

Background: Activation of p38 MAPK by poxviral A52 is TRAF6-dependent, but the mechanism is unknown.
Results: Disruption of either an identified TRAF6-binding motif or of A52 dimerization prevents TRAF6 oligomerization, TAK1 recruitment, and p38 activation.
Conclusion: A52 activates p38 MAPK activation by causing TRAF6 oligomerization, leading to TAK1 recruitment.
Significance: This work reveals the molecular basis for poxviral activation of p38 MAPK.
Keywords: NF-kappa B (NF-KB), p38 MAPK, Pox Viruses, Signal Transduction, Viral Protein, TRAF6
Abstract
Vaccinia virus encodes a number of proteins that inhibit and manipulate innate immune signaling pathways that also have a role in virulence. These include A52, a protein shown to inhibit IL-1- and Toll-like receptor-stimulated NFκB activation, via interaction with interleukin-1 receptor-associated kinase 2 (IRAK2). Interestingly, A52 was also found to activate p38 MAPK and thus enhance Toll-like receptor-dependent IL-10 induction, which was TRAF6-dependent, but the manner in which A52 manipulates TRAF6 to stimulate p38 activation was unclear. Here, we show that A52 has a non-canonical TRAF6-binding motif that is essential for TRAF6 binding and p38 activation but dispensable for NFκB inhibition and IRAK2 interaction. Wild-type A52, but not a mutant defective in p38 activation and TRAF6 binding (F154A), caused TRAF6 oligomerization and subsequent TRAF6-TAK1 association. The crystal structure of A52 shows that it adopts a Bcl2-like fold and exists as a dimer in solution. Residue Met-65 was identified as being located in the A52 dimer interface, and consistent with that, A52-M65E was impaired in its ability to dimerize. A52-M65E although capable of interacting with TRAF6, was unable to cause either TRAF6 self-association, induce the TRAF6-TAK1 association, or activate p38 MAPK. The results suggest that an A52 dimer causes TRAF6 self-association, leading to TAK1 recruitment and p38 activation. This reveals a molecular mechanism whereby poxviruses manipulate TRAF6 to activate MAPKs (which can be proviral) without stimulating antiviral NFκB activation.
Introduction
Immune detection of pathogens such as viruses relies on activation of cellular pattern recognition receptors (PRRs)2 by pathogen-associated molecular patterns, which leads to altered gene expression and appropriate immune responses. In particular, PRRs activate the transcription factors NFκB and IRF3 (IFN regulatory factor 3) causing induction of cytokines and type I IFNs. To evade and overcome such immune detection, viruses have evolved to encode proteins that can inhibit PRR signaling (1). In particular, poxviruses such as vaccinia virus (VACV) have been shown to encode a rich repertoire of immunomodulatory proteins, which act intracellularly to target both PRR complexes and downstream signaling components, often leading to inhibition of NFκB and IRF3 activation. Such poxviral PRR inhibitors include A46 (2), A52 (3), N1 (4), B14 (5), K7 (6) C6 (7), and A49 (8).
The Toll-like receptors (TLRs) were the first discovered and to date the best characterized family of PRRs. TLRs, which are localized on the plasma membrane, mainly detect microbial membrane components, including viral glycoproteins, whereas TLRs, which are localized in endosomes, detect viral nucleic acid (9). Signal transduction is initiated by ligand-induced dimerization of TLRs, leading to homotypic interactions between the intracellular receptor Toll/IL-1R (TIR) domains. TIR domain-containing adaptor proteins such as MyD88, Mal, TIR domain-containing adaptor inducing IFN-β (TRIF), and TRIF-related adaptor molecule are then recruited to a TIR domain signaling platform (10). MyD88 is the critical adaptor for signal transduction of all TLRs except for TLR3 (11). MyD88 contains a death domain at the N terminus, and it interacts with the downstream interleukin-1 receptor-associated kinase (IRAK) proteins, IRAK1, IRAK2, IRAK4, and IRAK-M via homotypic death domain interactions (12). IRAK4 is initially recruited to MyD88, and then IRAK1 and/or IRAK2 are phosphorylated by activated IRAK4 (13). IRAKs then mediate activation of MAP kinases and transcription factors, mainly via TNF receptor-associated factor 6 (TRAF6) (14).
The poxviral proteins A46 and A52 specifically disrupt TLR signaling and also affect IL-1 responses because the TLR and the IL-1R signaling pathways share common components that A46 and A52 target (2, 3, 15, 16). Importantly, non-redundant roles for A46 and A52 in VACV virulence have been demonstrated in earlier studies (2) (3). A46 disrupts TLR and IL-1R complexes by interacting with the conserved TIR domain found in both TLRs and the IL-1R (2), which in the case of TLR4 prevents receptor-adaptor interactions (15). Intracellular expression of A46 inhibits TLR- and IL-1R-mediated MAP kinase and transcription factor activation (2).
In contrast, A52, a 23-kDa, 190-amino acid intracellular protein encoded by the immediate early VACV gene A52R (17), acts further “downstream” of the receptor complex than A46, by binding to IRAK2, leading to inhibition of TLR- and IL-1R-mediated NFκB activation (18, 19). Similar to many viral immunomodulatory proteins, A52 has more than one function, and as well as exerting an inhibitory effect on IL-1R/TLR-mediated NFκB activation, A52 activates p38 MAP kinase in an IRAK2-independent manner (16). The ability of A52 to activate p38 MAP kinase depends on TRAF6 (16), a protein also required by IL-1, TLRs, and TGFβ for MAP kinase activation (20). A52-mediated p38 MAP kinase activation via cellular TRAF6 likely represents an immune subversion strategy whereby the virus gains a selective advantage by fine-tuning the signaling environment of an infected cell, for example to potentiate TLR-induced production of IL-10, a cytokine that inhibits inflammatory and cell-mediated immune responses and contributes to viral persistence (16). In fact, there are a number of examples of viral manipulation of signaling pathways via TRAFs for herpes viruses (21, 22) and for rotavirus (23). However, the mechanism whereby poxviral A52 subverts TRAF6 to activate p38 is unknown.
TRAF6 is an E3 ubiquitin ligase, which forms a complex with E2 ubiquitin-conjugating enzymes such as UBC13 and UEV1A (ubiquitin-conjugating enzyme variant 1A). For IL-1 and TLR signaling via TRAF6, the generation of polyubiquitin chains by TRAF6 is essential for the recruitment of the downstream kinase TGFβ-activated kinase 1 (TAK1), which is in a complex with the ubiquitin-binding TAK1-binding proteins, TAB1, -2, and -3 (24, 25). TAK1 subsequently activates distinct pathways leading to NFκB activation via the IκB kinase (IKK) complex, and MAP kinase activation via a MAP kinase cascade. For p38 activation, TAK1 activates MKK3 and MKK6, which then phosphorylate p38 (20).
Here we show that A52 has a non-canonical TRAF6 binding motif, which is essential for TRAF6 binding and p38 activation but dispensable for NFκB inhibition and IRAK2 binding. Furthermore, A52 directs TRAF6 to activate p38 by causing TRAF6 oligomerization and subsequent TAK1 recruitment. TRAF6 oligomerization, TAK1 recruitment, and p38 activation were all dependent on the ability of A52 to dimerize. This reveals a molecular mechanism of immune subversion by VACV whereby an A52 dimer sequesters TRAF6 to cause proviral p38 MAP kinase activation without antiviral NFκB activation.
EXPERIMENTAL PROCEDURES
Cell Culture
Wild-type and TRAF6−/− mouse embryonic fibroblasts (MEFs) were a gift from Z. J. Chen (University of Texas Southwestern Medical Center, Dallas, TX). HEK 293T cells were purchased from European Collection of Animal Cell Cultures (Salisbury, UK). HEK293 cells stably transfected with IL-1R (HEK293-R1) were a gift from Tularik (San Francisco, CA). HEK293 cells stably transfected with TLR4, MD2, and CD14 (HEK293-TLR4) were purchased from InvivoGen (San Diego, CA). Cells were maintained in DMEM containing 10% (v/v) FCS, 10 μg/ml ciproflaxin, and 2 mm l-glutamine. 10 μg/ml blasticidin (Sigma) and 50 μg/ml HygroGold (InvivoGen) were used as selection agents in the culture of HEK293-TLR4 cells.
Receptor Agonists
LPS from Escherichia coli (99.9% pure in respect to contaminating protein, DNA, and TLR2 agonists) was purchased from Alexis Biochemicals (Plymouth Meeting, PA). Interleukin-1α (IL-1α) was obtained from the National Cancer Institute (Frederick, MD).
Antibodies
Rabbit anti-Myc monoclonal, phospho-p38 MAP kinase (Thr-180/Tyr-182) and total p38 MAP kinase polyclonal antibodies were purchased from Cell Signaling Technology (Danvers, MA). Monoclonal anti-FLAG M2 antibody, mouse anti-Myc monoclonal antibody (clone 9E10), and monoclonal anti-β-actin antibody (clone AC-74) were all purchased from Sigma-Aldrich. The TRAF6 rabbit polyclonal antibody was purchased from Santa Cruz Biotechnology. For detection of endogenous IRAK2, a rabbit polyclonal antibody was generated using the following IRAK2 peptide as an antigen: (NHCOCH3)-CADVYRGHRHGKPFVFK-(CONH2) (Inbiolabs, Estonia). Mouse anti-HA monoclonal antibody was purchased from Covance (Princeton, NJ). Antibodies against GST-A52, encoded by a plasmid synthesized by inserting full-length A52 downstream of GST in the bacterial expression vector pGEX4T2 were raised as described (3). All HRP-conjugated secondary antibodies were purchased from Sigma-Aldrich.
Plasmids
Sources of expression plasmids were as follows: pCMV-Myc empty vector (Clontech), phRL-TK vector (Promega), pFR-luciferase reporter gene construct, CHOP-Gal4 and c-Jun-Gal4 (Stratagene), Myc-IRAK2 (M. Muzio, Mario Negri Institute, Milan, Italy), FLAG-TAK1 and dominant negative FLAG-TAK1 K63W (H. Sakurai, Tanabe Seiyaku Co., Osaka, Japan), Myc-MKK3 S189A/T193A (MKK3 DN), and Myc-MKK6 S207A/T211A (MKK6 DN, P. Cohen, University of Dundee, Scotland). The VACV ORF A52 was cloned previously by PCR amplification from Western reserve strain VACV DNA (18). The A52 TRAF6 binding mutant (A52ΔT6BM) and the A52 mutants M65E, R149A, E151A, K152A, and F154A were generated using the QuikChange mutagenesis kit (Stratagene). Myc-A52 and Myc-TRAF6 were synthesized by inserting full-length A52 or TRAF6 (FLAG-TRAF6 was originally obtained from Tularik, San Francisco, CA) into the mammalian expression vector pCMV-Myc. The NFκB luciferase reporter construct (18) and the human IL-10 promoter reporter plasmid (16) have been described previously.
Reporter Gene Assays
Cells (2 × 104 cells per well unless otherwise indicated) were seeded into 96-well plates and transfected 24 h later with expression vectors and luciferase reporter genes using GeneJuice (Novagen). In all cases, 20 ng/well of phRL-TK reporter gene was cotransfected to normalize data for transfection efficiency. For the NFκB assays, 60 ng of κB-luciferase reporter gene was used. For IL-10 promoter assays, 60 ng of the IL-10 promoter luciferase reporter gene was used. For the p38 MAP kinase reporter assay, the PathDetect SystemTM (Stratagene) was used, whereby 0.25 ng of a CHOP-Gal4 fusion vector was used in combination with 60 ng of pFR-luciferase reporter. For the JNK MAP kinase reporter assay, 0.25 ng of c-Jun-Gal4 fusion vector was used with 60 ng of pFR-luciferase reporter. The total amount of DNA per transfection was kept constant at 230 ng by addition of pCMV-Myc. Unless otherwise indicated, 100 ng of plasmid encoding A52 or A52 mutant were used. After 24 h, cells were stimulated with either 50 ng/ml IL-1α or 100 ng/ml LPS. After a further 8 h, cells were lysed in Passive Lysis Buffer (Promega), and whole cell lysates were analyzed for luciferase activity. IL-10 promoter assays were harvested 48 h after transfection. Firefly luciferase activity was normalized to Renilla luciferase activity, and data are expressed as the mean fold induction, relative to control levels, for a representative experiment from a minimum of three separate experiments, each performed in triplicate.
Immunoprecipitation and Immunoblotting
HEK293T cells were seeded into 15-cm dishes (3 × 106 cells) 24 h before transfection with GeneJuice. For co-immunoprecipitations, 4 μg of each construct was transfected. The total amount of DNA per transfection was kept constant by addition of pCMV-Myc plasmid. Cells were harvested after 48 h in 850 μl of lysis buffer (50 mm Hepes, pH 7.5, 250 mm NaCl, 1 mm EDTA, 10% glycerol, 1% Nonidet P-40 containing 0.01% aprotinin, 1 mm sodium orthovanadate, and 1 mm PMSF added freshly) for 30 min on ice. For immunoprecipitation of ubiquitinated proteins, lysis buffer was supplemented with 10 mm iodoacetamide. Whole cell lysates were clarified by centrifugation. 50 μl of cleared lysate was retained for analysis of protein expression (i.e. input lysate). For all co-immunoprecipitations, the appropriate antibodies, along with either protein A or protein G-Sepharose (Sigma), were incubated with the lysates for 2 h at 4 °C. The immune complexes were precipitated, washed four times in lysis buffer, and analyzed by SDS-PAGE and immunoblotting.
Determination of Cytokine Concentrations
HEK 293-TLR4 cells (2 × 104 cells per well) were seeded into 96-well plates and transfected 24 h later with 180 ng of either empty vector, A52- or F154A-expressing plasmid using GeneJuice (Novagen). The following day, the indicated wells were stimulated with 50 ng/ml LPS. The supernatants were collected 24 h post-stimulation and assessed for IL-8 production by ELISA (R&D Systems, Minneapolis, MN). Experiments were performed three times in triplicate, and data are expressed as mean ± S.D. from one representative experiment.
Native Gel Electrophoresis
TRAF6 self-association was examined by native PAGE. HEK293-TLR4 cells were seeded into 15-cm dishes (3 × 106 cells) 24 h before transfection with GeneJuice. Cells were stimulated with LPS 48 h after transfection and harvested by scraping. Cells were lysed for 1 h on ice in 200 μl of lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 30 mm NaF, 5 mm EDTA, 10% glycerol, 1 mm Na3VO4, 40 mm β-glycerophosphate, 1% Triton X-100 with 0.1 mm PMSF, 5 μg/ml leupeptin, pepstatin, and aprotinin freshly added). Whole cell lysates were clarified by centrifugation. 100 μl of cleared lysate was retained for analysis of protein expression (i.e. input lysate) by SDS-PAGE. Before sample loading, 7% polyacrylamide gels were pre-run for 60 min at 40 mA with 25 mm Tris, 192 mm glycine (pH 8.4) with and without 1% deoxycholate in the cathode and anode chamber, respectively. Cleared lysates in native sample buffer (10 mg protein, 62.5 mm Tris-Cl, pH 6.8, 15% glycerol, and 1% deoxycholate) were applied to the gels, and proteins were separated by electrophoresis for 2 h at 40 mA. Immunoblotting was performed using standard conditions.
Statistical Analysis and Densitometry
Statistical analysis was carried out using a paired Students t test. Densitometry was performed using ImageJ software, and values were normalized for protein loading as indicated.
RESULTS
A52 Has a Non-canonical TRAF6-binding Domain Required for p38 Activation
We previously showed that expression of A52 caused p38 MAPK activation, that this was inhibited by overexpression of dominant negative TRAF6, and that A52 could be co-immunoprecipitated with TRAF6 (16). Here, we confirmed that endogenous TRAF6 was required for A52-induced MAPK activation using wild-type and TRAF6−/− MEFs. When A52 was transfected into wild-type MEFs, increased phosphorylation of p38 was detected by Western blot, compared with cells transfected with empty vector (Fig. 1A). A52 also increased the phosphorylation of the transcription factor ATF-2, a substrate of p38, and of the MAP kinase JNK (Fig. 1A). A52-dependent phosphorylation of p38, ATF-2, and JNK were clearly TRAF6-dependent, as assessed by densitometric analysis of the protein bands detected in TRAF6−/− MEFs compared with wild-type cells (Fig. 1A). Activation of p38 was also measured by the PathDetect® CHOP trans-reporting system that is based on the ability of p38 MAP kinase to phosphorylate and activate the transcription factor CHOP. This is assayed by an increase in the ability of the Gal4-CHOP fusion protein to transactivate the pFR luciferase reporter, which contains Gal4 binding sites in its promoter. Fig. 1B shows that expression of A52 caused p38 MAPK activation in wild-type MEFs, which was absent in MEFs lacking TRAF6. Thus, A52 induced p38 MAPK activation is entirely dependent on the presence of TRAF6.
FIGURE 1.

A52 has a non-canonical TRAF6-binding domain required for p38 activation. A, MEFs from wild-type and TRAF6 knock-out mice were seeded into six-well plates (4 × 105 cells) 24 h before transfection with 2 μg of empty vector or Myc-A52. Where indicated, cells were stimulated with 50 ng/ml IL-1α for 30 min 24 h after transfections. Lysates were analyzed by SDS-PAGE, and immunoblotting was performed. Immunoblots were subjected to densitometric analysis with levels of P-ATF2 and P-JNK normalized to β-actin and levels of p-p38 normalized to p38. B, wild-type or TRAF6−/− MEFs were transfected with 150 ng of Myc-A52 or pCMV-Myc empty vector (EV), along with the pFR luciferase reporter gene and CHOP-Gal4. Luciferase activity was measured 24 h after transfection. C, presence of PxExx(Ar/Ac) motifs in TRAF6-binding proteins compared with A52. D–G, HEK293-R1 cells were transfected for 24 h with either the pFR luciferase reporter gene and CHOP-Gal4 (D and E) or the NFκB luciferase reporter gene (F and G). Cells were co-transfected with either 150 ng of Myc-A52, A52ΔT6BM, or pCMV-Myc (EV) (D and F) or 100 ng of Myc-A52 with 50 ng of FLAG-TRAF6 (E and G). Cells were stimulated with 50 ng/ml IL-1α for 6 h where indicated, and luciferase reporter gene activity was measured. The data are mean ± S.D. of triplicate samples and are representative of at least three separate experiments. *, p < 0.05; **, p < 0.005; or ***, p < 0.0005 compared with EV alone.
A52 has been shown to co-immunoprecipitate with the TRAF domain of TRAF6 (3). Many proteins that interact with TRAF6 via its TRAF domain have been shown to contain the motif, PXEXX(Ac/Ar) (where Ar is an aromatic and Ac an acidic residue, Fig. 1C) (26). Because the C-terminal 46 amino acids were required for the A52-TRAF6 association (16), this sequence of A52 was examined for the presence of potential TRAF6 binding motifs. One such putative TRAF6 binding motif was discovered at amino acid residues 149–154: RNEKLF (Fig. 1C). This differed from the canonical TRAF6 binding motif in having an Arg (Arg-149) rather than a Pro in the first position (Fig. 1C). However, other proteins have been demonstrated to bind TRAF6 through non-canonical TRAF6 binding domains, including motifs that lack a Pro in position 1 (27–29). Therefore, we tested whether residues 149–154 in A52 were required for TRAF6-dependent p38 activation by generating a construct encoding a deletion mutant of A52 (A52ΔT6BM) that lacked residues 149–154. Wild-type A52 and the A52ΔT6BM mutant protein were ectopically expressed in HEK293 cell stably expressing the IL-1R1 (HEK293-R1), and their ability to activate p38 was compared. Wild-type A52 activated p38 to a similar level as that observed when the cells were stimulated with IL-1 (Fig. 1D, left panel). In contrast, the A52ΔT6BM mutant was completely impaired in its ability to activate p38 (Fig. 1D, right panel), even though both proteins were expressed at similar levels (data not shown), indicating that this region of A52 is vital for its ability to drive MAPK activation. In agreement with these data, the presence of A52 enhanced TRAF6-dependent p38 activation (Fig. 1E). In contrast, the known ability of A52 to inhibit NFκB (16) was not affected by disrupting the putative TRAF6 binding motif because similar to wild-type A52 protein, expression of the A52ΔT6BM mutant inhibited IL-1-dependent NFκB activation (Fig. 1F). A52 could also impair NKκB activation stimulated by overexpression of TRAF6 (Fig. 1G). Thus, residues 149–154, which resemble a TRAF6 binding motif, are required for the ability of A52 to activate p38 but are dispensable for the ability of A52 to inhibit NFκB.
Phe-154 in A52 Is Essential for p38 Activation and TRAF6 Binding but Dispensable for NFκB Inhibition and IRAK2 Binding
To examine the relative importance of the individual residues within the putative TRAF6 binding domain for A52 function, four point mutations of A52 were generated: R149A, E151A, K152A, and F154A (Fig. 2A), all of which were found to be expressed at similar levels to wild-type A52 (data not shown). All of the mutants retained the ability to inhibit IL-1-induced NFκB activation, albeit to different levels (Fig. 2B). When the A52 point mutants were tested for their ability to activate p38 MAPK, R149A, E151A, and K152A activated p38 to the same extent as the wild-type protein (Fig. 2C). However, F154A lost the ability to activate p38 (Fig. 2C), indicating that this residue is critical to the ability of A52 to activate p38 via TRAF6. A52-induced JNK activation, which is also TRAF6-dependent (Fig. 1A), was also prevented by mutation of Phe-154 to Ala-154, as measured by the PathDetect® c-Jun trans-reporting system (Fig. 2D).
FIGURE 2.

A52 F154A cannot activate p38 MAPK activation nor bind TRAF6. A, four point mutations were made in the putative TRAF6-binding motif of A52. HEK293-R1 cells were transfected for 24 h with 150 ng of Myc-A52 or empty vector (EV), along with either the NFκB luciferase reporter gene (B) or the pFR luciferase reporter gene and CHOP-Gal4 (C). Cells were stimulated with 50 ng/ml IL-1α for 6 h, and luciferase reporter gene activity was measured. The data are presented as percentage remaining of fold induction (B) or percentage of fold induction (C) and are representative of at least three experiments. D, HEK293-R1 cells were transfected with the pFR luciferase reporter gene and c-Jun-Gal4 with either 150 ng of Myc-A52, A52-F154A, or pCMV-Myc (EV). Luciferase reporter gene activity was measured 24 h after transfection. E and F, HEK293T cells were transfected with Myc-A52 and Myc-A52 F154A. After 48 h, lysates were subject to immunoprecipitation, SDS-PAGE, and immunoblotting (IB) with the indicated antibodies. HC, heavy chain of anti-TRAF6 antibody. G, HEK293-TLR4 cells were transfected for 24 h with increasing amounts of Myc-A52 and Myc-A52-F154A, or empty vector (EV), along with the NFκB luciferase reporter gene. Cells were stimulated with 100 ng/ml LPS for 6 h, and luciferase reporter gene activity was measured. H, HEK293-TLR4 cells were transfected with Myc-A52, Myc-A52-F154A, or pCMV-Myc (EV). 24 h later, cells were stimulated with 50 ng/ml LPS for a further 24 h, after which the supernatants were assessed for IL-8 production by ELISA. The data are mean ± S.D. of triplicate samples and are representative of at least three separate experiments. *, p < 0.05; **, p < 0.005; or ***, p < 0.0005 compared with EV (B, D, G, and H) or A52 (C).
Given that Phe-154 was in the putative TRAF6 binding motif and was also essential for TRAF6-dependent p38 and JNK MAPK activation, an immunoprecipitation experiment was performed where the abilities of wild-type A52 and F154A to interact with endogenous TRAF6 were compared. A52 and F154A were expressed at similar levels (Fig. 2E, fourth panel). Although A52 interacted with endogenous TRAF6 (Fig. 2E, top panel, lane 1), F154A was unable to interact with TRAF6 (Fig. 2E, top panel, lane 2). This difference was not due to different amounts of TRAF6 being immunoprecipitated, as similar levels of TRAF6 were observed in the two immunoprecipitates (Fig. 2E, second panel). Earlier studies have demonstrated that A52 can interact with IRAK2 (3) and impairs NFκB by targeting IRAK2 (19). Consistent with the fact that F154A can still impair IL-1-induced NFκB signaling (Fig. 2B), F154A interacted with endogenous IRAK2 to the same extent as wild-type A52 (Fig. 2F, top panel). Thus, as would be expected, both wild-type A52 and F154A inhibited TLR4-dependent NFκB activation (Fig. 2G) and TLR4-dependent production of the NFκB-regulated chemokine IL-8 (Fig. 2H). In addition, both wild-type A52 and the F154A mutant were found to inhibit poly(I:C)/TLR3-induced NFκB activation (data not shown). Together, the data demonstrate that Phe-154 is essential for TRAF6 binding and p38 activation but dispensable for IRAK2 binding and NFκB inhibition.
A52, but Not Mutant F154A, Enhances TRAF6 Self-association
We next wondered how A52 binding to TRAF6 via Phe-154 would mobilize TRAF6 to cause p38 MAP kinase activation. Host signal transduction pathways that utilize TRAF6 in p38 activation variously cause TRAF6 oligomerization, TRAF6-dependent formation of polyubiquitin chains, and recruitment of downstream kinases such as TAK1 (20, 30–32). We therefore tested the ability of A52 versus F154A to cause these TRAF6-dependent events. Fig. 3A shows that, as reported previously (33), LPS stimulation of cells caused a fraction of TRAF6 within cells to oligomerize, as detected by the appearance of a TRAF6 band of reduced mobility on a native gel (top panel, compare lane 2 to lane 1). This correlated with the activation of p38, as measured by the appearance of phospho-p38 in cell lysates (Fig. 3A, middle panel, lane 2). When cells were transfected with A52, TRAF6 oligomerization was also observed (Fig. 3A, top panel, lane 3), as was the appearance of phospho-p38 (Fig. 3A, middle panel, lane 3). Interestingly, expression of equal amounts of the F154A mutant failed to cause TRAF6 oligomerization (Fig. 3A, top panel, lane 4) or p38 phosphorylation (Fig. 3A, second panel, lane 4). Thus, activation of p38 by A52 correlates with the induced TRAF6 self-assembly, both of which are dependent on Phe-154.
FIGURE 3.

A52, but not A52-F154A, enhances TRAF6 self-association and causes TRAF6-TAK1 association. A, HEK293-TLR4 cells were transfected with empty vector (EV), Myc-A52, or Myc-A52-F154A. Cells were stimulated with 100 ng/ml LPS for 30 min 48 h after transfection. Lysates were analyzed by either native gel electrophoresis or SDS-PAGE. Immunoblotting was performed using standard conditions. Immunoblots (IB) were subjected to densitometric analysis with levels of the TRAF6 oligomer and phospho-p38 normalized to total levels of TRAF6 monomer and p38, respectively. B and C, HEK293T cells were transfected for 24 h with 4 μg of A52, A52-F154A, IRAK2, or HA-ubiquitin (Ub) as indicated. Endogenous TRAF6 was immunoprecipitated from cell lysates, and levels of ubiquitinated TRAF6 were assessed by immunoblotting with anti-HA antibody. HC, heavy chain of anti-TRAF6 antibody. D, HEK293T cells were transfected with Myc-TRAF6, FLAG-TAK1, HA-A52, and HA-A52 F154A. After 48 h, lysates were subject to immunoprecipitation, SDS-PAGE, and immunoblotting with the indicated antibodies. E, HEK293-Ts were transfected with 50 ng of Myc-A52 or pCMV-Myc (EV) and 100 ng of either TAK1 DN, MKK3 DN, or MKK6 DN, along with the pFR luciferase reporter gene and CHOP-Gal4. Luciferase reporter gene activity was measured 24 h after transfection. The data are mean ± S.D. of triplicate samples and are representative of at least three separate experiments.
A52 Does Not Enhance TRAF6-associated Ubiquitination
TRAF6 is an E3 ubiquitin ligase, and TRAF6-associated polyubiquitination is a critical step in the activation of NFκB and p38 by TLRs (30). Previous studies have demonstrated that expression of wild-type IRAK2 is sufficient to trigger TRAF6-associated polyubiquitination and NFκB and p38 activation (19). Fig. 3B shows an ubiquitination assay where association of HA-ubiquitin with endogenous TRAF6 was clearly induced by IRAK2 expression (top panel, lane 7). As has been seen in an earlier published work (19), the detection of immunoprecipitated TRAF6 was reduced upon triggering of TRAF6 ubiquitination (Fig. 3B, lane 7, second panel), likely due to the presence of conjugated ubiquitin blocking antibody recognition of TRAF6 epitopes. In contrast to IRAK2, neither A52 nor F154A when overexpressed simulated TRAF6 ubiquitination (Fig. 3B, top panel, lanes 4 and 5), even though expression levels of IRAK2, A52, and F154A were similar (Fig. 3B, fourth and fifth panels, lanes 3–7). This was consistent with the fact that unlike IRAK2 expression, A52 expression does not stimulate NFκB and also suggests that TRAF6-associated polyubiquitination is not required for A52 to stimulate p38 MAPK activation. In fact, consistent with the ability of A52 to inhibit IRAK2 function, A52 caused some inhibition of IRAK-2-stimulated TRAF6 polyubiquitination (Fig. 3C, top panel, compare lane 6 with lane 5).
A52, but Not Mutant F154A, Triggers TRAF6-TAK1 Association
A critical signaling molecule downstream of TRAF6 is TAK1, a MAP3K that activates the MAPKKs, MKK3 and MKK6, which subsequently phosphorylate p38 (34). Because A52 requires TRAF6 to activate MAPKs, it was feasible that A52 might do this by stimulating the recruitment of TAK1 to TRAF6. To test this, TRAF6 and TAK1 were ectopically expressed in HEK293T cells. When expressed together, they did not interact with each other (Fig. 3D, top panel, lane 3). However, when A52 was also introduced, a TRAF6-TAK1 association was detected (Fig. 3D, top panel, lane 4). Importantly, mutant F154A, which did not associate with TRAF6 or cause TRAF6 self-association, did not induce the TRAF6-TAK1 interaction (Fig. 3D, top panel, lane 5).
Furthermore, the A52-stimulated p38 activation was inhibited by co-expression of dominant negative versions of TAK1, MKK3 or MKK6 (Fig. 3E), indicating the downstream involvement of these kinases in the A52-dependent MAPK pathway. Thus, A52 activated p38 via stimulation of recruitment of TAK1 to TRAF6 and subsequent signaling via MKKs.
A52 Dimerization Is Required to Stimulate a TRAF6-TAK1 Interaction and p38 Activation
A52 adopts an α-helical Bcl-2-like fold and forms a stable homodimer in solution (35). The dimer interface comprises the α-helices, α-1 and α-6, which are conserved among the dimeric Bcl-2 superfamily. Salt bridges, hydrogen bonds, and hydrophobic interactions are predicted to mediate A52 dimerization (Fig. 4A). The Phe-154 residue sits at the edge of the A52 dimer interface (Fig. 4B), suggesting that Phe-154 may be involved in dimer formation as well as TRAF6 binding. Indeed, co-immunoprecipitation experiments showed a clear self-association between Myc- and HA-tagged wild-type A52 (Fig. 4C, top panel, lane 1). However, A52-F154A was unable to self-associate under the same experimental conditions (Fig. 4C, top panel, lane 2).
FIGURE 4.

The dimerization mutant, A52-M65E, can still interact with TRAF6 but cannot cause TRAF6-TAK1 association or TRAF6 self-association. A, schematic diagram of residue interactions across the A52 dimer interface generated by PDBsum (48). Parallel lines represent non-bonded contacts, and solid lines represent hydrogen bonds. For non-bonded contacts, the width of the parallel line is proportional to the number of atomic contacts. B, dimer interface of A52 homodimer generated using ICM Molsoft browser. The Phe-154 and Met-65 residues are represented as red and blue stick models, respectively. The dimer interface comprises of the N terminus (N), α1, and α6 of the Bcl-2 fold domain. C, HEK293T cells were transfected with 4 μg each of Myc-A52 and HA-A52 (lane 1), Myc-A52 F154A, and HA-A52 F154A (lane 2), or Myc-A52 M65E, and HA-A52 M65E (lane 3). After 48 h, lysates were subject to immunoprecipitation, SDS-PAGE, and immunoblotting (IB) with the indicated antibodies. Immunoblots were subjected to densitometric analysis with levels of co-immunoprecipitated HA-A52, HA-A52 F154A, and HA-A52 M65E normalized to total levels of immunoprecipitated Myc-A52, Myc-A52 F154A, and Myc-A52 M65E, respectively. D, HEK293T cells were transfected with 4 μg each of HA-A52 M65E, Myc-TRAF6, and Myc-IRAK2. After 48 h, lysates were subject to immunoprecipitation, SDS-PAGE, and immunoblotting with the indicated antibodies. E, HEK293T cells were seeded into 10-cm dishes (1.5 × 106 cells) 24 h before transfection with 4 μg each of empty vector, Myc-A52, or Myc-A52-M65E. After 48 h, lysates were analyzed by either native gel electrophoresis or SDS-PAGE. Immunoblots were subjected to densitometric analysis with levels of TRAF6 oligomer normalized to total levels of TRAF6 monomer. F, HEK293T cells were transfected with the indicated amounts of Myc-TRAF6, FLAG-TAK1, HA-A52, and HA-A52 M65E. After 48 h, lysates were subject to immunoprecipitation, SDS-PAGE, and immunoblotting with the indicated antibodies. G–I, HEK293-R1 cells were transfected for 24 h with 150 ng of Myc-A52, Myc-A52 F154A, Myc-A52 M65E, or pCMV-Myc empty vector (EV), along with either the NFκB luciferase reporter gene (G), the pFR luciferase reporter gene and CHOP-Gal4 (H) or the IL-10 promoter luciferase reporter gene (I). MEK3 is the positive control for the PathdetectTM CHOP assay (H). Cells were stimulated with 50 ng/ml IL-1α for 6 h (G), and luciferase reporter gene activity was measured. The data are mean ± S.D. of triplicate samples and are representative of at least three separate experiments. *, p < 0.05; **, p < 0.005; or ***, p < 0.0005 compared with empty vector.
To determine whether A52 dimerization per se was necessary for p38 activation, we sought to determine whether we could generate an A52 mutant that would retain TRAF6 binding but be deficient in dimerization. The VACV protein B14, which, similar to A52, is a VACV Bcl-2 fold-containing protein, has been shown to exist in monomer-dimer equilibrium in vitro, and a Y35E mutation in B14 significantly disrupted dimerization (36). Analysis of the A52 sequence and dimer interface suggested that the equivalent A52 mutant that should be monomeric is A52-M65E (Fig. 4B) (35). When an M65E mutant was generated and expressed in cells, it was found to be expressed to an equal level as wild-type A52 and A52-F154A (Fig. 4C, third and fourth panels). Co-immunoprecipitation experiments demonstrated that M65E was severely impaired in its ability to dimerize, compared with A52 (Fig. 4C, top panel, compare lane 3 with lane 1). Densitometric analysis of the immunoblots indicate that the M65E self-association band was >90% less intense than that observed for wild-type A52; Fig. 4C). However, M65E did retain the ability to interact with both IRAK2 and TRAF6 (Fig. 4D), demonstrating that A52 dimerization was not required for either the IRAK2 or TRAF6 interaction.
Further experiments with M65E demonstrated that p38 activation was dependent on A52 dimerization: unlike A52, M65E could not stimulate TRAF6 self-association (Fig. 4E) and could not cause recruitment of TAK1 to TRAF6 (Fig. 4F). Consistent with these results, M65E retained the ability to inhibit NFκB (Fig. 4G), whereas similar to F154A, it also failed to activate p38 (Fig. 4H) and the p38-dependent promoter IL-10 (Fig. 4I).
Together, these data indicate that the ability of A52 to dimerize is not required for NFκB inhibition. However, for A52 to activate p38 MAPK, both A52-TRAF6 association and A52 dimerization are required, leading to TRAF6 oligomerization and recruitment of TAK1 to TRAF6.
DISCUSSION
TRAF proteins have emerged as important nodes of intracellular signaling pathways that viruses have evolved to manipulate to generate a signaling environment that is more favorable to the virus. The exact signaling outcome after TRAF engagement varies from virus to virus, but altered NFκB and MAP kinase activation are common consequences. For example, rotavirus protein VP4 engages TRAF2 to activate NFκB while inhibiting TRAF2-dependent JNK activation (23), whereas UL144 from human cytomegalovirus forms a complex with TRAF6 to activate NFκB (22). Epstein-Barr virus LMP-1 (latent membrane protein 1) also stimulates p38 activation via TRAF6 as well as activating NFκB via engaging TRAF2 and TRAF5 (21). NS5A from hepatitis C virus inhibits TRAF2-dependent NFκB activation while engaging TRAF2 to activate MAP kinase (JNK) (37). This strategy has similarities to A52 whereby NFκB activation-dependent on IRAK2-TRAF6 signaling is inhibited (3, 19), whereas engagement of A52 with TRAF6 activates MAP kinase (p38).
That A52 both inhibits NFκB and activates p38 is consistent with the known role of these two signaling proteins in VACV infection, and both activities of A52 likely contribute to the observation that a VACV deletion mutant lacking the A52 gene was attenuated compared with wild-type virus (3). The relative contribution of the NFκB inhibitory activity of A52 compared with its ability to stimulate TRAF6-mediated MAP kinase activation to the known role of A52 in virulence remains to be determined. Clearly, NFκB activation is strongly anti-poxviral because poxviruses such as VACV have evolved multiple non-redundant strategies to block NFκB (2–6, 8, 38, 39). However, p38 MAP kinase activation seems to favor VACV replication and gene expression (40) although much less is known about how VACV might manipulate this pathway, and to our knowledge, A52 is the only poxviral protein to date shown to directly activate p38.
Although it was known previously that A52-stimulated p38 activation was TRAF6-dependent and that A52 binds to TRAF6 via the TRAF6 TRAF domain (16), the mechanism whereby A52 manipulates TRAF6 to activate p38 (while not stimulating NFκB) was unclear. For IL-1 and TLR signaling, TRAF6 is required for activation of both p38 and NFκB (30). The generation of Lys-63-linked polyubiquitin chains associated with a TRAF6 complex and likely generated by the E3 ligase activity of TRAF6, is essential for downstream signaling (20, 24). The TAK1-associated protein TAB2 binds to Lys-63-linked polyubiquitin chains, resulting in autophosphorylation of TAK1 at Ser-187 and its subsequent activation (41). Activated TAK1 phosphorylates and activates MKK3/MKK6, which, in turn, activate p38 MAPK. TAK1 also activates the NFκB pathway by directly phosphorylating and activating IKK, which, in turn, phosphorylates IκB and induces its ubiquitination and degradation by the proteasome-dependent degradation machinery (42). Smad-independent activation of p38 by TGFβ also proceeds via TRAF6 and TAK1: the TGFβ receptor complex physically associates with TRAF6, promoting association between TRAF6 and TAK1. This results in Lys-63-linked ubiquitination and subsequent activation of TAK1, which is essential for p38 activation (31, 32). Other signaling pathways are also known to utilize a TRAF6-TAK1 axis for p38 activation (20).
This study demonstrates that the presence of A52 can cause TRAF6 self-association, the recruitment of TAK1 to TRAF6 and p38 phosphorylation, and that these activities are dependent on A52 dimerization. Similar to the VACV proteins N1, B14, and K7, A52 is an α helical Bcl-2-like protein (35, 43, 44). K7 consists of a bundle of six α-helices and was found to be a monomer in solution (44). This contrasts with N1, B14, and A52, which all form dimers via a common surface comprising α-helices 1 and 6 (35, 36, 43, 45). However, the relative contribution of their dimerization ability to their function as immunomodulators varies. Mutagenesis of the dimer interface of N1 only affected its inhibition of NFκB activation and not its anti-apoptotic activity (46). Benfield et al. (36) recently demonstrated that for B14, its ability to bind IKKβ and inhibit NFκB-dependent gene expression was not dependent upon B14 dimerization but rather that the dimerization and IKKβ-binding surfaces overlap. Their study showed that although B14 exists in monomer-dimer equilibrium in vitro, B14 monomers are fully functional and are likely to represent the physiologically relevant state of the protein (36). We have demonstrated that the ability of A52 to dimerise is not required for NFκB inhibition, as the A52 dimerization mutant, M65E, was as capable of inhibiting NFκB activation as wild-type protein (Fig. 4). However, for A52 to drive p38 MAPK activation, both A52-TRAF6 association and A52 dimerization were required. The Phe-154 residue of A52, which is required for TRAF6 association, is at the edge of the dimerization interface and may be part of a region where the dimerization and TRAF6-binding surfaces overlap. Met-65 in contrast lies at the very center of the dimeric interface, and its mutation did not prevent interaction with IRAK2 or TRAF6, demonstrating that A52 dimerization per se is essential for p38 activation.
Our data are consistent with the following model for A52 activation of p38 (Fig. 5): A52 interacts with TRAF6 via a non-canonical TRAF6 binding motif that contains the residue Phe-154. Because A52 exists as a dimer, two A52-associated TRAF6 molecules may come into close proximity and dimerize. Because TRAF6 dimerization is a known essential step in its ability to stimulate downstream signaling (47) and may also trigger further self-association leading to active oligomeric forms of TRAF6, A52-stimulated TRAF6 dimerization could be the key trigger that causes TAK1 recruitment to TRAF6, which would lead to TAK1 activation and subsequent phosphorylation of p38 MAPK via MKK3 and/or MKK6.
FIGURE 5.
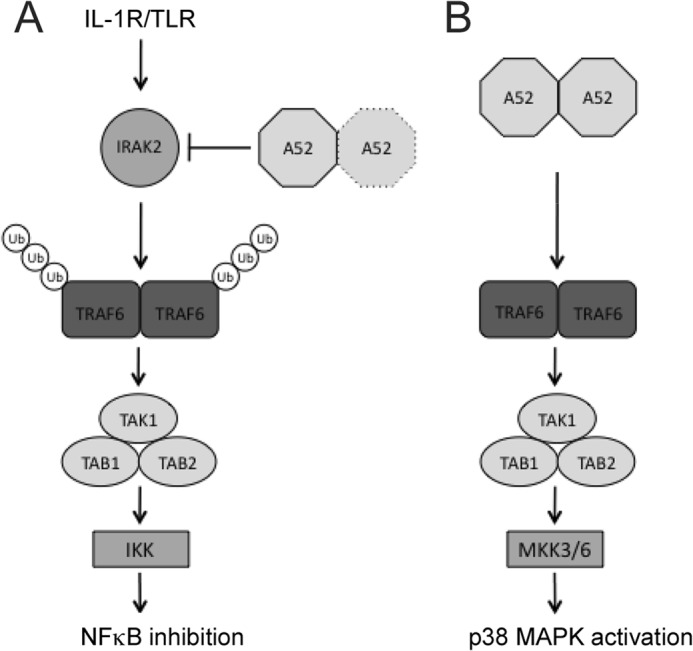
Model for manipulation of signaling by A52. A, upon TLR activation, IRAK2 associates with TRAF6. The interaction of IRAK2 with TRAF6 triggers the polyubiquitination of TRAF6 (19), which allows the subsequent recruitment of a complex containing TAB2, TAB1, and TAK1. This activates the kinase activity of TAK1 leading to the phosphorylation of IKKβ, culminating in the induction of NFκB. A52 inhibits IL-1R/TLR-dependent NFκB activation by interacting with IRAK2; its dimerization ability is not required for this function. B, as a dimer, A52 brings TRAF6 molecules together in the absence of TRAF6 polyubiquitination, inducing formation of the TRAF6-TAK1 complex. Activated TAK1 can then activate downstream kinases such as MKK3/6, leading to p38 phosphorylation. Ub, ubiquitin.
Why recruitment of TRAF6 and TAK1 by A52 does not lead to NFκB activation is not entirely clear, although LMP1 also recruits TRAF6 and TAK1, via its N-terminal transmembrane region, to activate p38 and not NFκB (21). Interestingly, activation of p38 by A52 did not require TRAF6-associated polyubiquitination (Fig. 3), which suggests that in certain circumstances, oligomerization of TRAF6 and recruitment of TAK1 without TRAF6-associated polyubiquitination may be sufficient for p38, but not NFκB activation (Fig. 5). Consistent with this, expression of IRAK2, which is known to activate both NFκB and p38 (19), did cause TRAF6-associated polyubiquitination (Fig. 3). Hence, A52 may have evolved to exploit a differential ability of TRAF6 to activate NFκB and p38 MAP kinase, to enable enhanced p38 MAP kinase activation in the absence of NFκB activation during VACV infections.
Acknowledgments
We thank Drs. Z.J. Chen, A. Mansell, M. Muzio, and H. Sakurai for the kind gifts of cell lines and expression plasmids.
This work was supported by Science Foundation Ireland Grants 07/IN1/B934 and 08/RFP/BIC980 and as part of the Science Foundation Ireland-funded Immunology Research Centre (Grant 07/SRC/B1144).
- PRR
- pattern recognition receptor
- VACV
- vaccinia virus
- CHOP
- C/EBP homologous protein
- IKK
- IκB kinase
- IRAK
- IL-1 receptor-associated kinase
- MEF
- murine embryonic fibroblast
- TAK1
- TGFβ activated kinase-1
- TIR
- Toll/IL-1R
- TLR
- Toll-like receptor
- TRAF
- tumor necrosis factor receptor-associated factor.
REFERENCES
- 1. Bowie A. G., Unterholzner L. (2008) Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stack J., Haga I. R., Schröder M., Bartlett N. W., Maloney G., Reading P. C., Fitzgerald K. A., Smith G. L., Bowie A. G. (2005) Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 201, 1007–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harte M. T., Haga I. R., Maloney G., Gray P., Reading P. C., Bartlett N. W., Smith G. L., Bowie A., O'Neill L. A. (2003) The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197, 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DiPerna G., Stack J., Bowie A. G., Boyd A., Kotwal G., Zhang Z., Arvikar S., Latz E., Fitzgerald K. A., Marshall W. L. (2004) Poxvirus protein N1L targets the I-κB kinase complex, inhibits signaling to NF-κB by the tumor necrosis factor superfamily of receptors, and inhibits NF-κB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 279, 36570–36578 [DOI] [PubMed] [Google Scholar]
- 5. Chen R. A., Ryzhakov G., Cooray S., Randow F., Smith G. L. (2008) Inhibition of IκB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 4, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schröder M., Baran M., Bowie A. G. (2008) Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKϵ-mediated IRF activation. EMBO J. 27, 2147–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Unterholzner L., Sumner R. P., Baran M., Ren H., Mansur D. S., Bourke N. M., Randow F., Smith G. L., Bowie A. G. (2011) Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 7, e1002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mansur D. S., Maluquer de Motes C., Unterholzner L., Sumner R. P., Ferguson B. J., Ren H., Strnadova P., Bowie A. G., Smith G. L. (2013) Poxvirus targeting of E3 ligase β-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog. 9, e1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blasius A. L., Beutler B. (2010) Intracellular toll-like receptors. Immunity 32, 305–315 [DOI] [PubMed] [Google Scholar]
- 10. O'Neill L. A., Bowie A. G. (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature reviews. Immunology 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 11. Takeuchi O., Akira S. (2002) MyD88 as a bottle neck in Toll/IL-1 signaling. Curr. Top. Microbiol. Immunol. 270, 155–167 [DOI] [PubMed] [Google Scholar]
- 12. Lin S. C., Lo Y. C., Wu H. (2010) Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawagoe T., Sato S., Matsushita K., Kato H., Matsui K., Kumagai Y., Saitoh T., Kawai T., Takeuchi O., Akira S. (2008) Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat. Immunol. 9, 684–691 [DOI] [PubMed] [Google Scholar]
- 14. Kawai T., Akira S. (2007) TLR signaling. Semin Immunol. 19, 24–32 [DOI] [PubMed] [Google Scholar]
- 15. Stack J., Bowie A. G. (2012) Poxviral protein A46 antagonizes Toll-like receptor 4 signaling by targeting BB loop motifs in Toll-IL-1 receptor adaptor proteins to disrupt receptor:adaptor interactions. J. Biol. Chem. 287, 22672–22682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maloney G., Schröder M., Bowie A. G. (2005) Vaccinia virus protein A52R activates p38 mitogen-activated protein kinase and potentiates lipopolysaccharide-induced interleukin-10. J. Biol. Chem. 280, 30838–30844 [DOI] [PubMed] [Google Scholar]
- 17. Assarsson E., Greenbaum J. A., Sundström M., Schaffer L., Hammond J. A., Pasquetto V., Oseroff C., Hendrickson R. C., Lefkowitz E. J., Tscharke D. C., Sidney J., Grey H. M., Head S. R., Peters B., Sette A. (2008) Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc. Natl. Acad. Sci. U.S.A. 105, 2140–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowie A., Kiss-Toth E., Symons J. A., Smith G. L., Dower S. K., O'Neill L. A. (2000) A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 97, 10162–10167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keating S. E., Maloney G. M., Moran E. M., Bowie A. G. (2007) IRAK-2 participates in multiple toll-like receptor signaling pathways to NFkappaB via activation of TRAF6 ubiquitination. The Journal of biological chemistry 282, 33435–33443 [DOI] [PubMed] [Google Scholar]
- 20. Landström M. (2010) The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 42, 585–589 [DOI] [PubMed] [Google Scholar]
- 21. Uemura N., Kajino T., Sanjo H., Sato S., Akira S., Matsumoto K., Ninomiya-Tsuji J. (2006) TAK1 is a component of the Epstein-Barr virus LMP1 complex and is essential for activation of JNK but not of NF-κB. J. Biol. Chem. 281, 7863–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poole E., King C. A., Sinclair J. H., Alcami A. (2006) The UL144 gene product of human cytomegalovirus activates NFκB via a TRAF6-dependent mechanism. EMBO J. 25, 4390–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. LaMonica R., Kocer S. S., Nazarova J., Dowling W., Geimonen E., Shaw R. D., Mackow E. R. (2001) VP4 differentially regulates TRAF2 signaling, disengaging JNK activation while directing NF-κB to effect rotavirus-specific cellular responses. J. Biol. Chem. 276, 19889–19896 [DOI] [PubMed] [Google Scholar]
- 24. Adhikari A., Xu M., Chen Z. J. (2007) Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 26, 3214–3226 [DOI] [PubMed] [Google Scholar]
- 25. Kumar H., Kawai T., Akira S. (2009) Pathogen recognition in the innate immune response. Biochem. J. 420, 1–16 [DOI] [PubMed] [Google Scholar]
- 26. Ye H., Arron J. R., Lamothe B., Cirilli M., Kobayashi T., Shevde N. K., Segal D., Dzivenu O. K., Vologodskaia M., Yim M., Du K., Singh S., Pike J. W., Darnay B. G., Choi Y., Wu H. (2002) Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443–447 [DOI] [PubMed] [Google Scholar]
- 27. Noels H., van Loo G., Hagens S., Broeckx V., Beyaert R., Marynen P., Baens M. (2007) A Novel TRAF6 binding site in MALT1 defines distinct mechanisms of NF-κB activation by API2·MALT1 fusions. J. Biol. Chem. 282, 10180–10189 [DOI] [PubMed] [Google Scholar]
- 28. Meads M. B., Li Z. W., Dalton W. S. (2010) A novel TNF receptor-associated factor 6 binding domain mediates NF-κB signaling by the common cytokine receptor β subunit. J. Immunol. 185, 1606–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gentry J. J., Rutkoski N. J., Burke T. L., Carter B. D. (2004) A functional interaction between the p75 neurotrophin receptor interacting factors, TRAF6 and NRIF. J. Biol. Chem. 279, 16646–16656 [DOI] [PubMed] [Google Scholar]
- 30. Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 [DOI] [PubMed] [Google Scholar]
- 31. Yamashita M., Fatyol K., Jin C., Wang X., Liu Z., Zhang Y. E. (2008) TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol. Cell 31, 918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sorrentino A., Thakur N., Grimsby S., Marcusson A., von Bulow V., Schuster N., Zhang S., Heldin C. H., Landström M. (2008) The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207 [DOI] [PubMed] [Google Scholar]
- 33. Yu X., Yi H., Guo C., Zuo D., Wang Y., Kim H. L., Subjeck J. R., Wang X. Y. (2011) Pattern recognition scavenger receptor CD204 attenuates Toll-like receptor 4-induced NF-κB activation by directly inhibiting ubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6. J. Biol. Chem. 286, 18795–18806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Katsoulidis E., Li Y., Mears H., Platanias L. C. (2005) The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J. Interferon Cytokine Res. 25, 749–756 [DOI] [PubMed] [Google Scholar]
- 35. Graham S. C., Bahar M. W., Cooray S., Chen R. A., Whalen D. M., Abrescia N. G., Alderton D., Owens R. J., Stuart D. I., Smith G. L., Grimes J. M. (2008) Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog. 4, e1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benfield C. T., Mansur D. S., McCoy L. E., Ferguson B. J., Bahar M. W., Oldring A. P., Grimes J. M., Stuart D. I., Graham S. C., Smith G. L. (2011) Mapping the IκB kinase beta (IKKβ)-binding interface of the B14 protein, a vaccinia virus inhibitor of IKKβ-mediated activation of nuclear factor κB. J. Biol. Chem. 286, 20727–20735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park K. J., Choi S. H., Choi D. H., Park J. M., Yie S. W., Lee S. Y., Hwang S. B. (2003) 1Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 278, 30711–30718 [DOI] [PubMed] [Google Scholar]
- 38. Shisler J. L., Jin X. L. (2004) The vaccinia virus K1L gene product inhibits host NF-κB activation by preventing IκBα degradation. J. Virol. 78, 3553–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ember S. W., Ren H., Ferguson B. J., Smith G. L. (2012) Vaccinia virus protein C4 inhibits NF-κB activation and promotes virus virulence. J. Gen. Virol. 93, 2098–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu N., Yu R., Shikuma C., Shiramizu B., Ostrwoski M. A., Yu Q. (2009) Role of cell signaling in poxvirus-mediated foreign gene expression in mammalian cells. Vaccine 27, 2994–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanayama A., Seth R. B., Sun L., Ea C. K., Hong M., Shaito A., Chiu Y. H., Deng L., Chen Z. J. (2004) TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol. Cell 15, 535–548 [DOI] [PubMed] [Google Scholar]
- 42. Liu S., Chen Z. J. (2011) Expanding role of ubiquitination in NF-κB signaling. Cell Res. 21, 6–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cooray S., Bahar M. W., Abrescia N. G., McVey C. E., Bartlett N. W., Chen R. A., Stuart D. I., Grimes J. M., Smith G. L. (2007) Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 88, 1656–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kalverda A. P., Thompson G. S., Vogel A., Schröder M., Bowie A. G., Khan A. R., Homans S. W. (2009) Poxvirus K7 protein adopts a Bcl-2 fold: biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J. Mol. Biol. 385, 843–853 [DOI] [PubMed] [Google Scholar]
- 45. Oda S., Schröder M., Khan A. R. (2009) Structural basis for targeting of human RNA helicase DDX3 by poxvirus protein K7. Structure 17, 1528–1537 [DOI] [PubMed] [Google Scholar]
- 46. Maluquer de Motes C., Cooray S., Ren H., Almeida G. M., McGourty K., Bahar M. W., Stuart D. I., Grimes J. M., Graham S. C., Smith G. L. (2011) Inhibition of apoptosis and NF-κB activation by vaccinia protein N1 occur via distinct binding surfaces and make different contributions to virulence. PLoS Pathog. 7, e1002430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yin Q., Lin S. C., Lamothe B., Lu M., Lo Y. C., Hura G., Zheng L., Rich R. L., Campos A. D., Myszka D. G., Lenardo M. J., Darnay B. G., Wu H. (2009) E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Laskowski R. A. (2009) PDBsum new things. Nucleic Acids Res. 37, D355–D359 [DOI] [PMC free article] [PubMed] [Google Scholar]