

Background: The bacteriophage P22 coat protein has an HK97-like fold with an additional, genetically inserted domain.
Results: The insertion domain provides thermodynamic stability to the full-length coat protein.
Conclusion: The insertion domain acts as an intramolecular chaperone with the help of a peptidyl prolyl isomerase.
Significance: This is the first study to establish the function of domain addition to a protein with the HK97 fold.
Keywords: Bacteriophage, Protein Folding, Protein Self-assembly, Protein Stability, Virus Assembly
Abstract
The bacteriophage P22 coat protein has the common HK97-like fold but with a genetically inserted domain (I-domain). The role of the I-domain, positioned at the outermost surface of the capsid, is unknown. We hypothesize that the I-domain may act as an intramolecular chaperone because the coat protein folds independently, and many folding mutants are localized to the I-domain. The function of the I-domain was investigated by generating the coat protein core without its I-domain and the isolated I-domain. The core coat protein shows a pronounced folding defect. The isolated I-domain folds autonomously and has a high thermodynamic stability and fast folding kinetics in the presence of a peptidyl prolyl isomerase. Thus, the I-domain provides thermodynamic stability to the full-length coat protein so that it can fold reasonably efficiently while still allowing the HK97-like core to retain the flexibility required for conformational switching during procapsid assembly and maturation.
Introduction
The proper folding of viral coat proteins is a crucial step in the virus life cycle that can drastically affect the subsequent assembly process and production of infectious particles. The coat proteins of viruses with icosahedral symmetry must retain sufficient flexibility for assembly and maturation. During assembly, identical subunits are “switched” into the slightly different conformations required to occupy quasi-equivalent sites in the icosahedron, a process known as conformational switching. Conformational flexibility is also necessary for the rearrangements during the maturation process of icosahedral capsids (1, 2). Thus, a fine balance is necessitated between conformational polymorphism and the stability of monomeric coat proteins. The HK97-like viruses (named after bacteriophage HK97), which include viruses with bacterial, archaeal, and eukaryotic hosts, have icosahedral symmetry and utilize structurally conserved coat proteins (3). Many viruses in the HK97-like structural group rely on host- or virus-encoded folding chaperonins for the proper folding of their viral coat proteins. One member of this group, bacteriophage P22, is an exception to this generality.
Bacteriophage P22, which infects Salmonella enterica serovar Typhimurium, has long been a model used for studying protein folding and assembly mechanisms of dsDNA icosahedral viruses (4). The assembly of bacteriophage P22 in vivo proceeds through a nucleation-limited reaction in which 415 copies of monomeric coat protein copolymerize with 60–300 scaffolding proteins, the dodecameric portal complex, and ejection proteins to form a precursor (“procapsid”) structure (5, 6). Scaffolding protein directs the assembly of the procapsid but is not found in mature phage and so acts as an assembly chaperone. Procapsids mature into virions as dsDNA is packaged through the portal complex (7) and scaffolding protein concomitantly exits (8). Maturation is accompanied by an increase in volume of the head and a change in capsid morphology from one that is nearly spherical to one that is faceted (9, 10).
The P22 coat protein (430 amino acids) contains 148 more residues than the mature HK97 coat protein (282 amino acids). A nearly complete, pseudoatomic model of the 47-kDa P22 coat protein based on three-dimensional image reconstruction and homology modeling (11) revealed that the additional residues were inserted into the primary amino acid sequence. The core (with the HK97-like fold) makes the bulk of the capsid contacts, with the additional domain positioned as a protrusion. The majority of the known temperature-sensitive-folding (tsf)2 substitutions in the P22 coat protein cluster in this additional domain (4, 14), indicating it is important for the folding and stability of coat protein monomers. Subsequently, a higher resolution reconstruction and model of the coat protein was published (12) that also revealed the domain. The authors suggested alternative hypotheses about the function of the I-domain in procapsid assembly and stability. The domain has been referred to as extra density domain (12, 13) or telokin-like domain (11), but we will henceforth refer to it as the “insertion” domain or I-domain.
Because there is evolutionary pressure on viruses to maintain the smallest possible genome, we reasoned there must be a vital purpose for the I-domain. When the N-terminal (residues 1–190) and C-terminal (residues 191–430) halves of the P22 coat protein are expressed separately, they form inclusion bodies. However, the C-terminal half can be refolded into soluble protein that exhibits β-sheet content by circular dichroism (15). The autonomous folding of the C-terminal half suggests that it contains the folding nucleus of the P22 coat protein. The I-domain may act as an intramolecular chaperone, thereby eliminating the need for cellular chaperones to aid in proper folding of the coat protein. Characterization of the isolated I-domain and the coat protein core was performed to elucidate the role of this domain.
EXPERIMENTAL PROCEDURES
Chemicals, Buffers, and Media
Ultrapure urea was purchased from ICN. All other chemicals were of reagent grade and purchased from common sources. All in vitro experiments described were done in 20 mm sodium phosphate buffer (unless otherwise specified) made using Na2HPO4, with the pH adjusted to 7.6 with H3PO4. Luria broth (LB, from Invitrogen) was used to support bacterial growth.
Bacteria
Salmonella enterica serovar Typhimurium strain DB7136 (leuA414am, hisC525am, su−) and the su+ derivative DB7155 (supE20 gln, leuA414am, hisC525am) have been described previously (16). S. enterica serovar Typhimurium strain SB300A#1 (17) and Escherichia coli strain BL21 (DE3) (18) were used for protein purification from plasmids under control of the T7 promoter (18).
Plasmids and Generation of Plasmid-encoded Variants
The I-domain plasmid (pID, comprising coat protein amino acids 223–345 with an N-terminal hexa-histidine tag) was constructed by PCR subcloning using primers designed to amplify coat protein amino acids 223–345, with the addition of the hexa-histidine tag, a stop codon, and appropriate restriction enzyme sites. The amplified region was digested with Nde1 and Sal1, ligated into a pET-30b expression vector, and transformed into E. coli strain BL21 (DE3) (18) by electroporation. Transformants were selected by kanamycin resistance, screened for the appropriate insert size by restriction digestion, and verified by DNA sequencing.
The core coat protein plasmid (pPC-Core, comprising coat protein amino acids 1–224 and 346–430, with a 5-glycine linker) was constructed from pPC, a pET-3a vector (Novagen) with an insert encoding for both the scaffolding and coat protein genes (19). Inverse PCR (20) was performed with 5′ phosphorylated PCR primers that also coded for the 5-glycine linker. Following amplification, the plasmids were recircularized by ligation and transformed into S. enterica serovar Typhimurium strain SB300A#1 (17) by electroporation. Transformants were selected by ampicillin resistance and verified by DNA sequencing. Protein expression from the various constructs was verified by induction of cultures with 1 mm isopropyl β-d-1-thiogalactopyranoside.
Purification of the I-domain
The recombinant I-domain was overexpressed in E. coli strain BL21 (DE3). Cells were grown at 37 °C to mid-log phase in LB medium containing 40 μg/ml kanamycin. Protein expression was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside for 16 h at 30 °C. After induction, the cells were harvested by sedimentation in a Sorvall SLC-6000 rotor at 5368 × g for 20 min. The cells were disrupted by resuspension in 20 mm sodium phosphate (pH 7.6) containing 0.1% w/v Triton X-100, lysozyme (200 μg/ml), and a 1:100 dilution of EDTA-free protease inhibitor mixture (Sigma), followed by lysis using a French press operating at 20,000 p.s.i. The lysate was brought to 5 mm MgSO4, 0.5 mm CaCl2, and 1 mm PMSF and treated with DNase and RNase (100 μg/ml each) for 30 min. Cell debris was removed by sedimentation in a Sorvall F18–12 × 50 rotor at 38,725 × g for 30 min. The supernatant was loaded onto a 15-ml Talon metal affinity column (Clontech, Mountain View, CA) for purification of the I-domain via the engineered N-terminal His6 tag. Fractions containing the I-domain were pooled, and the protein was precipitated with 57.5% ammonium sulfate at 4 °C. The purified I-domain was dialyzed three times against 20 mm phosphate buffer (pH 7.6).
Purification of the Core Coat Protein
The core coat protein was overexpressed in SB300A#1. Cells were grown at 30 °C to mid-log phase in LB medium containing 100 μg/ml ampicillin. Protein expression was induced with 0.2% L-arabinose (final concentration) for 4 h at 30 °C. After induction, the cells were harvested by sedimentation in a Sorvall SLC-6000 rotor at 5,368 × g for 20 min, and the resulting pellets were resuspended in buffer B (50 mm Tris-HCl, 25 mm NaCl, and 2 mm EDTA (pH 7.6)) with a 1:100 dilution of EDTA-free protease inhibitor mixture (Sigma). The cell suspension was sonicated for 6 min, and the pellets were collected by centrifugation in a Sorvall F18–12 × 50 rotor at 38,725 × g for 30 min. The pellet was resuspended with washing buffer (2 m urea, 2% Triton X-100, and 0.2% β-mercaptoethanol in buffer B). This procedure was repeated twice. To remove the detergent and denaturant, the pellets were washed twice with washing buffer without Triton X-100 and urea. The core coat protein was solubilized with extracting buffer (9 m urea and 0.2% β-mercaptoethanol in buffer B) and clarified by centrifugation in a Sorvall RP80AT3 rotor at 100,000 × g for 1 h, and the supernatant was reserved. The core coat protein was refolded by extensive dialysis against 20 mm phosphate buffer (pH 7.6) at 4 °C.
Purification of the WT Coat Protein
The WT coat protein was obtained from empty procapsid shells that had been prepared as described previously (21, 22). The ambers in gene 2 and gene 13 prevent DNA packaging and cell lysis, respectively. Empty procapsid shells were generated from the purified procapsids by incubation of the procapsids with 0.5 m guanidine hydrochloride, which extracts the scaffolding protein and the minor capsid proteins while leaving the coat protein lattice intact. Coat protein monomers were obtained from urea-denatured, empty procapsid shells. Briefly, urea-unfolded coat protein monomers were refolded by extensive dialysis against 20 mm phosphate buffer (pH 7.6 at 4 °C) and clarified by centrifugation at 175,000 × g at 4 °C for 20 min in a Sorvall RP80AT3 rotor.
Thermolysin Time Course of Digestion
Thermolysin from Bacillus thermoproteolyticus rokko (Sigma) was prepared in 2.5 m NaCl containing 10 mm CaCl2 (23). WT coat protein monomers, core coat protein, and the I-domain in 20 mm HEPES (pH 7.5) were digested using thermolysin at 20 °C with an enzyme:substrate ratio of 1:250. At each time point, a sample was removed and quenched with reducing sample buffer with extra SDS and EDTA (4.4% SDS and 75 mm EDTA), heated for 5 min at 95 °C, and analyzed by 16% Tricine-SDS-PAGE (24). The sequence of the ∼20-kDa peptide resulting from digestion of the WT coat protein was determined by in-gel digestion and liquid chromatography coupled with tandem mass spectrometry performed by the Vermont Genetics Network Proteomics Facility.
Tryptophan Fluorescence
Fluorescence spectra were taken with an SLM Aminco-Bowman 2 spectrofluorometer thermostated at 20 °C. The excitation wavelength was set at 295 nm, the emission was monitored from 310–400 nm, and the band passes were set to 1 and 8 nm, respectively. All proteins were at a final concentration of 2.1 μm.
Circular Dichroism
CD experiments were done with an Applied Photophysics (Leatherhead, Surrey, UK) Pi-Star 180 circular dichroism spectrapolarimeter with the 0.2-cm path-length quartz cuvette maintained at 20 °C with a circulating water bath. Refolded coat protein monomers were at a final concentration of 2.1 μm for the WT and 2.9 μm for the core. The I-domain was at a final concentration of 14 μm. All measurements were done in 20 mm sodium phosphate buffer (pH 7.6), with or without 6 m urea. Wavelength scans were done over 200–250 nm with the following settings: step resolution, 0.5 nm; bandwidth, 3.0 nm; and data averaging for 30 s/point, leading to a scan time of ∼52 min.
To determine the melting temperature of the I-domain, the temperature was ramped from 20–80 °C with a step size of 0.5 °C. At each temperature, the CD at 220 nm was averaged for 30 s. The raw data were normalized to show the fraction unfolded at each temperature.
Equilibrium Urea Titration
Solutions of the folded I-domain and the I-domain in 6 m urea were diluted to a final concentration of 14 μm (0.2 mg/ml). The solutions were then mixed with a Hamilton Microlab 50 titrator to final urea concentrations from 0–6 m. Samples were incubated at 20 °C for 3 h prior to measurement. Separate equilibrium titration experiments beginning from only the folded or unfolded I-domain were done to ensure the reversibility of the folding. The CD signal at 220 nm was averaged for 30 s/point with a slit width of 4 nm. A 0.2-cm path length quartz cuvette maintained at 20 °C with a circulating water bath was employed for all measurements. The thermodynamic stability (ΔG°N-U) and the sensitivity of the transition to denaturant (meq) were determined by fitting the equilibrium data, assuming a linear relationship between the free energy of unfolding for the transition and the denaturant (25).
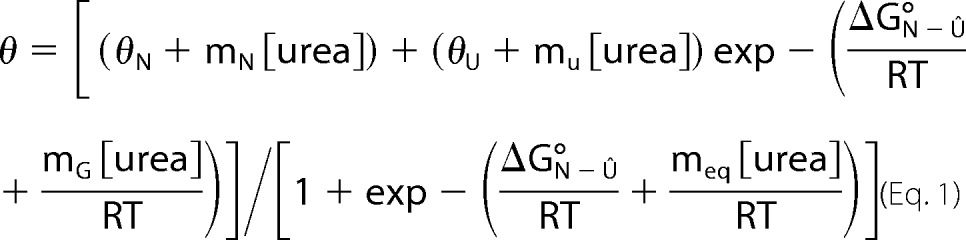 |
Kinetics of Unfolding and Refolding
Unfolding and refolding kinetic experiments were done with the folded and unfolded I-domain. In each experiment, the final concentration of the I-domain was 14 μm. To initiate an unfolding reaction, the folded I-domain at 800 μm was diluted (14 μm final) with buffered urea. The solution of the folded I-domain was placed in the bottom of the cuvette, and the buffered urea was pipetted onto the folded protein while stirring constantly. Refolded experiments were done similarly with the I-domain that had been denatured in 4.5 m urea. To initiate refolding, the unfolded I-domain at 400 μm was diluted (14 μm final) with buffered urea solutions. The final urea concentration was determined by measuring the refractive index. The constantly stirred reactions were monitored by CD at 220 nm using a 1.0-cm path length quartz cuvette maintained at 20 °C with a circulating water bath. This mixing method led to a dead time of ∼ 5–7 s.
Refolding kinetic experiments of the I-domain and coat protein were done in the presence of cyclophilin A (CyPA, Sigma-Aldrich) similar to the above. The unfolded I-domain at 400 μm was diluted (14 μm final) with buffer containing CyPA to give final CyPA concentrations (I-domain alone, 0 nm; 1:100, 0.14 μm; 1:50, 0.28 μm; and 1:25, 0.57 μm). The unfolded coat protein at 60 μm was diluted (2.1 μm final) with buffer containing CyPA. The final CyPA concentrations were as follows: coat protein alone, 0 nm; 1:100, = 21.4 nm; 1:50, 42.8 nm; and 1:25, 85.6 nm. For the coat protein, the reactions were monitored by CD at 222 nm. All kinetic traces were fit to a first-order reaction with either one (for the I-domain) or two (for the coat protein) exponentials using the Kaleidagraph program,
or
where θ(t) is the CD signal as a function of time, t, θf is the final signal, θ1 is the amplitude of the CD signal change because of the first relaxation, θ2 is the amplitude of the CD signal change because of the second relaxation, and τ is the relaxation time (or the inverse of the apparent rate constant, 1/k.
The relaxation times from the kinetic experiments were plotted, in a single chevron plot, against the urea concentration on a semilog plot. The urea dependence of the unfolding relaxation times was determined with Kaleidagraph using the equation (26),
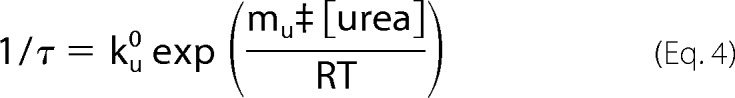 |
where ku0 is the apparent rate constant of the unfolding reaction in the absence of urea and mu‡ is the denaturant dependence on the unfolding reaction.
Double Jump Folding Kinetics by Pulse Proteolysis
The I-domain was unfolded in 20 mm HEPES (pH 7.5) with 8 m urea at 20 °C for 6 or 3600 s. It was then diluted into 20 mm HEPES and, at each indicated time after refolding was initiated, an aliquot was transferred to a tube with thermolysin (TLN) for 15 s. The final concentration of TLN was 0.4 μm, and of the I-domain was 10 μm, to give a TLN:I-domain molar ratio of 1:25. After the 15-s pulse, the samples were quenched with reducing sample buffer with extra SDS and EDTA (4.4% SDS and 75 mm EDTA), heated for 5 min at 95 °C, and analyzed by 16% Tricine-SDS-PAGE (24). The bands quantified by densitometry using Bio-Rad Quantity One software.
RESULTS
Cloning and Purification of the I-domain and the Coat Protein Core
Limited proteolysis of the C-terminal half of the coat protein (residues 191–430) results in a stable fragment identified as residues 223–349 (11). This contiguous, folded domain is linked to the HK97-like core of the coat protein via two strands of the β-hinge, a structurally conserved β-sheet at the center of the protein (4). To investigate the properties and role of the I-domain of P22 coat protein, we cloned the I-domain (residues 223–345, Fig. 1a) and the coat protein without its I-domain (core coat protein, residues 1–224 linked to 346–430, b). The N- and C-terminal sections of the core coat protein were connected with a 5-glycine linker, joining two strands of the β-hinge (Fig. 1, b and e). Distance measurements on the basis of the cryoelectron microscopy reconstructions (11, 12) indicate a distance between residues 225 and 345 of ∼10–13 Å. A typical staggered C-α to C-α bond is 3.63 Å (27). Therefore, a 5-glycine linker would span about 22 Å and should provide sufficient length and flexibility to accommodate the connection between the N- and C-terminal sections of the core coat protein.
FIGURE 1.

The purified I-domain and core coat protein. Shown is a diagram of constructs used to study the function of the I-domain. Illustrated is the I-domain by itself (a) and the coat protein without its I-domain (core coat protein) (b). c, 16% Tricine-SDS gel showing the purified I-domain. d, 12.5% SDS gel showing the purified core coat protein, WT coat protein, and scaffolding protein. In b and c, MWM indicates the molecular weight markers. Their masses in kDa are shown on the left. e, anticipated structure of the HK97-like fold of core coat protein joined by a short linker.
The I-domain was overexpressed, easily purified by a Talon metal affinity column (Clontech), and remained soluble at concentrations exceeding 40 mg/ml (Fig. 1c). Previous work indicated that, when separately expressed, the C-terminal 239 residues of the coat protein, which includes the I-domain, form a dimer in solution. The oligomeric state of the I-domain was assessed by size exclusion chromatography using a Superdex 200 column (GE Healthcare). The protein was loaded onto the column at 4 and 40 mg/ml. Both elution profiles, monitored by absorbance at 280 nm, showed a single peak (data not shown). On the basis of calibration of the column with standards of known molecular weight, the molecular mass of the I-domain (15 kDa) is consistent with a monomeric protein.
The core coat protein formed inclusion bodies when overexpressed in Salmonella cells, indicating that it does not fold properly in vivo without the I-domain. The inclusion bodies were denatured with urea, and the core coat protein was refolded by dialysis following a procedure similar to that used to refold WT coat protein monomers from urea-denatured empty procapsid shells (Fig. 1d).
Stability of the Core Coat Protein and I-domain Compared with the Full-length WT Coat Protein
The stability of the core and I-domain was first assessed using sensitivity to protease. TLN digestion of refolded WT and core coat protein monomers and the I-domain was analyzed over time. The core coat protein showed a substantial increase in digestion with time compared with the WT coat protein. Complete digestion by TLN of the full-length core coat protein occurred in 5 min, whereas the full-length coat protein required 30–60 min. Conversely, the I-domain was more protease-resistant compared with the WT coat protein, with a substantial amount of the full-length I-domain remaining at 60 min (Fig. 2) and longer (data not shown). Interestingly, digestion of the WT coat protein resulted in a well defined stable band at ∼ 20 kDa (Fig. 2, arrow). In-gel digestion and liquid chromatography coupled with tandem mass spectrometry revealed that it comprised residues 203–261. This protease-resistant peptide is the I-domain with N- and C-terminal extensions of 20 and 16 residues, respectively. These results indicate that the I-domain forms an independently folded domain within the WT coat protein. In addition, the I-domain has a high stability in both the isolated domain and in the context of the full-length WT coat protein.
FIGURE 2.
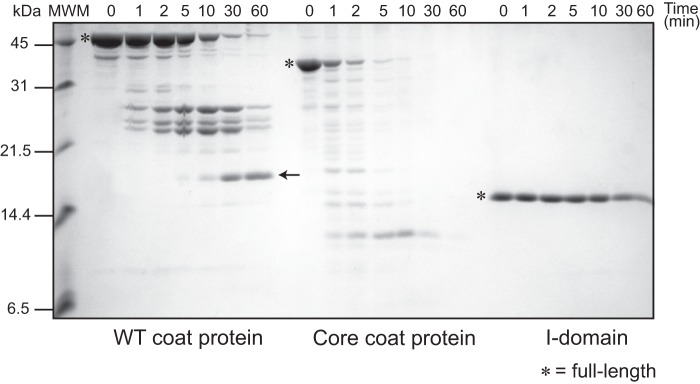
Kinetics of thermolysin digestion in native conditions. 16% Tricine-SDS gel showing the TLN digestion time course of the WT coat protein, refolded core coat protein, and I-domain. All protein concentrations were 10 μm, and the TLN concentration was 4 nm to give a TLN:protein molar ratio of 1:250. The full-length proteins are marked by asterisks. The TLN-resistant peptide comprising amino acids 203–361 is marked by an arrow. MWM indicates the molecular weight markers, shown on the left.
The Refolded Core Coat Protein Has More Random Coil than the WT Coat Protein
The enhanced protease sensitivity observed in the core coat protein could be due to improper folding or a decrease in stability. The folding of the full-length WT coat protein has been thoroughly investigated using CD for the secondary structure and tryptophan (Trp) fluorescence for the tertiary structure (21). The CD spectrum of the core coat protein was compared with the spectrum of WT coat protein monomers to determine whether the refolded core coat protein adopted a conformation with a regular secondary structure. The spectrum of the WT coat protein (Fig. 3a, gray) is consistent with a protein with high α-helical content mixed with β-sheets. The spectrum of the core coat protein (Fig. 3a, green) indicates a decrease in α-helical content and an increase in random coil. These data, along with the formation of inclusion bodies in vivo and enhanced protease sensitivity, indicate that the core coat protein cannot adopt the appropriate HK97-like fold (Fig. 1e) in the absence of the I-domain.
FIGURE 3.
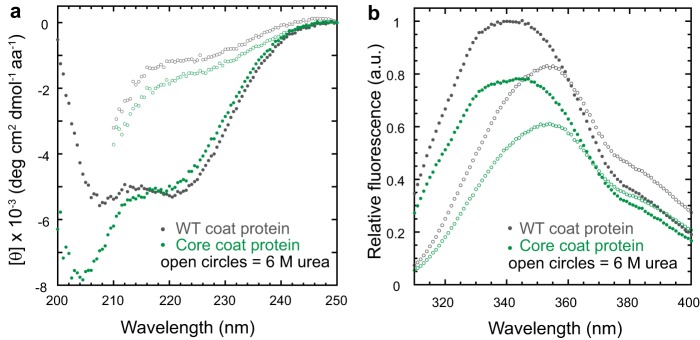
The core coat protein has significant random coil. a, circular dichroism spectra of the WT and core coat proteins in mean residue ellipticity (degrees cm2 dmol−1 amino acids−1). The spectra were obtained as described under “Experimental Procedures.” The protein concentrations were 0.1 mg/ml (2.1 μm for the WT coat protein and 2.9 μm for the core coat protein). b, fluorescence emission spectra of native (closed) and denatured (open) WT and core coat protein in arbitrary units (a.u.). The excitation wavelength was 295 nm. The concentrations of the WT and core coat protein were 2.1 μm.
The fluorescence of tryptophan residues can be used to probe for the presence of a hydrophobic core in proteins. Under native conditions, both the WT and core coat proteins displayed fluorescence emission maxima at 340 nm (Fig. 3b), indicating that some tryptophan residues are buried in a hydrophobic environment. Under denaturing conditions, the emission maxima of both proteins shifted to 355 nm, and the intensity decreased. Thus, the core coat protein likely adopts a compact conformation even though it is not properly folded.
The core coat protein deletion length (Δ225–345, Δ225–365, Δ233–338, and Δ236–334) and linker (5G, 3A, 3G, GGPGG, and GPG) were varied to verify that the inability to adopt the proper fold was not due to construct design. None of the core coat protein variations remained soluble when expressed in vivo or exhibited a WT-like secondary structure when refolded in vitro (data not shown).
The I-domain Is Spectroscopically Invisible
Next we set out to characterize the isolated I-domain in more detail. The I-domain had little CD signal (Fig. 4a). The I-domain has significant β-sheet content connected by long loops (random coil) that have opposing CD signals (28). In this peculiar instance, these CD signals counterbalance, leading to a CD spectrum with a signal near the base line. In addition, its single tryptophan was solvent-exposed (Fig. 4b). Because the I-domain is spectroscopically invisible at the concentrations and conditions used in previous studies of the WT coat protein, only indirect contributions from this domain were observed in our previous characterization of the WT coat protein (21). Also, because of these considerations, monitoring the unfolding transition of the I-domain using these conventional techniques is difficult. Although small, the change in CD signal upon I-domain unfolding was found to be reproducible. Therefore, the CD signal at 220 nm was used as a probe to monitor the folded state of the I-domain for many of the experiments described below.
FIGURE 4.
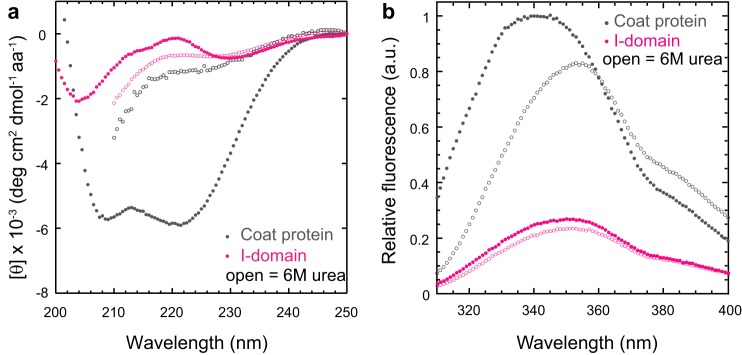
The I-domain is spectroscopically invisible. a, circular dichroism spectra of the native (closed) and denatured (open) coat protein and I-domain in mean residue ellipticity (degrees cm2 dmol−1 amino acid −1). The protein concentrations were 0.1 mg/ml (2.1 μm for the coat protein and 7.2 μm for the I-domain). b, fluorescence emission spectra of the native (closed) and denatured (open) coat protein and I-domain in arbitrary units (a.u.). The emission maximum of the I-domain is centered at 350 nm under both native and denaturing conditions. The excitation wavelength was 295 nm. The concentration of the coat protein and I-domain was 2.1 μm.
The I-domain Is More Stable to Heat and Denaturant than the Full-length Coat Protein
The I-domain shows a thermally induced decrease in the CD signal that can be used to monitor its melting transition. This was compared with the melting transition of the WT coat protein monitored by Trp fluorescence. Because the I-domain has only a single solvent-exposed tryptophan, changes in fluorescence upon unfolding of the WT coat protein only reflect denaturation of the HK97-like core (with no signal from the I-domain). The transition midpoint of the I-domain is ∼15 °C higher than the transition midpoint of the coat protein (Fig. 5a). This suggests that, for the full-length coat protein, the core may unfold first, followed by the I-domain. The heat-induced denaturation of the I-domain is ∼90% reversible, whereas the full-length coat protein aggregates with heat denaturation (data not shown).
FIGURE 5.
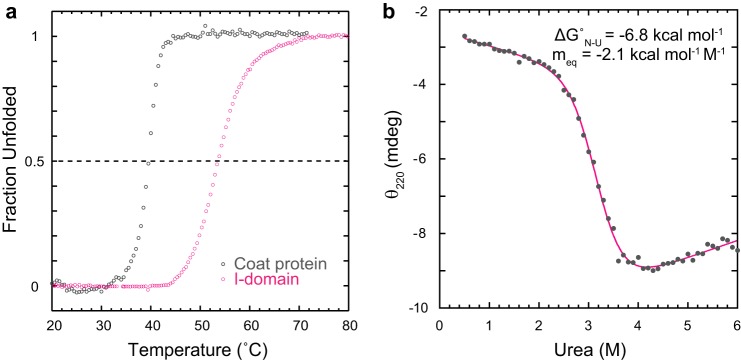
The I-domain has high thermal and thermodynamic stability. a, to determine the melting temperatures, the coat protein (2.1 μm) and I-domain (14 μm) were ramped from 20–80 °C. At each 0.5 °C increment in temperature, either the fluorescence emission at 340 nm (coat protein) or the CD at 220 nm (I-domain) was measured. The data were normalized to show the fraction unfolded at each temperature. b, the I-domain, either folded or unfolded in 6 m urea, was titrated with urea to give final concentrations between 0 and 6 m and incubated at 20 °C. The I-domain concentration was 14 μm. The solid magenta line is the fit of the data to a two-state model (see “Experimental Procedures”).
Equilibrium urea denaturation experiments were done with the I-domain to determine its thermodynamic stability (ΔGN-Uo). In these experiments, I-domain was incubated in 0–6 m urea until equilibrium was established, and CD at 220 nm was used to monitor the unfolding transition (Fig. 5b). Despite the small change in signal upon urea denaturation, it was possible to monitor the transition region and the pre- and post-transition base lines. The equilibrium data were fit to a standard two-state thermodynamic model to extract the ΔGN-Uo and the sensitivity of the unfolding transition to denaturant (meq). Randomly distributed residuals indicated a good fit to the two-state model (data not shown), with the I-domain having a ΔGN-Uo of −6.8 kcal/mol. The ΔGN-Uo for the full-length coat protein is −5.8 kcal/mol (21). Additionally, the I-domain is demonstrated to be well folded because its meq value is consistent with that of model proteins of similar size (29). Thus, a significant portion of the coat protein thermodynamic stability is due to the I-domain but was not observed because of its optical properties. We hypothesize that this domain may serve to add stability to the monomeric coat protein while still allowing the HK97-like core to retain the inherent flexibility required for conformational switching during procapsid assembly and maturation.
Refolding and Unfolding Kinetics of the I-domain
Single jump experiments were performed to determine whether the I-domain also enhances the refolding kinetics of the WT coat protein. For refolding jumps, the denatured I-domain, denatured in 4.5 m urea, was rapidly diluted into buffered solutions containing lower urea concentrations. For unfolding jumps, the folded I-domain was diluted with buffered urea solutions. For both refolding and unfolding reactions, the relaxation time to reach a new equilibrium was monitored by CD at 220 nm. The refolding and unfolding reactions of the I-domain were fit well by a first-order rate equation with a single exponential (Fig. 6, a and b). The residuals, the difference between the fit and the data, are randomly distributed above and below zero, indicating a good fit (30). In contrast, WT coat protein folding and unfolding kinetics have been shown previously to be the best fit to a first-order reaction with two exponentials (21).
FIGURE 6.

Kinetics of folding and unfolding monitored by CD. a, the I-domain was folded by rapidly diluting the unfolded I-domain in 4.5 m urea with a buffer containing various concentrations of urea. b, the native I-domain was rapidly unfolded by dilution into solutions with various concentrations of urea. Shown are representative scans of the raw data monitored by circular dichroism at 220 nm. The final I-domain concentration was 14 μm. The solid magenta line is the fit of the data to a first-order rate equation with one exponential (see “Experimental Procedures”). Shown below are the residuals of the fits. c, chevron plot showing the relaxation times for the folding and unfolding kinetics at varied urea concentrations. The rate of folding determined from the pulse proteolysis experiment described in Fig. 7 is indicated by the X. d, ball and stick model of the NMR structure of the isolated I-domain (Ref. 33 and unpublished data (see Footnote 3)) showing residues 309–311. The peptide bond preceding the proline at position 310 is in the cis conformation.
The relaxation times from the fits of the kinetic data obtained by CD were plotted against the urea concentration in a chevron plot (Fig. 6c) (31). The peak of the chevron does not occur at the urea concentration that corresponds to the midpoint in our equilibrium urea titration experiment (3.2 m urea, see Fig. 5b). In addition, the slope of the folding arm is relatively independent of urea concentration. The ΔGN-Uo determined from extrapolation of the τunfolding and τfolding to 0 m urea (ΔGN-Uo = −RT ln (τfo/τuo)) was calculated to be ∼−1.2 kcal/mol, which is also inconsistent with the ΔGN-Uo determined from the equilibrium titration. The deviations from the expected V-shaped chevron suggest an accumulation of partially folded intermediates (either on or off the productive folding pathway) or changes in rate-limiting steps (32). We independently fit the unfolding arm (see “Experimental Procedures”) to determine the τuo = 3.9 × 104 s and mu‡ = −8.7 kcal mol/M, where τuo is the apparent relaxation time of the unfolding reaction in the absence of urea and mu‡ is the denaturant dependence on the unfolding reaction.
Nuclear magnetic resonance studies to determine a high-resolution NMR structure of the isolated I-domain are currently underway (Ref. 33 and unpublished data).3 Preliminary results from these studies revealed that the peptide bond preceding the proline (peptidyl prolyl bond) at position 310 is in the cis conformation (Fig. 6d). A cis peptidyl prolyl bond can complicate the in vitro folding analysis because the incorrect trans form predominates in an unfolded protein, and the cis ⇌ trans isomerization is intrinsically slow. The slow refolding kinetics of the I-domain suggests the possibility that interconversion of the peptidyl prolyl bond at residue 310 from the nonnative trans isomer to the correct cis configuration is limiting the rate of refolding. Such rate-limiting steps for in vitro protein folding are well documented (34, 35). To determine whether the slow folding reaction of the I-domain is limited by proline isomerization, we performed several experiments, which are described below.
Double Jump Pulse Proteolysis Refolding Kinetics
As a protein unfolds, the peptidyl prolyl bonds become free to isomerize. Initially, the bond preceding Pro-310 will remain in its native cis configuration, and the protein should be in its “fast-folding” form. For longer unfolding times, isomerization of this bond will reach equilibrium, producing “slow-folding” species for which the prolyl bond at residue 310 is in the trans configuration. These slow-folding species require isomerization of this peptidyl prolyl bond prior to the formation of the native structure. A double jump experiment coupled with pulse proteolysis was performed to analyze this possibility (see the experiment schematic in Fig. 7a).
FIGURE 7.

The I-domain folds immediately when the prolines are in the correct isomeric state. a, the I-domain was unfolded in 8 m urea for 6 or 3600 s, diluted into buffer, and then allowed to refold for varying times before being subjected to a 15-s pulse of digestion using a relatively high concentration of the nonspecific protease thermolysin. b, samples were quenched with sample buffer, and the extent of proteolysis was observed by 16% Tricine-SDS-PAGE. c, the full-length I-domain was quantified using densitometry and plotted versus refolding time. The solid gray line is the fit of the 3600-s unfolding data to a first-order rate equation with one exponential (see “Experimental Procedures”). The relaxation time from these data is included in Fig. 6.
In the first jump, the I-domain is unfolded by rapid dilution into 8 m urea (for 6 s or 3600 s). The duration of the 6-s unfolding jump was chosen so that refolding initiates from a state that is fully unfolded (determined from extrapolation of the unfolding arm in the chevron plot, Fig. 6c) but contains native proline isomers because cis → trans prolyl isomerizations generally have relaxation times of tens to hundreds of seconds at 25 °C (36). In fact, the relaxation time for cis → trans prolyl isomerization when the preceding residue is a threonine (as in our case) was measured on a model peptide to be 370 s (37). The refolding jump is then initiated by dilution into buffer. Thermolysin pulse proteolysis, which exploits the change in proteolytic susceptibility between folded and unfolded proteins (38, 39), was used to measure the amount of native I-domain at varying times after refolding was initiated (Fig. 7b). The short incubation time and TLN concentration of the pulse proteolysis (15 s) were designed so that only unfolded protein is digested, leaving native protein intact. Quantification of the full-length I-domain (Fig. 7c) shows that the slow-folding species seen as a result of long-term denaturation (unfolded for 3600 s) is not present in the short-term denaturation sample (unfolded for 6 s). The double jump refolding experiments demonstrated that the short-term denatured I-domain with all proline residues in their native isomeric state folds to the native state within 6 s, likely because there are no slow isomerization reactions that need to occur. It was possible to fit the refolding pulse proteolysis data of the long-term denaturation sample. The relaxation time from pulse proteolysis is consistent with the CD kinetics data (Fig. 6c, X), indicating that both techniques were monitoring a global folding event. However, this folding event is rate-limited by peptidyl prolyl isomerization, for which direct evidence is provided below. Pulse proteolysis has been used previously to determine protein stability (38) and monitor unfolding kinetics (39). Here we show that this technique can also be modified to monitor refolding kinetics and in a double jump experiment.
Refolding with Cyclophilin A
In vivo the cis ⇌ trans isomerization of peptidyl prolyl bonds is catalyzed by a group of ubiquitous enzymes, the peptidyl prolyl isomerases (PPIases). The I-domain was refolded in the presence of the PPIase CyPA. This enzyme has been shown to catalyze peptidyl prolyl cis ⇌ trans isomerization reactions for model peptides and numerous proteins in vitro (36). Refolding jumps were performed by rapidly diluting I-domain denatured in 4.5 m urea for 60 min with buffer containing CyPA, and the relaxation time to reach a new equilibrium was monitored by CD at 220 nm. The refolding reactions of the I-domain were fit well by a first-order reaction with a single exponential (Fig. 8a, solid lines).
FIGURE 8.
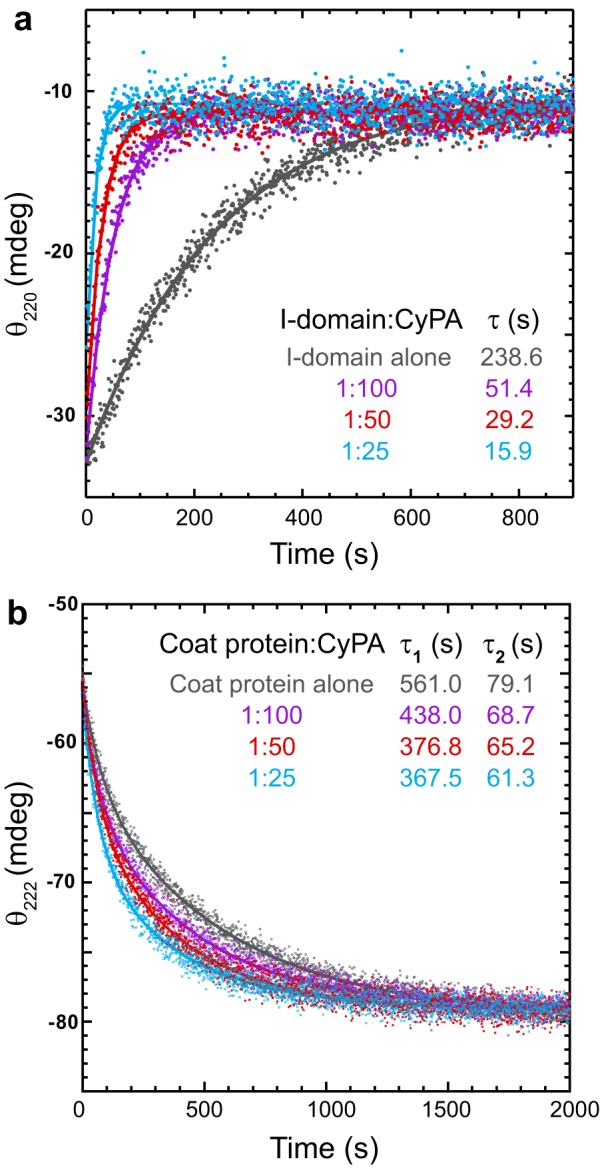
Kinetics of refolding in the presence of cyclophilin A. The proteins, denatured in 4.5 m urea, were refolded by rapid dilution with buffer containing increasing concentrations of CyPA. a, the I-domain:CyPA molar ratios are indicated in the legend. Refolding was monitored by circular dichroism at 220 nm. The final I-domain concentration was 14 μm. The CyPA concentrations were as follows: I-domain alone, 0 nm; 1:100, 0.14 μm; 1:50, 0.28 μm; and 1:25, 0.57 μm. The lines are the fit of the data to a first-order rate equation with one exponential (see “Experimental Procedures”). b, refolding of the WT coat protein was monitored by circular dichroism at 222 nm. The coat protein:CyPA molar ratios are indicated in the legend. The final coat protein concentration was 2.1 μm. The CyPA concentrations were as follows: coat protein alone, 0 nm; 1:100, 21.4 nm; 1:50, 42.8 nm; and 1:25, 85.6 nm. The solid lines are the fits of the data to a first-order rate equation with two exponentials (see “Experimental Procedures”).
As the CyPA concentration was increased, the rate of I-domain folding increased correspondingly (decrease in relaxation time). The observed relaxation time of I-domain refolding decreases almost 15-fold at the highest CyPA concentration tested (0.57 μm, Fig. 8a, cyan), providing direct evidence that proline isomerization is responsible for the slow folding kinetics. If the I-domain provides kinetic enhancement for coat protein folding, then CyPA should also increase the rate of coat protein folding. The full-length WT coat protein was refolded with CyPA to determine what effect increasing the rate of I-domain folding had on the rate of core folding. Refolding jumps were performed by rapidly diluting coat protein denatured in 4.5 m urea for 60 mn with buffer containing CyPA, and the relaxation time to reach a new equilibrium was monitored by CD at 222 nm. The refolding reactions of the coat protein were best fit well to a first-order reaction with two exponential (Fig. 8b, solid lines). Although not as drastic as with the isolated I-domain, CyPA decreased both the fast and slow relaxation times. Thus, rapid folding of the I-domain accelerates the folding of the core coat protein.
DISCUSSION
Efficient and rapid folding of viral coat proteins is vital to produce a supply of functional proteins for capsid assembly. However, there is a balance between the stability of the monomeric coat protein and the structural flexibility required for conformational switching during assembly and maturation. Bacteriophage P22 has a genetically inserted domain (I-domain) in its HK97-like core (11–13). Here we provide compelling evidence the I-domain acts as a folding nucleus and provides thermodynamic stability to the monomeric coat protein.
The I-domain Acts as an Intramolecular Chaperone
The role of a protein domain serving as a nucleator for the folding of multidomain proteins was proposed 30 years ago (40). The kinetic studies performed in the presence of CyPA suggest that folding of the coat protein initiates with proper conformational folding of the I-domain. Once in the native conformation, the I-domain contributes substantial thermodynamic stability, thereby allowing productive folding of the surrounding core. This domain is connected to the core via two strands of the β-hinge and possibly mediates the interactions between the N- and C-terminal halves of the core. Indeed, the core cannot adopt the proper HK97-like fold in the absence of the I-domain, as evidenced by the in vivo aggregation and in vitro destabilization of the core coat protein.
The coat proteins of phages HK97 and T4 require assistance from chaperonin complexes for proper folding both in vivo and in vitro. HK97 hijacks the GroEL-ES chaperone machinery of the host to properly fold its coat protein (41). T4 utilizes a hybrid chaperone complex composed of the bacterial chaperone GroEL and the phage-encoded gp31 (42). Conversely, the P22 coat protein does not require assistance from chaperonins for proper folding (43). The I-domain addition to the P22 core increases the stability of the monomeric coat protein, likely eliminating the chaperonin requirement seen with other HK97-like coat proteins. Another striking difference is that the coat proteins of HK97 and T4 are not stable as monomers but form pentamers and hexamers immediately after folding with the assistance of chaperonin complexes. HK97 and T4 assemble from these preformed capsomers, whereas P22 assembles from monomeric coat proteins (44). This alteration in the assembly pathways may also reflect the stability of the monomeric coat proteins.
There is no evidence that the coat protein of eukaryotic HSV-1 requires chaperones for proper folding. The upper domain of the HSV-1 coat protein has been proposed to function as a folding nucleus (45). The authors suggest that this stable, upper domain may act as an inherent chaperone providing structural stability for the HK97-like floor domain of the protein, which must be flexible to participate in procapsid assembly. We propose a similar role for the I-domain of the P22 coat protein. The additional stability provided by the I-domain does not restrict the necessary conformational plasticity of the core, which is essential because a decrease in flexibility of the core results in assembly defects such as polyhead formation (46). Therefore, I-domain insertion provides a means of stabilizing the core without disrupting its ability to participate in assembly or subsequent rearrangements during maturation. However, this intramolecular chaperone likely requires assistance from a host-encoded PPIase before conformational folding can proceed.
Coat Protein Likely Utilizes a Host-encoded PPIase for Folding
The peptide bond has a partial double-bond character and, therefore, two energetically preferred states: cis and trans. For non-prolyl peptide bonds, the trans conformation with dihedral angles (ω) of 180° is energetically favored because of steric constraints. However, peptide bonds N-terminal to proline residues (peptidyl prolyl bonds) are unique in that the trans conformation is only slightly energetically favored over the cis conformation. Consequently, in native, folded proteins, a higher occurrence of cis peptidyl prolyl bonds (∼6%) is found compared with < 0.1% for non-prolyl peptide bonds (47). The ribosome is thought to synthesize peptide bonds in the trans form and, therefore, trans → cis isomerization must occur for proteins with native cis peptidyl prolyl bonds. In vivo, the PPIases catalyze this cis ⇌ trans isomerization of peptidyl prolyl bonds. Cyclophilins are a structurally conserved family of enzymes with PPIase activity that have been found in both prokaryotic and eukaryotic organisms (48, 49). CyPA, a member of the cyclophilin family, was shown here to increase the rate of coat protein folding in vitro by catalyzing the isomerization of the peptidyl prolyl bond at residue 310 in the I-domain to its native cis conformation. Therefore, we propose that a member of the PPIase enzymes is involved in P22 coat protein folding in vivo, catalyzing native I-domain formation and, thereby, allowing its intramolecular chaperone activity.
Replacement of proline residues with alanine by site-directed mutagenesis is a classic means of detecting and characterizing proline-limited folding reactions. Substitutions of cis prolines are often strongly destabilizing because the cis conformation of the respective bond may be maintained after replacement (50, 51). Coincidently, an alanine substitution of the cis peptidyl prolyl bond at residue 310 in the coat protein was investigated unintentionally. The P22 coat protein has been used as a model to study protein folding for some time (4). Eighteen single amino acid substitutions in the coat protein, resulting from random mutagenesis of the phage, have been characterized that result in a tsf phenotype (14). One of the most severe of these tsf coat protein variants (lethal at 33 °C) is P310A (14, 52). This amino acid substitution results in significant coat protein misfolding and aggregation, even at the permissive temperature of 28 °C (14). PPIases do not catalyze isomerization of non-prolyl peptide bonds. Therefore, maintenance of a cis peptide bond at this position after alanine substitution would have a detrimental effect on folding. A strong decrease in the folding rate because of slow uncatalyzed trans → cis isomerization of this non-prolyl peptide bond would abolish the ability of this domain to serve as an intramolecular chaperone. The GroEL-ES complex rescues the folding of the tsf coat protein variants, including P310A, suggesting that encapsulation of the coat protein in the folding chamber of the chaperonin allows sufficient time for proper folding. In vitro characterization of the P310A coat protein is necessary to confirm the maintenance of the cis peptide bond and the effect on the protein folding mechanism, although a preliminary study indicated that CyPA had no effect on the kinetics of refolding of the P310A coat protein in vitro.3 A limited suppressor search of the P310A tsf phenotype identified five suppressors (53). Four of the five suppressors are located in the I-domain. The effect of these suppressor substitutions on the folding of coat protein has yet to be investigated.
Additions to the HK97-like Fold in Other Phage Coat Proteins
Other phages also have additional domains in their coat protein structure that are not part of the HK97-like fold, such as the Big2-like domain in φ29 (54) and a chitin-binding-like insertion domain in T4 (55). These additional domains are hypothesized to stabilize the assembled capsid by bridging the neighboring molecules within one capsomer (T4) or between neighboring capsomers (φ29). Cryoelectron microscopy reconstructions of the phage C1 (56) and phage P-SPP7 (57) capsids also revealed additional density (not attributed to the HK97-like fold) at the two-fold axes resulting from interactions across capsomers. Although the coat protein of HK97 does not have any additional domains, its capsid is uniquely stabilized through a network of covalent cross-links (58).
The process of protein folding is complex and unresolved. The folding mechanisms of large multidomain proteins are extremely complicated. Much advancement in understanding protein folding has come from studying small, isolated domains. Our work on the isolated I-domain of the P22 coat protein allows insight into its role in the bacteriophage life cycle that was not possible from studies of the WT coat protein. This domain provides thermodynamic stability to the monomeric coat protein and possibly contributes structural stability to assembled particles. In addition, it may abolish the requirement for host chaperonins for folding of the coat protein by serving as an intramolecular chaperone. Indeed, it may be possible to use the P22 insertion domain to stabilize HK97-like proteins or other, generally unstable proteins. A similar approach has been investigated. An unstable guest protein was inserted into a highly stable host protein (59). However, this approach tethers the N- and C termini and, therefore, constricts the flexibility of the protein. Using a stable insertion as an intramolecular chaperone may be preferred when conformational plasticity is required for assembly or function.
This work was supported, in whole or in part, by National Institutes of Health Grant GMS 076661 (to C. M. T.) and the IDeA Networks of Biomedical Research Program, NIGMS, National Institutes of Health Grant 8P20GM103449 (to the Vermont Genetics Network).
M. M. Suhanovsky and C. M. Teschke, unpublished data.
- tsf
- temperature-sensitive folding
- I-domain
- inserted domain
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- CyPA
- cyclophilin A
- TLN
- thermolysin
- PPIase
- peptidyl prolyl isomerase.
REFERENCES
- 1. Caspar D. L., Klug A. (1962) Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol. 27, 1–24 [DOI] [PubMed] [Google Scholar]
- 2. Johnson J. E., Speir J. A. (1997) Quasi-equivalent viruses. A paradigm for protein assemblies. J. Mol. Biol. 269, 665–675 [DOI] [PubMed] [Google Scholar]
- 3. Bamford D. H., Grimes J. M., Stuart D. I. (2005) What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 15, 655–663 [DOI] [PubMed] [Google Scholar]
- 4. Teschke C. M., Parent K. N. (2010) “Let the phage do the work.” Using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology 401, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. King J., Botstein D., Casjens S., Earnshaw W., Harrison S., Lenk E. (1976) Structure and assembly of the capsid of bacteriophage P22. Philos. Trans. R. Soc. Lond. B Biol. Sci. 276, 37–49 [DOI] [PubMed] [Google Scholar]
- 6. Prevelige P. E., Jr., King J. (1993) Assembly of bacteriophage P22. A model for ds-DNA virus assembly. Prog. Med. Virol. 40, 206–221 [PubMed] [Google Scholar]
- 7. Bazinet C., King J. (1988) Initiation of P22 procapsid assembly in vivo. J. Mol. Biol. 202, 77–86 [DOI] [PubMed] [Google Scholar]
- 8. Casjens S., King J. (1974) P22 morphogenesis. I. Catalytic scaffolding protein in capsid assembly. J. Supramol. Struct. 2, 202–224 [DOI] [PubMed] [Google Scholar]
- 9. Earnshaw W., Casjens S., Harrison S. C. (1976) Assembly of the head of bacteriophage P22. X-ray diffraction from heads, proheads and related structures. J. Mol. Biol. 104, 387–410 [DOI] [PubMed] [Google Scholar]
- 10. Prasad B. V., Prevelige P. E., Marietta E., Chen R. O., Thomas D., King J., Chiu W. (1993) Three-dimensional transformation of capsids associated with genome packaging in a bacterial virus. J. Mol. Biol. 231, 65–74 [DOI] [PubMed] [Google Scholar]
- 11. Parent K. N., Khayat R., Tu L. H., Suhanovsky M. M., Cortines J. R., Teschke C. M., Johnson J. E., Baker T. S. (2010) P22 coat protein structures reveal a novel mechanism for capsid maturation. Stability without auxiliary proteins or chemical crosslinks. Structure 18, 390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen D.-H., Baker M. L., Hryc C. F., DiMaio F., Jakana J., Wu W., Dougherty M., Haase-Pettingell C., Schmid M. F., Jiang W., Baker D., King J. A., Chiu W. (2011) Structural basis for scaffolding-mediated assembly and maturation of a dsDNA virus. Proc. Natl. Acad. Sci. U.S.A. 108, 1355–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang W., Li Z., Zhang Z., Baker M. L., Prevelige P. E., Jr., Chiu W. (2003) Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions. Nat. Struct. Biol. 10, 131–135 [DOI] [PubMed] [Google Scholar]
- 14. Gordon C. L., King J. (1993) Temperature-sensitive mutations in the phage P22 coat protein which interfere with polypeptide chain folding. J. Biol. Chem. 268, 9358–9368 [PubMed] [Google Scholar]
- 15. Kang S., Prevelige P. E., Jr. (2005) Domain study of bacteriophage p22 coat protein and characterization of the capsid lattice transformation by hydrogen/deuterium exchange. J. Mol. Biol. 347, 935–948 [DOI] [PubMed] [Google Scholar]
- 16. Winston F., Botstein D., Miller J. H. (1979) Characterization of amber and ochre suppressors in Salmonella typhimurium. J. Bacteriol. 137, 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McKinney J., Guerrier-Takada C., Galán J., Altman S. (2002) Tightly regulated gene expression system in Salmonella enterica serovar Typhimurium. J. Bacteriol. 184, 6056–6059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Studier F. W., Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 [DOI] [PubMed] [Google Scholar]
- 19. Kang S., Uchida M., O'Neil A., Li R., Prevelige P. E., Douglas T. (2010) Implementation of p22 viral capsids as nanoplatforms. Biomacromolecules 11, 2804–2809 [DOI] [PubMed] [Google Scholar]
- 20. Ochman H., Gerber A. S., Hartl D. L. (1988) Genetic applications of an inverse polymerase chain reaction. Genetics 120, 621–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anderson E., Teschke C. M. (2003) Folding of phage P22 coat protein monomers. Kinetic and thermodynamic properties. Virology 313, 184–197 [DOI] [PubMed] [Google Scholar]
- 22. Parent K. N., Suhanovsky M. M., Teschke C. M. (2007) Polyhead formation in phage P22 pinpoints a region in coat protein required for conformational switching. Mol. Microbiol. 65, 1300–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inouye K., Kuzuya K., Tonomura B. (1998) Sodium chloride enhances markedly the thermal stability of thermolysin as well as its catalytic activity. Biochim. Biophys. Acta 1388, 209–214 [DOI] [PubMed] [Google Scholar]
- 24. Schägger H., von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 25. Santoro M. M., Bolen D. W. (1988) Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 [DOI] [PubMed] [Google Scholar]
- 26. Gloss L. M., Matthews C. R. (1998) Mechanism of folding of the dimeric core domain of Escherichia coli trp repressor. A nearly diffusion-limited reaction leads to the formation of an on-pathway dimeric intermediate. Biochemistry 37, 15990–15999 [DOI] [PubMed] [Google Scholar]
- 27. Creighton T. E. (1992) Proteins. Structures and Molecular Properties, 2nd Ed., p. 5, Freeman, W. H. & Company, New York [Google Scholar]
- 28. Woody R. W. (1995) Circular dichroism. Methods Enzymol. 246, 34–71 [DOI] [PubMed] [Google Scholar]
- 29. Myers J. K., Pace C. N., Scholtz J. M. (1995) Denaturant m values and heat capacity changes. Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4, 2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zitzewitz J. A., Bilsel O., Luo J., Jones B. E., Matthews C. R. (1995) Probing the folding mechanism of a leucine zipper peptide by stopped-flow circular dichroism spectroscopy. Biochemistry 34, 12812–12819 [DOI] [PubMed] [Google Scholar]
- 31. Matthews C. R. (1987) Effect of point mutations on the folding of globular proteins. Methods Enzymol. 154, 498–511 [DOI] [PubMed] [Google Scholar]
- 32. Sánchez I. E., Kiefhaber T. (2003) Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J. Mol. Biol. 325, 367–376 [DOI] [PubMed] [Google Scholar]
- 33. Rizzo A. A., Fraser L. C., Sheftic S. R., Suhanovsky M. M., Teschke C. M., Alexandrescu A. T. (2013) NMR assignments for the telokin-like domain of bacteriophage P22 coat protein. Biomol. NMR Assign. 7, 257–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brandts J. F., Halvorson H. R., Brennan M. (1975) Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 14, 4953–4963 [DOI] [PubMed] [Google Scholar]
- 35. Kiefhaber T., Schmid F. X. (1992) Kinetic coupling between protein folding and prolyl isomerization. II. Folding of ribonuclease A and ribonuclease T1. J. Mol. Biol. 224, 231–240 [DOI] [PubMed] [Google Scholar]
- 36. Schmid F. X., Mayr L. M., Mücke M., Schönbrunner E. R. (1993) Prolyl isomerases. Role in protein folding. Adv. Protein Chem. 44, 25–66 [DOI] [PubMed] [Google Scholar]
- 37. Reimer U., Scherer G., Drewello M., Kruber S., Schutkowski M., Fischer G. (1998) Side-chain effects on peptidyl-prolyl cis/trans isomerisation. J. Mol. Biol. 279, 449–460 [DOI] [PubMed] [Google Scholar]
- 38. Park C., Marqusee S. (2005) Pulse proteolysis. A simple method for quantitative determination of protein stability and ligand binding. Nat. Meth. 2, 207–212 [DOI] [PubMed] [Google Scholar]
- 39. Na Y. R., Park C. (2009) Investigating protein unfolding kinetics by pulse proteolysis. Protein Sci. 18, 268–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wetlaufer D. B. (1973) Nucleation, rapid folding, and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. U.S.A. 70, 697–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie Z., Hendrix R. W. (1995) Assembly in vitro of bacteriophage HK97 proheads. J. Mol. Biol. 253, 74–85 [DOI] [PubMed] [Google Scholar]
- 42. van der Vies S. M., Gatenby A. A., Georgopoulos C. (1994) Bacteriophage T4 encodes a co-chaperonin that can substitute for Escherichia coli GroES in protein folding. Nature 368, 654–656 [DOI] [PubMed] [Google Scholar]
- 43. Nakonechny W. S., Teschke C. M. (1998) GroEL and GroES control of substrate flux in the in vivo folding pathway of phage P22 coat protein. J. Biol. Chem. 273, 27236–27244 [DOI] [PubMed] [Google Scholar]
- 44. Prevelige P. E., Jr., Thomas D., King J. (1993) Nucleation and growth phases in the polymerization of coat and scaffolding subunits into icosahedral procapsid shells. Biophys. J. 64, 824–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bowman B. R., Baker M. L., Rixon F. J., Chiu W., Quiocho F. A. (2003) Structure of the herpesvirus major capsid protein. EMBO J. 22, 757–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Suhanovsky M. M., Parent K. N., Dunn S. E., Baker T. S., Teschke C. M. (2010) Determinants of bacteriophage P22 polyhead formation. The role of coat protein flexibility in conformational switching. Mol. Microbiol. 77, 1568–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jabs A., Weiss M. S., Hilgenfeld R. (1999) Non-proline cis peptide bonds in proteins. J. Mol. Biol. 286, 291–304 [DOI] [PubMed] [Google Scholar]
- 48. Dornan J., Taylor P., Walkinshaw M. D. (2003) Structures of immunophilins and their ligand complexes. Curr. Top. Med. Chem. 3, 1392–1409 [DOI] [PubMed] [Google Scholar]
- 49. Howard B. R., Vajdos F. F., Li S., Sundquist W. I., Hill C. P. (2003) Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 10, 475–481 [DOI] [PubMed] [Google Scholar]
- 50. Mayr L. M., Willbold D., Rösch P., Schmid F. X. (1994) Generation of a non-prolyl cis peptide bond in ribonuclease T1. J. Mol. Biol. 240, 288–293 [DOI] [PubMed] [Google Scholar]
- 51. Dodge R. W., Scheraga H. A. (1996) Folding and unfolding kinetics of the proline-to-alanine mutants of bovine pancreatic ribonuclease A. Biochemistry 35, 1548–1559 [DOI] [PubMed] [Google Scholar]
- 52. Gordon C. L., King J. (1994) Genetic properties of temperature-sensitive folding mutants of the coat protein of phage P22. Genetics 136, 427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aramli L. A., Teschke C. M. (1999) Single amino acid substitutions globally suppress the folding defects of temperature-sensitive folding mutants of phage P22 coat protein. J. Biol. Chem. 274, 22217–22224 [DOI] [PubMed] [Google Scholar]
- 54. Morais M. C., Choi K. H., Koti J. S., Chipman P. R., Anderson D. L., Rossmann M. G. (2005) Conservation of the capsid structure in tailed dsDNA bacteriophages. The pseudoatomic structure of phi29. Mol. Cell. 18, 149–159 [DOI] [PubMed] [Google Scholar]
- 55. Fokine A., Chipman P. R., Leiman P. G., Mesyanzhinov V. V., Rao V. B., Rossmann M. G. (2004) Molecular architecture of the prolate head of bacteriophage T4. Proc. Natl. Acad. Sci. U.S.A. 101, 6003–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aksyuk A. A., Bowman V. D., Kaufmann B., Fields C., Klose T., Holdaway H. A., Fischetti V. A., Rossmann M. G. (2012) Structural investigations of a Podoviridae streptococcus phage C1, implications for the mechanism of viral entry. Proc. Natl. Acad. Sci. U.S.A. 109, 14001–14006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu X., Zhang Q., Murata K., Baker M. L., Sullivan M. B., Fu C., Dougherty M. T., Schmid M. F., Osburne M. S., Chisholm S. W., Chiu W. (2010) Structural changes in a marine podovirus associated with release of its genome into Prochlorococcus. Nat. Struct. Mol. Biol. 17, 830–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wikoff W. R., Liljas L., Duda R. L., Tsuruta H., Hendrix R. W., Johnson J. E. (2000) Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289, 2129–2133 [DOI] [PubMed] [Google Scholar]
- 59. Pierre B., Xiong T., Hayles L., Guntaka V. R., Kim J. R. (2011) Stability of a guest protein depends on stability of a host protein in insertional fusion. Biotechnol. Bioeng. 108, 1011–1020 [DOI] [PubMed] [Google Scholar]