

Background: AtAMY3 is an α-amylase implicated in leaf starch degradation.
Results: AtAMY3 releases small linear and branched glucans from starch under neutral-alkaline conditions and is subject to reductive activation by thioredoxins.
Conclusion: AtAMY3 is adapted for activity in the chloroplast and is a redox-regulated enzyme.
Significance: The unique properties of AtAMY3 among α-amylases provide new insight into the regulation of starch degradation in vivo.
Keywords: Arabidopsis, Carbohydrate Metabolism, Plant Biochemistry, Redox Regulation, Thioredoxin, α-Amylase, Amylopectin, Starch
Abstract
α-Amylases are glucan hydrolases that cleave α-1,4-glucosidic bonds in starch. In vascular plants, α-amylases can be classified into three subfamilies. Arabidopsis has one member of each subfamily. Among them, only AtAMY3 is localized in the chloroplast. We expressed and purified AtAMY3 from Escherichia coli and carried out a biochemical characterization of the protein to find factors that regulate its activity. Recombinant AtAMY3 was active toward both insoluble starch granules and soluble substrates, with a strong preference for β-limit dextrin over amylopectin. Activity was shown to be dependent on a conserved aspartic acid residue (Asp666), identified as the catalytic nucleophile in other plant α-amylases such as the barley AMY1. AtAMY3 released small linear and branched glucans from Arabidopsis starch granules, and the proportion of branched glucans increased after the predigestion of starch with a β-amylase. Optimal rates of starch digestion in vitro was achieved when both AtAMY3 and β-amylase activities were present, suggesting that the two enzymes work synergistically at the granule surface. We also found that AtAMY3 has unique properties among other characterized plant α-amylases, with a pH optimum of 7.5–8, appropriate for activity in the chloroplast stroma. AtAMY3 is also redox-regulated, and the inactive oxidized form of AtAMY3 could be reactivated by reduced thioredoxins. Site-directed mutagenesis combined with mass spectrometry analysis showed that a disulfide bridge between Cys499 and Cys587 is central to this regulation. This work provides new insights into how α-amylase activity may be regulated in the chloroplast.
Introduction
α-Amylases (EC 3.2.1.1) are endo-acting enzymes that specifically hydrolyze α-1,4 internal glucoside linkages in starch and related carbohydrates. α-Amylases occur widely among higher plants, animals, bacteria, and fungi (1, 2). In vascular plants, α-amylases can be classified into three subfamilies according to cellular localization and gene structure (3). Members of each family were shown to be present in representative monocotyledons, dicotyledons, and gymnosperms, and all share a conserved α-amylase active site at the C terminus of the protein. Family one α-amylases are characterized by having a signal peptide that targets the protein to the secretory pathway. Cereal grain α-amylases are classified within this family, and they are mainly active in the endosperm during mobilization of starch. Family two α-amylases have no predicted targeting peptide and are therefore thought to localize to the cytoplasm. The function of the members of this family remains largely unknown. Family three α-amylases are characterized by a large N-terminal extension, typically 400–500 amino acids in length, which contains a predicted chloroplast transit peptide and tandem carbohydrate-binding modules. Members of this family have been shown to participate in leaf starch breakdown (4).
Much work on plant α-amylases has focused on enzymes from the cereals due to their pivotal role in the degradation of endosperm starch upon seed germination. Barley (Hordeum vulgare L.) α-amylase 1 (HvAMY1) and HvAMY2 (both family one α-amylases) are among the most thoroughly described plant α-amylases and represent the only ones for which crystal structures are available (5). Although the two isozymes share 80% sequence identity, they differ in activity, stability, and natural abundance. Toward starch granules, HvAMY1 has higher activity than HvAMY2, although the latter has a higher turnover rate on soluble substrates (5). Antisense suppression of an AMY1-type α-amylase in the rice endosperm leads to delayed germination, confirming its importance in starch mobilization (6).
The importance of α-amylases in starch degradation in the chloroplasts and amyloplasts of living cells is not yet fully understood. Biochemical studies and analyses of protein sequences indicated that α-amylase is present inside plastids of some plant species (3, 7, 8). In rice, there are at least two type one α-amylase isoforms targeted to the leaf chloroplast via a Golgi-mediated secretory pathway. Notably, suppressed expression of one of the two isoforms (AMYI-1) resulted in increased starch accumulation in the young leaf tissue, suggesting that it may be involved in transitory starch degradation (6, 9).
In Arabidopsis, three genes encode α-amylase-like proteins (10). Of these genes, only α-amylase isozyme 3 from Arabidopsis thaliana (AtAMY3) has a chloroplast transit peptide, and previous studies have confirmed its localization in the chloroplast (10, 11). Compared with HvAMY1 and other plant α-amylases, AtAMY3 has a unique structure. Besides a highly conserved C-terminal catalytic domain, AtAMY3 has two N-terminal starch-binding domains from the carbohydrate-binding module family 45 (CBM45 (12)) that would enable interaction with the starch granule surface (10, 11). Interestingly, α-glucan water dikinase (GWD),4 which phosphorylates starch, also possesses two N-terminal CBM45 domains with high sequence similarity to those of AtAMY3 (11, 13). Despite being an active α-amylase, Arabidopsis plants lacking AMY3 degrade starch normally under standard growth conditions (4, 10). However, genetic analysis of mutant combinations has shown that there is a degree of redundancy in the pathway (14), such as when other starch-degrading enzymes are missing, AtAMY3 is required for the degradation of starch (4, 15, 16). In mutant plants lacking the glucan phosphatase STARCH-EXCESS4 (SEX4), AtAMY3 contributes to the degradation of starch at night and releases soluble phospho-oligosaccharides from the starch granule surface (4). Similarly, in plants lacking the two debranching enzymes, isoamylase 3 and limit dextrinase, AtAMY3 is induced and is responsible for releasing small branched malto-oligosaccharides from the granule surface (16, 17). These data support the hypothesis that AtAMY3 is part of the reaction network of starch degradation in the Arabidopsis chloroplast. To better understand the role of AtAMY3 in starch degradation, we took an in vitro approach to unravel the functional regulation of AtAMY3 and substrate specificity. We show that AtAMY3 is stimulated by β-amylase activity in vitro and suggest that both these activities can cooperate to achieve efficient starch degradation in the chloroplast. We also show that AtAMY3 is unique among other previously described α-amylases in terms of its pH optimum and redox regulation through a regulatory disulfide bridge. The significance of these findings in the context of Arabidopsis starch metabolism is discussed.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of AtAMY3 Recombinant Proteins
An AtAMY3 cDNA clone (At1g69830, RAFL08-08-F08) was obtained from the RIKEN Bioresource Center. The region encoding the full-length AtAMY3 protein (lacking the predicted chloroplast transit peptide; amino acids 1–55) was amplified with BamHI restriction sites using the following primers: 5′-CGCGGATCCATGAACAAAAGTCCCGTCGCCATTC-3′ and 5′-CGCGGATCCTTAAGATGTTTCCCACACCTTGTA-3′ and inserted into the pProEX HTb vector (Invitrogen) in-frame with the N-terminal His tag. Point mutations in the AtAMY3 gene were generated with the QuikChange site-directed mutagenesis kit (Agilent Technologies, Basel, Switzerland) according to the manufacturer's instructions. The region encoding the full-length AtBAM1 protein (lacking the predicted chloroplast transit peptide; amino acids 1–90) was amplified with EcoRI and SalI restriction sites using the following primers: 5′-CGGAATTCTATAGAGAAGGAGGGATTGG-3′ and 5′-CGGTCGACCTAGTGAGTGAGAGCCACTG-3′ and inserted into the pET28a+ vector (Invitrogen) in-frame with the N-terminal His tag. Recombinant proteins were expressed in Escherichia coli BL21 (DE3) CodonPlus cells (Stratagene, Basel, Switzerland) and purified from the lysate using Ni2+-nitrilotriacetic acid-agarose affinity chromatography as described previously (18, 19). The fractions containing AtAMY3 were pooled and stored in 50 mm Tris-HCl, pH 8, 10% (w/v) glycerol, and 2 mm DTT. Protein concentration was determined using the Bradford assay reagent (Bio-Rad).
α-Amylase Activity Assays
AtAMY3 amylolytic activity was assayed against the labeled substrate and blocked p-nitrophenyl maltoheptaoside (BPNP-G7; Ceralpha Method Assay; Megazyme, Bray, Ireland) (20). Premixed BPNP-G7 and thermostable α-glucosidase were purchased as the amylase high pH range (HR) reagent. Recombinant AtAMY3 (10 μg) was dissolved in 60 μl of 100 mm Tricine/NaOH, pH 7.9. The assay was started by adding the amylase HR reagent (60 μl) and incubated for 1 h (unless specified otherwise) at 37 °C. Reactions were stopped by adding 900 μl of 1% (w/v) tri-sodium phosphate, pH 11. Released para-nitrophenol was quantified spectrophotometrically (400 nm, extinction coefficient of 18.1 liters mmol−1 cm−1). For HvAMY1, the same assay was used but was conducted in 25 mm sodium acetate, pH 5.5, 5 mm CaCl2, using 10 ng of protein assayed for 20 min. The reduced amount of protein and shorter incubation time compensated for the higher specific activity of HvAMY1 as compared with AtAMY3.
To assay the AtAMY3 amylolytic activity on glucan substrates, purified recombinant protein (1 μg) was incubated at 25 °C in 200 μl of 50 mm HEPES-KOH, pH 8, 1 mm MgCl2, 5 mm DTT, with 1.5 mg of amylopectin (Sigma) or β-limit dextrin (Megazyme) predissolved in water by heating. Reactions were stopped after 1 h by adding 200 μl of 0.5 m NaOH. Blank digests were produced by substituting the enzyme with water. The MBTH (3-methyl-2-benzothiazolinone hydrazone) method was used to measure the reducing ends released during the digest (21). Briefly, 100 μl of the stopped reaction was mixed with 50 μl of MBTH reagent. The samples were heated at 80 °C for 15 min, after which 100 μl of 0.5% (w/v) (FeNH4(SO4)2)·12H2O, 0.5% (w/v) sulfamic acid, 0.5 m HCl was added. The samples were cooled to room temperature, and absorbance at 620 nm was measured. The amount of reducing ends released was determined against a standard curve consisting of 0–20 nmol of glucose.
For activity measurements against starch granules, starch was purified from whole rosettes of wild-type Arabidopsis plants as described previously (18). The dried granules were prehydrated in water for 1 h prior to the assay. An equivalent to 1.5 mg dry weight of granules was digested with the indicated amount of AtAMY3 and/or AtBAM1 in 100 mm Tricine/NaOH, pH 7.9, 5 mm DTT at room temperature with end-over-end mixing on a spinning wheel. The reaction was terminated after the indicated time points by pelleting the starch granules, and the supernatant was heated at 95 °C for 10 min. Soluble glucans released into the supernatant were quantified either using the MBTH method described above or by quantifying glucose equivalents after acid hydrolysis as follows. The supernatant was mixed with an equal volume of 2 m HCl and heated at 95 °C for 2 h. An appropriate volume of 1 m NaOH was added to neutralize the reaction (between pH 6 and 7). Glucose was quantified enzymatically using hexokinase and glucose-6-phosphate dehydrogenase as described previously (22).
Native PAGE and Native PAGE Blotting
Native affinity electrophoresis was performed on gels containing 7.5% (w/v) acrylamide, 9% (w/v) glycerol, 375 mm Tris-HCl, pH 8.8, and 0.1% (w/v) potato amylopectin or β-limit dextrin. The stacking gel contained 3.75% (w/v) acrylamide, 63 mm Tris-HCl, pH 6.8, and 0.1% (w/v) potato amylopectin or β-limit dextrin. Recombinant proteins in 50 mm Tris-HCl, pH 6.8, 3% (v/v) glycerol, 0.005% (w/v)bromophenol blue were loaded onto the gel.
Total soluble proteins were extracted from leaves of 4-week-old Arabidopsis plants by homogenizing in 100 mm MOPS, pH 6.8, 1 mm EDTA, 1 mm DTT, 10% (v/v) ethylene glycol, 1× Complete Protease Inhibitor Mixture (Roche Applied Science). Proteins in loading buffer (final concentrations: 5% (v/v) glycerol, 0.005% (v/v) bromphenol blue; 30 μg per lane) were loaded onto the gel and separated by electrophoresis. For a better separation of crude extracts, we added to the gel 5 mm MgCl2 and to the running buffer 2 mm MgCl2. After electrophoresis, gels were incubated for 2 h in 100 mm Tris-HCl, pH 8, 1 mm CaCl2, 1 mm MgCl2, 5 mm DTT, at 25 °C. Gels were stained in Lugol solution (Sigma).
To determine activity under reducing and oxidizing conditions, purified AtAMY3 was reduced or oxidized prior to electrophoresis as follows: purified protein (2 μg) was mixed with 50 mm HEPES-KOH, pH 8, 1 mm MgCl2, and either 5 mm DTT or 25 μm CuCl2 for reducing and oxidizing conditions, respectively. After incubation for 1 h at 25 °C, reactions were mixed with native PAGE loading buffer, and 1 μg of protein was loaded onto the gels. Reduced and oxidized samples were loaded on separate gels, and electrophoresis was carried out as described above. Gels containing the reduced protein were incubated in reducing incubation medium (described above). Gels containing the oxidized protein were incubated in an oxidizing medium containing 100 mm Tris-HCl, pH 8, 1 mm CaCl2, 1 mm MgCl2, 100 μm CuCl2. Gels were stained in Lugol solution. For immunodetection of AtAMY3, native gels were immersed in 2% SDS for 1 h, electroblotted onto polyvinylidene difluoride (PVDF) membrane, and probed with an antibody raised against AtAMY3 (10) or the commercial penta-His antibody (Qiagen).
Analysis of AtAMY3 Degradation Products by HPAEC-PAD
Starch was purified from whole rosettes of wild-type Arabidopsis plants as described previously (18). Prehydrated starch granules (1.5 mg dry weight) were digested at 25 °C with recombinant AtAMY3 (10 μg) or HvAMY1 (50 ng). At specified times, the starch was pelleted, and the supernatant was heated to 95 °C for 15 min to inactivate the enzymes. The soluble degradation products in the supernatant were analyzed by HPAEC-PAD. To debranch glucans, the supernatants were treated with isoamylase from Pseudomonas sp. (>500 units; Sigma) and pullulanase M1 from Klebsiella planticola (0.54 units; Megazyme) for 2 h at 37 °C in 50 mm sodium acetate, pH 4.8. For the β-amylase digest of degradation products, the combined supernatants were treated with barley β-amylase (180 units; Megazyme) for 2 h at 37 °C in 200 mm MES-KOH, 10 mm DTT, pH 6. Prior to HPAEC-PAD, samples were purified on sequential columns of Dowex 50W and Dowex 1 (Sigma) as described previously (4). The degradation products were separated on a CarboPac PA200 (3 × 250 mm) column (Dionex) following these gradient conditions: eluent A is 100 mm NaOH; eluent B is 150 mm NaOH, 500 mm sodium acetate: 0–13 min, a linear gradient from 95% A and 5% B to 60% A and 40% B; 13–50 min, a linear gradient to 15% A and 85% B; 50–70 min, step to 95% A and 5% B.
For predigestion of starch granules with β-amylase, an equivalent of 1.5 mg (dry weight) of prehydrated Arabidopsis starch granules was digested for 2 h at room temperature with recombinant AtBAM1 (7.5 μg) in 100 mm Tricine/NaOH, pH 7.9, 5 mm DTT. The starch was washed five times with 100 mm Tricine/NaOH, pH 7.9, 2% (w/v) SDS, followed by five washes in water. The starch was then digested with AtAMY3 as described above.
Determination of pH Optimum
pH-dependent activity curves were generated using 50 mm Britton-Robinson buffer in the pH range 5–9. Buffer stocks (150 mm) were prepared as described (23) by mixing equal volumes of 150 mm acetic acid, 150 mm boric acid, and 150 mm phosphoric acid and were titrated to the appropriate pH using NaOH. Recombinant AtAMY3 (10 μg) or HvAMY1 (10 ng) was preincubated in Britton-Robinson buffer at each pH for 15 min, after which activity was determined against BPNP-G7.
Oxidation/Reduction of AtAMY3 and Redox Titration
For redox modulation of AtAMY3 activity, the recombinant protein in 100 mm Tricine/NaOH, pH 7.9, was exposed to 5 mm DTT or 50 μm CuCl2 to fully reduce or oxidize the protein, respectively. After incubation at 25 °C for 2 h, the reactions were desalted into fresh Tricine buffer to remove DTT or CuCl2. For reactivation with DTT, both the reduced and oxidized proteins (10 μg) were incubated for 2 h with 20 mm DTT in Tricine buffer. Activity against BPNP-G7 was measured as described above. HvAMY1 was treated as described for AtAMY3, except all steps were carried out in 25 mm sodium acetate, pH 5.5. A lower amount of protein (10 ng) and a shorter incubation time (20 min) were used for the assay with BPNP-G7 to compensate for the higher specific activity of HvAMY1. For reactivation with thioredoxins (Trx), both the reduced and oxidized AtAMY3 proteins (10 μg) were incubated with 0.05 mm DTT and Trx at 5 μm (Trx from E. coli (Sigma), and the recombinant Arabidopsis Trx isoforms: f1, m1, m2, m3, m4, x, y1, and y2) in a total reaction volume of 60 μl. After incubation for 30 min at 25 °C, the enzyme was assayed with BPNP-G7.
For the redox titration, the fully oxidized AtAMY3 protein (10 μg) was incubated in 60 μl of Tricine buffer containing 20 mm total DTT with different ratios of dithiol/disulfide bridge. After incubating for 2 h at 25 °C, the activity was measured by adding 60 μl of amylase HR reagent. To calculate the midpoint redox potential, sigmoidal curves were fitted to the data using the graphing software Origin (version 8.5, OriginLab, Northampton, MA).
In-gel Redox Assay
Native PAGE was performed on recombinant AtAMY3 or crude extracts as described above. After electrophoresis, one lane was excised from the gel and immersed in reducing incubation medium, whereas the three remaining lanes were immersed in oxidizing medium. After incubation at room temperature for the specified time, one lane from the oxidizing medium and the lane incubated in the reducing medium were stained in Lugol solution. The remaining two lanes in the oxidizing medium were rinsed in washing medium (100 mm Tris-HCl, pH 8, 1 mm CaCl2, 1 mm MgCl2) to remove excess oxidizing agent. The lanes were split as follows: one lane was incubated for the specified amount of time in wash medium, and the other was incubated in reactivation medium (100 mm Tris-HCl, pH 8, 1 mm CaCl2, 1 mm MgCl2, 20 mm DTT). Both lanes were stained in Lugol solution.
MALDI-MS/MS Analysis of Reduced and Oxidized AMY3
Recombinant AMY3 was reduced/oxidized and subsequently buffer-exchanged into 100 mm Tricine/NaOH, pH 7.9, as described above. The protein (10 μg; in 45 μl) was digested at 37 °C overnight with LysC (100 ng/μl) and 6 μl of RapiGest surfactant (Waters, Baden, Switzerland). The reaction was stopped by adding 3 μl of HCl (1 m) and incubating at 37 °C for a further 30 min. Insoluble material was removed by centrifugation, and the supernatant was then spotted directly on the target with the matrix (0.7 mg/ml α-cyano-4-hydroxycinnamic acid in 85% acetonitrile, 0.1% trifluoroacetic acid). The spotted sample was then washed on the target with 0.1% TFA and covered with matrix. The mass spectrometry analysis was conducted using an UltrafleXtreme MALDI-TOF/TOF instrument (Bruker, Faellanden, Switzerland). MS/MS spectra were searched with Mascot software (Matrix Science) against the recombinant protein sequence.
Multiple Sequence Alignments of AtAMY3
Amino acid sequences of AtAMY3 protein orthologs from the following organisms were retrieved from the Phytozome database (24): Vitis vinifera (GSVIVT01020069001); Eucalyptus grandis (Eucgr.G02543.1); Ricinus communis (29736.m002033); Poplar trichocarpa (POPTR_0010s10300.1); Carica papaya (evm.model.supercontig_5.271); Arabidopsis lyrata (339218); Brassica rapa (Bra016262); Manihot esculenta (cassava4.1_001362m); Phaseolus vulgaris (Phvulv091010524m); and Glycine max (AMY3-1, Glyma02g02450.1; AMY3-2, Glyma08g40810.1). For HvAMY1, the sequence of the mature protein without the signal peptide (first 24 amino acids) was retrieved from the Uniprot database (Uniprot ID, P00693). Alignment was carried out with ClustalW (25) on the web server with the default parameters. Some sequences that aligned poorly with the other AMY3 sequences were removed; these were the Medicago trunculata, M. esculenta AMY3-2, and the Malus domestica sequences. These genes were either incomplete or had large sequence additions that disrupted alignment.
RESULTS
Catalytic Mechanism of AtAMY3 Is Highly Conserved
To study the biochemical properties of AtAMY3 in comparison with HvAMY1, the full-length AtAMY3 protein, excluding the chloroplast transit peptide (amino acids 56–887), was expressed in E. coli and purified to near homogeneity as described previously (11). The migration of the recombinant AtAMY3 on SDS-PAGE was consistent with the calculated molecular mass of 93.5 kDa (Fig. 1A). HvAMY1 was expressed in Pichia pastoris and purified as described previously (Fig. 1A) (26).
FIGURE 1.

Asp666 is essential for the amylolytic activity of AtAMY3. A, SDS-PAGE. Purified recombinant AtAMY3 (1 μg) and HvAMY1 (1 μg) were analyzed by SDS-PAGE in a 10% (w/v) gel stained with Coomassie Brilliant Blue. B and C, activity against BPNP-G7 of AtAMY3 WT and AtAMY3 D666N at pH 7.9 (B) and HvAMY1 at pH 5.5 (C). The purified proteins (10 μg of AtAMY3s and 10 ng of HvAMY1) were incubated for the indicated times with an excess of BPNP-G7 and α-glucosidase at 37 °C. Error bars indicate mean ± S.E. (n = 3) and are mostly smaller than the symbols. D, activity of AtAMY3 against amylopectin and β-limit dextrin. Recombinant protein (1 μg) was incubated for 30 min with 1.5 mg of solubilized substrate, and the amount of reducing ends released was assayed. Values represent the mean ± S.E. (n = 4). E, activity of HvAMY1 against amylopectin and β-limit dextrin. Recombinant protein (10 ng) was incubated for 10 min with 1.5 mg of solubilized substrate, and the amount of reducing ends released was assayed. Values represent the mean ± S.E. (n = 3). F, activity of AtAMY3 WT and D666N on native PAGE containing 7.5% acrylamide and 0.1% amylopectin or β-limit dextrin. Recombinant proteins (1 μg) were separated by native-PAGE for 3 h at 4 °C. Amylolytic activities were detected by staining with iodine solution after incubation for 1 h. Distinct AtAMY3 activities are indicated with arrows. One representative gel from five replicate gels is shown.
We assayed the purified recombinant proteins for amylolytic activity. First, we tested the activity of the proteins against the artificial substrate, blocked p-nitrophenyl maltoheptaoside (BPNP-G7, a chromogenic substrate specific for endoamylases (20)). AtAMY3 cleaved the BPNP-G7 substrate at a linear rate for 150 min (Fig. 1B). As described previously, HvAMY1 also displayed robust amylolytic activity toward the BPNP-G7 substrate. Thus, AtAMY3 is a functional α-amylase in vitro but is less active toward BPNP-G7 than HvAMY1 (Fig. 1, B and C), possibly due to a different specific activity. Within the HvAMY1 catalytic domain, the aspartic acid residue Asp204 is essential for activity (27). Protein sequence alignment of mature HvAMY1 with members of the AMY3-like family (as retrieved from the Phytozome database (24)) showed that the aspartic acid residue equivalent to Asp204 of HvAMY1 is highly conserved in all AMY3-like sequences (supplemental File 1). Mutation of the corresponding amino acid in AtAMY3 (Asp666) to an asparagine (D666N) abolished enzyme activity (Fig. 1B), suggesting that this aspartic acid is critical for AtAMY3 catalysis. It is likely that the catalytic mechanism between different families of plant α-amylases is conserved.
Next, we tested the ability of the recombinant AtAMY3 and HvAMY1 to degrade solubilized amylopectin and its derivative β-limit dextrin. After incubation for 1 h, both HvAMY1 and AtAMY3 were active on these substrates, as determined by the release of reducing sugars using the MBTH assay (Fig. 1, D and E (21)). The reaction times were such that the rate of hydrolysis was linear for both substrates (details not shown). AtAMY3 activity was 2-fold higher with β-limit dextrin compared with amylopectin (Fig. 1D). HvAMY1 showed very similar substrate preferences (Fig. 1E). When assayed using native PAGE in gels containing amylopectin, recombinant AtAMY3 migrated as four distinct activity bands with different electrophoretic mobility. All the detected activity bands could be attributed to AtAMY3, as revealed by immunoblotting and tandem mass spectrometry (LC-MS/MS) of excised bands (data not shown). The peptide coverage yielded from this experiment suggested that the multiple bands do not arise from degradation products of AtAMY3. It is likely that the multiple activity bands are due to molecular aggregation rather than biologically meaningful modifications or intermolecular interactions, because the native AtAMY3 protein in Arabidopsis leaf extracts migrated as a single band (as seen in Fig. 6D). As expected, AtAMY3 D666N mutant showed complete loss of activity against both substrates (Fig. 1F).
FIGURE 6.
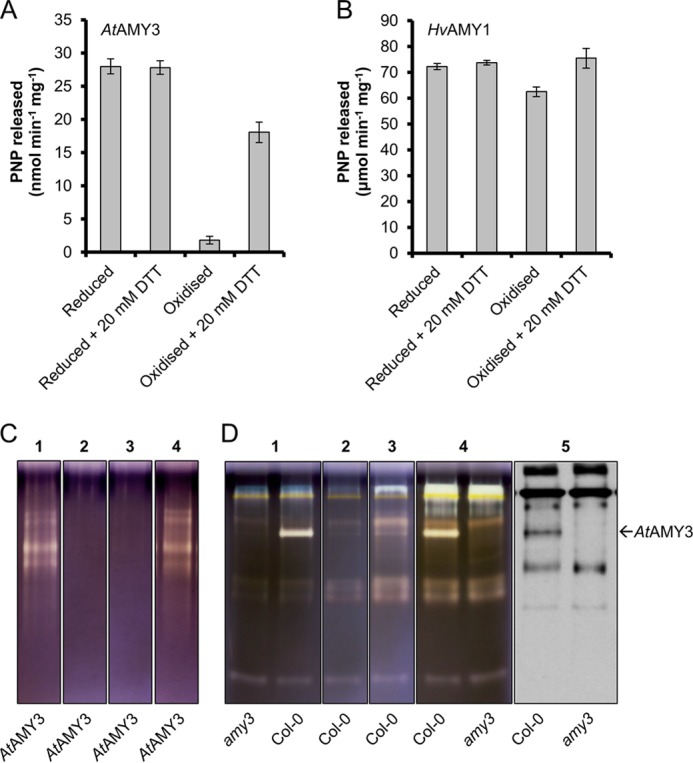
Redox regulation of AtAMY3 activity. A and B, redox sensitivity of AtAMY3 and HvAMY1. Purified recombinant proteins were exposed to 5 mm DTT or 50 μm CuCl2 for 2 h prior to activity assay. Reduced and oxidized proteins were then incubated for 2 h with or without 20 mm DTT prior to assaying activity. Values represent the mean ± S.E. (n = 4). Note the difference in the y axis scale between A and B. PNP, para-nitrophenol. C, in-gel reactivation of oxidized AtAMY3 protein. Recombinant AtAMY3 was run on a native polyacrylamide gels containing amylopectin. Lanes were incubated with 5 mm DTT (lane 1) or 25 μm CuCl2 (lane 2) for 1 h prior to staining in Lugol solution. Lanes incubated with 25 μm CuCl2 were rinsed in washing buffer and further incubated with (lane 4) or without (lane 3) 20 mm DTT for a further 3 h prior to staining in Lugol solution. D, in-gel redox regulation of native AtAMY3 enzyme. Arabidopsis leaf crude extracts from 4-week-old Col-0 wild-type and amy3 mutant plants were separated on native polyacrylamide gels containing amylopectin. Lanes were incubated with 5 mm DTT (lane 1) or 100 μm CuCl2 (lane 2) for 3 h prior to staining in Lugol solution. Lanes incubated with 100 μm CuCl2 were rinsed in washing buffer and further incubated overnight with (lane 4) or without (lane 3) 20 mm DTT prior to staining in Lugol solution. To exclude the possibility that other AtAMY3 bands with undetectable activity may be present, an immunoblot was probed with an anti-AtAMY3 antibody (lane 5).
AtAMY3 Releases Small Linear and Branched Glucans from the Starch Granule Surface in Vitro
The preference of AtAMY3 for a β-limit structure (polymers carrying short external chains), compared with amylopectin, suggests that during starch degradation in vivo, it may favor substrates generated by β-amylolysis. If so, AtAMY3 would be expected to release smaller branched glucans from the surface of starch granules pretreated with β-amylase.
To test this hypothesis, we investigated the activity of AtAMY3 on starch granules purified from Arabidopsis leaves and the impact of pretreating these granules with β-amylase. The starch granules were incubated with AtAMY3 recombinant protein, after which they were sedimented by centrifugation, and the reducing sugars in the supernatant were quantified. The same was done for HvAMY1. Significant starch breakdown by AtAMY3 or HvAMY1 alone was detected, showing that both enzymes can work directly on the granule surface (data not shown). We predigested the granules with recombinant Arabidopsis β-amylase (AtBAM1). After 15 min, AtBAM1 did not continue to release reducing ends, suggesting that the exterior chains had been digested to a β-limit structure (data not shown). AtAMY3 efficiently released reducing sugars from starch granules predigested with AtBAM1, but the rate of hydrolysis slowed down after 15 min of incubation (data not shown).
Analysis by HPAEC-PAD revealed that the reducing sugars released from starch by AtAMY3 or HvAMY1 were predominantly composed of 6, 7, and 8 glucose units (Fig. 2A (28)). Quantifications of the peak areas in these chromatograms are presented in Table 1. AtAMY3 released an increased amount of shorter glucans (with degree of polymerization (DP) of 3–5) from the starch granule compared with HvAMY1. When the starch was digested with AtAMY3 for longer periods (i.e. 120 min versus 30 min), a reproducible enrichment of short chains with DP of 3–6 was observed (Fig. 2, A and B). This was accompanied by a substantial decrease in the proportion of longer products (DP 7–11) after the extended digest. This trend was not as evident for HvAMY1, where we primarily observed an increase in the amount of glucan chains with DP of 6–7 over time (Fig. 2, A and C). These data suggest that, in addition to the granule surface itself, longer soluble malto-oligosaccharides produced during starch degradation were effectively further degraded by AtAMY3.
FIGURE 2.

AtAMY3 releases short linear and branched malto-oligosaccharides from the starch granule surface. Representative HPAEC-PAD chromatograms of malto-oligosaccharides released from isolated Arabidopsis starch by AtAMY3 or HvAMY1. A, starch (1.5 mg) was digested with AtAMY3 (10 μg) or HvAMY1 (50 ng), and the released glucans were analyzed. The amounts of protein used corresponded to equal amylolytic activity against starch granules. The DP is indicated on top of the chromatogram. B, difference plots in the chain length distribution of linear products of AtAMY3 against native starch granules between 120- and 30-min time points. Relative areas were calculated for each peak in chromatograms generated for A, yielding a percentage (relative to total peak area) for each malto-oligosaccharide species. Differences in this percentage between the 120- and 30-min time points were plotted. Values represent the mean ± S.E. (n = 3). C, same as B, but for HvAMY1.
TABLE 1.
Quantification of peak areas from Fig. 2A
Values represent relative peak areas from HPAEC-PAD chromatograms of malto-oligosaccharides released from Arabidopsis starch granules by AtAMY3 or HvAMY1. Starch (1.5 mg) was digested with AtAMY3 (10 μg) or HvAMY1 (50 ng) for 30 or 120 min, and the glucans released were analyzed. A representative chromatogram is presented in Fig. 2A. The relative peak area was calculated for each peak as a percentage of total peak area. Values represent the mean ± S.E. from chromatograms produced from three replicate digests.
| DP |
AtAMY3 |
HvAMY1 |
|||
|---|---|---|---|---|---|
| 30 min | 120 min | 30 min | 120 min | ||
| Linear glucans | 3 | 4.00 ± 0.09 | 5.71 ± 0.03 | 2.78 ± 0.05 | 4.28 ± 0.03 |
| 4 | 9.8 ± 0.1 | 12.28 ± 0.04 | 2.878 ± 0.008 | 3.78 ± 0.03 | |
| 5 | 7.00 ± 0.03 | 7.96 ± 0.01 | 1.191 ± 0.006 | 1.87 ± 0.02 | |
| 6 | 13.60 ± 0.06 | 16.07 ± 0.05 | 10.38 ± 0.02 | 13.0 ± 0.1 | |
| 7 | 22.76 ± 0.05 | 23.68 ± 0.01 | 34.1 ± 0.2 | 36.6 ± 0.3 | |
| 8 | 16.74 ± 0.05 | 13.70 ± 0.05 | 15.5 ± 0.2 | 13.66 ± 0.03 | |
| 9 | 10.19 ± 0.07 | 5.49 ± 0.06 | 7.96 ± 0.08 | 6.06 ± 0.01 | |
| 10 | 4.47 ± 0.07 | 1.09 ± 0.03 | 6.13 ± 0.06 | 4.33 ± 0.02 | |
| 11 | 1.03 ± 0.03 | 0.162 ± 0.008 | 4.42 ± 0.05 | 2.96 ± 0.01 | |
| 12 | 0.293 ± 0.007 | 0.096 ± 0.004 | 3.42 ± 0.04 | 2.22 ± 0.01 | |
| 13 | 0.186 ± 0.003 | 0.133 ± 0.001 | 2.60 ± 0.03 | 1.669 ± 0.007 | |
| 14 | 0.137 ± 0.002 | 0.162 ± 0.002 | 1.92 ± 0.03 | 1.245 ± 0.007 | |
| 15 | 0.119 ± 0.002 | 0.248 ± 0.002 | 1.31 ± 0.02 | 0.824 ± 0.002 | |
| Branched glucans | 8 | 0.08 ± 0.04 | 0.295 ± 0.002 | 0.009 ± 0.009 | 0.07 ± 0.04 |
| 9 | 0.31 ± 0.02 | 0.82 ± 0.01 | 0.303 ± 0.003 | 0.546 ± 0.006 | |
| 10 | 0.838 ± 0.007 | 1.64 ± 0.01 | 0.639 ± 0.004 | 0.942 ± 0.007 | |
| 11 | 1.30 ± 0.01 | 2.19 ± 0.02 | 0.537 ± 0.004 | 0.881 ± 0.007 | |
| 12 | 1.539 ± 0.004 | 2.17 ± 0.01 | 0.550 ± 0.005 | 0.91 ± 0.01 | |
| 13 | 1.596 ± 0.004 | 1.965 ± 0.007 | 0.622 ± 0.003 | 0.96 ± 0.01 | |
| 14 | 1.534 ± 0.005 | 1.66 ± 0.001 | 0.635 ± 0.005 | 0.941 ± 0.009 | |
| 15 | 1.345 ± 0.008 | 1.275 ± 0.002 | 0.545 ± 0.002 | 0.763 ± 0.004 | |
Some of the minor malto-oligosaccharides detected by HPAEC-PAD did not co-elute with linear malto-oligosaccharides standards, indicating that they were branched. This was confirmed by treatment of the digested samples with Pseudomonas isoamylase and Klebsiella pullulanase. The minor peaks disappeared, and malto-oligosaccharides with a smaller average size and co-eluting with linear standards increased in abundance (Fig. 3A). Treatment of digested samples with AtBAM1, which cannot metabolize branched malto-oligosaccharides, also confirmed that both AtAMY3 and HvAMY1 released branched glucans during starch hydrolysis. The major peaks disappeared, with a concomitant increase in maltose, although the minor peaks remained (Fig. 3A). When the starch granules were predigested with AtBAM1, recombinant AtAMY3 released a higher proportion of branched glucans with DP of 11–14 (Fig. 3B). This suggests that after β-amylase treatment, there are fewer external chains remaining for AtAMY3, which then begins to cleave glucosidic bonds between branch points. Taken together, these data show that AtAMY3 can act on both soluble and insoluble glucan substrates to release small linear and branched malto-oligosaccharides (16, 17).
FIGURE 3.
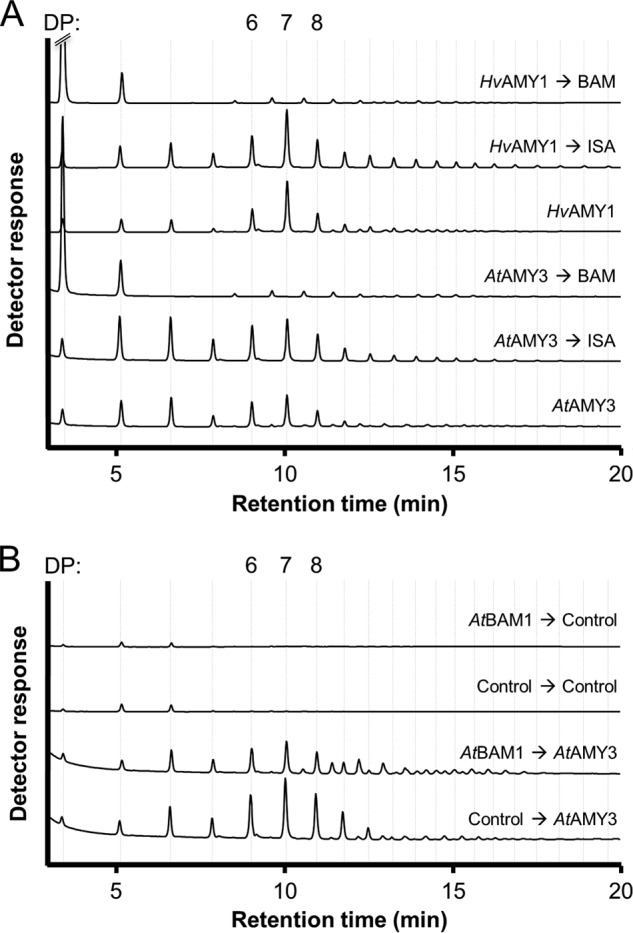
AtAMY3 releases more short branched malto-oligosaccharides from starch granules pretreated with β-amylase. Arabidopsis starch granules were digested as described in Fig. 2. A, treatment of the released glucans with Pseudomonas isoamylase and Klebsiella pullulanase resulted in smaller, linear malto-oligosaccharides, showing that the glucans were branched. Treatment with β-amylase resulted in the accumulation of maltose, maltotriose, and branched glucans. The order of enzymatic treatments is indicated with arrows. B, products released by AtAMY3 from starch pretreated with β-amylase. The order of treatments is indicated with arrows.
AtAMY3 Works Synergistically with β-Amylase toward Efficient Starch Degradation
We hypothesized that the ability of AtAMY3 to efficiently degrade β-limit structures may allow it to work synergistically with β-amylase during starch degradation. We tested this by degrading starch granules with AtAMY3 and AtBAM1, both individually and together (Fig. 4). To quantify the total amount of soluble glucans released from the starch granule, we acid-hydrolyzed the glucans in the soluble fraction after the digest, and we quantified the amount of glucose present. After an initial phase of rapid starch degradation (0–30 min), the rate of glucans released from the granule decreased for AtAMY3 (Fig. 4). This decrease probably occurs as the enzyme reaches more crystalline areas of the granule that are difficult to degrade. A similar trend was observed for AtBAM1, although we presume that β-amylase activity slows as a β-limit structure is approached. However, when both enzymes were added together, the amount of soluble glucans released from the starch granules was much higher than expected from the added individual activities of AtAMY3 and AtBAM1 alone. Furthermore, a steady release of glucans into the soluble fraction was sustained for up to 120 min. These data suggest that efficient starch degradation is achieved when both enzymes are present.
FIGURE 4.
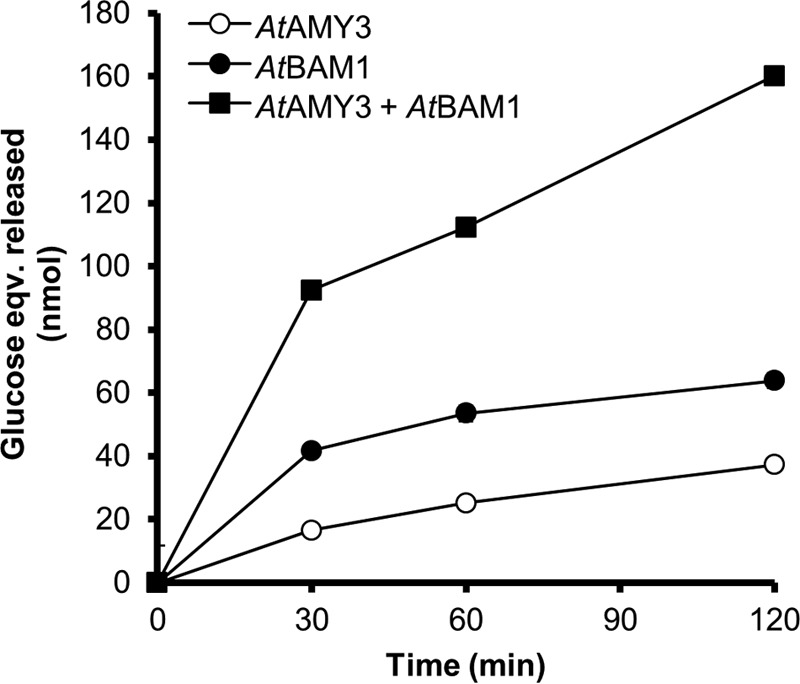
AtAMY3 and AtBAM1 degrade starch synergistically in vitro. Recombinant proteins AtAMY3 (10 μg) and/or AtBAM1 (2.5 μg) were incubated at 25 °C with purified Arabidopsis starch (1.5 mg). After the indicated time points, soluble glucans in the supernatant were removed and subjected to acid hydrolysis. Glucose was subsequently quantified. Values represent the mean ± S.E. (n = 3). Error bars not visible are smaller than the symbols.
AtAMY3 Has a pH Optimum Suited for Activity in the Chloroplast
We assayed AtAMY3 against the BPNP-G7 substrate at various pH values (Fig. 5A). The pH optimum of AtAMY3 was ∼7.5. However, a broad peak in catalytic activity (>90% of the maximum) was observed between pH 7 and 8.5. A similar pH-activity curve was obtained when amylopectin was used as a substrate (data not shown). In contrast, HvAMY1 showed a pH optimum at around 5.5, and retained only 13% of its maximum activity at pH 8 (Fig. 5B (27)). The differences in optimal pH between the chloroplastic AtAMY3 and the secreted barley AMY1 matched their respective subcellular localizations. The physiological pH range in the chloroplast ranges from neutral (pH 7) at night to slightly alkaline (pH 8) during the day (29). Thus, AtAMY3 would be highly active at all stromal pH conditions. AtAMY3 represents a rare example of plant α-amylase that is more active under alkaline than acidic conditions.
FIGURE 5.
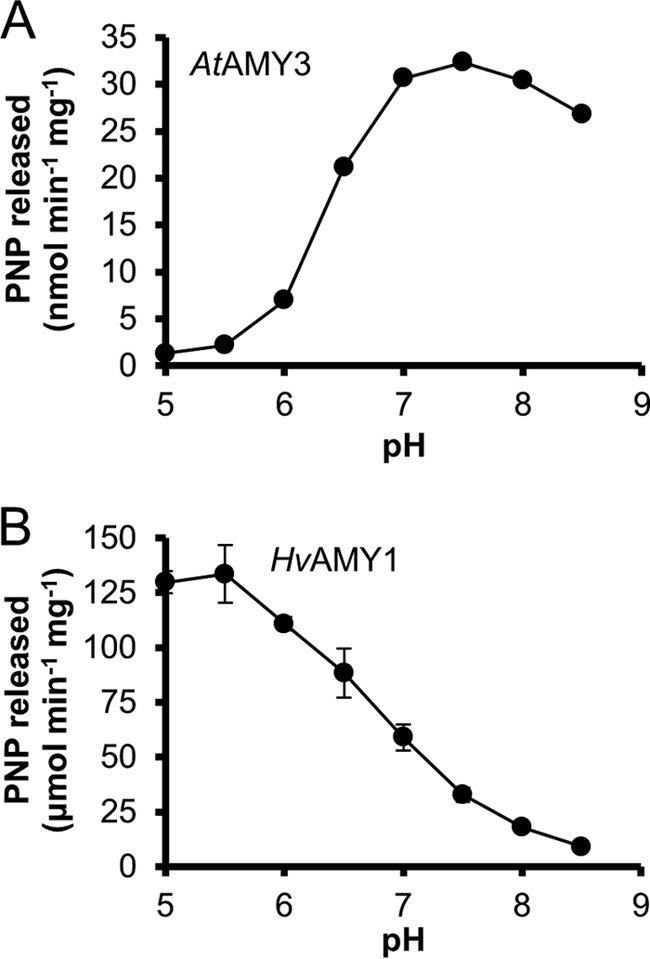
Effect of pH on AtAMY3 and HvAMY1 activity. The enzyme activity of AtAMY3 (A, 10 μg) and HvAMY1 (B, 10 ng) was assayed in Britton-Robinson buffer under the standard assay conditions, where pH of the mixture was titrated as indicated (pH 5–8.5). The reactions were stopped after 60 min for AtAMY3 and 20 min for HvAMY1. Values represent the mean ± S.E. (n = 3). Error bars not visible are smaller than the symbols. Note the difference in the y axis scale between A and B. PNP, para-nitrophenol.
AtAMY3 Activity Is Redox-regulated
Several enzymes involved in starch metabolism have been shown to be sensitive to changes in redox potentials in vitro (30–35). Recently, AtAMY3 was reported to be dependent on the reducing agent DTT for activity against amylopectin, suggesting that it could be redox-regulated (35). We investigated this hypothesis further. We found that AtAMY3 could be inactivated by exposing the enzyme to increasing concentrations of the oxidants copper chloride (CuCl2) or hydrogen peroxide (H2O2). Amylolytic activity against the BPNP-G7 substrate was efficiently suppressed after a 1-h treatment with 25–50 μm CuCl2 or 2–4 mm H2O2 (data not shown). We then investigated whether the loss of activity under oxidizing conditions was reversible. Fully reduced or fully oxidized AtAMY3 was desalted to eliminate excess DTT or CuCl2 and then incubated for 2 h with or without 20 mm DTT (Fig. 6A). Incubation with DTT had no effect on the activity of the reduced enzyme, which showed a consistent specific activity of 28 ± 1 nmol of para-nitrophenol released min−1 mg−1. In contrast, upon reduction of fully oxidized inactive AtAMY3 with an excess of DTT, activity was restored to about 65% of the original activity (Fig. 6A). This indicates that AtAMY3 could be reversibly inactivated and activated depending on its redox state, even though a fraction of the protein seems to have been irreversibly oxidized. For comparison, we exposed HvAMY1 to the same treatment. In contrast to AtAMY3, HvAMY1 lost only 14% of its fully reduced activity after treatment with 50 μm CuCl2 for 2 h (Fig. 6B). Given that CuCl2 is a thiol-oxidizing agent, we concluded that HvAMY1 was virtually redox-insensitive. To our knowledge, no other α-amylase has been previously reported to be redox-regulated. Thus, redox sensitivity is not a common feature of plant α-amylases but rather one specific to AtAMY3. The redox regulation of AtAMY3 can be seen as another adaptation for activity in the chloroplast.
To test whether changes in the redox conditions affected AtAMY3 activity toward amylopectin, we developed an in-gel redox assay using native gels (see “Experimental Procedures” for details). After electrophoresis of purified AtAMY3, individual lanes were treated with 5 mm DTT or 25 μm CuCl2 for 1 h prior to staining with iodine solution. AtAMY3 activity was detected only in lanes that had a reducing treatment, consistent with the BPNP-G7 assay results (Fig. 6C). Lanes treated with CuCl2 were rinsed to remove excess oxidizing agents and then re-incubated with or without 20 mm DTT for 3 h before staining with iodine. DTT treatment restored the amylolytic activity of the protein. Hence, the activity of recombinant AtAMY3 toward amylopectin is modulated by changes in redox potential. A redox-sensitive amylolytic activity corresponding to AtAMY3 was also observed when crude extracts from Arabidopsis leaf were analyzed using amylopectin-containing native gels exposed to the same treatment as described above (Fig. 6D), suggesting that native AtAMY3 from leaf extracts is similarly redox-regulated.
To investigate whether AtAMY3 activity responds to physiologically meaningful redox potentials, we conducted a redox titration analysis in which defined mixtures of DTTreduced and DTToxidized were used as a redox equilibration buffer. Enzyme activity against BPNP-G7 was assayed after the oxidized AtAMY3 was allowed to equilibrate for 2 h at the different redox potentials. At pH 7.9, AtAMY3 shifted from an active form at −400 mV to an inactive form at −280 mV, confirming that the protein was mostly active under reducing conditions (Fig. 7). We estimated the midpoint redox potential (Em) by fitting the Nernst equation to the data and obtained a value for AtAMY3 of −329 ± 2 mV. This is within the range reported for other redox-regulated chloroplast enzymes, including the β-amylase BAM1 (−350 mV at pH 7.9 (32)), and the glucan, water dikinase GWD (−310 mV at pH 7.9 (31)). Hence, the redox sensitivity of AtAMY3 appeared to be within a physiologically meaningful range.
FIGURE 7.
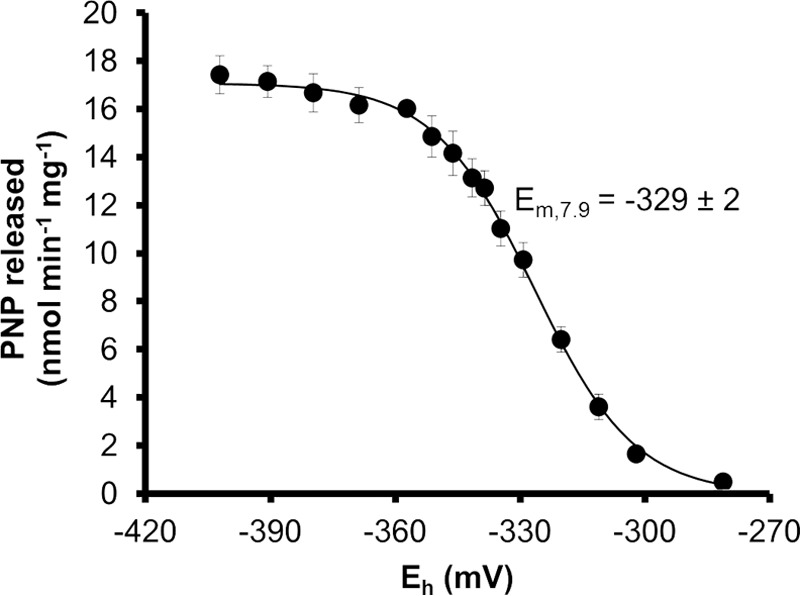
Midpoint redox potential of AtAMY3 at pH 7. 9. The ambient redox potential (Eh), adjusted by defined ratios of DTToxidized/DTTreduced, ranged from −270 to −420 mV. Preoxidized AtAMY3 protein (10 μg) was incubated in Tricine/NaOH buffer containing 20 mm total DTT in different dithiol/disulfide ratios for 2 h prior to assaying activity. The midpoint redox potential at this pH (Em, 7.9) was calculated by fitting the Nernst equation, and the error is the S.E. calculated from three replicate experiments. PNP, para-nitrophenol.
Chloroplastic Thioredoxins Can Reactivate the Oxidized AtAMY3 in Vitro
In plants, Trxs directly reduce their target redox-regulated enzymes by performing disulfide exchange reactions (36). We determined the ability of AtAMY3 to interact with Trxs in vitro. The isoforms tested included a commercially available recombinant Trx from E. coli, as well as chloroplastic Trxs from Arabidopsis, which were obtained as recombinant proteins (37, 38). Co-incubation of 5 μm Trx with 0.05 mm DTT in the reactivation treatment resulted in a drastic stimulation of AtAMY3 activity compared with DTT treatment alone (Table 2). The activity reached a maximum after 30 min of reduction (details not shown). These data revealed that AtAMY3 is efficiently reduced by Trxs. The chloroplast Trxs, however, showed selectivity in their interaction with AtAMY3. Trx f1 proved most effective, displaying a similar reactivation efficiency (59%) to that observed using 20 mm DTT for 2 h (65%, Fig. 6A). This is consistent with previous observations that the majority of Trx-regulated enzymes in starch metabolism are most efficiently activated by Trx f. Given that Trx can only reduce its target protein through a thiol/disulfide exchange mechanism (36), our results implicate the reduction of a disulfide bridge between different cysteine residues in the process of AtAMY3 activation.
TABLE 2.
Reactivation of oxidized AtAMY3 protein by different isoforms of Arabidopsis chloroplastic thioredoxins (AtTrx)
10 μg of recombinant AtAMY3 was initially oxidized by 50 μm CuCl2 and then incubated with various AtTrx isoforms (5 μm) in the presence of 0.05 mm DTT for 30 min before assaying the activity. Trx f, m, x, and y isoforms from Arabidopsis were purified recombinant proteins expressed in E. coli. Values represent the mean ± S.E. (n = 3).
| DTT | Thioredoxin (5 μm) | PNP released | % of reduced activity | |
|---|---|---|---|---|
| mm | nmol min−1 mg−1 | |||
| Reduced protein | 0 | 26.2 ± 0.3 | ||
| 20 | 26.3 ± 0.7 | 100 | ||
| Oxidized protein | 0 | 1.22 ± 0.05 | 5 | |
| 0.05 | 3.8 ± 0.3a | 14 | ||
| Trx f1 | 15.4 ± 0.3a | 59 | ||
| Trx m1 | 12.3 ± 0.4a | 47 | ||
| Trx m2 | 11.8 ± 0.5a | 45 | ||
| Trx m3 | 6.6 ± 0.3a | 25 | ||
| Trx m4 | 9.4 ± 0.4a | 36 | ||
| Trx x | 9.0 ± 0.4a | 34 | ||
| Trx y1 | 11.1 ± 0.6a | 42 | ||
| Trx y2 | 13.6 ± 0.8a | 52 | ||
| E. coli Trx | 13.9 ± 0.3a | 53 |
a Data denote the significant difference from oxidized protein without DTT or Trx with a two-tailed t test (p < 0.05).
Mechanism of AtAMY3 Redox-dependent Reactivation
Nine cysteine residues are present in the amino acid sequence of AtAMY3. Of these residues, four (Cys118, Cys285, Cys310, and Cys363) are located in the N-terminal extension and five (Cys499, Cys587, Cys652, Cys743, and Cys832) in the α-amylase domain (supplemental File 1). We performed site-directed mutagenesis of these cysteines to investigate the importance of each residue for disulfide formation in AtAMY3. Each mutated AtAMY3 variant, carrying a single cysteine to serine substitution, was produced in E. coli and purified as a soluble product.
AtAMY3 Cys mutants were all active under reducing conditions, but the level of activity varied. AtAMY3 C587S showed lower activity against both BPNP-G7 and amylopectin compared with the wild-type AtAMY3 (Fig. 8A and Table 3). AtAMY3 C285S and C652S also had slightly reduced activity, whereas C499S was virtually inactive. In contrast, the C363S mutant displayed higher activity. It is possible that these mutations change the stability or conformation of the proteins, altering their catalytic activity. Notably, under oxidizing conditions, C587S was the only AtAMY3 Cys mutant to retain amylolytic activity (Fig. 8B and Table 3). All other site-directed mutants were inactivated under oxidizing conditions, and all of them could be re-activated under reducing conditions (Fig. 8, C and D). These findings indicate that Cys587, in addition to being important for the optimal catalytic rate, is also involved in the redox regulation of AtAMY3 activity.
FIGURE 8.
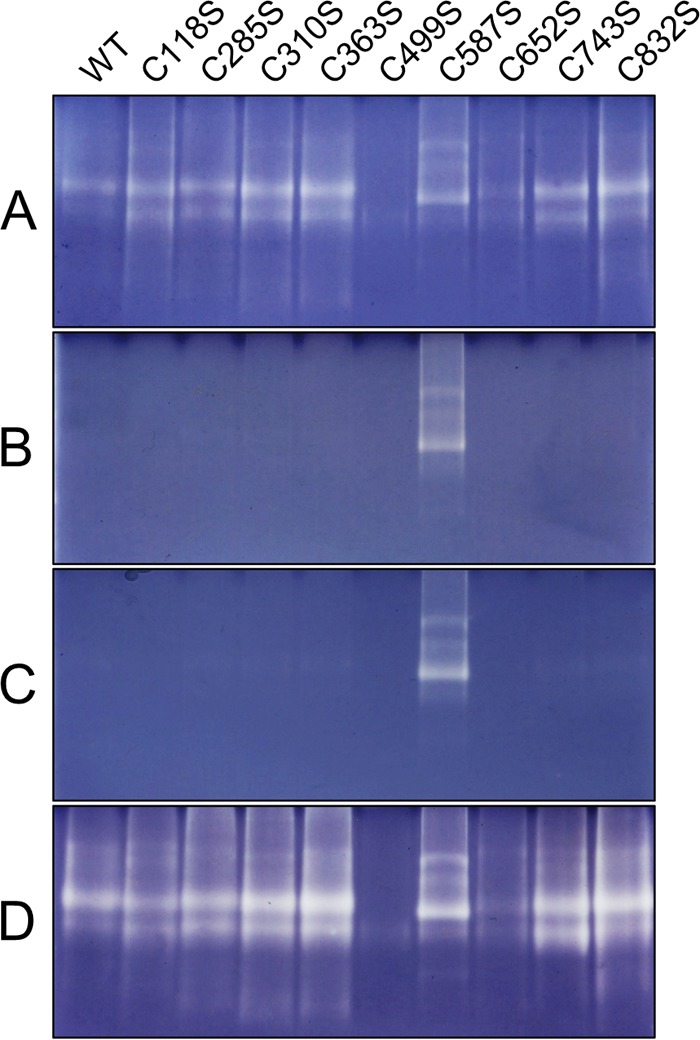
C587S site-specific mutant of AtAMY3 is redox-insensitive. Purified AtAMY3 and its cysteine mutant variants (1 μg each) were electrophoresed on native polyacrylamide gels containing amylopectin. Gels were incubated with 5 mm DTT (A) or 50 μm CuCl2 (B) for 3 h prior to staining in Lugol solution. Gels incubated with 50 μm CuCl2 were rinsed in washing medium and further incubated overnight with (D) or without (C) 20 mm DTT prior to staining in Lugol solution.
TABLE 3.
Activity of AtAMY3 and its cysteine mutant variants against BPNP-G7 under reducing or oxidizing conditions
Purified recombinant proteins (10 μg) were exposed to 5 mm DTT or 50 μm CuCl2 for 1 h prior to activity assay. Values represent the mean ± S.E. (n = 3).
| Reduced activity | Oxidized activity | ||
|---|---|---|---|
| nmol min−1 mg−1 | nmol min−1 mg−1 | % of reduced activity | |
| WT | 31.6 ± 0.5 | 0.06 ± 0.03 | <1%a |
| C118S | 27.7 ± 0.5 | 0.06 ± 0.03 | <1%a |
| C285S | 11.1 ± 0.6 | 0.14 ± 0.05 | 1%a |
| C310S | 21.7 ± 0.4 | 0.03 ± 0.03 | <1%a |
| C363S | 44.4 ± 0.4 | 0.13 ± 0.03 | <1%a |
| C499S | 2.4 ± 0.1 | 0 ± 0 | 0%a |
| C587S | 3.9 ± 0.2 | 3.3 ± 0.2 | 85% |
| C652S | 17.2 ± 0.3 | 0.06 ± 0.03 | <1%a |
| C743S | 29.1 ± 0.5 | 0.6 ± 0.3 | 2%a |
| C832S | 33.3 ± 0.7 | 0 ± 0 | 0%a |
a Data denote significant difference (p < 0.05) between reduced and oxidised activities with a two-tailed t test.
Structural Mechanism for the Redox Regulation of AtAMY3
Site-directed mutagenesis revealed Cys587 as an essential residue for the redox regulation of AtAMY3. However, two cysteines are required to form an inhibitory disulfide bridge. We explored the possibility that AtAMY3 may form intermolecular disulfide linkages via Cys587 residues using nonreducing PAGE. However, no high molecular weight bands corresponding to joined AtAMY3 proteins were detected under oxidizing conditions (data not shown). Because this finding rather suggested an intramolecular disulfide linkage, we used mass spectrometry to compare peptides generated after a proteolytic digest of reduced and oxidized AtAMY3. Following digest with the protease LysC, peptides were analyzed by MALDI-MS/MS, and the resulting spectra were searched against the recombinant protein sequence with Mascot software. The sequence coverage obtained was 53.2 and 43.5%, respectively, for reduced and oxidized proteins. A peptide containing Cys587 was identified at 1679.9 m/z and confirmed by MS/MS with a high Mascot score (Fig. 9A and Table 4). We found that the abundance of this peptide was dramatically reduced in the digested oxidized protein in comparison with the reduced protein (Fig. 9A). This is consistent with the hypothesis that Cys587 is involved in a redox-dependent modification. We also found that the abundance of a Cys499-containing peptide at 2361.1 m/z showed a similar change depending on redox state (Fig. 9B and Table 4). These data suggest that a disulfide bridge may form between Cys499 and Cys587.
FIGURE 9.
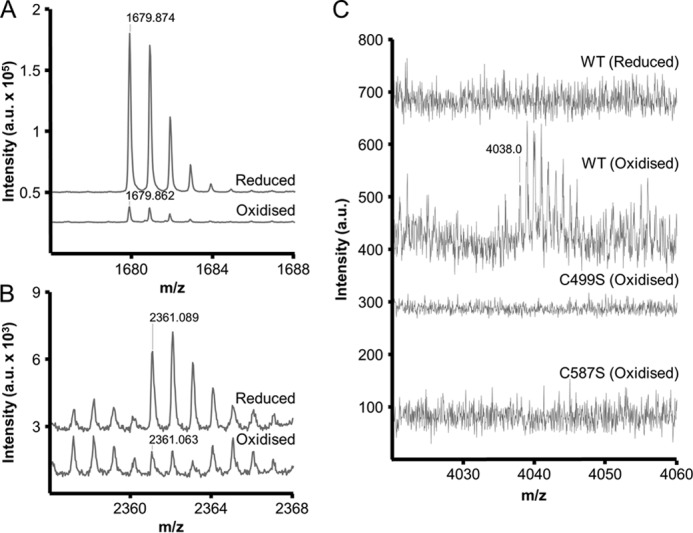
Disulfide linkage between Cys499 and Cys587 identified using mass spectrometry. Reduced and oxidized proteins were digested in solution with LysC, and the resulting peptides were analyzed by MALDI-MS/MS. A and B, mass spectra of two peptides generated from the digest of reduced and oxidized wild-type proteins. The y axis represents intensity in arbitrary units (a.u.). The peptides at 1679.9 m/z (A) and 2361.1 m/z (B) correspond to Cys587- and Cys499-containing peptides, respectively (Table 4). C, mass spectra generated from digested reduced and oxidized wild-type proteins, as well as oxidized C499S and C587S variants. A disulfide-linked peptide at the additive mass of peptides shown in A and B can be observed at 4038.0 m/z, exclusively in the oxidized wild-type protein.
TABLE 4.
AtAMY3 peptides confirmed by MALDI-MS/MS
Peptides generated after in-solution digest with LysC were visualized by MALDI-MS (Fig. 8), and the corresponding MS/MS spectra were searched with Mascot against the recombinant protein sequence. Mascot scores are from spectra generated with the reduced wild type protein.
| m/z | Peptide | Score |
|---|---|---|
| 1679.9 | VLGDAVLNHRC*AHFK (* is Cys587) | 70 |
| 2361.1 | ISSGTGSGFEILC*QGFNWESNK (* is Cys499) | 91 |
We then investigated whether a joined peptide linked by Cys499 and Cys587 could be detected after digesting the oxidized protein. We identified a peptide at 4038 m/z, corresponding to the additive mass of the Cys587- and Cys499-containing peptides. This peptide was only found after digesting the oxidized wild-type protein and not the reduced protein (Fig. 9C). Additionally, the peptide was also not detected after digesting the oxidized C499S and C587S site-directed mutants, confirming that both cysteines are involved in forming this mass. Furthermore, the peptide produced a typical fragmentation pattern for disulfide-linked peptides in the MS/MS spectrum (data not shown). These data suggest that the structural basis of AtAMY3 redox regulation is a disulfide bridge formed between Cys499 and Cys587 that inhibits catalysis. It should be noted that aside from this regulatory role, both cysteines are likely to play an important role in the activity of the enzyme, possibly by contributing to structural stability. The activity toward both amylopectin and BPNP-G7 was reduced or almost undetectable for C587S and C499S mutants, respectively.
DISCUSSION
Despite Conserved Catalytic Mechanism, AtAMY3 Is Adapted for Chloroplastic Activity
In this study, we have characterized some biochemical aspects of chloroplastic α-amylase 3 from A. thaliana (AtAMY3). Our results show that the purified heterologous AtAMY3 protein was active against the artificial substrate BPNP-G7, against soluble substrates such as amylopectin (Fig. 1, B, D, and F), and against the starch granule surface (Figs. 2–4). Site-directed mutagenesis of the aspartic acid residue Asp666 in AtAMY3 led to complete loss of activity toward all tested substrates (Fig. 1, B and F). This aspartic acid is located within the active site of other α-amylases like HvAMY1 (27) and is highly conserved among members of the entire α-amylase superfamily, including debranching enzymes and glucosidases from bacteria and animals (2, 39). Thus, Asp666 likely represents the catalytic nucleophile of AtAMY3. Interestingly, HvAMY1 was 3 orders of magnitude more active than AtAMY3 (Fig. 1, B and C), the molecular basis of which is unclear. This difference in kinetics may reflect the differences in starch metabolism between the tissues in which each enzyme is active. Cereal α-amylases are involved in rapid endosperm starch hydrolysis, and higher activities lead to better germination vigor (40). The involvement of β-amylase in this process is unclear (41). In contrast, the degradation of starch in leaf mesophyll cells is a highly regulated process. Leaf starch is degraded at a rate such that reserves accumulated during the day are sufficient for the whole night. Premature exhaustion of starch would lead to starvation at night and inhibition of growth (42, 43). It is possible that the slower kinetics of AtAMY3 is an inevitable consequence of the regulatory features described here and is necessary to control the degradation of transitory starch in leaves. Furthermore, despite the slower kinetics, we demonstrated that AtAMY3 is capable of efficient starch degradation together with β-amylase activity (Fig. 4), which is essential for leaf starch degradation (44).
β-Limit Dextrin at the Starch Granule Surface Is a Substrate of AtAMY3 in Vitro
During starch breakdown in leaf chloroplasts, reversible glucan phosphorylation allows continuous degradation by β-amylases until they reach the branch points at the root of each amylopectin cluster. Recent circumstantial evidence from in vivo studies suggests that the resulting β-limit dextrin structure at the granule surface is a suitable substrate for AtAMY3. Although AtAMY3 cannot hydrolyze α-1,6-linkages, it can access chains beyond the branch points, thereby releasing branched malto-oligosaccharides into the chloroplast stroma for subsequent metabolism by the debranching enzymes isoamylase 3 and limit dextrinase. In isa3lda mutants, soluble branched glucans accumulate during the night but are absent in the amy3isa3lda triple mutant (16, 17).
In accordance with this model, when tested against amylopectin and β-limit dextrin, AtAMY3 had a strong preference for β-limit dextrin (Fig. 1, D and F). A likely explanation for this is that the shorter external chains provide less steric hindrance to endoamylolytic cleavage sites. When tested against purified Arabidopsis starch granules, linear glucans with DP 6–8 were the predominant degradation products (Fig. 2A). However, when starch granules were pretreated with AtBAM1 (to generate a β-limit dextrin structure at the granule surface), AtAMY3 released a higher proportion of small branched glucans from the starch granule surface (Fig. 3B). Intriguingly, after an initial period of rapid activity, the release of reducing sugars by AtAMY3 from β-amylase-pretreated granules decreased (data not shown). In contrast, the release of reducing sugars by AtAMY3 was sustained for longer when acting against granules that had not been pretreated. The reason for this is unclear. It is possible that the activity of AtAMY3 slows when faced with more crystalline regions inside the granules (as observed in Fig. 4) and that such regions are reached more rapidly by AtAMY3 after the pretreatment of the granules by AtBAM1. However, efficient starch degradation was sustained for extended periods when both AtAMY3 and AtBAM1 were present in the digest (Fig. 4). We suggest that AtAMY3 stimulates β-amylolytic activity at the granule surface by cleaving past branch points and releasing small branched glucans for further degradation by debranching enzymes or AtAMY3 itself (Figs. 2 and 3). Meanwhile, β-amylolytic activity stimulates AtAMY3 by generating β-limit structures at the granule surface, which AtAMY3 can degrade. This observed synergistic effect between the enzymes provides insight into the role of AtAMY3 in the reaction network of starch degradation in the Arabidopsis chloroplast.
Regulatory Role of Cys499 and Cys587 in AtAMY3 Redox-dependent Activity
In this work, we showed that AtAMY3 has the characteristics of a redox-regulated enzyme. Both the recombinant protein and the native protein in crude extracts had a similar redox sensitivity (Fig. 6, C and D), suggesting that the endogenous enzyme has a similar chemistry in vivo. We observed that the shift between the active, reduced form and the inactive, oxidized form occurs at physiologically relevant redox potentials. The ability of the chloroplast thioredoxins to reactivate AtAMY3 strongly suggests that the redox regulation occurs at physiologically relevant redox potentials via a disulfide exchange (Fig. 7 and Table 2). Our data also provide evidence that an intramolecular disulfide bridge between residues Cys499 and Cys587 exclusively forms under oxidizing conditions. It is likely that the formation of this disulfide bridge leads to the inactivation of the enzyme by altering the tertiary structure of the protein in a way that blocks the active site. This type of mechanism was documented in the Calvin cycle enzyme, phosphoribulokinase (45). Also, in the phosphoglucan phosphatase SEX4, the catalytic Cys198 is involved in disulfide formation (34). A similar mechanism is likely for AtAMY3 due to the close proximity of Cys587 to the active site, which can be inferred from two lines of evidence. First, Cys587 is highly conserved among α-amylases (supplemental File 1), and the corresponding cysteine residue in HvAMY1 has been previously shown to be important for catalysis (46). Second, Cys587 is located two residues away from His585 of AtAMY3, which aligns with His117 of HvAMY1 (or His93 of mature protein) and functions as a transition state stabilizer. Substitution of His93 by Asn (H93N) leads to a sharp decrease in activity (27). The close proximity of Cys587 to the active site may also explain why the C587S mutant was less active than the wild-type protein (Fig. 8 and Table 3). Meanwhile, the site-directed mutant, C499S, was almost completely inactive against all substrates tested. It is possible that the Cys499 residue, like Cys587, also has a role in catalysis. Because Cys587 is located close to the active site, it is likely that Cys499 is located nearby, because disulfide bridges occur between two proximal cysteines. Other site-directed mutants with low activities included C285S and C652S, suggesting they may also contribute to the activity or stability of the protein.
Interestingly, Cys499 is generally conserved in most but not all AMY3 sequences examined. All examined sequences from species belonging to the order Malpighiales (R. communis, M. esculenta, and P. trichocarpa) instead had a leucine residue at this position (supplemental File 1). It should be noted that there are differences in how starch is metabolized between different species, and some plants do not store and degrade starch diurnally as in Arabidopsis (47). Therefore, it is possible that a loss of AMY3 redox regulation in the above-mentioned species may reflect altered regulatory requirements in their starch metabolism. Alternatively, although Cys499 was the only Cys residue that formed disulfide bridges with Cys587 in our experiments with AtAMY3, AMY3 in these Malpighiales species may be redox-regulated via a disulfide bridge between Cys587 and a different partner cysteine residue. All species that have a leucine residue instead of Cys499 also have an additional cysteine residue 11 amino acids upstream. This cysteine is not present in most AMY3 sequences, including AtAMY3. Because of its close proximity to Cys499 on the primary structure, we speculate that this may be an alternative partner cysteine for a disulfide bridge.
Redox regulation via disulfide exchange allows the reversible inactivation of the enzyme, and the inactive oxidized AtAMY3 was substantially reactivated by reduction. The loss of the 35% of AtAMY3 amylolytic activity during reactivation is most likely due to the irreversible oxidation of either regulatory cysteine to the sulfinic acid form (Cys-SO2H), which cannot be reduced by Trx or DTT included in the assay mixture. The fact that AtAMY3 is redox-regulated contrasted markedly with HvAMY1, which we found to be redox-insensitive under the same conditions (Fig. 6B). However, there is some evidence that α-amylases in the endosperm of cereal grains could be indirectly influenced by redox status (48). Therefore, redox-dependent α-amylase activity in germinating grains cannot be ruled out, and further investigations will be necessary to answer this question.
Significance of a Redox-regulated α-Amylase in Transitory Starch Degradation
In chloroplasts, the paradigm for redox regulation of target enzymes is that regulation is light-dependent, mediated by thioredoxins using electrons derived from photosystem I during photosynthesis (49). Light-activated Trxs directly reduce their target enzymes, thereby linking the availability of light to the activity of numerous enzymes (50). According to this view, redox-sensitive enzymes in chloroplasts are mainly active during the day and inhibited during the night. The light-dependent redox activation of enzymes of the Calvin cycle and starch biosynthesis by reduced Trx is intuitively relevant, as it ensures coordination between photosynthesis and accumulation of starch in response to light (51). Conversely, redox regulation of enzymes involved in starch degradation, such as GWD, SEX4, and BAM1 (31, 32, 34), is counterintuitive, as it would imply that these enzymes would be more active during periods of starch synthesis and not degradation.
In the case of AtAMY3, the broad pH optimum and the Trx-mediated redox regulation suggest that AtAMY3 activity may be promoted during the day. However, with the available evidence, it is difficult to imagine AtAMY3 playing a role in starch synthesis during the day. The amy3 mutant synthesizes the same amount of starch as wild-type plants (10), and knockouts of amy3 in combination with other enzymes involved in starch degradation result in more starch accumulation rather than less (4, 15, 16). Also, in rice, the antisense suppression of the chloroplastic AMYI-1 isoform leads to more starch in leaves (6) suggesting that chloroplastic α-amylases participate in starch breakdown.
These observations lead to speculation about the biological meaning of redox regulation of enzymes involved in the starch degradation pathway. First, redox-regulation is not necessarily a diurnal on/off switch, and pH and redox potentials are not independent of each other. The midpoint potential for GWD was −310 mV at pH 7.9, but it was calculated to shift to about −255 mV at pH 7.0, assuming the slope of Em versus pH to be 59 mV/pH unit (31). Because −255 mV is the most positive known Em value of any enzyme involved in starch degradation, the protein may not be fully oxidized during the night. Similarly, the midpoint redox potential of AtAMY3 would shift from −329 mV at pH 7.9 (Fig. 5) to −276 mV at pH 7. A previous study found that at pH 7 and −276 mV, Trx f is not completely oxidized (52). Thus, even during the night, AtAMY3 might also not be completely oxidized, and its redox regulation may be considered as a fine-tuning of activity. Second, a light-independent plastidial NADP-thioredoxin reductase (NTRC) could be involved in the redox regulation of starch-degrading enzymes at night. Unlike Trxs, NTRC uses NADPH produced via the oxidative pentose phosphate pathway as a source of reducing power (53). BAM1 was shown to be reactivated by NTRC in vitro, although it appears that NTRC is not as effective as Trx f at reactivating the enzyme (54). Thus, it will be interesting to test whether NTRC can also reactivate AtAMY3 in vitro. Finally, the presence of a redox-regulated, stress-induced starch degradation pathway has been proposed as an explanation for the Trx-induced activation of BAM1 (54). Although BAM1 has a marginal role in starch degradation in mesophyll chloroplasts (44), BAM1 expression and activity are induced by osmotic stress, and bam1 mutants fail to degrade starch in response to this stress (54). BAM1 may then induce starch degradation in the light to produce maltose as an active response against the osmotic stress (54, 55). Starch degradation in the light involving β-amylase activity has also been demonstrated under photorespiratory conditions in Arabidopsis (56). Similarly to bam1, the amy3 mutant also has a wild-type phenotype under standard growth conditions (10). However, AtAMY3 expression is increased after cold shock, and amy3 mutants accumulate more starch and less soluble sugars than in wild-type plants in response to this stress (57). Thus, redox regulation of AtAMY3 activity may become significant during stress responses.
Acknowledgments
We gratefully acknowledge Simone Wüthrich, Yolanda Joho-Auchli, and Dr. Peter Hunziker from the Functional Genomics Center Zürich for their assistance with the design, conduct, and the analysis of the MALDI-MS experiments; Dr. Sebastian Streb for critical reading of the manuscript; Dr. Sylvain Bischof and Burak Bali for assistance with preliminary experiments; and Dr. Richard Visser (Wageningen University, Netherlands) for the amylose-free potato starch granules. We also thank two anonymous reviewers for their helpful suggestions.
This work was supported by the Swiss-South African Joint Research Program Grant IZ LS Z3122916 (to D. Santelia and S. C. Z.), SNSF Marie Heim-Vögtlin Grant PMPDP3_139645 (to D. Santelia), and a Novartis-ETH Zurich Excellence Scholarship (to D. Seung).

This article contains supplemental file 1.
- GWD
- α-glucan water dikinase
- BPNP-G7
- p-nitrophenyl maltoheptaoside
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- MBTH
- - methyl-2-benzothiazolinone hydrazone
- Trx
- thioredoxin
- HPAEC-PAD
- high performance anion-exchange chromatography with pulsed amperometric detection
- DP
- degree of polymerization
- NTRC
- NADP-thioredoxin reductase.
REFERENCES
- 1. Søgaard M., Abe J., Martin-Eauclaire M. F., Svensson B. (1993) α-Amylases: structure and function. Carbohydr. Polym. 21, 137–146 [Google Scholar]
- 2. MacGregor E. A., Janecek S., Svensson B. (2001) Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim. Biophys. Acta 1546, 1–20 [DOI] [PubMed] [Google Scholar]
- 3. Stanley D., Fitzgerald A., Farnden K., MacRae E. (2002) Characterisation of putative α-amylases from apple (Malus domestica) and Arabidopsis thaliana. Biologia 57, 137–148 [Google Scholar]
- 4. Kötting O., Santelia D., Edner C., Eicke S., Marthaler T., Gentry M. S., Comparot-Moss S., Chen J., Smith A. M., Steup M., Ritte G., Zeeman S. C. (2009) STARCH-EXCESS4 is a laforin-like phosphoglucan phosphatase required for starch degradation in Arabidopsis thaliana. Plant Cell 21, 334–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robert X., Haser R., Gottschalk T. E., Ratajczak F., Driguez H., Svensson B., Aghajari N. (2003) The structure of barley α-amylase isozyme 1 reveals a novel role of domain C in substrate recognition and binding: A pair of sugar tongs. Structure 11, 973–984 [DOI] [PubMed] [Google Scholar]
- 6. Asatsuma S., Sawada C., Itoh K., Okito M., Kitajima A., Mitsui T. (2005) Involvement of α-amylase I-1 in starch degradation in rice chloroplasts. Plant Cell Physiol. 46, 858–869 [DOI] [PubMed] [Google Scholar]
- 7. Ziegler P. (1988) Partial purification and characterization of the major endoamylase of mature pea leaves. Plant Physiol. 86, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okita T. W., Greenberg E., Kuhn D. N., Preiss J. (1979) Subcellular localization of the starch degradative and biosynthetic enzymes of spinach leaves. Plant Physiol. 64, 187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kitajima A., Asatsuma S., Okada H., Hamada Y., Kaneko K., Nanjo Y., Kawagoe Y., Toyooka K., Matsuoka K., Takeuchi M., Nakano A., Mitsui T. (2009) The rice α-amylase glycoprotein is targeted from the Golgi apparatus through the secretory pathway to the plastids. Plant Cell 21, 2844–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu T.-S., Zeeman S. C., Thorneycroft D., Fulton D. C., Dunstan H., Lue W.-L., Hegemann B., Tung S.-Y., Umemoto T., Chapple A., Tsai D.-L., Wang S.-M., Smith A. M., Chen J., Smith S. M. (2005) α-Amylase is not required for breakdown of transitory starch in Arabidopsis leaves. J. Biol. Chem. 280, 9773–9779 [DOI] [PubMed] [Google Scholar]
- 11. Glaring M. A., Baumann M. J., Abou Hachem M., Nakai H., Nakai N., Santelia D., Sigurskjold B. W., Zeeman S. C., Blennow A., Svensson B. (2011) Starch-binding domains in the CBM45 family; low-affinity domains from glucan, water dikinase, and α-amylase involved in plastidial starch metabolism. FEBS J. 278, 1175–1185 [DOI] [PubMed] [Google Scholar]
- 12. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mikkelsen R., Suszkiewicz K., Blennow A. (2006) A novel type carbohydrate-binding module identified in α-glucan, water dikinases is specific for regulated plastidial starch metabolism. Biochemistry 45, 4674–4682 [DOI] [PubMed] [Google Scholar]
- 14. Zeeman S. C., Kossmann J., Smith A. M. (2010) Starch: its metabolism, evolution, and biotechnological modification in plants. Annu. Rev. Plant Biol. 61, 209–234 [DOI] [PubMed] [Google Scholar]
- 15. Streb S., Delatte T., Umhang M., Eicke S., Schorderet M., Reinhardt D., Zeeman S. C. (2008) Starch granule biosynthesis in Arabidopsis is abolished by removal of all debranching enzymes but restored by the subsequent removal of an endoamylase. Plant Cell 20, 3448–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Streb S., Eicke S., Zeeman S. C. (2012) The simultaneous abolition of three starch hydrolases blocks transient starch breakdown in Arabidopsis. J. Biol. Chem. 287, 41745–41756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delatte T., Umhang M., Trevisan M., Eicke S., Thorneycroft D., Smith S. M., Zeeman S. C. (2006) Evidence for distinct mechanisms of starch granule breakdown in plants. J. Biol. Chem. 281, 12050–12059 [DOI] [PubMed] [Google Scholar]
- 18. Kötting O., Pusch K., Tiessen A., Geigenberger P., Steup M., Ritte G. (2005) Identification of a novel enzyme required for starch metabolism in Arabidopsis leaves. The phosphoglucan, water dikinase. Plant Physiol. 137, 242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santelia D., Kötting O., Seung D., Schubert M., Thalmann M., Bischof S., Meekins D. A., Lutz A., Patron N., Gentry M. S., Allain F. H., Zeeman S. C. (2011) The phosphoglucan phosphatase Like Sex Four2 dephosphorylates starch at the C3-position in Arabidopsis. Plant Cell 23, 4096–4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCleary B. V., Sheehan H. (1987) Measurement of cereal α-amylase: A new assay procedure. J. Cereal Sci. 6, 237–251 [Google Scholar]
- 21. Anthon G. E., Barrett D. M. (2002) Determination of reducing sugars with 3-methyl-2-benzothiazolinonehydrazone. Anal. Biochem. 305, 287–289 [DOI] [PubMed] [Google Scholar]
- 22. Smith A. M., Zeeman S. C. (2006) Quantification of starch in plant tissues. Nat. Protoc. 1, 1342–1345 [DOI] [PubMed] [Google Scholar]
- 23. Ficarra R., Cutroneo P., Aturki Z., Tommasini S., Calabrò M. L., Phan-Tan-Luu R., Fanali S., Ficarra P. (2002) An experimental design methodology applied to the enantioseparation of a non-steroidal anti-inflammatory drug candidate. J. Pharm. Biomed. Anal. 29, 989–997 [DOI] [PubMed] [Google Scholar]
- 24. Goodstein D. M., Shu S., Howson R., Neupane R., Hayes R. D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N., Rokhsar D. S. (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thompson J. D., Higgins D. G., Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bak-Jensen K. S., André G., Gottschalk T. E., Paës G., Tran V., Svensson B. (2004) Tyrosine 105 and threonine 212 at outermost substrate binding subsites −6 and +4 control substrate specificity, oligosaccharide cleavage patterns, and multiple binding modes of barley α-amylase 1. J. Biol. Chem. 279, 10093–10102 [DOI] [PubMed] [Google Scholar]
- 27. Søgaard M., Kadziola A., Haser R., Svensson B. (1993) Site-directed mutagenesis of histidine 93, aspartic acid 180, glutamic acid 205, histidine 290, and aspartic acid 291 at the active site and tryptophan 279 at the raw starch binding site in barley α-amylase 1. J. Biol. Chem. 268, 22480–22484 [PubMed] [Google Scholar]
- 28. Nielsen J. W., Kramhøft B., Bozonnet S., Abou Hachem M., Stipp S. L., Svensson B., Willemoës M. (2012) Degradation of the starch components amylopectin and amylose by barley α-amylase 1: Role of surface binding site 2. Arch. Biochem. Biophys. 528, 1–6 [DOI] [PubMed] [Google Scholar]
- 29. Heldt W. H., Werdan K., Milovancev M., Geller G. (1973) Alkalization of the chloroplast stroma caused by light-dependent proton flux into the thylakoid space. Biochim. Biophys. Acta 314, 224–241 [DOI] [PubMed] [Google Scholar]
- 30. Hendriks J. H., Kolbe A., Gibon Y., Stitt M., Geigenberger P. (2003) ADP-glucose pyrophosphorylase is activated by posttranslational redox-modification in response to light and to sugars in leaves of Arabidopsis and other plant species. Plant Physiol. 133, 838–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mikkelsen R., Mutenda K. E., Mant A., Schürmann P., Blennow A. (2005) α-Glucan, water dikinase (GWD): A plastidic enzyme with redox-regulated and coordinated catalytic activity and binding affinity. Proc. Natl. Acad. Sci. U.S.A. 102, 1785–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sparla F., Costa A., Lo Schiavo F., Pupillo P., Trost P. (2006) Redox regulation of a novel plastid-targeted β-amylase of Arabidopsis. Plant Physiol. 141, 840–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sokolov L. N., Dominguez-Solis J. R., Allary A.-L., Buchanan B. B., Luan S. (2006) A redox-regulated chloroplast protein phosphatase binds to starch diurnally and functions in its accumulation. Proc. Natl. Acad. Sci. U.S.A. 103, 9732–9737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Silver D. M., Silva L. P., Issakidis-Bourguet E., Glaring M. A., Schriemer D. C., Moorhead G. B. (2013) Insight into the redox regulation of the phosphoglucan phosphatase SEX4 involved in starch degradation. FEBS J. 280, 538–548 [DOI] [PubMed] [Google Scholar]
- 35. Glaring M. A., Skryhan K., Kötting O., Zeeman S. C., Blennow A. (2012) Comprehensive survey of redox sensitive starch metabolising enzymes in Arabidopsis thaliana. Plant Physiol. Biochem. 58, 89–97 [DOI] [PubMed] [Google Scholar]
- 36. Meyer Y., Buchanan B. B., Vignols F., Reichheld J.-P. (2009) Thioredoxins and glutaredoxins: unifying elements in redox biology. Annu. Rev. Genet. 43, 335–367 [DOI] [PubMed] [Google Scholar]
- 37. Collin V., Issakidis-Bourguet E., Marchand C., Hirasawa M., Lancelin J.-M., Knaff D. B., Miginiac-Maslow M. (2003) The Arabidopsis plastidial thioredoxins: new functions and new insights into specificity. J. Biol. Chem. 278, 23747–23752 [DOI] [PubMed] [Google Scholar]
- 38. Collin V., Lamkemeyer P., Miginiac-Maslow M., Hirasawa M., Knaff D. B., Dietz K.-J., Issakidis-Bourguet E. (2004) Characterization of plastidial thioredoxins from Arabidopsis belonging to the new y-type. Plant Physiol. 136, 4088–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuriki T., Imanaka T. (1999) The concept of the α-amylase family: Structural similarity and common catalytic mechanism. J. Biosci. Bioeng. 87, 557–565 [DOI] [PubMed] [Google Scholar]
- 40. Williams J. F., Peterson M. L. (1973) Relations between α-amylase activity at and growth of rice seedlings. Crop Sci. 13, 612–615 [Google Scholar]
- 41. Ziegler P. (1999) Cereal β-Amylases. J. Cereal Sci. 29, 195–204 [Google Scholar]
- 42. Scialdone A., Mugford S. T., Feike D., Skeffington A., Borrill P., Graf A., Smith A. M., Howard M. (2013) Arabidopsis plants perform arithmetic division to prevent starvation at night. eLife 2, e00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stitt M., Zeeman S. C. (2012) Starch turnover: pathways, regulation and role in growth. Curr. Opin. Plant Biol. 15, 282–292 [DOI] [PubMed] [Google Scholar]
- 44. Fulton D. C., Stettler M., Mettler T., Vaughan C. K., Li J., Francisco P., Gil M., Reinhold H., Eicke S., Messerli G., Dorken G., Halliday K., Smith A. M., Smith S. M., Zeeman S. C. (2008) β-AMYLASE4, a noncatalytic protein required for starch breakdown, acts upstream of three active β-amylases in Arabidopsis chloroplasts. Plant Cell 20, 1040–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brandes H. K., Larimer F. W., Hartman F. C. (1996) The molecular pathway for the regulation of phosphoribulokinase by thioredoxin f. J. Biol. Chem. 271, 3333–3335 [DOI] [PubMed] [Google Scholar]
- 46. Mori H., Bak-Jensen K. S., Gottschalk T. E., Motawia M. S., Damager I., Møller B. L., Svensson B. (2001) Modulation of activity and substrate binding modes by mutation of single and double subsites +1/+2 and −5/−6 of barley α-amylase 1. Eur. J. Biochem. 268, 6545–6558 [DOI] [PubMed] [Google Scholar]
- 47. Zeeman S. C., Smith S. M., Smith A. M. (2007) The diurnal metabolism of leaf starch. Biochem. J. 401, 13–28 [DOI] [PubMed] [Google Scholar]
- 48. Wong J. H., Kim Y.-B., Ren P.-H., Cai N., Cho M.-J., Hedden P., Lemaux P. G., Buchanan B. B. (2002) Transgenic barley grain overexpressing thioredoxin shows evidence that the starchy endosperm communicates with the embryo and the aleurone. Proc. Natl. Acad. Sci. U.S.A. 99, 16325–16330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Buchanan B. B., Balmer Y. (2005) Redox regulation: a broadening horizon. Annu. Rev. Plant Biol. 56, 187–220 [DOI] [PubMed] [Google Scholar]
- 50. Lemaire S. D., Michelet L., Zaffagnini M., Massot V., Issakidis-Bourguet E. (2007) Thioredoxins in chloroplasts. Curr. Genet. 51, 343–365 [DOI] [PubMed] [Google Scholar]
- 51. Schürmann P., Buchanan B. B. (2008) The ferredoxin/thioredoxin system of oxygenic photosynthesis. Antioxid. Redox Signal. 10, 1235–1274 [DOI] [PubMed] [Google Scholar]
- 52. Hirasawa M., Schürmann P., Jacquot J. P., Manieri W., Jacquot P., Keryer E., Hartman F. C., Knaff D. B. (1999) Oxidation-reduction properties of chloroplast thioredoxins, ferredoxin:thioredoxin reductase, and thioredoxin f-regulated enzymes. Biochemistry 38, 5200–5205 [DOI] [PubMed] [Google Scholar]
- 53. Serrato A. J., Pérez-Ruiz J. M., Spínola M. C., Cejudo F. J. (2004) A novel NADPH thioredoxin reductase, localized in the chloroplast, which deficiency causes hypersensitivity to abiotic stress in Arabidopsis thaliana. J. Biol. Chem. 279, 43821–43827 [DOI] [PubMed] [Google Scholar]
- 54. Valerio C., Costa A., Marri L., Issakidis-Bourguet E., Pupillo P., Trost P., Sparla F. (2011) Thioredoxin-regulated β-amylase (BAM1) triggers diurnal starch degradation in guard cells, and in mesophyll cells under osmotic stress. J. Exp. Bot. 62, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaplan F., Sung D. Y., Guy C. L. (2006) Roles of β-amylase and starch breakdown during temperatures stress. Physiol. Plant. 126, 120–128 [Google Scholar]
- 56. Weise S. E., Schrader S. M., Kleinbeck K. R., Sharkey T. D. (2006) Carbon balance and circadian regulation of hydrolytic and phosphorolytic breakdown of transitory starch. Plant Physiol. 141, 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kaplan F., Guy C. L. (2005) RNA interference of Arabidopsis β-amylase 8 prevents maltose accumulation upon cold shock and increases sensitivity of PSII photochemical efficiency to freezing stress. Plant J. 44, 730–743 [DOI] [PubMed] [Google Scholar]