

Background: Reversible lysine acetylation regulates the fatty acid oxidation enzyme long-chain acyl-CoA dehydrogenase (LCAD).
Results: Residues Lys-318 and Lys-322 are responsible for these effects.
Conclusion: Acetylation of Lys-318/Lys-322 alters the conformation of the LCAD active site. Sirtuin 3 (SIRT3) deacetylates these lysines and restores function.
Significance: Acetylation of LCAD Lys-318/Lys-322 can disrupt fatty acid oxidation and contribute to metabolic disease.
Keywords: Electron Transfer, Enzyme Catalysis, FAD, Fatty Acid Oxidation, Mitochondria, Posttranslational Modification, Sirtuins, Acyl-CoA Dehydrogenase, Lysine Acetylation
Abstract
Long-chain acyl-CoA dehydrogenase (LCAD) is a key mitochondrial fatty acid oxidation enzyme. We previously demonstrated increased LCAD lysine acetylation in SIRT3 knockout mice concomitant with reduced LCAD activity and reduced fatty acid oxidation. To study the effects of acetylation on LCAD and determine sirtuin 3 (SIRT3) target sites, we chemically acetylated recombinant LCAD. Acetylation impeded substrate binding and reduced catalytic efficiency. Deacetylation with recombinant SIRT3 partially restored activity. Residues Lys-318 and Lys-322 were identified as SIRT3-targeted lysines. Arginine substitutions at Lys-318 and Lys-322 prevented the acetylation-induced activity loss. Lys-318 and Lys-322 flank residues Arg-317 and Phe-320, which are conserved among all acyl-CoA dehydrogenases and coordinate the enzyme-bound FAD cofactor in the active site. We propose that acetylation at Lys-318/Lys-322 causes a conformational change which reduces hydride transfer from substrate to FAD. Medium-chain acyl-CoA dehydrogenase and acyl-CoA dehydrogenase 9, two related enzymes with lysines at positions equivalent to Lys-318/Lys-322, were also efficiently deacetylated by SIRT3 following chemical acetylation. These results suggest that acetylation/deacetylation at Lys-318/Lys-322 is a mode of regulating fatty acid oxidation. The same mechanism may regulate other acyl-CoA dehydrogenases.
Introduction
SIRT3, SIRT4, and SIRT5 are mitochondrial-localized members of the sirtuin family that catalyze NAD+-dependent lysine deacylation (1–4). Of these, SIRT3 is the best characterized, with numerous confirmed target proteins in the mitochondria (1–4). SIRT3 is recognized as the major mitochondrial lysine deacetylase, whereas SIRT5 functions as a desuccinylase and demalonylase (5, 6). SIRT4 was thought to be an ADP-ribosyltransferase with weak deacetylase activity (7, 8). However, recent reports have challenged the ADP-ribosyltransferase activity of SIRT4, and it has been shown to regulate mitochondrial malonyl-CoA decarboxylase via deacetylation (9, 10). Although little is known about the extent of mitochondrial protein succinylation, malonylation, and ADP-ribosylation, lysine acetylation has become recognized as a widespread posttranslational modification that impacts mitochondrial function (1–4). As much as 35% of all mitochondrial proteins are subject to some degree of lysine acetylation (1).
Levels of mitochondrial protein acetylation are responsive to nutritional manipulations, including fasting, caloric restriction, high-fat diet, and ethanol feeding (11–15). Proteomics surveys indicate that mitochondrial protein acetylation disproportionately clusters among enzymes in metabolic pathways, suggesting that acetylation/deacetylation serves to regulate metabolic flux (12, 16–18). Indeed, we have demonstrated previously that SIRT3 regulates the fatty acid β-oxidation (FAO)2 pathway (19). FAO is a critical energy-producing pathway, particularly in heart, muscle, liver, and brown adipose tissue, all of which show a reduced capacity for FAO in SIRT3 knockout mice. We further identified long-chain acyl-CoA dehydrogenase (LCAD), a key FAO enzyme, as a target of SIRT3 (19). In mouse liver, the level of LCAD acetylation is dependent upon the level of SIRT3. For example, fasting increases SIRT3 expression and decreases LCAD acetylation (19), whereas high-fat feeding down-regulates SIRT3 and leads to LCAD hyperacetylation (11). Lysine acetylation suppresses LCAD enzymatic activity, and SIRT3 deacetylation increases activity (11, 19).
LCAD is one of five acyl-CoA dehydrogenases (ACADs) involved in FAO, and there are four additional ACADs that participate in branched-chain amino acid catabolism (20). Although substrate specificity varies among the nine ACADs, the reaction mechanism is shared (21). All have non-covalently bound FAD cofactors, and all have a catalytic glutamate required for dehydrogenation of acyl-CoA substrates. All ACADs are reoxidized by interaction with electron transferring flavoprotein (ETF). Finally, all nine ACADs as well as ETF are acetylated on multiple lysines in vivo (22). In this study, we sought to determine which lysines on LCAD are targeted by SIRT3 and the mechanism by which acetylation/deacetylation modulates enzymatic activity. Using a novel in vitro method, we have identified LCAD Lys-318 and Lys-322 as SIRT3-targeted lysines. LCAD Lys-318 and Lys-322 appear to play a role in coordinating FAD in the active site and are conserved in medium-chain acyl-CoA dehydrogenase (MCAD) and acyl-CoA dehydrogenase 9 (ACAD9), two other ACADS in the FAO pathway. These findings suggest a molecular mechanism by which reversible lysine acetylation controls FAO flux.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification, and Site-directed Mutagenesis
The mouse LCAD, human SIRT3, SIRT3-H248Y, human MCAD, human ACAD9, and human isovaleryl-CoA dehydrogenase (IVD) protein expression constructs have been described previously (19, 23–25). Briefly, the cDNAs for SIRT3 and LCAD, with mitochondrial leader sequences removed, were cloned into the expression vector pTrcHis2B (Invitrogen), expressed as His-tagged fusion proteins in Escherichia coli, and purified with nickel-nitrilotriacetic acid-agarose (Qiagen, Valencia, CA) by the batch method. Purity was > 95%, as determined by SDS-PAGE and Coomassie staining. Amino acid substitutions in LCAD were introduced using either the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) or overlapping PCR (26) and verified by sequencing prior to expression and purification. MCAD, ACAD9, and IVD were expressed as untagged proteins and purified from E. coli crude lysates by fast protein liquid chromatography as described (23–25).
Chemical Acetylation
For initial experiments, purified LCAD protein (2.0 mg/ml) was prepared in 100 mm KPO4 buffer (pH 9.0). Acetic anhydride (Sigma) was added to a concentration equal to the amount of lysine in the sample, as calculated from the protein concentration and known lysine content of LCAD (27 lysines/monomer). The reaction was incubated for 1 h at room temperature and then placed on ice. Tris-HCl (pH 8.0) was added to a concentration of 50 mm to quench the reaction. The protein was dialyzed against 50 mm Tris-HCl (pH 8.0) overnight and used for subsequent experiments. Control reactions were the same but with water substituted for acetic anhydride. Later experiments with LCAD, MCAD, ACAD9, and IVD utilized sulfo-NHS-acetate (Thermo Scientific, Rockford, IL). The proteins were prepared in 25 mm KPO4 (pH 8.0), and sulfo-NHS-acetate was added in a 5:1 molar excess on the basis of the calculated lysine content of each protein. After 1 h, the reactions were quenched with Tris-HCl and dialyzed overnight. Control proteins were treated similarly but with water substituted for sulfo-NHS-acetate.
Western Blotting
Protein samples were heat-denatured for 5 min and subjected to electrophoresis on 10% Criterion SDS-polyacrylamide gels (Bio-Rad). Separated proteins were transferred to nitrocellulose and probed with either rabbit anti-LCAD antiserum (1:1000) or rabbit anti-acetyllysine antibody (Cell Signaling Technology, Beverly, MA). After incubation with HRP-conjugated secondary antibodies (1:3000), the blots were visualized with chemiluminescence.
LC-MS/MS and Isobaric Tags for Relative and Absolute Quantification (iTRAQ)
For qualitative studies of LCAD acetylation sites, control and acetylated LCAD protein bands were excised from an SDS-PAGE gel and prepared for LC-MS/MS as described (27). Briefly, gel bands were destained, reduced with 2.5 mm Tris (2-carboxyethyl) phosphine and alkylated with 3.75 mm of iodoacetamide, followed by in-gel digestion with 13 ng/μl of trypsin (Promega, Madison, WI) overnight at 37 °C. Peptide digests were analyzed by nano-LC-MS/MS on a Thermo Fisher LTQ OrbitrapVelos connected to a Waters Acquity ultra performance liquid chromatography system (Waters Corp., Milford, MA). Settings were for a full trypsin digest with two missed cleavages, one static modification (carbamidomethylation of cysteines), and two dynamic modifications (oxidation of methionines and acetylation of lysines). The mass tolerance was set at 10 ppm for precursor mass and 0.5 Da for collision-induced dissociation fragment ion masses.
For iTRAQ, two samples of LCAD protein were acetylated in parallel, and each was split four ways for a total of eight samples. Four of these were then deacetylated with SIRT3 as described above and four were subjected to the same incubation but with water substituted for SIRT3. The reactions were snap-frozen in liquid nitrogen and stored at −80 °C until the time of the iTRAQ analysis. Prior to iTRAQ labeling, 10 μl of each sample was mixed with 6 μl of 0.5% RapiGest SF surfactant (Waters Corp.)/2.5 mm Tris(2-carboxyethyl) phosphine hydrochloride/0.5 m triethylammonium bicarbonate buffer and heated at 65 °C for 15 min. Samples were alkylated using 3.75 mm iodoacetamide, followed by digestion with 1 μg of trypsin for 24 h at 37 °C. The digests were labeled with the 8-plex iTRAQ reagent kit following the instructions of the manufacturer (Applied Biosystems). Labeling was quenched by addition of 5% formic acid in 5% acetonitrile. Samples were incubated at 4 °C overnight to degrade the RapiGest SF surfactant, then dried down and reconstituted to 100 μl with 0.1% formic acid in water. The labeled digests were checked using MALDI TOF/TOF MS/MS for the presence of reporter ions. Following cleanup with PepClean C-18 spin columns (Thermo Fisher Scientific), the peptide samples were again evaporated and reconstituted with 100 μl of 0.1% formic acid. LC-MS/MS was performed as described above.
For each sample of LCAD in the iTRAQ analysis, peptide abundances were normalized to the abundance of the internal peptide IFSSEHDIFR, which contains no lysines, to correct for differences in the amount of LCAD protein across the samples. Normalized peptide abundance values were compared between groups (SIRT3-treated and control, n = 4) by Student's t test.
LCAD Enzymatic Activity and Kinetics
The anaerobic ETF fluorescence reduction assay was performed as described previously (28). In this assay, the fluorescence of porcine ETF, which is the natural electron acceptor for LCAD, is quenched as it accepts electrons. Briefly, ∼150 ng of purified LCAD and 1 μm purified ETF were added by needle to a sealed, degassed quartz cuvette at 32 °C in a heated cuvette block, and the enzymatic reaction was started by the addition of a saturating concentration (25 μm) of palmitoyl-CoA (Sigma). The decrease in ETF fluorescence was followed for 1 min using a Jasco fluorescence spectrophotometer. Specific activity is calculated from the slope and Y-intercept and expressed as milliunits (mU) of activity per milligram of LCAD protein. All measurements were made in triplicate. For kinetic experiments, the ETF concentration was varied from 0.5–5.0 μm. Curve fitting and determination of Vmax and Km were done using GraphPad Prism 6.0. Catalytic efficiency was calculated as described (29).
LCAD Substrate Titrations
Acetylated LCAD was prepared with sulfo-NHS-acetate as described above. Samples of protein at 1 mg/ml were degassed with alternating cycles of argon and vacuum. Palmitoyl-CoA was dissolved to 1.66 mm in 2 mm sodium acetate (pH 5) and titrated into the LCAD protein 1 μl at a time using a 50-μl Hamilton syringe attached to an automatic dispenser. Ten seconds of equilibration time were allowed after mixing before the sample was scanned at 300- to 800-nm conditions using a Jasco V-650 spectrophotometer. Final substrate concentrations varied as indicated in figure legends. All data were adjusted for the dilution resulting from substrate addition.
Molecular Modeling
Molecular modeling was performed on a Silicon Graphics Fuel workstation (Mountain View, CA) using the Insight II 2005 software package (Accelrys Technologies, San Diego, CA), and the published atomic coordinates of recombinant human MCAD were complexed with human ETF E165A (PDB code 2A1T, Ref. 30), with MCAD being the template for LCAD.
SIRT3 deacetylation Assays
Deacetylation of chemically acetylated ACAD proteins was determined by following the cleavage of [14C]NAD+ (PerkinElmer Life Sciences) in the presence of recombinant SIRT3 (31). The [14C[NAD+ is cleaved to [14C]NAM on a 1:1 molar ratio with the lysines being deacetylated. A stock of radiolabeled NAD+ was made by mixing one part of [14C]NAD+ with nine parts of cold NAD+. Reactions were performed as described (32) but with substitution of the [14C]NAD+ mixture for NAD+. In initial experiments, the reaction time was varied, and it was determined that the assay was linear beyond 40 min, with chemically acetylated LCAD as the substrate. Subsequent experiments used 20 min of deacetylation at 37 °C in a 20-μl reaction volume. Following the deacetylation reaction, [14C]NAM was separated from excess unmetabolized [14C]NAD+ as follows. 10 μl of 1 mm sodium borate (pH 8.0) was added, and the reactions were placed on ice for 2 min. Then, 1 ml of water-saturated ethyl acetate was added, and the mixture was vortexed for 2 min at maximum speed. Following centrifugation for 5 min at 16,000 × g, the top layer containing the [14C]NAM was removed for scintillation counting. Negative controls consisted of identical reactions with no acetylated protein added. The specific activity of the NAD+ in the reaction mixture was determined for each experiment by scintillation-counting an aliquot of the reaction buffer without any protein added. The specific activity was used to calculate picomoles of NAD cleaved per minute.
FAD Microplate Assay
The FAD content of purified recombinant LCAD proteins was determined using an assay kit (Sigma) following the instructions of the manufacturer. Data were expressed as nanomoles of FAD per microgram of protein.
Selective Reaction Monitoring Mass Spectrometry (SRM-MS) Proteomics
Mitochondrial samples from five wild-type mice and five SIRT3 knockout mice were pooled, and acetyllysine-containing peptides were enriched as described previously (22). Samples were analyzed by nano-LC-SRM/MS on a 5500 QTRAP hybrid triple quadrupole/linear ion trap mass spectrometer (AB SCIEX, Foster City, CA). Chromatography was performed on a NanoLC-Ultra 2D LC system (Eksigent, Dublin, CA) with buffer A (0.1% (v/v) formic acid) and buffer B (90% acetonitrile in 0.1% formic acid). Digests were separated on a 15 cm-long, 75-μm, reversed phase C18 column (3 μm, 120 Å) (Eksigent) in a cHiPLC nanoflex chip configuration at a flow rate of 300 nl/min. The gradient was 3% buffer B from 0–5 min, increased to 7% buffer B over 3 min, increased to 25% buffer B over the next 28 min, and increased to 40% buffer B over the next 7 min. Peptides were ionized using a PicoTip emitter (20 μm, 10-μm tip, New Objective, Woburn, MA). Data acquisition was performed using Analyst 1.5.1 (AB SCIEX) with an ion spray voltage of 2350 V, curtain gas of 20 p.s.i., nebulizer gas of 7 p.s.i., and an interface heater temperature of 150 °C.
Four transitions were assayed per peptide. The declustering potential and collision energy for each transition were optimized using a synthetic peptide corresponding to Lys-318 on LCAD (KAFGK m/z - 296.67632+) or a stable heavy isotope-labeled synthetic peptide corresponding to Lys-42 on LCAD (LETPSAKK* m/z - 458.2612+), where K is N-acetyllysine and the C-terminal K* is 13C615N2-Lys. Retention time scheduling was also used, with a retention time window of 240 s and a target scan time of 2.3 s. A value of 40 was used as the collision cell exit potential for all transitions. SRM transitions were acquired at unit resolution both in the first and third quadrupoles (Q1 and Q3). Skyline post-acquisition software was used to process all SRM data (33). Samples were analyzed in duplicate with 25 fmol of the LETPSAKK* heavy peptide added. Each transition was individually integrated to generate peak areas, and the sum of the top three most intense transitions was used for analysis after normalization to the heavy peptide.
Statistical Analyses
Statistical comparisons between groups were done with Student's t test, with p < 0.05 considered to be statistically significant.
RESULTS
Chemically Acetylated Recombinant LCAD Is a Substrate for SIRT3
To determine the mechanism by which reversible acetylation regulates LCAD function, we developed an in vitro system for investigating LCAD-SIRT3 interactions. First, recombinant His-tagged mouse LCAD (hereafter called LCAD) was incubated with the chemical acetylating agent acetic anhydride at a ratio of 1:1 anhydride to lysine, as calculated on the basis of the known 27 lysine residues per LCAD monomer (34). LCAD became highly acetylated, as detected by Western blotting with anti-acetyllysine antibodies (Fig. 1a), and mass spectrometry analysis of triplicate samples of acetic anhydride-treated LCAD detected 22 acetylated lysines with 88% total coverage of the LCAD protein. Of the 15 lysines reported previously to be acetylated in mouse liver LCAD (19, 22), 13 were acetylated by acetic anhydride. The exceptions were Lys-151, which was detected by LC-MS/MS but showed no modification by acetic anhydride, and Lys-189 which was not detected in our survey because of incomplete coverage.
FIGURE 1.

Chemically acetylated recombinant LCAD is a substrate for SIRT3. a, anti-acetyllysine Western blotting of acetic anhydride (Anh)- versus mock-treated recombinant LCAD protein, 20 ng/lane. Ctrl, control. b, 2.0 μm chemically acetylated LCAD was deacetylated by SIRT3 for the indicated time periods. Deacetylation was measured by following degradation of [14C]NAD+. Data points represent the average of duplicate assays. c, chemically acetylated LCAD and BSA were used as substrates for in vitro deacetylation assays with recombinant human SIRT3. Data points represent the average of duplicate assays. conc., concentration. d, anhydride acetylation of LCAD significantly reduces enzymatic activity. Deacetylation of anhydride-treated LCAD with SIRT3, but not the inactive mutant SIRT3-H248Y, partially restores enzymatic activity. Error bars represent mean ± S.D. of triplicate assays. e and f, sulfo-NHS-acetate was tested as an alternative to anhydride, which is chemically unstable. Incubation of LCAD with increasing concentrations of sulfo-NHS-acetate both increased the rate of SIRT3 deacetylation (e) and reduced the enzymatic activity of LCAD (f). Subsequent experiments utilized sulfo-NHS-acetate as an acetylating agent.
Acetic anhydride-acetylated LCAD was tested as a substrate for recombinant human SIRT3 (hereafter called SIRT3) by following the rate of [14C]NAD+ cleavage to [14C]NAM. SIRT3 deacetylated acetic anhydride-treated LCAD in a reaction that was linear for at least 40 min (Fig. 1b). Subsequently, using a 20-min reaction time and a constant amount of SIRT3, the amount of acetylated LCAD was varied from 0–25 μm. Despite the complexity of the LCAD substrate, with 22 acetylated lysines that could potentially be deacetylated by SIRT3, the reaction followed Michaelis-Menten kinetics with a Km of 3.9 μm (Fig. 1c). BSA chemically acetylated by the same method was not a substrate for SIRT3, demonstrating the specificity of the reaction. Further, acetylated LCAD was not a substrate for recombinant human SIRT5 because no cleavage of [14C]NAD+ was detected (not shown). Previous studies with mouse models and mammalian cells have suggested that increased acetylation causes LCAD enzymatic activity to decrease (11, 19). This effect of acetylation was confirmed by the observation that acetic anhydride treatment reduced LCAD activity by 75% (Fig. 1d). The loss of activity was not due to a loss of FAD cofactor because the absorbance ratio of the FAD peak (∼445 nm) to the protein peak (280 nm) did not change after chemical acetylation (data not shown). Incubation with SIRT3, but not an inactive H248Y SIRT3 mutant, led to a partial rescue of activity.
During the course of these experiments, we observed a high degree of variability in protein acetylation outcomes that we attributed to the inherent chemical instability of acetic anhydride. We therefore tested the effects of sulfo-NHS-acetate on LCAD. Sulfo-NHS-acetate is commonly used to acetylate and block surface lysines for proteomics studies and is known to be more stable than acetic anhydride under physiologically relevant conditions (34). Aliquots of LCAD were acetylated with increasing concentrations of sulfo-NHS-acetate and used as a substrate for SIRT3 deacetylation assays. A linear relationship was found between the concentration of sulfo-NHS-acetate used and the rate of deacetylation (Fig. 1e). Similarly, LCAD activity declined with increasing sulfo-NHS-acetate concentration (Fig. 1f). For subsequent studies, with the exception of the iTRAQ proteomics, which had already been initiated using acetic-anhydride treated LCAD, an optimized 5:1 sulfo-NHS-acetate:lysine molar ratio was used to chemically acetylate LCAD.
Acetylation Affects the Active Site of LCAD
As shown in Fig. 1d, acetylation reduces LCAD activity. This could be due to disruption of the reductive half-reaction, in which the catalytic glutamate introduces a double bond into the acyl-CoA substrate and passes a hydride to FAD, or the oxidative half-reaction, in which ETF reoxidizes the FAD. The reductive half-reaction can be monitored by titration of the purified enzyme with acyl-CoA substrate in the absence of electron acceptors. The characteristic FAD absorbance peak at ∼450 nm becomes quenched as the enzyme binds the substrate. At the same time, a broad peak forms in the region of 570 nm, representing the “charge-transfer complex” of the enzyme with the tightly bound enoyl-CoA product (21). Equal amounts of LCAD and chemically acetylated LCAD were titrated with 0–28 μm palmitoyl-CoA substrate (Fig. 2, a and b). Acetylation interfered with the transfer of hydride from the substrate to FAD and with formation of the charge-transfer complex. The change in absorbance of the FAD peak was plotted and used to calculate Δmax (maximum change in FAD absorbance) and Kf, the substrate concentration at which FAD quenching is half-maximal (Fig. 2c). Kf is an indicator of substrate-enzyme affinity (25). Acetylation of LCAD was found to reduce Δmax with little effect on Kf. This suggests that acetylation interferes with the transfer of hydride from the substrate to FAD and formation of the charge-transfer complex but does not reduce the binding affinity between LCAD and palmitoyl-CoA. The oxidative half-reaction is similarly affected by acetylation. In kinetic experiments with increasing concentrations of ETF (0–5 μm) and constant LCAD/substrate concentrations, acetylation of LCAD had little effect on the Km for ETF but reduced Vmax 4-fold (Fig. 2d). As a result, the calculated catalytic efficiency was reduced 5-fold, from 34.2 to 6.7 μm/s.
FIGURE 2.

Acetylation affects the active site of LCAD. Recombinant LCAD was either mock-treated (a) or acetylated with sulfo-NHS-acetate (Ac-LCAD) (b) and then titrated with palmitoyl-CoA under anaerobic conditions to study the reductive half-reaction. After each addition of substrate, the enzyme was allowed to stabilize for 10 s and then scanned on a spectrophotometer from 300–800 nm. The characteristic FAD peak (444 nm) becomes reduced, and the charge-transfer complex peak (∼570 nm) increases with increasing substrate concentrations. c, the absorbance (Abs.) of the FAD peak (444 nm) from the curves in a and b were plotted, fit with nonlinear regression, and used to calculate Δmax (maximum change in absorbance) and Kf (apparent substrate binding constant, in micromolar). d, the oxidative half-reaction was studied by kinetic assays with increasing concentrations of the physiological electron acceptor ETF. Data points represent the average of duplicate assays. The curves were fit with non-linear regression (Michaelis-Menten) and used to calculate Km, Vmax, and catalytic efficiency.
LCAD Residues Lys-318 and Lys-322 Are Targets of SIRT3 and Modulate Enzymatic Activity
The results of Fig. 2 suggested the involvement of one or more lysines near the LCAD active site in mediating the effects of acetylation and SIRT3 on enzymatic activity. To identify SIRT3-targeted lysines, chemically acetylated LCAD was deacetylated by SIRT3 and subjected to iTRAQ quantitative proteomics. Quadruplicate samples of acetylated LCAD ± SIRT3 treatment were trypsin-digested, labeled with 8-plex iTRAQ reagents, and analyzed by mass spectrometry. Peptide ratios (SIRT3-treated:control) were obtained for 40 peptide sequences covering 52.6% of the protein, including 13 acetylated LCAD lysine residues. Statistical comparisons of peptide abundance between SIRT3-treated and control revealed significant changes (p < 0.05) for eight sequences. These sequences and related neighboring peptides are illustrated in Fig. 3a.
FIGURE 3.

LCAD residues Lys-318 and Lys-322 are targets of SIRT3 and modulate enzymatic activity. a, chemically acetylated LCAD was either mock-treated or SIRT3-treated in quadruplicate. The protein samples were digested with trypsin, labeled with 8-plex iTRAQ isobaric tags, and subjected to LC-MS/MS. The relative abundance of eight peptides changed significantly with SIRT3 incubation. Three of these were acetylated peptides (Lys-42, Lys-318, and Lys-322), and five were unmodified peptides. Some peptides with non-significant change are shown for context. *, p < 0.05; SIRT3-treated versus control; N/A, not applicable, this peptide was not quantified in the iTRAQ assay. b, the three lysines identified by iTRAQ proteomics (Lys-42, Lys-318, and Lys-322) were mutated to either alanine (A) or arginine (R) and expressed in E. coli. Crude lysates (5 μg) were Western-blotted with anti-LCAD antibody. 10 ng of purified LCAD was run as positive control (PC). Subsequently, six of these mutant proteins were purified to homogeneity (K42R, K318A, K318R, K322A, K322R, and K318R/K322R), and enzymatic activity was measured in triplicate. Activities are shown as a percentage of WT LCAD activity below the Western blot. N/A, not applicable. These proteins were not purified and thus were not assayed. c, the purified arginine mutant LCAD proteins were acetylated with sulfo-NHS-acetate and used as substrates for SIRT3 deacetylation assays. Shown is the mean ± S.D. of triplicate 20-min deacetylation assays using 19 μm LCAD monomer. *, p < 0.001 versus the wild type. d, samples of wild-type LCAD and arginine mutants were acetylated with sulfo-NHS-acetate and tested for enzymatic activity versus mock-treated enzyme. For ease of comparison, the activity of each mock-treated enzyme was set to 1.0. Note that only K318R/K322R had reduced starting activity compared with WT (b). Shown is the mean ± S.D. of triplicate activity assays. *, p < 0.001, acetylated enzyme versus the corresponding non-acetylated control; #, p < 0.001 acetylated mutant LCAD versus acetylated wild-type LCAD.
Five of the eight peptide sequences showing significant changes in abundance are clustered in the region surrounding residues Lys-318 and Lys-322. The sequence KAFGKTVAHIQTVQHK, doubly acetylated at residues Lys-318 and Lys-322, decreased 2.1-fold in SIRT3-treated LCAD. The singly acetylated peptides KAFGK and AFGKTVAHIQTVQHK both decreased 1.7-fold. Consistent with increased trypsinization at Lys-318 and Lys-322, which would occur with deacetylation at these sites, the non-acetylated “daughter” peptides AFGK and TVAHIQTVQHK increased 3.7- and 2.5-fold, respectively. Together, this pattern of changes strongly suggested SIRT3 targeting of Lys-318 and Lys-322. Only one other acetylated peptide decreased significantly upon SIRT3 treatment, which was the sequence LETPSAKK acetylated at Lys-42. However, unlike the region surrounding Lys-318 and Lys-322, the changes in expected daughter peptides were either non-significant or changed in the wrong direction; i.e. a significant 2.0-fold decrease was detected in the peptide LTDGIRR (Fig. 3a). Finally, the peptide THICVTR showed a modest (1.2-fold) but statistically significant decrease. The nearest lysine residue is Lys-338, which was not quantified in the iTRAQ assay, although it was found to be modified by anhydride in our initial survey. Lys-338 has not been reported to be acetylated in vivo and was not further pursued in this study.
To further evaluate Lys-42, Lys-318, and Lys-322 as potential SIRT3 target sites, alanine and arginine substitutions were introduced, including double substitutions at Lys-318/Lys-322. Arginine substitutions retain the positive charge of the lysine residues while eliminating acetylation as a posttranslational modification, whereas alanine substitutions remove the positive charge at these positions. Relative expression of the eight mutant LCAD enzymes was measured by Western blotting crude E. coli lysates (Fig. 3b). Expression levels were comparable with the wild-type LCAD enzyme, except for the double alanine mutant Lys-318A/Lys-322A, which was expressed weakly. We next purified the Lys-318 and Lys-322 mutant enzymes as well as K42R (K42A was not attempted). K318A/K322A could not be purified to homogeneity, whereas the double arginine mutant K318R/K322R was stable but found to have reduced enzymatic activity (53% of wild-type activity). The importance of charged residues at Lys-318 and Lys-322 is highlighted by the dramatic loss of activity in the K318A and K322A mutants (12 and 20% of wild-type activity, respectively) compared with the K318R and K322R mutants (93 and 106% activity, respectively).
The K42R, K318R, K322R, and K318R/K322R mutant LCAD enzymes were chemically acetylated with sulfo-NHS-acetate and tested as substrates with SIRT3. None of the single mutations significantly reduced the rate of deacetylation, whereas double substitution at Lys-318/Lys-322 reduced the rate by half (Fig. 3c). Finally, we tested whether the arginine substitutions would confer protection against acetylation-induced loss of LCAD activity. LCAD K318R and K322R both demonstrated significant resistance to the effects of lysine acetylation (Fig. 3d). Acetylation reduced the activity of wild-type LCAD by 80% and K42R by 90%, as compared with only a 37% reduction for K318R and 23% for K322R. The K318R/K322R double mutant, which had 53% of wild-type activity prior to chemical acetylation (Fig. 3b), was completely resistant to any further loss of activity.
Acetylation at Lys-318 and Lys-322 Is Predicted to Alter the Conformation of the LCAD Active Site
Lys-318 and Lys-322 are conserved between mouse and human LCAD (Fig. 4a) and localize to the loop connecting α-helices G and H. MCAD and ACAD9 also have lysines at the equivalent positions to Lys-318 and Lys-322, whereas the remaining ACAD family members do not (Fig. 4a). Importantly, Lys-318 and Lys-322 flank residues Arg-317 and Phe-320, which are invariant across the ACAD enzyme family. Three-dimensional modeling indicates that Arg-317 and Phe-320 are crucial for FAD′ binding (the prime designation indicates a residue or ligand of the second LCAD subunit, Fig. 4b). The Arg-317 guanidinium group is within hydrogen bonding distance from Pro-175′ carbonyl oxygen as well as from oxygen atoms of the two phosphate moieties of the FAD′. Pro-175′ is part of the loop connecting β-strand 1′ and 2′, which forms part of the acyl-CoA binding site. The Phe-320 phenyl group is within a π-π interacting distance from the adenine of the FAD′. Although Lys-318 and Lys-322 do not directly interact with the FAD′ or acyl-CoA substrate, acetylation at these residues may alter the positioning of Arg-317 and Phe-320 and, subsequently, shift the alignment of the flavin moiety at the active site. Subtle perturbation of the flavin in the active site could explain the effects of acetylation on the formation of the charge-transfer complex, as noted in Fig. 2. An impaired ability to bind the FAD could also contribute to the relative instability of the K318A/K322A double mutant LCAD enzyme. To determine whether the positive charges at Lys-318 and Lys-322 play a role in FAD binding, we measured the FAD content of purified recombinant LCAD and the mutants K318R, K318A, K322R, K322A, and K318R/K322R. The conservative arginine substitutions did not alter the FAD content of LCAD (Fig. 4c). However, the K322A mutant was found to have significantly less FAD. Loss of FAD would contribute to the reduced enzymatic activity seen for this mutant (20% of the wild type). The K318A mutant had no change in FAD content yet had only 12% of wild-type activity, possibly because of an FAD conformation change.
FIGURE 4.

Acetylation at Lys-318 and Lys-322 is predicted to alter the conformation of the LCAD active site. a, amino acid sequence alignment of the region containing Lys-318 and Lys-322. All ACADs contain an invariant arginine (position 317 in LCAD) and phenylalanine (position 320 in LCAD). Mouse LCAD, human LCAD, human MCAD, and human ACAD9 have lysines at the positions equivalent to 318 and 322. Mouse MCAD and ACAD9 also have lysines at these positions (not shown). Other species besides mouse and human were not examined. VLCAD, very long-chain acyl-CoA dehydrogenase; SCAD, short-chain acyl-CoA dehydrogenase. b, ribbon and stick representation of the interface of LCAD and ETF. The model was generated from the published atomic coordinates of MCAD complexed with ETF (PDB code 2A1T, Ref. 30). The ribbon shown in jade is for LCAD monomer A of the tetramer, whereas the white ribbon is for LCAD monomer B. The tan ribbon is for the α subunit of ETF. The substrate modeled and shown is an octanoyl-CoA. c, purified mutant enzymes with substitutions at Lys-318 and Lys-322 were evaluated for FAD content using a commercial kit. Shown is the mean ± S.D. of triplicate assays. *,p < 0.001 versus wild-type LCAD.
LCAD Lys-318 Is a SIRT3 Target Site in Vivo
Previous quantitative mass spectrometry studies comparing wild-type and SIRT3−/− mouse liver mitochondrial proteins have identified significantly increased acetylation at three sites on LCAD: Lys-81, Lys-322, and Lys-358 (18, 22). Additionally, Rardin et al. (22) observed significant increases for the singly acetylated peptide KAFGKTVAHIQTVQHK, which encompasses both Lys-318 and Lys-322. Acetylation of this peptide could not be unequivocally assigned to either Lys-318 or Lys-322. For this study, we employed SRM-MS proteomics to specifically detect the peptide KAFGK (Fig. 3a), acetylated at Lys-318 in mouse liver mitochondrial protein samples. Monitoring KAFGK singles out a subpopulation of LCAD that is acetylated at Lys-318 but not Lys-322. Samples were supplemented with an isotopically labeled peptide acetylated at Lys-42 (LETPSAKK*, where K* is 13C6,2N15-Lys) as an internal standard for quantification. As reported previously, the levels of Lys-42 acetylation did not vary between wild-type and SIRT3−/− mice (Fig. 5a). The acetylated peptide KAFGK was increased 2.8-fold in SIRT3−/− mice, confirming Lys-318 as a SIRT3 target in vivo.
FIGURE 5.
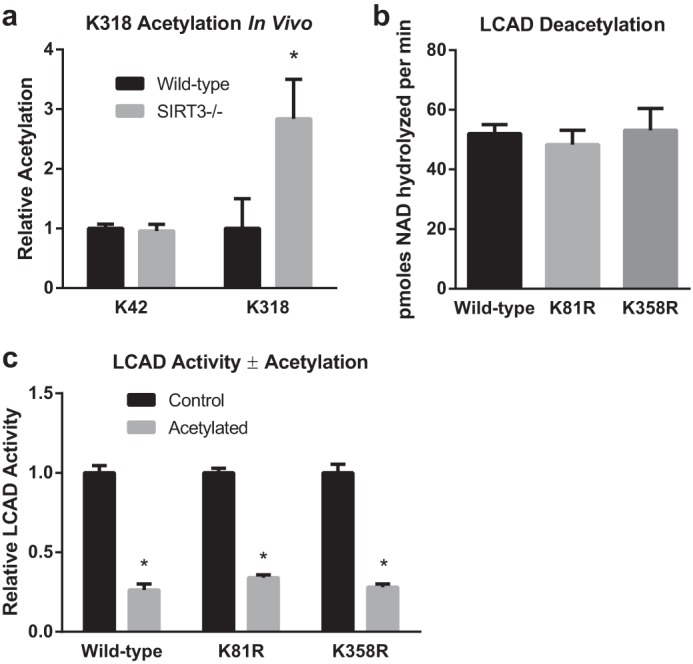
LCAD Lys-318 is a SIRT3 target site in vivo, whereas Lys-81 and Lys358 do not appear to play a role in the regulation of LCAD activity by acetylation. a, SRM proteomics were used to determine the abundance of the acetylated peptide KAFGK (acetylated at Lys-318) in liver mitochondria isolated from wild-type and SIRT3 knockout mice (n = 5). As a control, the abundance of the acetylated peptide LETPSAKK was determined. Samples were supplemented with isotopically labeled LETPSAKK* (acetylated at Lys-42, and where the C-terminal residue is 13C6,15N2-lysine), and the resulting heavy peptide signal was used for normalization. Shown is the mean ± S.D. *, p < 0.05. b and c, the importance of Lys-81 and Lys-358 as potential SIRT3-targeted sites was evaluated by creating K81R and K358R mutant LCAD proteins, chemically acetylating them with sulfo-NHS-acetate, and testing the resulting acetylated proteins as substrates for SIRT3 (b) and for enzymatic activity (c). The K81R and K358R substitutions did not affect the rate of deacetylation or provide protection against acetylation-induced loss of activity. Shown is the mean ± S.D. of triplicate assays. *, p < 0.001 for acetylated versus control proteins.
Next, we evaluated Lys-81 and Lys-358 as potential additional SIRT3 target sites using our chemical acetylation method. Arginine substitutions were made at both residues, and the mutant enzymes were purified to homogeneity. Both mutant enzymes had normal enzymatic activity and FAD content (not shown). LCAD K81R and K358R were chemically acetylated and tested as substrates for SIRT3. Neither mutant showed any reduction in the rate of deacetylation (Fig. 5b). Finally, neither mutant demonstrated protection against acetylation-induced loss of activity (Fig. 5c).
Chemically Acetylated MCAD and ACAD9 Are Also Substrates for SIRT3
MCAD and ACAD9 have lysine residues in the positions equivalent to Lys-318 and Lys-322 (Fig. 4a) and, thus, may also be subject to regulation by reversible acetylation in the same manner as LCAD. In contrast, the related enzyme IVD, which is involved in leucine catabolism, has no lysines in this region. Purified recombinant human MCAD, ACAD9, and IVD were chemically acetylated with sulfo-NHS-acetate at a molar ratio of 5:1 and tested as substrates for SIRT3 (Fig. 6). Like LCAD, the deacetylation reactions with increasing concentrations of MCAD and ACAD9 followed Michaelis-Menten kinetics with calculated Km constants of 16.1 μm and 8.7 μm, respectively (monomer concentrations). In contrast, IVD did not exhibit saturable deacetylation kinetics (Fig. 6c) and, thus, may possess one or more low-affinity SIRT3 target sites. Further work is necessary to confirm that the equivalent lysines to Lys-318 and Lys-322 are the primary SIRT3 sites on MCAD and ACAD9.
FIGURE 6.

Chemically acetylated MCAD and ACAD9 are also substrates for SIRT3. a–c, purified MCAD, ACAD9, and IVD were chemically acetylated with sulfo-NHS-acetate and used as substrates in kinetic deacetylation assays with purified SIRT3. MCAD and ACAD9, which have lysines at positions equivalent to Lys-318/Lys-322, both displayed Michaelis-Menten kinetics with SIRT3, whereas IVD, which does not have lysines at these positions, was not saturable out to 55 μm protein concentration. Each data point represents the average of duplicate assays.
DISCUSSION
In this study, we developed a method for using chemically acetylated recombinant proteins to identify sirtuin recognition sites and used this method to demonstrate that SIRT3 targets two lysines on the LCAD protein, Lys-318 and Lys-322. Site-directed mutagenesis further showed that Lys-318 and Lys-322 are necessary and sufficient for the suppression of LCAD enzymatic activity after treatment with chemical acetylating agents. Three-dimensional molecular modeling and FAD content analysis of recombinant mutant LCAD enzymes indicated that Lys-318 and Lys-322, in particular Lys-322, can influence the FAD cofactor within the LCAD active site. Mutation K322A resulted in a significant loss of the FAD cofactor, whereas the double substitution K318A/K322A destabilized the protein. We propose a model in which acetylation at Lys-318 and Lys-322 shifts the conformation of neighboring residues Arg-317 and Phe-320, both of which directly interact with the FAD cofactor. Additionally, Arg-317 is directly in contact with the loop connecting β-strand 1′ and 2′, which is crucial for substrate binding. Conformational changes in the loop containing Arg-317 and Phe-320 are predicted to result in altered substrate binding and a non-optimal coordination of the flavin moiety with the acyl-CoA substrate and catalytic glutamate, resulting in an impaired formation of the charge-transfer complex. This is evidenced by reduced Δmax in the substrate titration assays (impaired reductive half-reaction) and reduced Vmax in kinetic assays with ETF (impaired oxidative half-reaction).
As with other characterized SIRT3 targets, such as 3-hydroxy-3-methylglutaryl-CoA synthase (35), LCAD appears to have many acetylation sites that are not regulated by SIRT3. A total of 15 different LCAD lysine residues have been reported as acetylated in mouse liver mitochondria (18, 19, 22, 36). Including the SRM-MS results for Lys-318 presented here, four of these 15 lysines show significantly increased acetylation in SIRT3 knockout mice: Lys-81, Lys-318, Lys-322, and Lys-358 (18, 22). Finally, of these four, only Lys-318 and Lys-322 appear to contribute to the regulation of LCAD function on the basis of our site-directed mutagenesis studies. Interestingly, none of the single arginine substitutions altered the rate of SIRT3 deacetylation of chemically acetylated LCAD. The double substitution K318R/K322R reduced the rate of deacetylation only by half. Therefore, there may be other SIRT3-targeted lysines that we missed in our analysis. The failure of either the K318R or K322R single mutations to alter the rate of deacetylation may be a function of the complex kinetics of SIRT3 binding to two lysines that are close together, i.e. they may be recognized as a single target site, with either Lys-318, Lys-322, or both residues being deacetylated in a single LCAD-SIRT3 interaction. Also, supraphysiological chemical acetylation may also have revealed low-affinity lysine targets that manifested as residual deacetylation activity with the K318R/K322R mutant. The dose-dependent deacetylation of IVD, which showed no sign of saturation out to 55 μm enzyme concentration (Fig. 6c), highlights the potential for low-affinity lysine targets in our recombinant protein system. Interestingly, the non-mitochondrial protein BSA showed no such interaction with SIRT3 (Fig. 1c), suggesting that SIRT3 may interact with IVD because of its structural similarity to LCAD and the other ACADs (20). In summary, Lys-81 and Lys-358 may indeed be targeted by SIRT3, with the effects of the individual mutations K81R and K358R masked by the limitations of our deacetylation assay. However, Lys-81 and Lys-358 do not appear to modulate LCAD activity, and the ramification of SIRT3 regulating acetylation at these sites is not obvious. When evaluating SIRT3 target proteins, a residue-by-residue analysis of acetylation sites is clearly required to identify the subset of acetylated lysines that are of functional and physiological relevance.
LCAD and the FAO pathway produce acetyl-CoA from fatty acids, and acetyl-CoA is the presumed donor of acetyl groups for lysine acetylation. Thus, we speculate that suppression of FAO by lysine acetylation is a form of feedback inhibition. Although we previously demonstrated an association between increased mitochondrial protein acetylation and reduced FAO (19), this work provides a direct mechanism. Because arginine substitutions at Lys-318 and Lys-322 were sufficient to protect recombinant LCAD against acetylation-induced loss of activity, we postulate that acetylation at these sites causes the reduction in LCAD enzymatic activity seen in the livers of SIRT3 knockout mice (11). LCAD activity was reduced by about 50% in SIRT3−/− mice (11). Here, from the effects of chemical acetylation on the activity of the individual K318R and K322R mutant enzymes (Fig. 3d), it can be estimated that acetylation of Lys-318 alone can reduce LCAD activity by 23% (equal to the degree of residual activity suppression observed for the K322R mutant), whereas acetylation at Lys-322 can reduce activity by 37% (equal to the degree of activity suppression observed for the K318R mutant). There is likely some synergy when both sites are acetylated because we routinely observed 75–80% loss of activity for the wild-type enzyme after chemical acetylation, which is greater than the sum of the estimated individual contributions of Lys-318 and Lys-322 noted above (60%). Now that we have identified the mechanism of SIRT3 regulation of LCAD, future studies can be directed at determining the contribution of this mechanism to the overall loss of FAO capacity seen in SIRT3-deficient animals and in disease states such as obesity and metabolic syndrome. Lys-318 and Lys-322 are both conserved in human LCAD, and, thus, the mechanism described here is anticipated to extend to human biology. Likewise, our findings may extend to the regulation of MCAD and ACAD9, two other ACADs that also have lysines flanking the invariant residues analogous to Arg-317 and Phe-320.
The methods we have developed should be useful for screening other proteins of interest for SIRT3 target sites. Chemical acetylation is technically simple and provides large quantities of acetylated protein that can be compared with the non-acetylated starting material. Expression of acetylated recombinant proteins in E. coli has been accomplished previously by adding nicotinamide to the culture medium (37, 38). For LCAD, this method induced only very low-level acetylation, and the resulting purified protein was inconsistent as a substrate for SIRT3 (data not shown). Chemically acetylating after purification of the protein allows for an objective evaluation of all solvent-accessible lysines. Combining in vitro deacetylation assays with assays of enzyme function and site-directed mutagenesis can separate physiologically relevant SIRT3 target sites from artifactual deacetylation sites caused by supraphysiological modification of the target protein.
We conclude that LCAD is regulated by reversible lysine acetylation at Lys-318 and Lys-322. Acetylation at these sites deactivates and potentially destabilizes the enzyme, and SIRT3-mediated deacetylation restores activity. This mechanism may be relevant to disease states characterized by suppressed FAO. Finally, chemical acetylation is an inexpensive, efficient means of screening SIRT3 target proteins. Identified target proteins can then be subjected to proteomics analysis and site-directed mutagenesis to determine the specific lysine residues involved.
Footnotes
- FAO
- fatty acid β-oxidation
- LCAD
- long-chain acyl-CoA dehydrogenase
- ACAD
- acyl-CoA dehydrogenase
- ETF
- electron-transferring flavoprotein
- MCAD
- medium-chain acyl-CoA dehydrogenase
- ACAD9
- acyl-CoA dehydrogenase-9
- IVD
- isovaleryl-CoA dehydrogenase
- iTRAQ
- isobaric tags for relative and absolute quantification
- SRM
- selective reaction monitoring.
REFERENCES
- 1. Anderson K. A., Hirschey M. D. (2012) Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 52, 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He W., Newman J. C., Wang M. Z., Ho L., Verdin E. (2012) Mitochondrial sirtuins. Regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 23, 467–476 [DOI] [PubMed] [Google Scholar]
- 3. Newman J. C., He W., Verdin E. (2012) Mitochondrial protein acylation and intermediary metabolism. Regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 287, 42436–42443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hirschey M. D. (2011) Old enzymes, new tricks. Sirtuins are NAD+-dependent de-acylases. Cell Metab. 14, 718–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou Y., Zhang H., He B., Du J., Lin H., Cerione R. A., Hao Q. (2012) The bicyclic intermediate structure provides insights into the desuccinylation mechanism of human sirtuin 5 (SIRT5). J. Biol. Chem. 287, 28307–28314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., Lin H. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahuja N., Schwer B., Carobbio S., Waltregny D., North B. J., Castronovo V., Maechler P., Verdin E. (2007) Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 282, 33583–33592 [DOI] [PubMed] [Google Scholar]
- 8. Haigis M. C., Mostoslavsky R., Haigis K. M., Fahie K., Christodoulou D. C., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Karow M., Blander G., Wolberger C., Prolla T. A., Weindruch R., Alt F. W., Guarente L. (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell 126, 941–954 [DOI] [PubMed] [Google Scholar]
- 9. Laurent G., German N. J., Saha A. K., de Boer V. C., Davies M., Koves T. R., Dephoure N., Fischer F., Boanca G., Vaitheesvaran B., Lovitch S. B., Sharpe A. H., Kurland I. J., Steegborn C., Gygi S. P., Muoio D. M., Ruderman N. B., Haigis M. C. (2013) SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 50, 686–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Du J., Jiang H., Lin H. (2009) Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry 48, 2878–2890 [DOI] [PubMed] [Google Scholar]
- 11. Hirschey M. D., Shimazu T., Jing E., Grueter C. A., Collins A. M., Aouizerat B., Stančáková A., Goetzman E., Lam M. M., Schwer B., Stevens R. D., Muehlbauer M. J., Kakar S., Bass N. M., Kuusisto J., Laakso M., Alt F. W., Newgard C. B., Farese R. V., Jr., Kahn C. R., Verdin E. (2011) SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 13. Fritz K. S., Galligan J. J., Hirschey M. D., Verdin E., Petersen D. R. (2012) Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 11, 1633–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chalkiadaki A., Guarente L. (2012) Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 8, 287–296 [DOI] [PubMed] [Google Scholar]
- 15. Qiu X., Brown K., Hirschey M. D., Verdin E., Chen D. (2010) Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667 [DOI] [PubMed] [Google Scholar]
- 16. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., Denu J. M., Coon J. J. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., Grueter C. A., Harris C., Biddinger S., Ilkayeva O. R., Stevens R. D., Li Y., Saha A. K., Ruderman N. B., Bain J. R., Newgard C. B., Farese R. V., Jr., Alt F. W., Kahn C. R., Verdin E. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Swigonová Z., Mohsen A. W., Vockley J. (2009) Acyl-CoA dehydrogenases. Dynamic history of protein family evolution. J. Mol. Evol. 69, 176–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thorpe C., Kim J. J. (1995) Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 9, 718–725 [DOI] [PubMed] [Google Scholar]
- 22. Rardin M. J., Newman J. C., Held J. M., Cusack M. P., Sorensen D. J., Li B., Schilling B., Mooney S. D., Kahn C. R., Verdin E., Gibson B. W. (2013) Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 110, 6601–6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ensenauer R., He M., Willard J. M., Goetzman E. S., Corydon T. J., Vandahl B. B., Mohsen A. W., Isaya G., Vockley J. (2005) Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial β-oxidation of unsaturated fatty acids. J. Biol. Chem. 280, 32309–32316 [DOI] [PubMed] [Google Scholar]
- 24. Kormanik K., Kang H., Cuebas D., Vockley J., Mohsen A. W. (2012) Evidence for involvement of medium chain acyl-CoA dehydrogenase in the metabolism of phenylbutyrate. Mol. Genet. Metab. 107, 684–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Volchenboum S. L., Mohsen A. W., Kim J. J., Vockley J. (2001) Arginine 387 of human isovaleryl-CoA dehydrogenase plays a crucial role in substrate/product binding. Mol. Genet. Metab. 74, 226–237 [DOI] [PubMed] [Google Scholar]
- 26. Heckman K. L., Pease L. R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 [DOI] [PubMed] [Google Scholar]
- 27. Halfter W., Candiello J., Hu H., Zhang P., Schreiber E., Balasubramani M. (2013) Protein composition and biomechanical properties of in vivo-derived basement membranes. Cell Adh. Migr. 7, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goetzman E. S. (2009) The regulation of acyl-CoA dehydrogenases in adipose tissue by rosiglitazone. Obesity 17, 196–198 [DOI] [PubMed] [Google Scholar]
- 29. Goetzman E. S., Mohsen A. W., Prasad K., Vockley J. (2005) Convergent evolution of a 2-methylbutyryl-CoA dehydrogenase from isovaleryl-CoA dehydrogenase in Solanum tuberosum. J. Biol. Chem. 280, 4873–4879 [DOI] [PubMed] [Google Scholar]
- 30. Toogood H. S., van Thiel A., Basran J., Sutcliffe M. J., Scrutton N. S., Leys D. (2004) Extensive domain motion and electron transfer in the human electron transferring flavoprotein.medium chain Acyl-CoA dehydrogenase complex. J. Biol. Chem. 279, 32904–32912 [DOI] [PubMed] [Google Scholar]
- 31. Landry J., Sternglanz R. (2003) Enzymatic assays for NAD-dependent deacetylase activities. Methods 31, 33–39 [DOI] [PubMed] [Google Scholar]
- 32. Vaquero A., Sternglanz R., Reinberg D. (2007) NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 26, 5505–5520 [DOI] [PubMed] [Google Scholar]
- 33. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline. An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo X., Bandyopadhyay P., Schilling B., Young M. M., Fujii N., Aynechi T., Guy R. K., Kuntz I. D., Gibson B. W. (2008) Partial acetylation of lysine residues improves intraprotein cross-linking. Anal. Chem. 80, 951–960 [DOI] [PubMed] [Google Scholar]
- 35. Shimazu T., Hirschey M. D., Hua L., Dittenhafer-Reed K. E., Schwer B., Lombard D. B., Li Y., Bunkenborg J., Alt F. W., Denu J. M., Jacobson M. P., Verdin E. (2010) SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Still A. J., Floyd B. J., Hebert A. S., Bingman C. A., Carson J. J., Gunderson D. R., Dolan B. K., Grimsrud P. A., Dittenhafer-Reed K. E., Stapleton D. S., Keller M. P., Westphall M. S., Denu J. M., Attie A. D., Coon J. J., Pagliarini D. J. (2013) Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. J. Biol. Chem. 288, 26209–26219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirschey M. D., Shimazu T., Huang J. Y., Verdin E. (2009) Acetylation of mitochondrial proteins. Methods Enzymol. 457, 137–147 [DOI] [PubMed] [Google Scholar]
- 38. Schwer B., Bunkenborg J., Verdin R. O., Andersen J. S., Verdin E. (2006) Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. U.S.A. 103, 10224–10229 [DOI] [PMC free article] [PubMed] [Google Scholar]