

Background: The heparin activation mechanism of antithrombin as a factor IXa and Xa inhibitor is not established.
Results: Mutations adjacent to helix D result in full activation of antithrombin without heparin.
Conclusion: Activation is largely dependent on Tyr-131 and Ala-134 and minimally on reactive center loop hinge expulsion.
Significance: This changes the understanding of the activation mechanism of antithrombin.
Keywords: Antithrombin, Blood Coagulation Factors, Circular Dichroism (CD), Coagulation Factors, Enzyme Kinetics, Fluorescence, Heparin, Mutagenesis, Protease Inhibitor, Serine Protease
Abstract
Allosteric conformational changes in antithrombin induced by binding a specific heparin pentasaccharide result in very large increases in the rates of inhibition of factors IXa and Xa but not of thrombin. These are accompanied by CD, fluorescence, and NMR spectroscopic changes. X-ray structures show that heparin binding results in extension of helix D in the region 131–136 with coincident, and possibly coupled, expulsion of the hinge of the reactive center loop. To examine the importance of helix D extension, we have introduced strong helix-promoting mutations in the 131–136 region of antithrombin (YRKAQK to LEEAAE). The resulting variant has endogenous fluorescence indistinguishable from WT antithrombin yet, in the absence of heparin, shows massive enhancements in rates of inhibition of factors IXa and Xa (114- and 110-fold, respectively), but not of thrombin, together with changes in near- and far-UV CD and 1H NMR spectra. Heparin binding gives only ∼3–4-fold further rate enhancement but increases tryptophan fluorescence by ∼23% without major additional CD or NMR changes. Variants with subsets of these mutations show intermediate activation in the absence of heparin, again with basal fluorescence similar to WT and large increases upon heparin binding. These findings suggest that in WT antithrombin there are two major complementary sources of conformational activation of antithrombin, probably involving altered contacts of side chains of Tyr-131 and Ala-134 with core hydrophobic residues, whereas the reactive center loop hinge expulsion plays only a minor additional role.
Introduction
It is well established that, in the absence of heparin, antithrombin is a poor inhibitor of the blood coagulation proteinases factor IXa and factor Xa and that the binding of a high affinity heparin pentasaccharide (H5)3 is sufficient to enhance by over a hundredfold the rate at which each proteinase can be inhibited (1). It is also clear from solution studies using a variety of techniques, including fluorescence (2–4), CD (5), and NMR (6–8) spectroscopies, that heparin binding causes significant conformational changes in antithrombin, some or all of which are likely to be required for the increased rates of inhibition of factor IXa and factor Xa.
An early proposal for the link between conformational change and activation, based on x-ray structures of α1-proteinase inhibitor and antithrombin, was that heparin directly promotes the extension of helix D, which in turn results in expulsion of the partially inserted hinge of the reactive center loop (RCL) from β-sheet A and so renders the P1 arginine in the RCL more accessible to target proteinase (Fig. 1) (9). A number of attempts have been made to test this idea. Thus, removal of one to four residues from the region 134–137 (10) or introduction of a proline at position 133 (11) eliminated most of the heparin-induced fluorescence enhancement, suggesting that expulsion of the RCL hinge had been blocked. However, in both studies it was found that, although heparin enhancements of the rate of inhibition of factor Xa had been reduced from that observed with WT antithrombin, there were still rate enhancements of 13–22-fold. One explanation is that residue(s) between the end of helix D and residue 133 might be responsible for this residual heparin-inducible activation, even though the RCL hinge is no longer expellable. In keeping with this, a more recent report on a Y131L antithrombin variant found that, in the absence of heparin, the variant was partially activated against factor Xa and was activated further by heparin binding, to a rate even higher than for WT antithrombin (12). In that study, however, no fluorescence enhancements were reported that might have indicated whether the RCL hinge was expelled, either before or after heparin binding.
FIGURE 1.
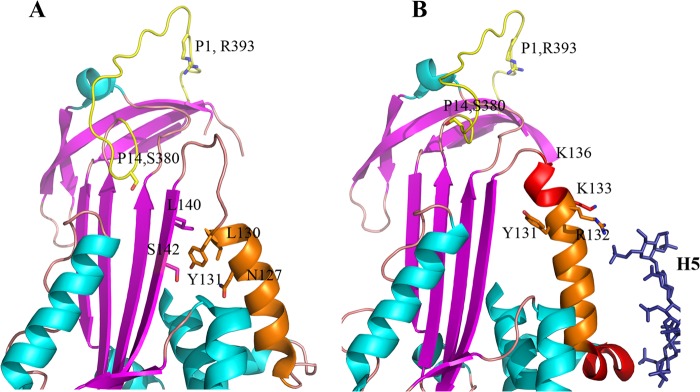
Structural changes in and around helix D of antithrombin upon heparin binding. A, heparin binding region of antithrombin in the heparin-free state (Protein Data Bank code 1E05) (35), showing helix D, the unstructured conformation of residues 131–136 and residues 379 and 380 (P15 and P14) of the reactive center loop inserted into β-sheet A. B, heparin binding region of antithrombin with heparin pentasaccharide bound (Protein Data Bank code 1E03) (35), showing the extension of helix D involving residues 131–136, formation of helix P, and expulsion of residues 379 and 380 of the reactive center loop from β-sheet A.
To more fully examine the role of Tyr-131 and of residues C-terminal to this in the heparin activation mechanism, we have created a new, more extensively mutated antithrombin variant in which helix-promoting residues were introduced at all positions in the stretch 131–136, including leucine at position 131, as well as another variant with the same mutations, although without the Y131L mutation, and we compared the properties to both WT and the recreated Y131L variant. We found that the 131–136 mutations result in dramatic activation as an inhibitor of both factor IXa and factor Xa in the absence of heparin, with rates close to those of fully heparin-activated WT. H5 binding subsequently gives only minor additional rate enhancement but to rates that surpass those of WT by ∼3-fold. The mutations also induced major conformational changes in the variant, observed by perturbations of CD and NMR spectra. Variants with the Y131L mutation alone or else the 132–136 mutations alone each gave lower activation in the absence of heparin, with values that, when combined, were similar to the full 131–136 set of mutations. Basal fluorescence of all three variants was similar to WT and gave enhancements of 23–36% upon binding heparin, consistent with the RCL hinge being inserted in the absence of heparin and expelled upon heparin binding. These findings support a three-part contribution to rate acceleration, with Tyr-131 and Ala-134 (the latter as a result of D helix extension) independently contributing the bulk of the rate enhancements and expulsion of the RCL hinge an additional ∼3-fold increase. These components are likely to be similar in the normal heparin-induced activation of WT antithrombin.
EXPERIMENTAL PROCEDURES
Materials
Human prothrombin and antithrombin were isolated from outdated plasma by previously described procedures (13, 14). Prothrombin was activated to thrombin and purified as described (15). Human factor Xa and factor IXa were from Hematologic Technologies, Inc., or Enzyme Research. Proteinase concentrations were determined by chromogenic substrate assays that had been standardized with active site-titrated proteinase (16). Synthetic high affinity pentasaccharide was from Sanofi-Synthelabo LLC. Heparin concentrations were determined by stoichiometric fluorescence titrations of plasma α-antithrombin.
Expression and Purification of Wild-type and Variant Antithrombins
Recombinant wild-type antithrombin and its variants were produced in the Baculovirus expression system (Invitrogen) similarly to past studies (17). Antithrombin cDNA was cloned into the pFastBac donor plasmid to carry out site-specific transposition of an expression cassette into a baculovirus vector (bacmid) propagated in Escherichia coli. High molecular weight mini-prep DNA isolated from selected E. coli clones containing the recombinant plasmid was used to transfect insect cells. Conditioned serum-free medium harvested 3–4 days post-infection of High 5 cells was clarified by centrifugation and used for antithrombin purification by affinity chromatography on a heparin-Sepharose column and ion-exchange chromatography on a Q-Sepharose column. The final yield of pure protein was 1–2 mg from a liter of cultivated medium.
Determination of Stoichiometry of Inhibition
Stoichiometries of inhibition of human thrombin, factor Xa, and factor IXa in the presence and absence of heparin were measured by titration of proteinase with inhibitor monitored spectroscopically by the chromogenic assay of residual thrombin, factor Xa, and factor IXa activity with substrates S-2238, Spectrozyme FXa, and Spectrozyme FIXa, respectively. Thrombin and factor Xa reactions were carried out in either 20 mm sodium phosphate, 0.1 m NaCl, or 20 mm Tris-hydrochloride, 0.25 m NaCl, buffers at pH 7.4 each containing 0.1 mm EDTA and 0.1% PEG 8000, whereas factor IXa reactions were carried out in 0.1 m Hepes, 93.5 mm NaCl, 5 mm CaCl2, 0.1% PEG 8000, pH 7.4, all at 25 °C. For the reactions in the absence of heparin, the buffer system contained 50 μg/ml Polybrene to complex any trace heparin contamination. Increasing amounts of antithrombin (10–120 nm) were added to proteinase (100 nm), and reactions, with or without added H5 (200 nm), were allowed to proceed to completion. Residual enzyme activity was measured by chromogenic assay in reaction buffer, with factor IXa assays being supplemented with 33% ethylene glycol to enhance activity. The inhibition stoichiometries were determined by plotting the residual proteinase activity against the ratio of inhibitor to enzyme, based on antithrombin concentrations determined by absorbance (18).
Kinetic Measurements
The rates of inhibition of thrombin, factor IXa, and factor Xa by antithrombin alone and H5-activated antithrombin were measured under pseudo first-order conditions of 50–2500 nm antithrombin and 1–250 nm proteinase in the reaction buffers and with chromogenic substrates described above. For H5-catalyzed reactions with thrombin and factor IXa, H5 was added in 1.5–2-fold molar excess over antithrombin to saturate the inhibitor. For reactions with factor Xa subsaturating levels of H5 were used, although with antithrombin concentrations (100 nm) that ensured full complexation of the added H5. The pseudo first-order constants (kobs) for all reactions, except H5-catalyzed reactions with factor Xa, were obtained by nonlinear regression fitting of residual enzyme activity against time of incubation, by a single exponential function. A non-zero end point was assumed for factor IXa reactions to account for small amounts of degraded proteinase (<10%) less susceptible to inhibition. kobs for H5-catalyzed antithrombin-factor Xa reactions was obtained by fitting residual enzyme activity measured in fixed time reactions (1–5 min) against H5 concentration by a single exponential function. The second-order rate constant in the absence of heparin or in the presence of saturating H5 was obtained by dividing kobs by the concentration of antithrombin. For the H5-catalyzed antithrombin-factor Xa reaction, the second-order rate constant (kapp) was determined by dividing kobs by the fixed reaction time (17). This method yields equivalent results as the independent time course method with saturating H5. All kapp values were found to be independent of the concentration of antithrombin or antithrombin-H5 complex over the range of concentrations examined, consistent with second-order conditions. All reactions were carried out in polyethylene glycol-coated cuvettes. For calculation of the effective second-order rate constant, the value of kapp was multiplied by SI.
CD Spectroscopy
Circular dichroism spectra were obtained on a Jasco J-710 spectrometer at room temperature in 2-mm cuvettes. For far-UV spectra, 5–10 μm samples of antithrombin, in the presence or absence of pentasaccharide at a 3:1 ratio with antithrombin in 10 mm potassium phosphate buffer, pH 7.5, 0.1 m NaCl, and 0.1 mm EDTA, were scanned from 250 to 200 nm. For near-UV spectra, 25–30 μm samples of antithrombin in the same buffer system were scanned from 310 to 250 nm. Heparin was added to antithrombin at 6:1 ratio. Base lines were recorded by scanning either buffer alone or with the presence of appropriate amount of heparin.
Fluorescence Spectroscopy
Emission spectra were recorded on an SLM 8000 spectrofluorimeter (excitation at 280 nm). Slits of 4 nm were used for excitation and 16 nm for emission, and integration time was 5 s. 100 nm samples of antithrombin were in 20 mm sodium phosphate buffer, pH 7.4, 100 mm NaCl, 0.1 mm EDTA, 0.1% PEG 8000. Following acquisition of duplicate spectra of antithrombin, the sample was titrated with H5 to an end point by adding at least 3 molar eq of H5. Duplicate spectra of H5-complexed antithrombin were then acquired. Spectra were corrected for buffer and dilution and averaged. Observed binding stoichiometries obtained by fitting titrations by the quadratic equilibrium binding function were within 10% of equimolar, ensuring that equivalent concentrations of free and H5-complexed WT and variant antithrombins were being compared. Errors represent ± 2 S.E. from at least four titrations.
NMR Spectroscopy
1H NMR spectra were recorded on a Bruker Avance spectrometer operating at 900 MHz and equipped with a cryoprobe. Antithrombin samples at 10–20 μm were dialyzed against D2O buffer containing 10 mm potassium phosphate and 0.1 m NaCl, pH 7.7. Heparin pentasaccharide was added to antithrombin at a 5:1 molar ratio to ensure saturation.
Heparin Affinity
Dissociation constants for the binding of control and variant antithrombins to H5 were determined at I = 0.45, to weaken affinity and hence improve accuracy, by monitoring the enhancement of the intrinsic tryptophan fluorescence of antithrombin upon titration with a heparin stock solution as described previously (12, 20). 100 nm antithrombin in 20 mm sodium phosphate buffer, pH 7.4, 0.4 m NaCl, 0.1 mm EDTA, 0.1% PEG 8000 was titrated with 25–2000 nm heparin with excitation and emission at 280 and 340 nm, respectively. The excitation bandwidth was 4 nm, and the emission bandwidth was 16 nm. Kd values were determined by computer fitting to the quadratic equilibrium binding equation assuming a 1:1 stoichiometry.
RESULTS
Choice of Residues for the Helix D-Extension Variant hD(131–136)
In wild-type antithrombin helix D forms a major part of the heparin pentasaccharide-binding site. Immediately following helix D are the residues 131–136, which do not take part in binding the pentasaccharide. In the absence of heparin, these residues are unstructured but adopt a helical conformation upon pentasaccharide binding. Residues 131–136 have the sequence YRKANK (YRKAQK in the N135Q background used). Using the Chou-Fasman predictive algorithm (19), this has a helix-forming potential (Pα) of only 0.95, where a value of 1 represents a frequency the same as for the average residue in a chain. To promote helix formation in the absence of heparin, these residues were changed to LEEAAE, which has a Chou-Fasman Pα of 1.47, i.e. close to the maximum possible. Although the hD(131–136) variant differs in sequence from WT at 5 of the 6 positions, individual changes at 4 of these positions have been made without affecting basal rates of reaction with factor Xa or the enhancements seen upon heparin binding. Thus, none of the individual mutations R132M, K133M (20), N135Q (21), N135A (22), or K136A/K136N (23) were perturbing.
hD(131–136) Antithrombin Is Highly Activated as an Inhibitor of Factor IXa and Factor Xa
hD(131–136) antithrombin reacted normally with thrombin in the absence of heparin with a very similar rate and SI to those of WT antithrombin (Table 1), suggesting that the extensive mutations had not had any adverse effect on its ability to function as an inhibitor of this target proteinase. Saturating H5 modestly increased the second-order rate constant for both the hD(131–136) variant and WT (Table 1).
TABLE 1.
Second order rate constants and SI values of WT and variant antithrombins
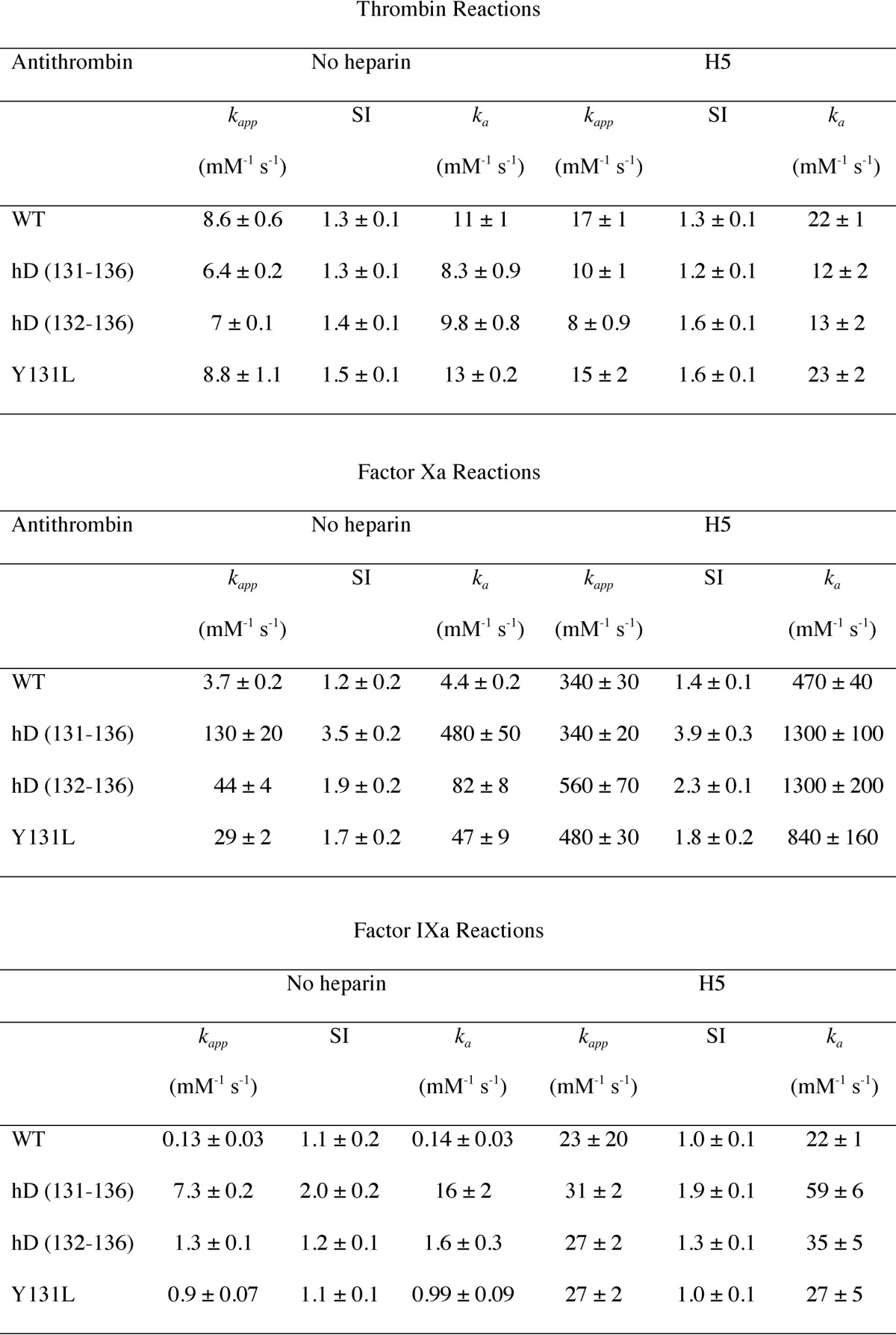
In dramatic contrast to the small effects that the 131–136 mutations had on either second-order rate constants or SI for reaction with thrombin, the effects for reaction with either factor Xa or factor IXa, in the absence of heparin, were very large increases in rate constants and an increase in SI. For factor Xa, the rate constant in the absence of H5 was increased by ∼110-fold, whereas for factor IXa the increase was ∼114-fold, compared with WT. The SI values increased from 1.2 and 1.1 for WT reacting with factor Xa and factor IXa, respectively, to 3.5 and 2.0. Whereas H5 binding to WT resulted in an ∼110- and ∼160-fold increase in rate constant for reaction with factor Xa and factor IXa, respectively, H5 binding caused only an additional 2.7- and 3.7-fold increase, respectively, with little further change in SI. The magnitude of the changes for the variant means that, in the absence of H5, the hD(131–136) variant is about as reactive as fully heparin-activated WT. Because H5 does still cause modest further increases, the H5-bound hD(131–136) variant is actually about 3-fold faster that the equivalent H5-bound WT antithrombin for both factor IXa and factor Xa.
Enhancements Are Approximately Additive
Of the five mutations in residues 131–136, the change of Y131L, present as part of hD(131–136), has been previously reported and partly characterized (12). It is known from that study that the Y131L mutation results in partial activation against factor Xa, but not thrombin, with no data reported for factor IXa. To determine whether the mutations at 131 and 132–136 contributed in an additive or more synergistic way to the rate enhancements for factors IXa and Xa, we made both the same Y131L variant as before, as well as a new variant in which only the residues 132–136 were changed (to the same EEAAE as in hD(131–132)). We refer to this variant as hD(132–136).
As with the hD(131–136) variant, neither the Y131L nor the hD(132–136) variant showed much difference in rates of reaction with thrombin compared with WT antithrombin (Table 1). For reaction with factor Xa in the absence of H5, Y131L and hD(132–136) variants gave ka ∼11- and ∼18-fold increases, respectively, compared with WT. This represents a close to additive contribution compared with the hD(131–136) variant (11 × 18 = 198-fold versus 110-fold observed for hD(131–136)). Similar behavior was observed for reaction with factor IXa: increases in ka in the absence of H5 of 7- and 11-fold were found for Y131L and hD(132–136) variants, respectively (Table 1), which would predict an increase of ∼77-fold if the contributions were simply additive, compared with the observed 114-fold increase.
For reaction with both proteinases, the effect of binding H5 to either variant was a much greater increase in ka than for the hD(131–136) variant, although the absolute values obtained were similar to those for the H5-bound hD(131–136) variant (Table 1), indicating that whatever activating changes were present in the hD(131–136) variant but absent from the Y131L or hD(132–136) variants were made up by binding H5. For reactions with both factor Xa and factor IXa, the final value of ka for hD(132–136) represented a higher rate of reaction than for H5-bound WT.
hD(131–136) Antithrombin Has Undergone Large Conformational Changes
Activation of WT antithrombin by the binding of heparin results in major conformational changes that are seen not only in x-ray structures but also in changes in solution CD and NMR spectra. We therefore examined both CD and one-dimensional 1H NMR spectra to determine whether the mutations had caused significant conformational changes and also to determine what changes were subsequently brought about by H5 binding.
Although far-UV CD can only give a measure of overall secondary structure and of changes in it, near-UV CD can report sensitively on changes around aromatic residues (24). Both far- and near-UV CD spectra were recorded for WT and hD(131–136) variant antithrombins in the absence and presence of H5 (Fig. 2). The spectra of the WT antithrombin were the same as reported previously, with far-UV spectra reporting a modest but significant alteration in ellipticity and near-UV spectra showing very large changes throughout (Fig. 2, A and B) (2, 5). In contrast, the far-UV spectrum of hD(131–136) antithrombin was distinct from either of the wild-type spectra and showed no change upon H5 binding (Fig. 2A). Similar behavior was found in the near-UV, where the non-heparin-bound state gave a spectrum distinct from either of the wild-type spectra, although closer to that of heparin-bound wild type, and changes upon binding H5 that were small relative to those caused by pentasaccharide binding to the WT control (Fig. 2B). In contrast to the large differences between hD(131–136) and WT antithrombin, the Y131L variant gave near-UV CD spectra that were very similar to WT in the absence of H5 and underwent similar changes to WT upon H5 binding (Fig. 2C).
FIGURE 2.
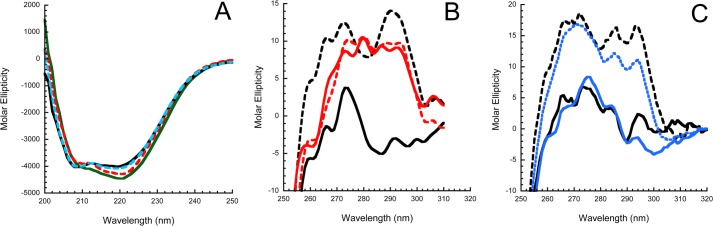
Near- and far-UV CD spectra of antithrombins. A, far-UV CD spectra of wild-type antithrombin alone (solid green line) and with 3 eq of H5 (red dashed line) and of hD(131–136) antithrombin alone (black solid line) and with 3 eq of heparin pentasaccharide (blue line). B, near-UV CD spectra of WT antithrombin alone (solid black line) and with 3 eq of H5 (dashed black line) and of hD(131–136) antithrombin alone (red line) and with 3 eq of H5 (red dashed line). C, near-UV CD spectra of WT antithrombin alone (solid black line) and with 3-fold excess H5 (dashed black line) and of Y131L antithrombin alone (solid blue line) and with 6-fold excess of H5 (dashed blue line).
For WT antithrombin, the aromatic region of the one-dimensional 1H NMR spectrum (6–8 ppm) showed good dispersion reflecting the extensive β-sheet and many buried perturbing aromatic side chains (Fig. 3A), as has been seen earlier in studies on plasma antithrombin (6–8). Because aromatic resonances have chemical shifts that depend exquisitely on the precise geometry relative to other proximal aromatic residues that perturb them, they are extremely sensitive reporters of conformational changes that affect the hydrophobic core. As documented previously, the conformational changes brought about by H5 binding are very extensive in the aromatic region and result in large magnitude difference spectra (Fig. 3A), which affect a major fraction of the aromatic side chains of the protein (7), suggesting that the heparin-induced changes alter the packing in the hydrophobic core. The corresponding spectra for hD(131–136) antithrombin differ in two major ways. First, the spectrum of hD(131–136) antithrombin in the absence of H5 is quite distinct from WT antithrombin (Fig. 3B). Second, although H5 binding does cause further spectral perturbations, the magnitude is very much smaller than for WT. These two basic differences imply that the mutations of residues 131–136 have already caused a major alteration in the hydrophobic core of the protein and that the additional changes brought about by heparin binding cause only minor conformational modifications that probably do not involve the core.
FIGURE 3.
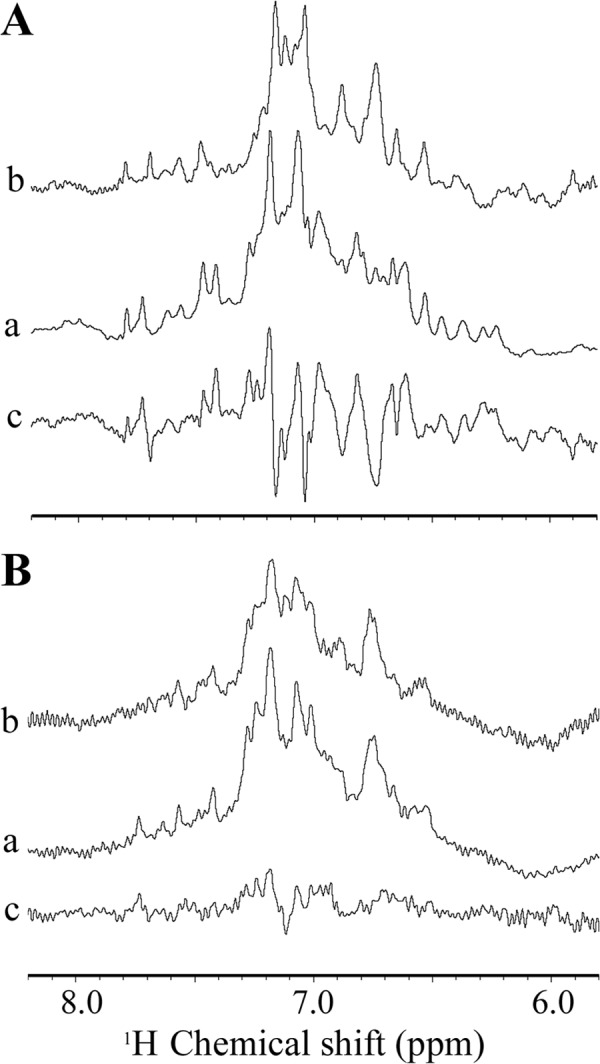
900 MHz 1H NMR spectra of antithrombins. A, wild-type antithrombin; B, hD(131–136) antithrombin. In each panel, spectrum a represents the heparin-free protein; spectrum b represents the heparin-complexed antithrombin, and trace c represents the difference spectrum of a and b. Only the aromatic region of the complete spectrum, which is extremely sensitive to conformation, is shown. H5 does not contribute resonances in the region shown.
Heparin-induced Fluorescence Changes
The hallmark for the presence of an inserted RCL hinge in antithrombin that is expelled by heparin binding is a basal tryptophan fluorescence that is increased 30–40% by heparin binding. This enhancement arises predominantly from Trp-225, which is very close to the buried P14 side chain, and from Trp-307 (4). To determine whether the helix D mutations had also resulted in expulsion of the RCL hinge in the absence of heparin, we recorded fluorescence emission spectra of the hD(131–136) variant in the absence and presence of H5, as well as for WT and the two variants with sub-sets of mutations.
For the hD(131–136) variant, the basal protein fluorescence was the same as for WT and gave a 23 ± 1% enhancement upon H5 binding, compared with 42 ± 1% enhancement for WT under the same conditions (Fig. 4). For the Y131L variant, the basal fluorescence was about 13% higher than for WT, but it showed a further increase of about 35% upon H5 binding, to give final fluorescence spectrum ∼8% higher than for H5-bound WT, whereas for the hD(132–136) variant the basal fluorescence was 3% lower than for WT, but it gave a 36% increase to a level ∼6% higher than that for the longer hD(131–136) (Fig. 4 and Table 2).
FIGURE 4.
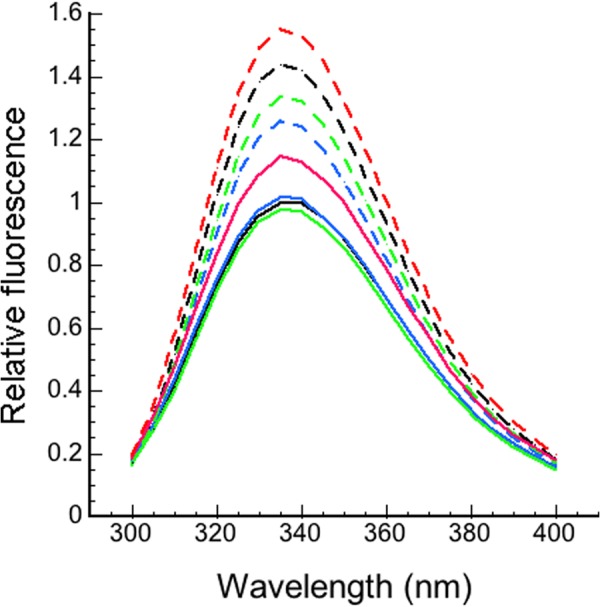
Fluorescence emission spectra of antithrombins. WT antithrombin without (solid black line) and with (dashed black line) 3 eq of H5, hD(131–136) variant antithrombin without (solid blue line) and with (dashed blue line) 3 eq of H5, hD(132–136) antithrombin without (solid red line) and with (dashed red line) 3 eq of H5, and Y131L antithrombin without (solid green line) and with (dashed green line) 3 eq of H5. Antithrombin concentration was 100 nm. Excitation was at 280 nm.
TABLE 2.
Fluorescence emission enhancements for WT and variant antithrombins
| −H5a | +H5 | % increase | |
|---|---|---|---|
| WT | 1.00 | 1.42 ± 0.01 | 42 ± 1 |
| hD(131–136) | 1.01 ± 0.01 | 1.24 ± 0.01 | 23 ± 1 |
| Y131L | 1.13 ± 0.01 | 1.53 ± 0.01 | 35 ± 1 |
| hD(132–136) | 0.97 ± 0.05 | 1.32 ± 0.03 | 36 ± 2 |
a Values are relative to WT set as 1.00.
Heparin Affinities Are Perturbed
High affinity pentasaccharide binds to antithrombin in two steps. In the first, it associates weakly, without major conformational change. Subsequently, major conformational changes occur, including extension of helix D, formation of helix P, and expulsion of the RCL hinge, which result in an overall 1000-fold tightening of the binding (25). Because none of the 131–136 mutations lies directly within the interaction site with H5, changes in heparin affinity resulting from these mutations are likely to reflect allosteric changes in antithrombin conformation. Furthermore, because the inserted residues of the RCL hinge are thought to stabilize the protein, RCL hinge expulsion in any of the variants should translate into a large increase in heparin affinity, as has been seen for the P14 S380E variant (26).
For the hD(131–136) and hD(132–136) variants, only a modest increase in affinity was observed at I = 0.45 (KD values of 55 ± 23 and 56 ± 17 nm, respectively, compared with 103 ± 17 nm for WT). Somewhat surprisingly, the less extensively modified Y131L bound more tightly (KD 12 ± 2 nm at I = 0.45).
DISCUSSION
Our rationale for examining the hD(131–136) variant was to test whether extension of helix D directly resulted in activation of antithrombin as an inhibitor of factors IXa and Xa. By changing the helix-forming potential of the region 131–136 from indifferent (Pα of 0.95) to very strongly helix-promoting (Pα of 1.47), we expected that, even in the absence of H5, the second-order rate constants for inhibition of factors IXa and Xa would be increased, whereas those of thrombin, for which H5-induced conformational changes do not normally give more than a 2-fold rate increase, would be almost unaltered. Not only were these expectations borne out, but the enhancements for inhibition of factors IXa and Xa were so large that the rate constants were similar to those of fully H5-activated WT antithrombin. Furthermore, H5 binding caused modest further increases in rate constants, such that the H5-bound hD(131–136) variant reacted with factors IXa and Xa about three times faster than H5-bound WT. These observations suggest that the mutations did induce helix extension and that such helix extension is responsible for most, if not all, of the rate enhancements normally seen in WT antithrombin only upon binding H5.
Although previous studies have suggested that the role of helix extension is simply to expel the RCL hinge and that it is the latter that is the sine qua non for activation, such a role does not fit with our results. Thus, the basal fluorescence of the hD(131–136) variant is indistinguishable from that of WT and is enhanced 23 ± 1% upon H5 binding (compared with 42 ± 1% observed here for WT). This suggests that the hinge of the RCL is buried similarly to WT and is expelled only upon H5 binding, with the altered fluorescence enhancement resulting from conformational alteration of the variant that is sensed in the H5-bound state. If the effect of the mutations had simply been to cause a major shift in the population of RCL-expelled species, one would have expected an increased fluorescence in the absence of heparin and a rate of inhibition of factors IXa and Xa in the presence of H5 the same as that for H5-bound WT, rather than the much higher rates that are observed. In addition, the equilibrium shift model predicts a very large increase in heparin affinity, such as was found for a hinge-expelled P14 S380E antithrombin variant (26), where the KD value was found to be decreased ∼100-fold,4 rather than the ∼2-fold decrease in the KD values observed here for the hD(131–136) variant and the somewhat larger change (∼10-fold) for the less activated Y131L variant.
Other pieces of our data are also not explicable by a simple shift in equilibrium between hinge-inserted and hinge-expelled conformations. These include the CD and NMR spectra of the hD(131–136) variant, which both imply a different conformation for the variant compared with WT and only minimal further changes induced by H5 binding. In addition, the major increases in SI values for inhibition of factor Xa and factor IXa by the hD(131–136) variant argue for a distinct novel conformation.
Overall, these findings suggest that the hD(131–136) variant is highly activated as a result of conformational changes induced by helix extension and by the Y131L mutation, which are largely independent of RCL hinge expulsion (Fig. 5). This requires that the hD mutations have decoupled RCL hinge expulsion from these conformational changes. Although this may seem surprising given the importance attached by many to the role of RCL expulsion in activation, it is in keeping with results from other variants where such decoupling is more obviously present. These variants include one in which residues 134–136 were removed (10) and another in which Lys-133 was mutated to proline (11). Both had properties consistent with an inability to expel the RCL upon H5 binding, yet with the ability of H5 to cause significant rate enhancements for factor Xa.
FIGURE 5.
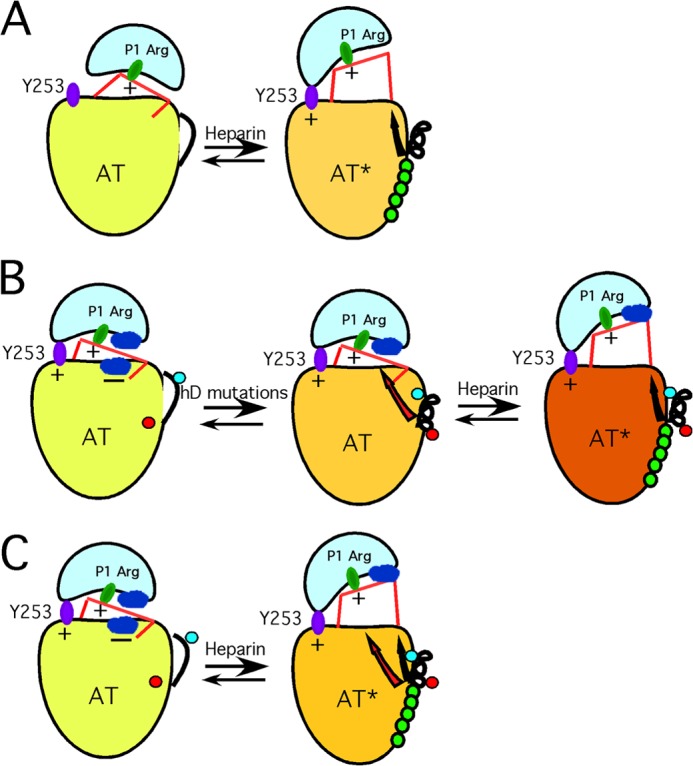
Proposed new model of antithrombin activation. A, schematic of the traditional view of how H5 results in activation of antithrombin as a factor IXa and factor Xa inhibitor. The low activity state (left), with inserted RCL hinge, reacts poorly because of the inability of the proteinase to dock with an exosite represented by Tyr-253. The sole mechanistic role of H5 binding is to extend helix D, thereby expelling the RCL hinge and allowing the proteinase (IXa or Xa) to engage the Tyr-253 exosite. B, schematic of how we envision the 131–136 mutations cause activation without the need for RCL hinge expulsion. The left-hand structure represents an altered view of how WT antithrombin is configured in a low activity state, based on evidence presented in an earlier publication (36). Here, low activity results not from an inability of the proteinase to engage the Tyr-253 exosite but from nonoptimal interactions at the interface between proteinase and the antithrombin surface (shown as blue patches on each protein and signified as unfavorable by a minus sign). Introduction of the 131–136 mutations (middle structure) leads to the following: (i) change in conformation of Tyr-131 (red dot) from buried to exposed, and (ii) extension of helix D such that the previously exposed Ala-134 (blue dot) is buried in the protein core. Together, these changes give rise to a long range conformational change (red arrow) sensed at the site of the unfavorable interaction between antithrombin and the proteinase, thereby giving rise to the massive activation that normally requires H5 binding. Because RCL hinge expulsion has been decoupled by introduction of the 131–136 mutations, H5 binding is required to give the final RCL hinge expulsion (black arrow) and results in a final small augmentation in rate (right structure). C, represents our new proposed mechanism of how H5 activates WT antithrombin, based on the findings of the 131–136 and related variants. Here, because RCL hinge expulsion and the other conformational changes are no longer decoupled, both sets of conformational changes are induced by H5 binding. Again, the reduction in unfavorable interactions (red arrow) results in most of the rate enhancement, although the RCL hinge expulsion (black arrow) may give additional small enhancement by allowing the proteinase to move further away from the antithrombin surface.
If our novel hD(131–136) variant is indeed highly activated against factors IXa and Xa without RCL hinge expulsion, the question is how it accomplishes this. Here, both the K133P and Δ134–136 variants, as well as the Y131L and hD(132–136) variants, shed light on the question. For the former two variants it seems clear that blocking extension of helix D beyond residue 133 prevents the RCL hinge from being expelled, yet still permits significant, although not full, H5-induced activation against factor Xa. Because different ways were used to bring about blocking helix extension (shortening the polypeptide or introducing a helix-blocking residue), this requires that the subsequent, similar H5-induced rate enhancements must arise from residues preceding residue 133. The potential importance of Tyr-131 has been suggested by Bock and co-workers (12). They noted that the Tyr-131 side chain has intimate contacts with several hydrophobic core residues in the absence of H5 (Leu-130 and Leu-140), but undergoes a large conformational change upon H5 binding such that the tyrosine side chain flips to an outward-pointing conformation (Fig. 1). This alters the packing of the core in the vicinity of where the Tyr-131 side chain had been and could thus be the means of long range transmission of conformational change to the proteinase-binding site. Given the magnitudes of the second-order rate constants for the Y131L variant in the absence of H5 and the K133P and Δ134–136 variants in the presence of H5, such altered packing might contribute ∼11–22-fold to rate enhancement against factor Xa and ∼7-fold against factor IXa. Significantly, Leu-130 and Leu-140 are conserved in all sequenced antithrombins (with the exception of a Val-140 in guinea pig) (27), although neither of these residues is one of the “conserved core 51 hydrophobics” present in most serpins (28). Similarly, Tyr-131 is conserved in all antithrombins (except in frog, where it is Phe) but is variable in other serpins (27, 28). In the Y131L variant the replacement of Tyr with Leu might give a side chain that preferentially adopts the out-pointing conformation and so mimics the activation resulting from Tyr-131 in the H5-bound state. Here, it is perhaps pertinent that the heparin affinity of the Y131L is enhanced ∼10-fold, suggesting that in WT one of the critical energy-requiring aspects of H5 activation is extraction of the Tyr-131 side chain, which is not necessary in the Y131L variant. In contrast, the mutations of residues 132–136 alone have only a 2-fold effect on heparin affinity, suggesting that helix extension in this region is not linked to H5 binding and in WT may be triggered by the H5 binding-induced release of the Tyr-131 side chain. One complication with this explanation is that the presence of both Y131L and the 132–136 mutations (in hD(131–136)) results in only a 2-fold change in heparin affinity. This, however, may be linked to the decoupling of RCL hinge expulsion for these variants (see below).
Clearly, however, such changes to the side chain of Tyr-131, either through mutation to Leu or through H5 binding, are insufficient to account for the much larger activation seen for WT upon H5 binding or, here, for the hD(131–136) variant in the absence of H5. Given that the hD(132–136) variant, in the absence of H5, also shows large activations against both factor Xa and factor IXa, compared with WT, this suggests that there is a second component to the conformational activation resulting separately from the residues 132–136. Because the hD(132–136) variant shows activations as an inhibitor of factor Xa and factor IXa of ∼18 and ∼11-fold, respectively, compared with WT, conformational changes in this region, brought about either by helix-inducing mutations (as in the hD(132–136) or hD(131–136) variants) or through H5 binding to WT and induction of helix in this region, may thus contribute such additional rate enhancements, through transmission through the core to the proteinase-binding site.
The likely mediator of such altered conformational interaction in the 132–136 region is Ala-134. This residue has a side chain pointing out into solution in WT antithrombin, but, as a result of H5-induced extension of the D helix, makes contacts with the body of antithrombin in the H5-bound state, being drawn into close proximity to the core residues Leu-140 and Ile-279. Significantly, in both the hD(131–136) and hD(132–136) variants, residue 134 is Ala, as in WT. In the two variants, the effect of the flanking helix-promoting residues would then be to induce helix around Ala-134 even in the absence of H5, although in WT H5 binding itself induces helix formation in this region. Both the Ala-134 and the contact residues Leu-140 and Ile-279 show high conservation in antithrombins (27) but not in other serpins (28).
Such a role for Ala-134 would also account for the altered properties of the antithrombin deletion variants and the K133P variant in which either removal of Ala-134 and two or three additional residues or altering the ability of Ala-134 to adopt the same conformation relative to the antithrombin body might eliminate or diminish the conformational activation contribution of Ala-134, while still leaving the contribution from Tyr-131 upon H5 binding. This would leave the bulk of the conformational activation of WT antithrombin arising in roughly equal measures from the following: (i) removal of the Tyr-131 side chain from close association with Leu-130, Leu-140, and Ser-142, and (ii) formation of new contacts between the side chain of Ala-134 with Leu-140 and Ile-279. Each of these alterations in packing with core residues could then result in long range conformational alterations, mediated through the hydrophobic core, that are finally sensed at the critical docking site between antithrombin and proteinase and so give allosteric rate enhancement. Whether the transmitted changes result in alteration in the ability of the proteinase to interact with the known exosites (17, 29–31) or other changes in surface electrostatics is not yet known. Finally, from the roughly 3-fold additional rate enhancements (for both factors IXa and Xa) seen with the hD(131–136) variant upon H5 binding and coincident with large fluorescence enhancements indicative of, finally, RCL hinge expulsion, an additional third minor role can be ascribed to such hinge expulsion in the activation mechanism.
Two remaining, and possibly related, questions are as follows. (i) Why, if the conformation of H5-bound hD(131–136) is not identical to that of H5-bound WT antithrombin (see Figs. 2–4), is hD(131–136) so successfully activated against factors Xa and IXa? (ii) How has a likely successful attempt to induce helix D extension failed to result in RCL hinge expulsion and large increase in heparin affinity (true for both hD(131–136) and hD(132–136))? Although one can only speculate at this point as to how this decoupling might have occurred and why these two variants are not identical to H5-bound WT, it is noteworthy that, although the side chain of Lys-133 points into solution in heparin-free antithrombin (32), it points inward to make a salt bridge with Glu-414 in all H5-bound antithrombin complexes examined in which the RCL hinge has been expelled, including antithrombin-H5 (32), antithrombin-H5-anhydrothrombin (33), antithrombin-H5-S195A factor Xa (30), and antithrombin-H5-S195A factor IXa (31). In these complexes the lysine side chain is further flanked by another acidic residue (Asp-278). Because each of the hD variants contains a glutamate at position 133, it is likely that, although the various mutations do induce helix extension, the new helical extension does not adopt exactly the same orientation as in H5-WT, because that would juxtapose an acidic Glu-133 with Glu-414 and Asp-278. In this way Ala-134 might make even more beneficial contacts with the core in the hD variants than in WT (recall the higher ultimate activations for both factor Xa and factor IXa inhibition). Finally, with regard to the decoupling of activation from RCL hinge expulsion, a potentially critical change between H5-free and H5-bound antithrombin might be a salt bridge between the side chains of Glu-374 in strand s5A and Lys-222 in strand s3A that forms in the latter, but is absent in the former. In heparin-free antithrombin, strands s5A and s3A are held apart by the partially inserted RCL hinge such that the backbone Cα separation between Glu-374 and Lys-222 is 9.4 Å. In the H5-bound state of WT antithrombin, expulsion of the RCL hinge allows the strands to come together and hydrogen bond, while the side chains form a new salt bridge. Between Lys-222 and the cluster of Lys-133, Glu-414, and Asp-278 lie additional charged residues engaged in salt bridge interactions that switch between the H5-free and H5-bound states (34). It is therefore quite possible that in WT antithrombin the route to H5-induced RCL hinge expulsion is through alteration in this salt bridge network that promotes the interaction of the side chains of Glu-374 and Lys-222 and thereby squeezes out the intervening hinge. In the hD variants examined here, the replacement of Lys-133 with Glu might, in the absence of heparin, fail to alter the charge network appropriately and so fail to expel the RCL hinge until H5 is bound. This may also be the basis for the surprising lack of large change in H5 affinities for both variants that contain Glu-133. Consistent with important roles for each of these charged residues (Lys-133, Lys-222, Asp-278, Glu-374, as well as the additional intervening residues Lys-139 and Glu-195) is their conservation in almost all antithrombins (27) and yet extensive variability in other serpins (28).
This work was supported, in whole or in part, by National Institutes of Health Grants R37 HL49234 (to P. G. W. G.) and R37 HL 39888 (to S. T. O.).
R. Roth and S. Olson, unpublished results.
- H5
- high affinity synthetic heparin pentasaccharide
- RCL
- reactive center loop
- SI
- stoichiometry of inhibition.
REFERENCES
- 1. Olson S. T., Richard B., Izaguirre G., Schedin-Weiss S., Gettins P. G. (2010) Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin superfamily proteinase inhibitors. Biochimie 92, 1587–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Björk I., Danielsson Å., Fish W. W., Larsson K., Lieden K., Nordenman B. (1979) in The Physiological Inhibitors of Coagulation and Fibrinolysis (Collen D., Wiman B., Verstraete M., eds) pp. 67–84, Elsevier/North-Holland, Amsterdam [Google Scholar]
- 3. Olson S. T., Shore J. D. (1981) Binding of high affinity heparin to antithrombin III. Characterization of the protein fluorescence enhancement. J. Biol. Chem. 256, 11065–11072 [PubMed] [Google Scholar]
- 4. Meagher J. L., Beechem J. M., Olson S. T., Gettins P. G. (1998) Deconvolution of the fluorescence emission spectrum of human antithrombin and identification of the tryptophan residues that are responsive to heparin binding. J. Biol. Chem. 273, 23283–23289 [DOI] [PubMed] [Google Scholar]
- 5. Stone A. L., Beeler D., Oosta G., Rosenberg R. D. (1982) Circular dichroism spectroscopy of heparin-antithrombin interactions. Proc. Natl. Acad. Sci. U.S.A. 79, 7190–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horne A., Gettins P. (1992) 1H NMR spectroscopic studies on the interactions between human plasma antithrombin III and defined low molecular weight heparin fragments. Biochemistry 31, 2286–2294 [DOI] [PubMed] [Google Scholar]
- 7. Gettins P., Choay J. (1989) Examination by 1H NMR spectroscopy of the binding of a synthetic, high affinity heparin pentasaccharide to human antithrombin III. Carbohydr. Res. 185, 69–76 [DOI] [PubMed] [Google Scholar]
- 8. Gettins P. (1987) Antithrombin III and its interaction with heparin. Comparison of the human, bovine, and porcine proteins by 1H NMR spectroscopy. Biochemistry 26, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 9. van Boeckel C. A., Grootenhuis P. D., Visser A. (1994) A mechanism for heparin-induced potentiation of antithrombin III. Nat. Struct. Biol. 1, 423–425 [DOI] [PubMed] [Google Scholar]
- 10. Meagher J. L., Olson S. T., Gettins P. G. (2000) Critical role of the linker region between helix D and strand 2A in heparin activation of antithrombin. J. Biol. Chem. 275, 2698–2704 [DOI] [PubMed] [Google Scholar]
- 11. Belzar K. J., Zhou A., Carrell R. W., Gettins P. G., Huntington J. A. (2002) Helix D elongation and allosteric activation of antithrombin. J. Biol. Chem. 277, 8551–8558 [DOI] [PubMed] [Google Scholar]
- 12. dela Cruz R. G., Jairajpuri M. A., Bock S. C. (2006) Disruption of a tight cluster surrounding tyrosine 131 in the native conformation of antithrombin III activates it for factor Xa inhibition. J. Biol. Chem. 281, 31668–31676 [DOI] [PubMed] [Google Scholar]
- 13. Ngai P. K., Chang J.-Y. (1991) A novel one-step purification of human α-thrombin after direct activation of crude prothrombin enriched from plasma. Biochem. J. 280, 805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olson S. T., Björk I. (1991) Predominant contribution of surface approximation to the mechanism of heparin acceleration of the antithrombin-thrombin reaction. Elucidation from salt concentration effects. J. Biol. Chem. 266, 6353–6364 [PubMed] [Google Scholar]
- 15. Miletich J. P., Broze G. J., Jr., Majerus P. W. (1980) The synthesis of sulfated dextran beads for isolation of human plasma coagulation factors II, IX, and X. Anal. Biochem. 105, 304–310 [DOI] [PubMed] [Google Scholar]
- 16. Chuang Y.-J., Swanson R., Raja S. M., Bock S. C., Olson S. T. (2001) The antithrombin P1 residue is important for target proteinase specificity but not for heparin activation of the serpin. Characterization of P1 antithrombin variants with altered proteinase specificity but normal heparin activation. Biochemistry 40, 6670–6679 [DOI] [PubMed] [Google Scholar]
- 17. Izaguirre G., Olson S. T. (2006) Residues Tyr253 and Glu255 in strand 3 of β-sheet C of antithrombin are key determinants of an exosite made accessible by heparin activation to promote rapid inhibition of factors Xa and IXa. J. Biol. Chem. 281, 13424–13432 [DOI] [PubMed] [Google Scholar]
- 18. Nordenman B., Nyström C., Björk I. (1977) The size and shape of human and bovine antithrombin III. Eur. J. Biochem. 78, 195–203 [DOI] [PubMed] [Google Scholar]
- 19. Chou P. Y., Fasman G. D. (1974) Conformational parameters for amino acids in helix, β sheet and random coil regions calculated for proteins. Biochemistry 13, 211–222 [DOI] [PubMed] [Google Scholar]
- 20. Meagher J. L., Huntington J. A., Fan B., Gettins P. G. (1996) Role of arginine 132 and lysine 133 in heparin binding to, and activation of, antithrombin. J. Biol. Chem. 271, 29353–29358 [DOI] [PubMed] [Google Scholar]
- 21. Turko I. V., Fan B., Gettins P. G. (1993) Carbohydrate isoforms of antithrombin variant N135Q with different heparin affinities. FEBS Lett. 335, 9–12 [DOI] [PubMed] [Google Scholar]
- 22. Ersdal-Badju E., Lu A., Peng X., Picard V., Zendehrouh P., Turk B., Björk I., Olson S. T., Bock S. C. (1995) Elimination of glycosylation heterogeneity affecting heparin affinity of recombinant human antithrombin III by expression of a β-like variant in baculovirus-infected insect cells. Biochem. J. 310, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arocas V., Turk B., Bock S. C., Olson S. T., Björk I. (2000) The region of antithrombin interacting with full-length heparin chains outside the high-affinity pentasaccharide sequence extends to Lys-136 but not to Lys-139. Biochemistry 39, 8512–8518 [DOI] [PubMed] [Google Scholar]
- 24. Rogers D. M., Hirst J. D. (2004) First-principles calculation of protein circular dichroism in the near ultraviolet. Biochemistry 43, 11092–11102 [DOI] [PubMed] [Google Scholar]
- 25. Olson S. T., Björk I., Sheffer R., Craig P. A., Shore J. D., Choay J. (1992) Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J. Biol. Chem. 267, 12528–12538 [PubMed] [Google Scholar]
- 26. Futamura A., Gettins P. G. (2000) Serine 380 (P14) → glutamate mutation activates antithrombin as an inhibitor of factor Xa. J. Biol. Chem. 275, 4092–4098 [DOI] [PubMed] [Google Scholar]
- 27. Backovic M., Gettins P. G. (2002) Insights into the function of antithrombin from an expanded database of sequences. J. Proteome Res. 1, 367–373 [DOI] [PubMed] [Google Scholar]
- 28. Irving J. A., Pike R. N., Lesk A. M., Whisstock J. C. (2000) Phylogeny of the serpin superfamily: Implications of patterns of amino acid conservation for structure and function. Genome Res. 10, 1845–1864 [DOI] [PubMed] [Google Scholar]
- 29. Izaguirre G., Zhang W., Swanson R., Bedsted T., Olson S. T. (2003) Localization of an antithrombin exosite that promotes rapid inhibition of factors Xa and IXa on heparin activation of the serpin. J. Biol. Chem. 278, 51433–51440 [DOI] [PubMed] [Google Scholar]
- 30. Johnson D. J., Li W., Adams T. E., Huntington J. A. (2006) Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 25, 2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson D. J., Langdown J., Huntington J. A. (2010) Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7Å structure of the ternary complex. Proc. Natl. Acad. Sci. U.S.A. 107, 645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Skinner R., Abrahams J.-P., Whisstock J. C., Lesk A. M., Carrell R. W., Wardell M. R. (1997) The 2.6Å structure of antithrombin indicates a conformational change at the heparin binding site. J. Mol. Biol. 266, 601–609 [DOI] [PubMed] [Google Scholar]
- 33. Dementiev A., Petitou M., Herbert J. M., Gettins P. G. (2004) The ternary complex of antithrombin-anhydrothrombin-heparin reveals the basis of inhibitor specificity. Nat. Struct. Mol. Biol. 11, 863–867 [DOI] [PubMed] [Google Scholar]
- 34. Whisstock J. C., Pike R. N., Jin L., Skinner R., Pei X. Y., Carrell R. W., Lesk A. M. (2000) Conformational changes in serpins: II. The mechanism of activation of antithrombin by heparin. J. Mol. Biol. 301, 1287–1305 [DOI] [PubMed] [Google Scholar]
- 35. McCoy A. J., Pei X. Y., Skinner R., Abrahams J. P., Carrell R. W. (2003) Structure of β-antithrombin and the effect of glycosylation on antithrombin's heparin affinity and activity. J. Mol. Biol. 326, 823–833 [DOI] [PubMed] [Google Scholar]
- 36. Gettins P. G., Olson S. T. (2009) Activation of antithrombin as a factor IXa and Xa inhibitor involves mitigation of repression rather than positive enhancement. FEBS Lett. 583, 3397–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]