in a diffraction-limited spot (< 0.5 μm × 0.5 μm in the plane of focus, marked by an X in Fig. 1), CFP fluorescence intensity dropped, and YFP (FRET) intensity increased in the surrounding cytoplasm of the targeted cell, resulting in a rapid increase of the FRET/CFP ratio (Fig. 1A). These dynamic fluorescence changes were blunted by intracellular calcium chelation with BAPTA/AM (Fig. 1B). Results show that a measurable and local calcium mobilization occurs in response to the damage. However, this highly focal damage did not result in cell exfoliation, so imposing damage on several cells was subsequently evaluated.
FIGURE 1.
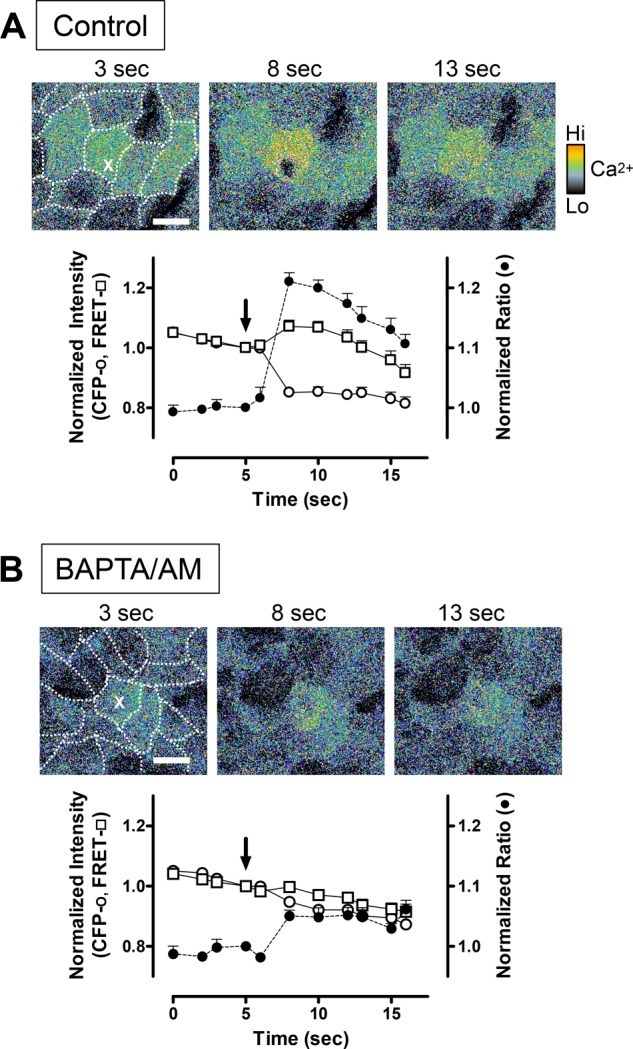
Using YC3.0 transgenic mice to detect calcium mobilization by two-photon FRET after damage to single gastric surface epithelial cells. Shown are a representative time course of images and two-photon fluorescence data collected in control conditions (A, n = 7) or after 250 μm BAPTA/AM incubation (B, n = 9). Pseudocolor FRET/CFP ratio images were collected at the indicated times, with damage imposed at 5 s. The first image in each series has an overlay showing the outlines of individual gastric surface epithelial cells. A single cell in the image was stimulated at 5 s by high-power, two-photon light at a single pixel (X). Scale bars = 10 μm. Shown are fluorescence intensity values (mean ± S.E.) extracted from the damaged cell and normalized to values before imposing photodamage (arrows) of CFP (○) and FRET fluorescence (□) as well as calculated FRET/CFP ratio values (●).
Mobilization of Intracellular Ca2+ via Signaling Cascades during Epithelial Wounding
Fig. 2A shows pseudocolored images of the FRET/CFP ratio and qualitative effects of PD to 3–5 cells caused by (10 s) high-intensity, 840-nm excitation within the red rectangle region. We observed damage progression followed by restitution, qualitatively similar to previous findings using 700–730 nm PD (3, 20, 21). The size of damage was quantified by digital comparison of the area occupied by all cells (identified by reflectance) versus areas of dead/dying cells (no YC protein), and the outcomes are compiled in Fig. 2B. The damage area was maximal at 102 ± 15 s after imposing damage (n = 6 animals, 1908 ± 195 μm2 maximal damage area) and was almost fully repaired at ∼1200 s (70 ± 43 μm2). The restitution rate (4.2 ± 0.5 10−3/s) was calculated as described under “Experimental Procedures.”
FIGURE 2.

Gastric surface repair and intracellular calcium (CaIN) mobilization after photodamage. Experiments were performed using YC transgenic mice. U-73122or indomethacin (Indo) was applied 1 h before PD. Averaged results are mean ± S.E., *, p < 0.05 versus control. A, pseudocolor FRET/CFP ratio images (2 × 2 median-filtered) collected at the indicated times from a representative time course experiment. Gastric surface cells were photodamaged (red rectangle) at time zero. Measurements were made in viable cells adjacent to the damaged area (near, white circles) or in viable cells 100 μm from damage area (far, white squares). Scale bar = 50 μm. B, the time course of damage size observed after inducing PD at the indicated time (arrow). The inset shows the expanded time scale. Values are mean ± S.E. (n = 6 mice). C, the FRET/CFP ratio was measured in viable near or far cells as indicated. The time of PD is indicated by the arrow. The inset shows the expanded time scale. n = 6 mice. D, exponential curve fits to results such as those in B were used to measure rates of repair, as described under “Experimental Procedures.” Resulting rate constants are presented in the absence of drugs (control, n = 6) or in the presence of U-73122 (n = 5) or indomethacin (n = 5). E, using the animals evaluated in D, the FRET/CFP ratio was calculated from cells adjacent to damage in the absence or presence of drugs as indicated. F, results showing the calculated difference between the FRET/CFP ratio at time zero and the indicated time point. Results are shown for the absence of drug (near and far cells as defined above, n = 6), or near cells in the presence of U-73122 (n = 5) or indomethacin (n = 5).
The FRET/CFP ratio (Cain) was measured in the restituting viable epithelial cells adjacent to the damaged area (Fig. 2A, white circles) as well as cells > 100 μm from the damaged area (Fig. 2A, white squares). As shown qualitatively in Fig. 2A and quantitatively in C, the FRET/CFP ratio selectively and rapidly increased in the cells surrounding the damaged region but was unchanged in cells far from damage. Statistical analyses evaluate the ratio change observed during the first 3 min, as compiled in Fig. 2F. In subsequent analyses, multiple ratio time points are averaged to simplify graphical presentation and reduce experimental noise.
To find out whether Ca2+ was released nonspecifically by cellular damage or specifically by activation of signaling pathways, we investigated effects of the phospholipase C inhibitor U-73122 or COX inhibitor indomethacin. As shown in Fig. 2D, the wound repair rates after administration of U-73122 or indomethacin (1.1 ± 0.2 10−3/s (n = 5), or 0.7 ± 0.5 10−3/s (n = 5), respectively) were significantly slower (p < 0.05) than the control. Either U-73122 or indomethacin significantly blocked the intracellular Ca2+ increase during PD-induced wounding (Fig. 2, E and F). We further tested the effect of the 1,4,5-triphosphate IP3 receptor antagonist 2-APB on gastric repair (Fig. 3). 2-APB significantly slowed gastric repair (Fig. 3, A and B, n = 4) and the intracellular Ca2+ increase after PD-induced damage (C and D).
FIGURE 3.

Effects of 2-APB on gastric surface repair. Compiled results (mean ± S.E.) with YC transgenic mice (control, n = 5; 2-APB, n = 4) showing the time course of the damaged area (A), which was used to derive the calculated repair rate (B). Under the same conditions, the FRET/CFP ratio (CaIN) (C) was calculated from cells adjacent to the damage (C) or as the difference calculated between the FRET/CFP ratio at time zero and the indicated time point (D). *, p < 0.05 versus control.
Extracellular Ca2+ Mobilization in the Gastric Lumen during Epithelial Wounding
The ratio of Fura-Red in response to alternating 458 and 488 nm excitation (F458/F488) was dependent on Ca2+ concentration (28), and dye responsiveness was equivalent when the pH was > 3.8 (supplemental Fig. S2, A –C). Because we demonstrated previously that the pH at the gastric surface was > 3.8 when the gastric lumen was perfused with pH 5 solutions (29, 30), we used pH 5 superfusates in all subsequent experiments.
When the gastric surface of C57Bl/6 mice was exposed to a luminal pH 5 solution containing Fura-Red but no added Ca2+, PD caused a local and transient increase in the F458/F488 ratio (CaLU) adjacent to the site of damage, indicative of a local increase in luminal Ca2+ fueled by endogenous Ca2+. Fig. 4A shows a representative time course of F458/F488 ratio images. Simultaneously collected images of NAD(P)H autofluorescence confirm the site and extent of epithelial damage (Fig. 4A, arrows). The sharply bounded region of dye observed at the bottom of all ratio images in Fig. 4A is due to dye within a concave portion of the surface at the opening of a gastric pit. Compiled results from such experiments (Fig. 4B) document the localized extracellular Ca2+ increase as being larger in the luminal space near damage (< 50 μm, Fig. 4A, white circles), with the peak Ca2+ level occurring 5–6 min after imposing damage. On the basis of a Fura-Red calibration curve (supplemental Fig. S2A), calcium increased by 651 ± 173 nm at 5 min. U-73122 or indomethacin pretreatment inhibited the increase of luminal Ca2+ during gastric wounding (Fig. 4, C and D), suggesting that nonspecific cell destruction was not the source of mobilized extracellular Ca2+.
FIGURE 4.

Extracellular Ca2+ (CaLU) mobilization adjacent to gastric surface damage. Experiments were performed using C57Bl/6 mice. A, representative experiment with PD imposed directly after time zero (at the site shown by the arrows), and NAD(P)H autofluorescence and Fura-Red F458/F488 ratio images were collected at the indicated times. Calcium levels in the ratio images are pseudocolored according to the color bar. Scale bar = 50 μm. B, time course of the measured damage area (left y axis scale) and F458/F488 ratio (CaLU), right y axis scale) measured in the luminal space directly adjacent (< 50 μm) from the PD site (Near, white circles in A) or > 100 μm from the damage site (Far, white squares in A). Values are mean ± S.E. n = 6. The time course of the F458/F488 ratio (CaLU) (C) and values directly prior to damage (Basal) and 5 min after damage (D) are shown in the absence of drug (n = 6) or the presence of U-73122 (n = 7) or indomethacin (n = 4) as applied in Fig. 2. *, p < 0.05 versus basal control; †, p < 0.05 versus control at 5 min.
Endogenous Ca2+ Requirement for Epithelial Wound Repair
Intracellular Ca2+ chelation by exposure to BAPTA/AM significantly inhibited both the repair of gastric damage (Fig. 5, A and B) and the rise in intracellular free Ca2+ reported in YC transgenic mice after PD (Fig. 5, C and D). Extracellular Ca2+ chelation by luminal HEDTA significantly inhibited the repair of gastric damage (Fig. 5, A and B), and the luminal Ca2+ increase reported by Fura-Red after PD (E and F). In addition, the PD-induced intracellular Ca2+ increase was modestly blunted by HEDTA at later time points (Fig. 5, C and D), but BAPTA/AM completely blocked the observed extracellular Ca2+ increase at all time points (E and F). Control in vitro experiments (supplemental Fig. S2D) confirmed that HEDTA did not reduce the free Ca2+ in fresh superfusate solution, suggesting that the solution contained submicromolar [Ca2+]. The results suggest that endogenous Ca2+ is essential to promote repair via the interdependence between the mobilization of intracellular and extracellular Ca2+.
FIGURE 5.

Effects of calcium chelation on gastric surface repair and calcium mobilization. Experiments tested the effects of luminal HEDTA (▿ or light hatched bar) or BAPTA/AM (■ or dark hatched bar) on the response to PD compared with the absence of drug (control). Compiled results (mean ± S.E.) with YC transgenic mice (control, n = 4; HEDTA, n = 4; BAPTA/AM, n = 4) showing the time course of damaged area (A), which was used to derive the calculated repair rate (B). Under the same conditions, the FRET/CFP ratio (CaIN) (C) was calculated from cells adjacent to the damage, and the difference was calculated between the FRET/CFP ratio at time zero and the indicated time point (D). Using C57Bl/6 mice and Fura-Red in luminal perfusates (control, n = 6; HEDTA, n = 4; BAPTA/AM, n = 5), the time course of the F458/F488 ratio (CaLU) (E) and values directly prior to damage (basal) and 5 min after damage (F) are shown. *, p < 0.05 versus control; †, p < 0.05 versus control at 5 min. The symbols in C and E are the same as in A, whereas in D and F they are the same as in B.
Routing of the Endogenous Ca2+ Needed for Restitution
We asked whether cellular Ca2+ uptake was a source of mobilized Ca2+ in response to damage. Voltage-activated calcium channels are involved in Ca2+ homeostasis of multiple gastric epithelial cells (26, 31), including enterochromaffin-like cells (32), D cells (33), and G cells (10). A voltage-activated calcium channel blocker, verapamil, significantly inhibited the speed of gastric repair (Fig. 6A). As shown in Fig. 6, B and C, verapamil blocked the increase of intracellular and extracellular Ca2+ in response to PD. Given the confirmed absence of superfusate Ca2+ in this protocol, the results suggest that basolateral Ca2+ uptake may be required for the Ca2+ mobilization that promotes the repair of damage.
FIGURE 6.
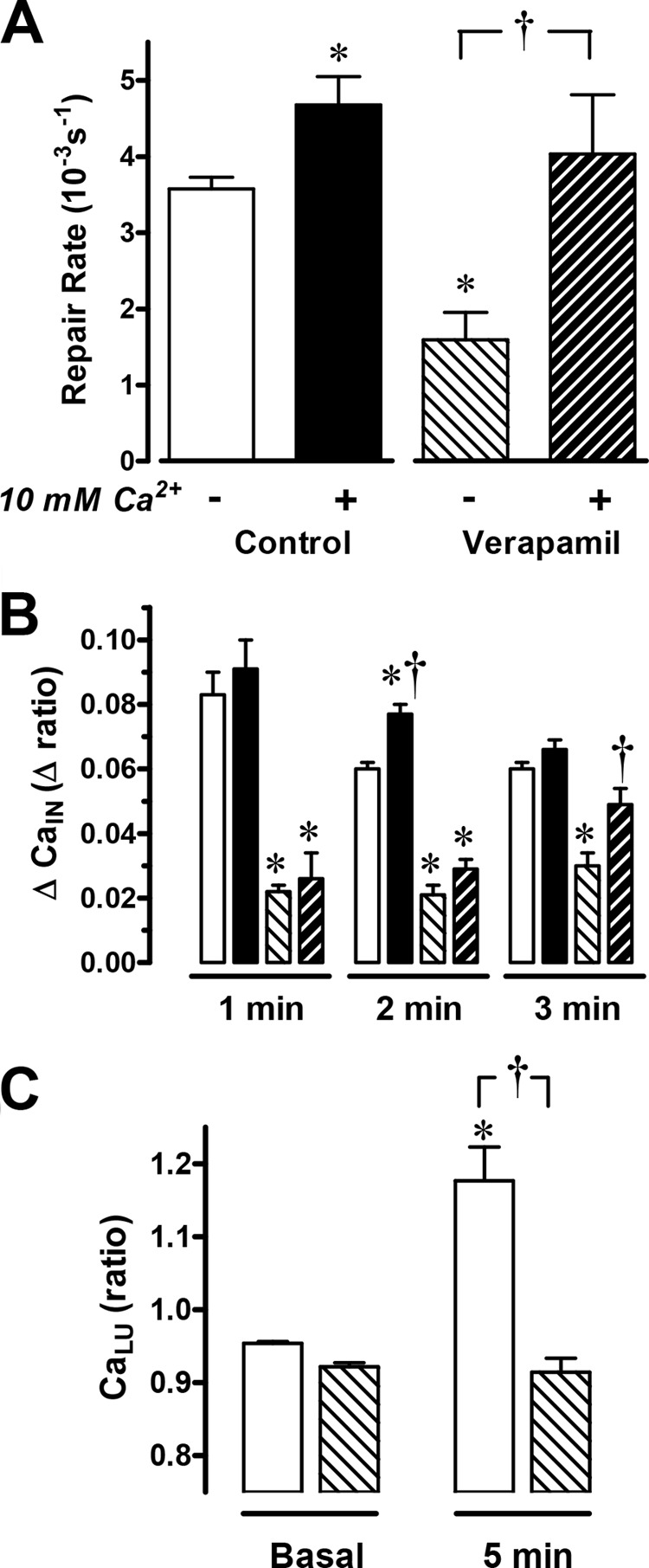
The effects of verapamil on gastric surface repair. Verapamil was given 1 h before PD was applied at time zero. The gastric mucosa were superfused either with conventional perfusate solution with no added Ca2+ (control, n = 6; verapamil, n = 8) or with a solution containing 10 mm Ca2+ (control, n = 4; verapamil, n = 6). A, compiled results from YC transgenic mice show the calculated repair rate (*, p < 0.05 versus no Ca2+ control; †, p < 0.05 versus absence of verapamil). B, the change in FRET/CFP ratio (ΔCaIN) calculated from cells adjacent to damage where the basal ratio value before damage is subtracted from the ratio at the indicated time point (*, p < 0.05 versus time control. †, p < 0.05 versus absence of verapamil) (B). C, using C57Bl/6 mice and Fura-Red in luminal perfusates (n = 6), F458/F488 ratio values before (basal) and 5 min after PD. *, p < 0.05 versus basal control; †, p < 0.05 versus control at 5 min. The symbols in B and C are the same as in A.
Raising luminal Ca2+ has been shown to enhance the repair of extensive gastric damage, but the mechanism has not been examined (4). The addition of 10 mm Ca2+ into the luminal perfusate also significantly accelerated the repair of microscopic gastric lesions (4.7 ± 0.2 10−3/s, n = 4) compared with the absence of added luminal Ca2+ (3.6 ± 0.4 10−3/s, n = 6) (Fig. 6A). Further, the inhibition of gastric restitution by verapamil (1.5 ± 0.2, n = 8) was rescued in the presence of 10 mm luminal Ca2+ (4.0 ± 0.8 10−3/s, n = 6) (Fig. 6A). In contrast, intracellular Ca2+ mobilization was not increased consistently by the extracellular Ca2+ supplement in either the absence or presence of verapamil (Fig. 6B). The results suggest that luminal Ca2+ can stimulate repair by mechanisms that are downstream of intracellular Ca2+ mobilization.
We explored the mechanisms that could mediate the raise of extracellular Ca2+ after damage. The gene families of sodium/calcium exchangers (NCX) and PMCA mediate Ca2+ extrusion from mammalian cells (34). In the stomach muscle layers, the expression and function of these transporters is well established, but there is no corresponding information about their distribution or roles in other regions of the gastric mucosa (35–37). To evaluate localized expression of mRNA for NCX and PMCA isoforms, four regions of the gastric tissue were collected separately using laser capture microdissection: surface epithelium (mucous cell region), mid-gland (parietal cell region), basal gland (chief cell region), and muscle layer (Fig. 7A). Using reverse transcriptase PCR (Fig. 7B) or real-time PCR (C) applied to cDNA created from pooled mRNA from each tissue region, we confirmed RNA quality (ubiquitously expressed GAPDH and NHE1 mRNA) and cellular heterogeneity among microdissected regions (mucin MUC5ac mRNA was detected only in the surface epithelial layer, and the parietal cell-specific H,K-ATPase α subunit was detected in the middle and basal gland layers).
FIGURE 7.

Site-specific expression of mRNA for PMCA, NCX, and IP3 receptor isoforms in stomach tissue. Laser capture microdissection collected four stomach regions: surface epithelium (S, < 10 μm from the lumen), middle (Mi, midway through the glandular region), bottom (B, < 50 μm from the base of gastric glands), and muscle (Mu). A, image showing the four gastric regions collected. B, conventional PCR and agarose gel separation evaluated 14 genes as indicated. The brain section was the positive control, and no cDNA (water) was a negative control. [single dagger symbol]. C, real-time PCR was performed to compare the expression levels among the four gastric regions. Data were normalized to GAPDH expression using ΔΔCT calculations. Data are mean ± S.E., n = 3. ND, no detection.
NCX1 mRNA was only observed in the basal gland and muscle layers. The PMCA2, PMCA3, NCX2, and NCX3 genes did not have measurable mRNA expression in the stomach (Fig. 7B). Because 2-APB inhibited gastric repair of damage (Fig. 3), we checked IP3 receptor subtype expression. We detected only IP3 receptor isoform 3 expressed in the gastric surface epithelium (Fig. 7B). PMCA1 mRNA was found in all regions, whereas PMCA4 was expressed in the muscle layer and, less prominently, in the basal gland layer (Fig. 7, B and C). Immunohistochemistry confirmed the site of PMCA expression in the gastric mucosa. Using an antibody that recognizes all PMCA isoforms (36, 38), PMCA was observed in the WT gastric surface epithelium, lamina propria, and muscle layer (Fig. 8B, a, d, g, and j). PMCA colocalized with a basolateral membrane protein (NHE1) in the gastric surface epithelium (Fig. 8A). The results suggest that PMCA1 is a candidate Ca2+ extrusion transporter in the gastric surface epithelium, although PMCA4 and NCX1 may play a role in other gastric epithelial cell types.
FIGURE 8.
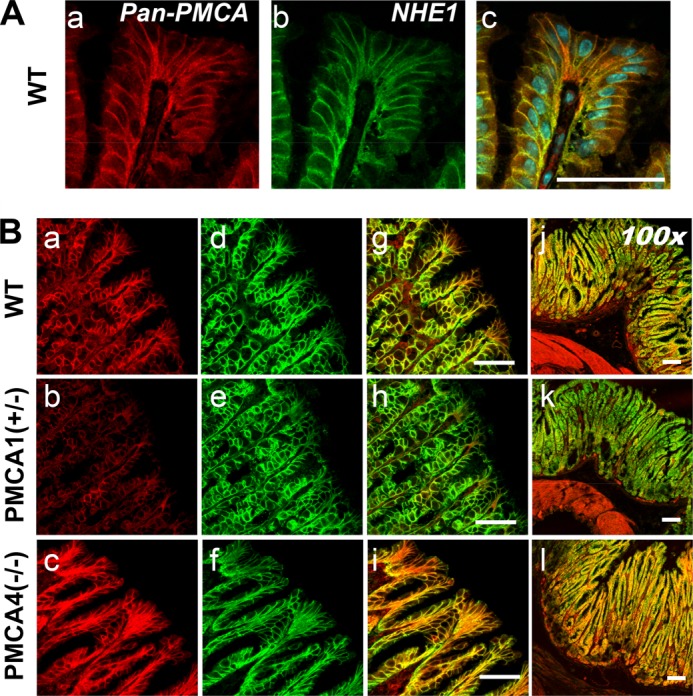
PMCA localization in mice with selective PMCA isoform knockout. A, C57Bl/6 mouse stomach stained with primary antibodies that recognize all PMCA isoforms (a), NHE1 (b), or a merged image with nuclei stain (c). B, WT, PMCA1+/−, or PMCA4−/− mouse stomach reacted with primary antibodies that recognize all PMCA isoforms (a–c) or NHE1 (d–f). These images were merged to compare labeling patterns (g–i) and were compared with a lower-magnification merged image that shows muscle layers (j–l). Scale bars = 50 μm.
Because PMCA1−/− mice die before birth (37), we examined gastric PMCA protein expression in PMCA1+/− mice and PMCA4−/− mice. Under identical imaging conditions (and compared with WT stomach), PMCA staining of the gastric epithelium appeared diminished in PMCA1+/− mice (Fig. 8B, b) and elevated in PMCA4−/− mice (c). A PMCA4-specific antibody showed staining only in the lamina propria and in the muscle layer, and this signal was absent in PMCA4−/− mouse stomach (Fig. 9), suggesting that the pan-PMCA antibody was detecting another PMCA isoform elevated in the gastric epithelium of PMCA4−/− mouse stomach.
FIGURE 9.
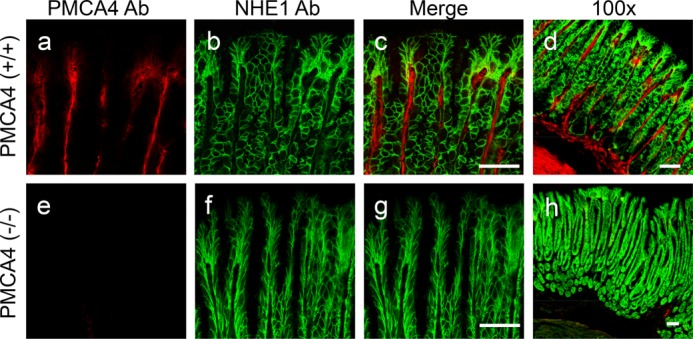
PMCA4 localization in mouse stomach. The WT or PMCA4−/− mouse stomach was reacted with primary antibodies for PMCA4 (a and e) or NHE1 (b and f). These images were merged to compare labeling patterns (c and g) and were also compared with a lower-magnification merged image that shows muscle layers (d and h). Scale bars = 50 μm.
Western blot analysis confirmed that PMCA protein (∼140 kDa, Refs. 36, 37) was reduced in PMCA1+/− gastric mucosal homogenates, whereas it was increased in corresponding PMCA4−/− tissue. A representative blot is shown in Fig. 10A, and a densitometric analysis from multiple experiments is shown in B. Additionally, PMCA1 mRNA was increased in PMCA4−/− gastric mucosa, and this increase was diminished in PMCA1+/−/PMCA4−/− mouse stomach (Fig 10C). In PMCA1+/− mice, we observed a reduction of PMCA protein but not PMCA1 mRNA compared with the WT. This is consistent with previous Northern blot findings in PMCA1+/− mouse stomach (37). The results confirm that PMCA1+/− mice have reduced amounts of the Ca2+ pump and suggest that PMCA4−/− mice have adaptive up-regulation of alternative PMCA isoforms (likely PMCA1) in the surface gastric epithelium.
FIGURE 10.
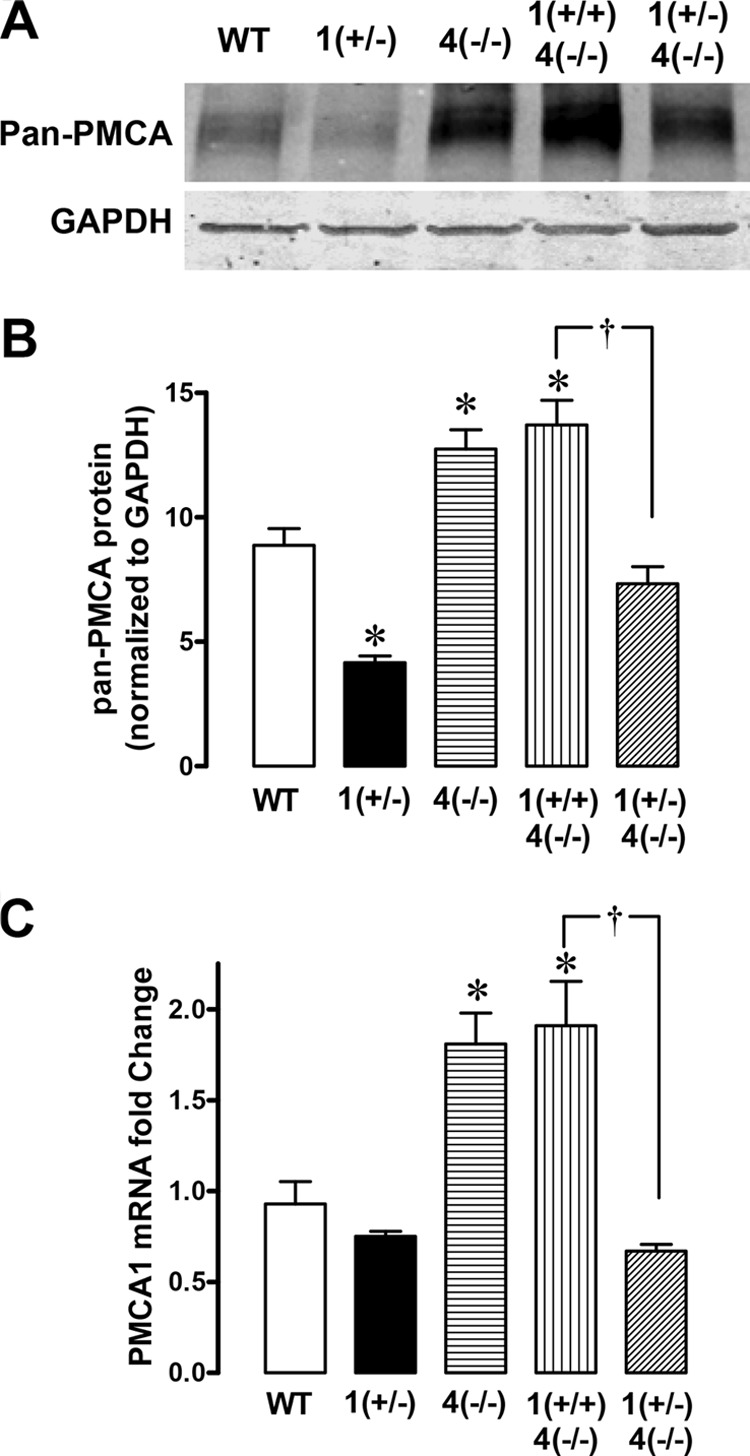
PMCA protein and mRNA levels in mice with selective PMCA isoform knockout. A, representative Western blot analysis comparing extracts from gastric mucosal scrapings (30 μg). The PMCA protein band was ∼140 kDa, and GAPDH was ∼37 kDa. B, results compiling densitometry evaluation of the PMCA band normalized to GAPDH. Data are mean ± S.E., n = 4 mice. *, p < 0.05 versus WT. †, p < 0.05 versus PMCA1+/−/PMCA4−/−. C, real-time PCR was performed to compare PMCA1 mRNA expression levels in extracts from gastric mucosal scrapings. Data were normalized to wild-type whole stomach (including muscle) PMCA1 expression using ΔΔCT calculations. Data are mean ± S.E., n = 4–6. *, p < 0.05 versus WT. †, p < 0.05 versus PMCA1+/+/PMCA4−/−.
The Role of PMCA1 in Epithelial Restitution
The rate of gastric restitution was similar in PMCA4−/− and WT mice but was significantly slowed in PMCA1+/− mice (Fig. 11A). In PMCA1+/− mice, the luminal Ca2+ level prior to damage was significantly lower compared with WT mice and was not significantly elevated after damage (Fig. 11B). The results with PMCA1+/− mice suggest that diminishing the amount of this transporter in heterozygotes reduces Ca2+ flux into the gastric lumen. Conversely, in PMCA4−/− mice, the resting gastric luminal Ca2+ level was elevated prior to damage compared with WT mice and remained elevated after PD (Fig. 11B). These effects in PMCA4−/− mice were eliminated in PMCA1+/−/PMCA4−/− double knockout mice (Fig. 11, C and D). Up-regulation of PMCA1 in surface cells of PMCA4−/− mouse stomach may explain the modified Ca2+ mobilization after damage in this tissue compared with the WT. The results suggest that Ca2+ efflux via PMCA1 is required to generate the luminal Ca2+ increase that promotes wound healing.
FIGURE 11.

The effect of PMCA gene deletion on gastric surface repair and luminal Ca2+ mobilization. Experiments with WT (n = 5), PMCA1 +/− (n = 8), or PMCA4−/− (n = 6) mice using luminal Fura-Red to report juxtamucosal Ca2+ levels. Compiled results show the calculated repair rate (A) and Fura-Red F458/F488 ratio values before (basal) and 5 min after photodamage (B). Data are mean ± S.E. *, p < 0.05 versus WT basal; †, p < 0.05 versus WT at 5 min. Experiments were performed using PMCA1+/+/PMCA4−/− (n = 5), or PMCA1+/−)/PMCA4−/− (n = 5) mice. Compiled results (mean ± S.E.) are shown. C, calculated repair rate. *, p < 0.05 versus PMCA1+/+/PMCA4−/−. D, F458/F488 values before (basal) and after (5 min) PD. The symbols in D are the same as in C. *, p < 0.05 versus PMCA1+/+/PMCA4−/− basal; †, p < 0.05 versus PMCA1+/+/PMCA4−/− at 5 min.
DISCUSSION
Observations of the beneficial effects of Ca2+ on epithelial repair (4, 11–13) have been limited because of the lack of mammalian models to examine Ca2+ dynamics in vivo. This paper establishes the conditions necessary for monitoring intracellular Ca2+ mobilization during gastric restitution in vivo in mice. Miyawaki et al. (18) introduced YC proteins as genetically encoded calcium sensors in 1997. There is only one prior report using isolated mouse cardiomyocytes that successfully used the YC3.0 transgene as a FRET-based calcium reporter in mouse tissue (39). Limitations of the YC3.0 sensor have been noted (low Ca2+ affinity, small FRET changes detected in wide field imaging), as have limitations of the YC transgenic mouse (mosaic expression of YC in some tissues) (19, 40, 41). Although genetically encoded calcium sensors have been improving continuously, in vivo reports remain limited. This work extends the outcomes to measure gastric intracellular Ca2+ dynamics in vivo and benefits from the relatively homogenous expression of the gastric YC protein and the increased FRET signal resolved by two-photon excitation. We noted that higher magnification studies of immobilized tissue (Fig. 1) resolve cell boundaries precisely but do not allow for cell exfoliation because of limited free space above the epithelium and the required small size of damage. In contrast, when imaging and damaging a larger field of view with a clearly open perfusion space and unrestricted tissue (as in Fig. 2 and all subsequent work), the imaging noise and even modest tissue motion eliminate single cell resolution. In this imaging mode, required to see exfoliation and restitution, it was not possible to study a single cell continuously and reliably.
The Ca2+ mobilization we observed after microscopic damage is not derived from the nonspecific release of Ca2+ from dead/dying cells. Either a phospholipase C inhibitor (U-73122), IP3 receptor antagonist (2-APB), or COX inhibitor (indomethacin) slowed gastric repair while simultaneously inhibiting the mobilization of intracellular Ca2+. It has been observed that prostaglandins produced from COX activity regulate gastric mucosal defenses via the EP1 receptor that utilizes a Ca2+-dependent, verapamil-sensitive signal transduction pathway (26). All inhibitor effects are likely to be mediated by inhibiting the activation of the G protein, Gq, that regulates intracellular Ca2+ metabolism mediated by IP3 turnover (23, 24). Further, we observed that intracellular Ca2+ selectively increased in the viable cells adjacent to fatally damaged cells but not in the cells located far away from the damage. This is consistent with work using cultured rabbit gastric epithelial cells, which showed elevated Ca2+ levels in migrating cells at the edge of an imposed wound (17). We reported previously that the repair of two photon-induced gastric damage was inhibited in COX-1 knockout or trefoil factor 2 knockout mice and, conversely, was stimulated by exogenous application of prostaglandin or trefoil factor (20, 21). Although it is unclear how these factors increase in response to damage, evidence suggests that trefoil factor 2 and prostaglandins are candidate agonists to stimulate intracellular Ca2+ via the phospholipase C/IP3 pathway. Results suggest that the signaling pathways activated by microscopic damage in vivo converge on the Ca2+ mobilization pathway(s) as a common downstream target to promote tissue repair (Fig. 12).
FIGURE 12.
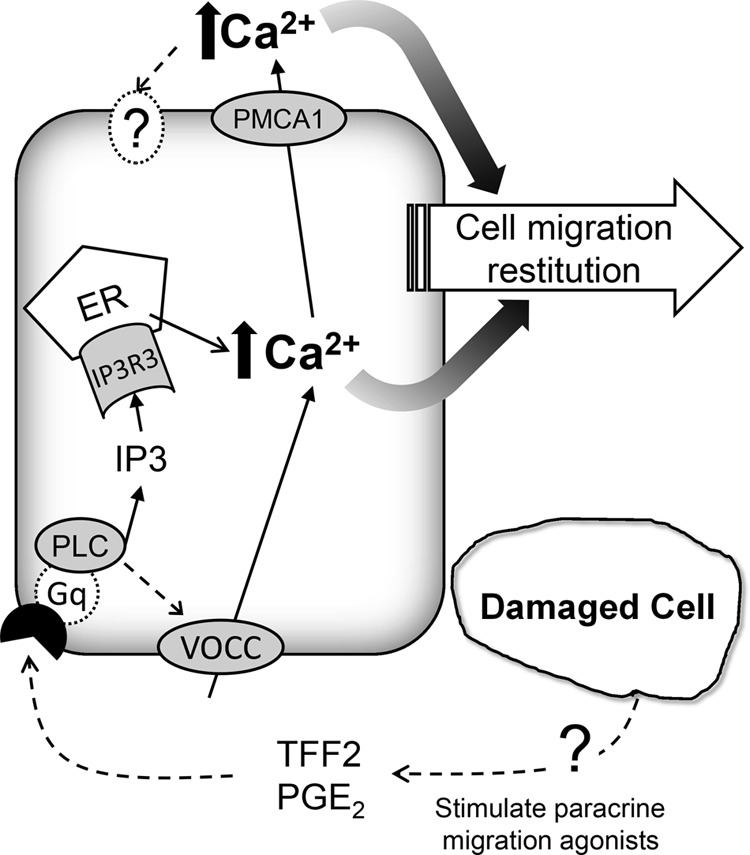
Schematic of calcium mobilization in response to two-photon damage. Dotted lines indicate more speculative pathways. VOCC, voltage operated Ca2+ channel; TFF2, trefoil factor 2; PGE2, prostaglandin E2; PLC, phospholipase C; ER, endoplasmic reticulum; IP3R3, IP3 receptor isoform 3; PMCA1, plasma membrane Ca-ATPase isoform 1.
Evidence suggests that the intracellular Ca2+ mobilization occurring after damage is required to observe subsequent changes in extracellular Ca2+ in the juxtamucosal space of the lumen. Four compounds that are expected to have independent effects primarily on intracellular Ca2+ mobilization (the Ca2+ channel blocker verapamil, the COX inhibitor indomethacin, the phospholipase C inhibitor U-73122, and the intracellular Ca2+ chelator BAPTA) all blunt intracellular Ca2+ mobilization, as predicted, but also inhibit the subsequent luminal Ca2+ rise. After damage, the rapid increase in intracellular Ca2+ precedes the slower luminal Ca2+ change, and mice with reduced PMCA1 have diminished luminal Ca2+ after damage. Combined, these results suggest that the raised intracellular Ca2+ is a source and stimulus for PMCA1-mediated Ca2+ efflux, which could be the transport route by which endogenous Ca2+ reaches the lumen. Ca2+ efflux via basolateral PMCA1 in surface cells would need to access the lumen via the paracellular pathway (and, likely, the epithelial breach caused by damage). Alternatively, Ca2+ efflux via apical PMCA1 in gastric gland cells (7) could access the lumen via transit up the gland lumen, but this alternative would also require a localized stimulation that created a luminal Ca2+ increase at the site of damage. Independent of the mechanism, our results suggest that a linkage between intracellular and extracellular Ca2+ mobilization is important to promote efficient gastric repair.
The results suggest that the mobilization of endogenous Ca2+ is essential to observe efficient repair. In overview, Ca2+ chelation slows repair, and luminal Ca2+ supplementation accelerates repair. It is possible that some of the observed Ca2+ could promote damage expansion through toxic overloading of mitochondria and other intracellular stores. However, a variety of experimental conditions causing reduced or buffered calcium mobilization utilized in our study did not lead to reduction in maximum damage size (supplemental Fig. S3). These results strongly suggest that the mobilized Ca2+ is not promoting the expansion of damage to adjacent cells. Because reduced Ca2+ did delay the time to reach maximum damage size (supplemental Fig. S3), the results suggest that Ca2+ has a role in accelerating the cell migration that leads to repair and, potentially, execution/expulsion of cells already fated to die. Our work extends the early findings of Critchlow et al. (4), Takeuchi et al. (11, 12), and Koo et al. (13), who demonstrated that, after a variety of insults causing pervasive surface damage, the luminal application of Ca2+ improved the gastric repair process, whereas Ca2+ chelation delayed it. Our results extend these results to a focal damage model and provide new information from directly measuring both intracellular and extracellular Ca2+ levels. Tested conditions did not selectively affect Ca2+ in only one of these spaces, so the results support interactions between Ca2+ signals observed in the two environments. These results also make it difficult to separate the roles of Ca2+ in the intracellular versus extracellular domain. However, HEDTA did not affect the early intracellular Ca2+ signal dynamics and, therefore, the observed repair rate in the presence of HEDTA (∼40% of normal) may indicate the portion that is stimulated by this intracellular Ca2+ signal. Conversely, supplementing Ca2+ in the lumen accelerated repair in normal tissue ∼30%, despite only a small (one time point significant) increase in intracellular Ca2+ levels after damage. Finally, when intracellular Ca2+ mobilization is blocked by verapamil, supplemental luminal Ca2+ is able to fully rescue repair, despite only partial restoration of intracellular Ca2+ levels toward normal values at later time points. The results support the speculation that there are separate beneficial effects of intracellular and extracellular Ca2+ to promote repair (Fig. 12).
There are only a few in vitro reports implicating the PMCA gene family of P-type ATPases in cell migration or wound healing. Dreval et al. (42) reported that vanadate, a nonspecific ATPase inhibitor, impairs the migration of MDCK-F cells, which express only PMCA1, whereas Talarico (43) used siRNA knockdown to show that the PMCA4 protein is necessary for rabbit corneal epithelial cell migration during wound healing. In this study, results from PMCA1+/− mice suggest that PMCA1 plays an important role in gastric surface restitution and in regulation of extracellular Ca2+ following damage. We were surprised to observe such a pronounced phenotype in a PMCA1 heterozygote. In contrast, full ablation of PMCA4 did not affect the speed of gastric surface restitution, although this is the only other PMCA isoform detectable in any region of the gastric mucosa. We confirmed that PMCA protein was reduced in the gastric mucosa in (43) and, therefore, the results can be explained by the heterozygote cells dropping below the threshold of Ca2+ extrusion capacity that is required to fully activate migration of the restituting epithelial cells. Because the PMCA1−/− homozygote does not survive to birth, we could not evaluate the effect of full PMCA1 ablation.
The results suggest that PMCA contributes to luminal Ca2+ levels even in undamaged tissue. Prior to imposing damage, basal Ca2+ levels were higher in PMCA4−/− mice that had a demonstrated increase in basolateral PMCA in the surface epithelium (most likely up-regulated PMCA1). We confirmed in PMCA1+/−/PMCA4−/− mice that this high basal luminal Ca2+ level reversed, in parallel with changes in PMCA total protein and PMCA1 mRNA abundance. As stated earlier, when talking about the response to damage, the basolateral Ca2+ exported from surface cells by PMCA1 would need paracellular transit to reach the lumen (Fig. 11). The work from Hofer and co-workers (7) shows that PMCA (isoform unspecified) colocalizes with the H,K-ATPase found in the apical membrane of parietal cells but also confirms our findings that PMCA is basolateral in surface epithelial cells, and they observed that PMCA can affect Ca2+ levels in the gastric gland lumen of healthy tissue (7).
In conclusion, our results provide insight into the molecular and cellular mechanisms whereby endogenous Ca2+ benefits gastric epithelial repair in vivo. The results reveal unanticipated Ca2+ signals in the damage microenvironment that are required to repair epithelial function. Our results suggest that signal transduction pathway(s) activated in response to damage drive a PMCA-1 mediated interaction between intracellular and extracellular Ca2+ signals that is necessary for efficient repair of damage.
Acknowledgments
We thank Drs. Varda Lev-Ram and Roger Y. Tsien (University of California, San Diego) for supplying breeding pairs of the YC transgenic mice and Dr. Lev-Ram for critical reading of the manuscript. We also thank Richard J. Paul, Ph.D., for the gift of specific PMCA4 antibody; Andrea Matthis, Ph.D., for breeding and maintaining the YC mice colony; and Chet Closson for critical two-photon confocal microscopy technical support.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1-DK-54940 (to M. H. M.), R01-DK050594 (to G. E. S.), and P30-DK078392. This work was also supported by university research council funding from the University of Cincinnati (to E. A.).

This article contains supplemental Figs. S1–S3.
- PMCA
- plasma membrane Ca-ATPase
- HEDTA
- N-(2-hydroxyetheyl)-ethylendiamin-N,N,N′-triacetic acid trisodium
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- homopipes
- homopiperazine- N,N′-bis-2-(ethane sulfonic acid)
- 2-APB
- 2-aminoethoxydiphenylborane
- YC
- yellow cameleon 3.0
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- PD
- photodamage
- COX
- cyclooxygenase
- NCX
- sodium/calcium exchanger
- IP3
- inositol 1,4,5-trisphosphate.
REFERENCES
- 1. Belkacemi L., Bédard I., Simoneau L., Lafond J. (2005) Calcium channels, transporters and exchangers in placenta. A review. Cell Calcium 37, 1–8 [DOI] [PubMed] [Google Scholar]
- 2. Hofer A. M. (2005) Another dimension to calcium signaling. A look at extracellular calcium. J. Cell Sci. 118, 855–862 [DOI] [PubMed] [Google Scholar]
- 3. Demitrack E. S., Soleimani M., Montrose M. H. (2010) Damage to the gastric epithelium activates cellular bicarbonate secretion via SLC26A9 Cl−/HCO3−. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Critchlow J., Magee D., Ito S., Takeuchi K., Silen W. (1985) Requirements for restitution of the surface epithelium of frog stomach after mucosal injury. Gastroenterology 88, 237–249 [DOI] [PubMed] [Google Scholar]
- 5. Cheng A. M., Morrison S. W., Yang D. X., Hagen S. J. (2001) Energy dependence of restitution in the gastric mucosa. Am. J. Physiol. Cell Physiol. 281, C430–438 [DOI] [PubMed] [Google Scholar]
- 6. Allen A., Flemström G., Garner A., Kivilaakso E. (1993) Gastroduodenal mucosal protection. Physiol. Rev. 73, 823–857 [DOI] [PubMed] [Google Scholar]
- 7. Caroppo R., Gerbino A., Debellis L., Kifor O., Soybel D. I., Brown E. M., Hofer A. M., Curci S. (2001) Asymmetrical, agonist-induced fluctuations in local extracellular [Ca2+] in intact polarized epithelia. EMBO J. 20, 6316–6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caroppo R., Gerbino A., Fistetto G., Colella M., Debellis L., Hofer A. M., Curci S. (2004) Extracellular calcium acts as a “third messenger” to regulate enzyme and alkaline secretion. J. Cell Biol. 166, 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Negulescu P. A., Machen T. E. (1993) Ca transport by plasma membrane and intracellular stores of gastric cells. Am. J. Physiol. 264, C843–851 [DOI] [PubMed] [Google Scholar]
- 10. Ray J. M., Squires P. E., Meloche R. M., Nelson D. W., Snutch T. P., Buchan A. M. (1997) L-type calcium channels regulate gastrin release from human antral G cells. Am. J. Physiol. 273, G281–288 [DOI] [PubMed] [Google Scholar]
- 11. Takeuchi K., Kato S., Konaka A., Sugawa Y. (1999) Luminal calcium in regulation of nitric oxide release and acid secretion in rat stomachs after damage. Dig. Dis. Sci. 44, 515–522 [DOI] [PubMed] [Google Scholar]
- 12. Takeuchi K., Nobuhara Y., Okabe S. (1985) Role of luminal Ca2+ on normal and damaged gastric mucosa in the rat. Dig. Dis. Sci. 30, 1072–1078 [DOI] [PubMed] [Google Scholar]
- 13. Koo M. W. (1994) The effects of milk and calcium on ethanol-induced gastric mucosal damage. Pharmacol. Res. 29, 217–224 [DOI] [PubMed] [Google Scholar]
- 14. Wallace J. L. (2008) Prostaglandins, NSAIDs, and gastric mucosal protection. Why doesn't the stomach digest itself? Physiol. Rev. 88, 1547–1565 [DOI] [PubMed] [Google Scholar]
- 15. Miller T. A., Kokoska E. R., Smith G. S., Banan A. (2001) Role of calcium homeostasis in gastric mucosal injury and protection. Life Sci. 69, 3091–3102 [DOI] [PubMed] [Google Scholar]
- 16. deFoneska A., Kaunitz J. D. (2010) Gastroduodenal mucosal defense. Curr. Opin. Gastroenterol. 26, 604–610 [DOI] [PubMed] [Google Scholar]
- 17. Ranta-Knuuttila T., Kiviluoto T., Mustonen H., Puolakkainen P., Watanabe S., Sato N., Kivilaakso E. (2002) Migration of primary cultured rabbit gastric epithelial cells requires intact protein kinase C and Ca2+/calmodulin activity. Dig. Dis. Sci. 47, 1008–1014 [DOI] [PubMed] [Google Scholar]
- 18. Miyawaki A., Llopis J., Heim R., McCaffery J. M., Adams J. A., Ikura M., Tsien R. Y. (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882–887 [DOI] [PubMed] [Google Scholar]
- 19. Nyqvist D., Mattsson G., Köhler M., Lev-Ram V., Andersson A., Carlsson P. O., Nordin A., Berggren P. O., Jansson L. (2005) Pancreatic islet function in a transgenic mouse expressing fluorescent protein. J. Endocrinol. 186, 333–341 [DOI] [PubMed] [Google Scholar]
- 20. Xue L., Aihara E., Podolsky D. K., Wang T. C., Montrose M. H. (2010) In vivo action of trefoil factor 2 (TFF2) to speed gastric repair is independent of cyclooxygenase. Gut 59, 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Starodub O. T., Demitrack E. S., Baumgartner H. K., Montrose M. H. (2008) Disruption of the Cox-1 gene slows repair of microscopic lesions in the mouse gastric epithelium. Am. J. Physiol. Cell Physiol. 294, C223–232 [DOI] [PubMed] [Google Scholar]
- 22. Dong X., Smoll E. J., Ko K. H., Lee J., Chow J. Y., Kim H. D., Insel P. A., Dong H. (2009) P2Y receptors mediate Ca2+ signaling in duodenocytes and contribute to duodenal mucosal bicarbonate secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G424–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi T. J., Liu S. X., Hammarberg H., Watanabe M., Xu Z. Q., Hökfelt T. (2008) Phospholipase Cβ3 in mouse and human dorsal root ganglia and spinal cord is a possible target for treatment of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 105, 20004–20008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hou C., Kirchner T., Singer M., Matheis M., Argentieri D., Cavender D. (2004) In vivo activity of a phospholipase C inhibitor, 1-(6-((17β-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-dione (U73122), in acute and chronic inflammatory reactions. J. Pharmacol. Exp. Ther. 309, 697–704 [DOI] [PubMed] [Google Scholar]
- 25. Peng T., Shen E., Fan J., Zhang Y., Arnold J. M., Feng Q. (2008) Disruption of phospholipase Cγ1 signalling attenuates cardiac tumor necrosis factor-α expression and improves myocardial function during endotoxemia. Cardiovasc. Res. 78, 90–97 [DOI] [PubMed] [Google Scholar]
- 26. Takeuchi K., Yagi K., Kato S., Ukawa H. (1997) Roles of prostaglandin E-receptor subtypes in gastric and duodenal bicarbonate secretion in rats. Gastroenterology 113, 1553–1559 [DOI] [PubMed] [Google Scholar]
- 27. Chu J., Chu S., Montrose M. H. (2002) Apical Na+/H+ exchange near the base of mouse colonic crypts. Am. J. Physiol. Cell Physiol. 283, C358–372 [DOI] [PubMed] [Google Scholar]
- 28. Lohr C. (2003) Monitoring neuronal calcium signalling using a new method for ratiometric confocal calcium imaging. Cell Calcium 34, 295–303 [DOI] [PubMed] [Google Scholar]
- 29. Baumgartner H. K., Kirbiyik U., Coskun T., Chu S., Montrose M. H. (2002) Endogenous cyclo-oxygenase activity regulates mouse gastric surface pH. J. Physiol. 544, 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Demitrack E. S., Aihara E., Kenny S., Varro A., Montrose M. H. (2012) Inhibitors of acid secretion can benefit gastric wound repair independent of luminal pH effects on the site of damage. Gut 61, 804–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ishihara T., Tanaka K., Tashiro S., Yoshida K., Mizushima T. (2010) Protective effect of rebamipide against celecoxib-induced gastric mucosal cell apoptosis. Biochem. Pharmacol. 79, 1622–1633 [DOI] [PubMed] [Google Scholar]
- 32. Lindström E., Eliasson L., Björkqvist M., Håkanson R. (2001) Gastrin and the neuropeptide PACAP evoke secretion from rat stomach histamine-containing (ECL) cells by stimulating influx of Ca2+ through different Ca2+ channels. J. Physiol. 535, 663–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. DelValle J., Wakasugi J., Takeda H., Yamada T. (1996) Linkage of [Ca2+]i in single isolated D cells to somatostatin secretion induced by cholecystokinin. Am. J. Physiol. 270, G897–901 [DOI] [PubMed] [Google Scholar]
- 34. Prasad V., Okunade G., Liu L., Paul R. J., Shull G. E. (2007) Distinct phenotypes among plasma membrane Ca2+-ATPase knockout mice. Ann. N.Y. Acad. Sci. 1099, 276–286 [DOI] [PubMed] [Google Scholar]
- 35. Liu L., Ishida Y., Okunade G., Pyne-Geithman G. J., Shull G. E., Paul R. J. (2007) Distinct roles of PMCA isoforms in Ca2+ homeostasis of bladder smooth muscle. Evidence from PMCA gene-ablated mice. Am. J. Physiol. Cell Physiol. 292, C423–431 [DOI] [PubMed] [Google Scholar]
- 36. Liu L., Ishida Y., Okunade G., Shull G. E., Paul R. J. (2006) Role of plasma membrane Ca2+-ATPase in contraction-relaxation processes of the bladder. Evidence from PMCA gene-ablated mice. Am. J. Physiol. Cell Physiol. 290, C1239–1247 [DOI] [PubMed] [Google Scholar]
- 37. Okunade G. W., Miller M. L., Pyne G. J., Sutliff R. L., O'Connor K. T., Neumann J. C., Andringa A., Miller D. A., Prasad V., Doetschman T., Paul R. J., Shull G. E. (2004) Targeted ablation of plasma membrane Ca2+-ATPase (PMCA) 1 and 4 indicates a major housekeeping function for PMCA1 and a critical role in hyperactivated sperm motility and male fertility for PMCA4. J. Biol. Chem. 279, 33742–33750 [DOI] [PubMed] [Google Scholar]
- 38. Caride A. J., Filoteo A. G., Enyedi A., Verma A. K., Penniston J. T. (1996) Detection of isoform 4 of the plasma membrane calcium pump in human tissues by using isoform-specific monoclonal antibodies. Biochem. J. 316, 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsien R. Y. (2003) Breeding molecules to spy on cells. Harvey Lect. 99, 77–93 [PubMed] [Google Scholar]
- 40. Fan G. Y., Fujisaki H., Miyawaki A., Tsay R. K., Tsien R. Y., Ellisman M. H. (1999) Video-rate scanning two-photon excitation fluorescence microscopy and ratio imaging with cameleons. Biophys. J. 76, 2412–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hasan M. T., Friedrich R. W., Euler T., Larkum M. E., Giese G., Both M., Duebel J., Waters J., Bujard H., Griesbeck O., Tsien R. Y., Nagai T., Miyawaki A., Denk W. (2004) Functional fluorescent Ca2+ indicator proteins in transgenic mice under TET control. PLoS Biol. 2, e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dreval V., Dieterich P., Stock C., Schwab A. (2005) The role of Ca2+ transport across the plasma membrane for cell migration. Cell Physiol. Biochem. 16, 119–126 [DOI] [PubMed] [Google Scholar]
- 43. Talarico E. F., Jr. (2010) Plasma membrane calcium-ATPase isoform four distribution changes during corneal epithelial wound healing. Mol. Vis. 16, 2259–2272 [PMC free article] [PubMed] [Google Scholar]