

Background: Genetic variations in voltage-gated calcium (CaV) channels can alter function leading to disease.
Results: A variant from migraine patients was identified in the synprint site of the CaV2.1 channel which increased function.
Conclusion: This gain-of-function occurs via alterations in inactivation and SNARE protein regulation of the CaV2.1 channel.
Significance: Increased channel activity caused by this variant may contribute to migraine pathophysiology.
Keywords: Calcium Channels, Genetics, Neurological Diseases, Protein-Protein Interactions, SNARE Proteins, Migraine
Abstract
Mutations in the CACNA1A gene, which encodes the pore-forming α1A subunit of the CaV2.1 voltage-gated calcium channel, cause a number of human neurologic diseases including familial hemiplegic migraine. We have analyzed the functional impact of the E1015K amino acid substitution located in the “synprint” domain of the α1A subunit. This variant was identified in two families with hemiplegic migraine and in one patient with migraine with aura. The wild type (WT) and the E1015K forms of the GFP-tagged α1A subunit were expressed in cultured hippocampal neurons and HEK cells to understand the role of the variant in the transport activity and physiology of CaV2.1. The E1015K variant does not alter CaV2.1 protein expression, and its transport to the cell surface and synaptic terminals is similar to that observed for WT channels. Electrophysiological data demonstrated that E1015K channels have increased current density and significantly altered inactivation properties compared with WT. Furthermore, the SNARE proteins syntaxin 1A and SNAP-25 were unable to modulate voltage-dependent inactivation of E1015K channels. Overall, our findings describe a genetic variant in the synprint site of the CaV2.1 channel which is characterized by a gain-of-function and associated with both hemiplegic migraine and migraine with aura in patients.
Introduction
Mutations in the CACNA1A gene encoding the α1A subunit of the human CaV 2.14 Ca2+ channel cause a group of dominantly inherited human neurological disorders including familial hemiplegic migraine (FHM1-OMIM 141500) (1, 2), episodic ataxia type-2 (3, 4), and spinocerebellar ataxia type 6 (5).
CaV2.1 channels are located mainly in nerve terminals where they form clusters in specialized subdomains of the presynaptic membrane (the active zones). Here, they play an important role in fast release of classical neurotransmitters like glutamate, acetylcholine, and GABA by mediating depolarization induced calcium entry into synaptic boutons (6). Extensive studies indicate that CaV2.1 activity is modulated by a complex network of interactors that includes protein kinase C, the β and γ subunits of the heterotrimeric G protein, and presynaptic proteins of the active zones (7–10). The first presynaptic proteins shown to be involved in protein-protein interactions with CaV2.1 were the t-SNAREs syntaxin 1A and SNAP-25. They bind directly to the “synaptic protein interaction” (synprint) site (of 245–314 amino acids) present in the cytoplasmic loop (LII–III) connecting the II and the III domain of the pore-forming α1A (11, 12). This protein complex (also called excitosome) (13) plays an important role in the fast release of neurotransmitters by localizing the Ca2+ channels at the presynaptic terminals near the docked synaptic vesicles. Furthermore, the t-SNARE proteins affect channel activity, and studies reported that co-expression of syntaxin 1A and SNAP-25 with CaV2.1 reduces channel availability by shifting their voltage dependence of steady-state inactivation toward more negative membrane potentials (9, 10, 14).
Although many of the CaV2.1 mutations in the transmembrane or C-terminal domains of the channel that cause hemiplegic migraine (HM) have been characterized, there is little information on how mutations in the synprint site of CaV2.1 impact channel function. In this study, we identify a missense variant (E1015K) associated with HM and migraine with aura (MA) in Italian families that occurs in the synprint site of CaV2.1 and characterize how it affects localization and function of the channel.
EXPERIMENTAL PROCEDURES
Patients and Genetic Analysis
Family 1
The 8-year-old proband (II.2) suffers from HM attacks. Her 13-year-old brother (II.1) also had HM attacks associated with paresthesia. The father (I.1) shows migraine without aura, whereas the mother (I.2) experienced a migraine attack with hemiplegia.
Family 2
the 41-year-old proband (II.1) has suffered, since age 25, from two or three attacks per year, lasting all day, showing frontal headache pain, preceded by arm paresthesia, peribuccal paresthesia, language difficulties, flashing lights, and confusion. Her 48-year-old sister (II.3) reports headache attacks preceded by flashing lights and hand paresthesia. Another sister (II.2) shows migraine without aura, and the father (I.1) manifests common headache.
Family 3
The 43-year-old proband has suffered, since age 16, from two or three attacks of MA per month, lasting 2–10 days. She reported a similar phenotype in other relatives, without referring to family structure.
For genetic testing, a patient's DNA was extracted from blood leukocytes using the Biorobot EZ1 Extractor (Qiagen), according to the standard protocol. Coding region and flanking intron sequences of the CACNA1A gene were amplified by PCR with specific primers, for a total of 51 fragments (range 120–430 bp). PCR products were analyzed directly on denaturing HPLC (Transgenomic), after a heteroduplex formation cycle. Samples with an abnormal elution profile were sequenced to determine the nature and the position of the variation. The PCR products and sequencing reactions were purified on Multiscreen 96 PCR plates (Millipore) and G50 Multiscreen TM-HV plates (Millipore), respectively, using the automated liquid handling system Biomeck FX (Beckmann Coulter). Dye-terminator cycle sequencing reactions were set up following the manufacturer's instructions and loaded on a ABI Prism 3730 DNA Analyzer (Applied Biosystems). In addition to CACNA1A, we also analyzed the other two genes associated with FHM, ATP1A2 (FHM2-OMIM 602481) and SCN1A (FHM3-OMIM 609634) by direct Sanger sequencing, as described previously (15, 16). Called sequences were assembled and compared with the reference sequences in the Locus Reference Genomic databases (CACNA1A, LRG_7 NCBI_NM_001127221.1; ATP1A2, LRG_6 NCBI NM_000702.2; SCN1A, LRG_8 NCBI NM_006920.4).
Reagents and Antibodies
The protease inhibitor cocktails, antibodies against β3, syntaxin 1A, actin, and microtubule-associated protein 2 (MAP2) anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase came from Sigma-Aldrich; the antibodies against synaptobrevin-2 and SNAP-25 came from SynapticSystem; the antibody against transferrin receptor was from Zymed Laboratories Inc. The sulfosuccinimidyl-2-(biotinamido)ethyl-1,3′-dithiopropionate (EZ-LinkTM, Sulfo-NHS-SS-Biotin) was from Thermo Scientific (Milan, Italy), and streptavidin Plus UltraLink Resin was from Pierce. The fluorescein-, rhodamine-, or Cy3-conjugated anti-mouse or rabbit IgGs were purchased from Jackson ImmunoResearch Laboratories. The cDNA encoding for the human CaV2.1 α1A subunit (CaV2.1-pCMV), the rat CaVβ3, and the rabbit α2δ subunits were a generous gift from Dr. J. Striessnig (4). An antibody was raised in rabbit using a synthetic peptide corresponding to the sequence of human α1A between amino acids 2226 and 2247 (cg-KDRYAQERPDHGRARARDQRWS) coupled to keyhole limpet hemocyanin. The antibodies (from here on referred to as anti-hα1A) were tested for their specificity by Western blotting and immunofluorescence after preadsorption with antigen.
Molecular Biology and Site-directed Mutagenesis
The human full-length cDNA encoding for the human CaV2.1 α1A subunit was cloned in pEGFP-C1 (Takara Bio Europe/Clontech) expression vectors to allow for channel identification after expression. The p.E1015K variant was introduced into α1A by applying a PCR-based, site-directed mutagenesis approach. For this purpose we used the QuikChange site-directed mutagenesis XL kit (Stratagene), according to the supplier's recommendation, forward (5′-GCTCCAGCCACGTACAAGGGGGACGCG-3′) and reverse (5′-GCGCGTCCCCCTTGTACGTGGCTGGAG-3′) primers. We then used resulting plasmids to transform XL10-Gold ultracompetent bacterial cells (Stratagene), selected colonies, and extracted DNA using the PEG preparation of plasmid DNA protocol. The construct was sequenced to verify the presence of the desired variant and rule out additional substitutions.
Cell Culture and Transfection
HEK293 cells were maintained at 37 °C in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% glutamine. HEK293 cells were plated on glass coverslips and transiently transfected with plasmids encoding the wild type (EGFP-α1AWT) or the mutant (EGFP-α1AE1015K) α1A subunit combined with accessory rat CaVβ3 and rabbit α2δ subunits in a 1:2:2 molar ratio using jetPEI reagent (PolyplusTransfection) or Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction. In some experiments, cells were also co-transfected with CaV2.1 subunits (as described above) and syntaxin 1A (1:2:2:1 molar ratio) or SNAP-25 (1:2:2:1 molar ratio). Primary cultures of hippocampal neurons were prepared from the cerebral cortex of 18-day-old rat embryos and maintained in neurobasal medium supplemented with B27 as described previously (17). For hippocampal neuron transfection, plasmids encoding EGFP-α1AWT or EGFP-α1AE1015K were transfected into 6 days in vitro cultures using the calcium phosphate method. Neurons were analyzed by immunofluorescence 5–8 days after transfection.
Electrophysiology
Whole cell CaV2.1 currents were recorded from EGFP-positive HEK293 cells bathed in an external solution containing 145 mm tetraethylammonium chloride, 10 mm HEPES, and 10 mm BaCl2 or 10 mm CaCl2, pH 7.4, with tetraethylammonium hydroxide. Patch pipettes with resistances measuring 2–3 megohms were filled with internal recording solution (120 mm CsMeSO4-, 10 mm CsCl, 2 mm MgCl2, 1 mm EGTA, 10 mm HEPES, 4 mm Mg-ATP, and 3 mm Tris-GTP, pH 7.2, with CsOH), and peak whole cell Ba2+ or Ca2+ currents were measured using an Axon Axopatch 200B amplifier (Molecular Devices) interfaced to a PC via a Digidata 1320 (Molecular Devices). Data were acquired at 5–10 kHz with leak and capacitative transients subtracted online with a P/4 protocol using pClamp 10.0 software. Series resistance was routinely compensated at 60–75%, and access resistance was monitored continually during the experiments such that cells with uncompensated voltage errors >5 mV were excluded from analysis. Current-voltage (I-V), activation, and inactivation curves were fit with modified Boltzmann functions, and inactivation kinetics were determined as described previously (18). Recovery from inactivation was measured using a double-pulse protocol consisting of a 4-s depolarizing voltage prepulse to 20 mV followed by a 20-ms test depolarization to the same voltage at increasing time intervals. The current amplitude evoked by each test potential was then normalized to the maximum current during the prepulse and plotted against the interval time. Data are expressed as the mean ± S.E. of n experiments with statistical significance determined at the p level indicated using either an unpaired t test or analysis of variance as appropriate.
Immunofluorescence
HEK293 cells and neurons were fixed with 4% paraformaldehyde in phosphate buffer, pH 7.3, containing 4% sucrose at 37 °C and permeabilized for 5 min at room temperature in PBS containing 0.3% Triton X-100. After immunostaining as described previously (19), images were recorded using a Zeiss LSM510 Meta or a MRC-1024 laser-scanning microscope (Bio-Rad) equipped with a 60× or 40× objective. To compare the double-stained patterns, images from the fluorescein or rhodamine channels were acquired separately and superimposed. The images were processed using Photoshop (Adobe Systems).
Cell Extracts
HEK293 cells were lysed in buffer A (150 mm NaCl, 2 mm EGTA, 50 mm Tris-HCl, pH 7.5, and a Sigma-Aldrich protease inhibitor mixture diluted 1:1000) containing 1% Triton X-100, 0.5% saponin, and a protease inhibitor mixture. Extracts were then centrifuged at 20,000 × g for 20 min at 4 °C. Protein concentrations were determined in the clear supernatants by means of the Bradford protein assay.
Biotinylation Assay
HEK293 cells transiently expressing EGFP-α1AWT or EGFP-α1AE1015K with the accessory subunits were incubated with 0.3 mg/ml sulfo-NHS-SS-Biotin (Thermo Scientific) dissolved in PBS with 0.1 mm CaCl2 and 1 mm MgCl2 for 30 min at 4 °C. The labeled cells were then washed three times for 10 min with 50 mm glycine in TBS (25 mm Tris, 85 mm NaCl, 5 mm KCl, 1 mm CaCl2, 1 mm MgCl2) to quench free biotin and with ice-cold PBS containing 0.1 mm CaCl2 and 1 mm MgCl2 followed by lysis in buffer A as described above. The clear supernatants containing equal amounts of protein were then incubated with streptavidin beads to isolate the biotinylated proteins. Following extensive washes in buffer A, proteins were eluted from the streptavidin beads and analyzed by SDS-PAGE followed by Western blotting.
Immunoisolation
Isolation of protein complexes was performed using a μMACSTM GFP Isolation kit (Miltenyi Biotec). Stably transfected HEK293 cell lines expressing β3 and α2δ were transfected for GFP or co-transfected with EGFP-α1AWT or EGFP-α1AE1015K and syntaxin 1A. Cells were lysed by incubation for 1 h at 4 °C with precooled (4 °C) lysis buffer (1% Triton, 150 mm NaCl, 2 mm EGTA, 50 mm Tris-HCl, pH 7.5, protease inhibitors) and centrifuged for 10 min at 10,000 rpm at 4 °C). Cell extracts (0.8 mg) were incubated with 50-μl microBeads on a rotating wheel overnight at 4 °C. Columns were prepared applying 200 μl of the lysis buffer. Cell lysate was applied onto the column, and then the column was washed with 3 × 500 μl of cell lysis buffer and 1 × 100 μl of wash buffer (20 mm Tris-HCl, pH 7.5). Immunocomplexes were eluted with 80 μl of elution buffer. The eluates were analyzed by Western blotting.
Western Blotting
Total cell extracts, biotinylated proteins, or immunocomplexes isolated as described above were analyzed by SDS-PAGE followed by immunoblotting, using anti-rabbit IgG light chains or anti-mouse IgG conjugated to peroxidase (diluted 1:50,000) as secondary antibodies. The peroxidase was revealed using a chemiluminescent substrate (Pierce). For quantitative analysis, unsaturated autoradiograms were acquired using an ARCUS II scanner (Agfa-Gevaert, Mortsel, Germany), and the density of bands corresponding to α1A or syntaxin 1A was quantified using National Institutes of Health ImageJ software. Data expressed as arbitrary units were collected from five independent experiments and expressed as the ratio between EGFP-α1AWT or EGFP-α1AE1015K and syntaxin 1A (α1A density/syntaxin 1A density) in each experiment.
Statistical Analysis
We analyzed the data with Student's t test by using GraphPad Prism software, and we used the customary threshold of p < 0.05 to declare statistical significance.
RESULTS
Clinical and Genetic Spectrum in Studied Subjects
Clinical analysis classified affected individuals as HM, fulfilling the International Headache Society criteria (IHS II) in Families 1 and 2 (Family 1: II.2, II.1; Family 2: II.1) (Fig. 1, C and D). In Family 3, a nonhemiplegic migraine with aura phenotype was diagnosed (Family 3: proband). The CACNA1A c.3043G>A variant was detected by Sanger sequencing in three probands and two affected relatives, who were heterozygotes (Fig. 1, A and B). The variant is reported in 1000 genomes (rs16024) as a rare variant, with minor allele frequency = 0.002 (4/2184), and is not present in 380 chromosomes from the general Italian population. In all of the analyzed genes, only common variants (minor allele frequency range 16–28%) classified as polymorphisms were present whereas none of the mutations known to cause FHM or neither new nor rare variants was detected (data not shown).
FIGURE 1.

Genetic analysis. A, denaturing HPLC analysis of CACNA1A, exon 19. Chromatograms at 68 °C shows different profiles corresponding to wild type and variant samples. B, Sanger sequence analysis of CACNA1A exon 19, showing a sample heterozygous for the c. 3043G>A (E1015K) variant. C, Family 1 pedigree and segregation of the variant with the hemiplegic migraine phenotype. D, Family 2 pedigree. Segregation analysis was not feasible. Black symbol, hemiplegic migraine; gray symbol, migraine without aura; hatched symbol, common headache; plus (+) sign indicates a subject heterozygous for the E1015K variant; minus (−) sign indicates a subject without the variant.
Expression and Trafficking of E1015K Variant
The E1015K variant converts a negative to a positive charge in the synprint domain of the α1A subunit. This mutation was introduced in the EGFP-tagged α1A subunit, and the WT and mutated proteins have been expressed together with α2δ1 and β3 in HEK293 cells. It has been reported that β subunits may differently affect the channel properties (20, 21). The β3 subunit was selected for our studies because it associates mainly with CaV2.1 (21), and it is highly expressed in mammalian brains where it appears to contribute more to presynaptic channels than other β isoforms at least in hippocampal neurons (22–24).
As even small structural changes in CaV2 channel α1 subunits can affect α1 protein expression, we tested the expression, the cell surface localization, and trafficking of EGFP-tagged WT and E1015K CaV2.1 α1A after expression in HEK293 or in hippocampal neurons. Initially, the specificity of our affinity-purified antibody for the α1A subunit was tested by immunofluorescence or Western blotting. As shown in Fig. 2A, the anti-hα1A antibody immunolabeled EGFP-tagged hα1A. The immunostaining was completely abolished after preincubation of the antibody with antigen (Fig. 2B), and it was not detected in cells expressing EGFP (Fig. 2C). In Western blotting analysis (Fig. 2D), the anti-hα1A recognized two prominent bands of ∼160–180 kDa and 230–240 kDa. Immunolabeling of these bands was abolished when the antibody was preincubated with the antigen. From these data we concluded that the antibody specifically recognizes hα1A and that the bands detected in Western blots correspond to two forms of the EGFP-tagged α1A. The polypeptide of ∼240 kDa corresponds to the full-length, mature form of EGFP-α1A. This conclusion was also supported by the following observations: (i) the band at ∼240 kDa is close to that expected for mature hα1A (∼209 kDa) (4) together with the EGFP tag (∼ 27 kDa); (ii) in biotinylation experiments (see below) the ∼240-kDa biotinylated polypeptide was recognized by the antibody. Taking into account these results, in our following experiments we followed the band of 240 kDa.
FIGURE 2.
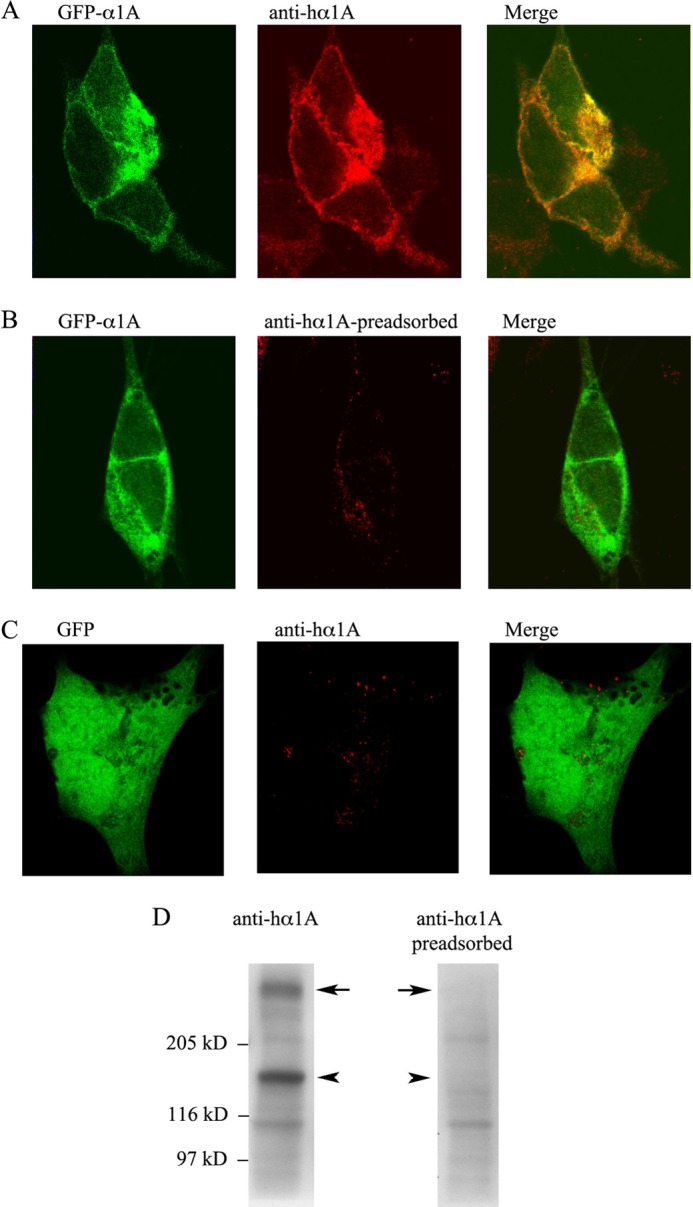
Specificity of the anti-hα1A antibody. A–C, representative images of SY5Y cells transfected with EGFPα1AWT/β3/α2δ or EGFP-α1AE1015K/β3/α2δ subunits and immunostained with affinity-purified anti-hα1A antibody before (A and C) or after preincubation (B) with the peptide used for immunization (preadsorbed). D, representative Western blots of total cell extracts (40 μg of proteins) from HEK293 cells transfected with WT channels and labeled with anti-hα1A or anti-hα1A antibodies preincubated with antigen.
After confirming the specificity of the anti-hα1A antibody, we used immunofluorescence analysis to compare α1A expression levels. A consistent number of cells (∼40%) expressed detectable levels of both EGFPα1AWT and the EGFP-α1Ae1015K variant (Fig. 3A). The full-length form of the EGFP-tagged α1A protein was detected by Western blot analysis in both WT and mutated α1A-transfected cells, suggesting that this mutation does not underlie misfolding and proteolytic degradation of the mutated subunit (Fig. 3B). Next, we evaluated whether both proteins were delivered to the plasma membrane. To this aim, cell surface proteins were labeled with biotin and isolated by means of streptavidin beads. Western blot analysis revealed that WT and E1015K α1A subunits were biotinylated with similar efficiency, thus suggesting that both proteins were similarly delivered to the cell surface (Fig. 3C).
FIGURE 3.
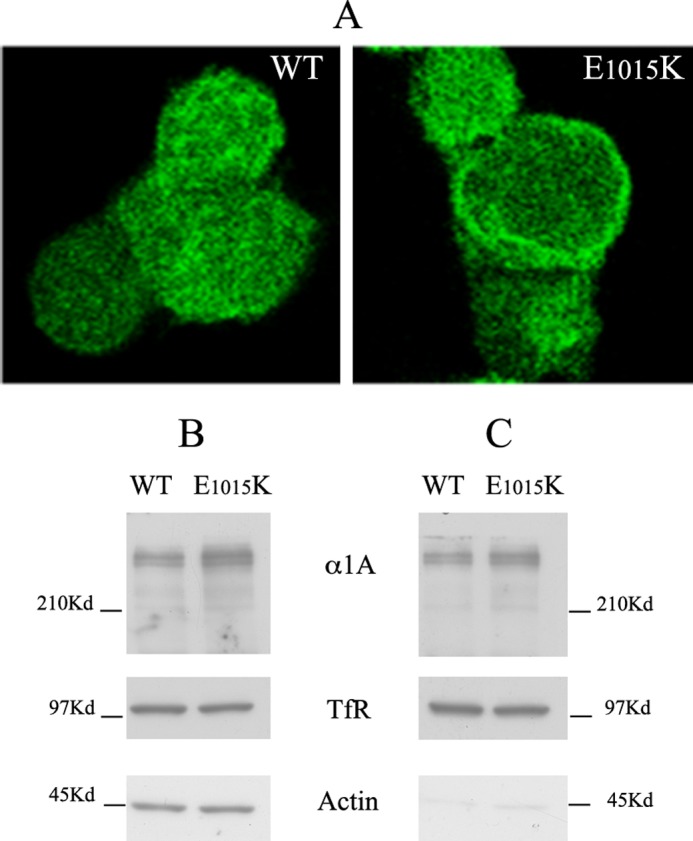
Expression and cell surface delivery of WT and E1015K CaV2. 1 in HEK cells. A, representative images of HEK293 cells transfected with EGFPα1AWT or EGFP-α1AE1015K subunits. B and C, representative Western blots of total cell extracts (B, 40 μg of protein, input) and biotinylated proteins isolated with streptavidin beads (C, 400 μg of total protein was used for each experiment) from HEK293 cells transfected with either WT or mutated (E1015K) channels and labeled with biotin at 4 °C for 30 min. Blots were probed with antibodies against human α1A, transferrin receptor (TfR) or actin. Note that aliquots of both WT and E1015Kα1A are biotinylated and isolated with streptavidin beads, indicating their expression at the cell surface.
Next, we evaluated the distribution of WT and E1015K CaV2.1 in cultured hippocampal neurons, a model system in which the properties and trafficking of CaV2 WT channels and mutants have been deeply analyzed (25). In addition, these studies demonstrated that hippocampal neurons are able to synthesize and insert CaV2.1 channels into membranes at ∼5 times over their normal level without introducing auxiliary subunits. Therefore, in our experiments neurons (6 days in vitro) were transfected with only the α1A subunits and analyzed after 5 or 8 days (11–14 days in vitro) to allow neurons to differentiate and to form synapses. As previously reported, overexpressed EGFP-tagged WTα1A was detected in both somato-dendritic and in presynaptic compartments as revealed by co-staining with MAP2 or synaptobrevin-2 (Fig. 4). Modifications of the synprint domain have been proposed to alter the presynaptic distribution of CaV2.1 (26). In the case of the E1015K variant, no differences were observed in the distribution in neurons (Fig. 4). Altogether, these results indicated that expression and cellular distribution of the EGFP-α1A E1015K variant are not altered.
FIGURE 4.
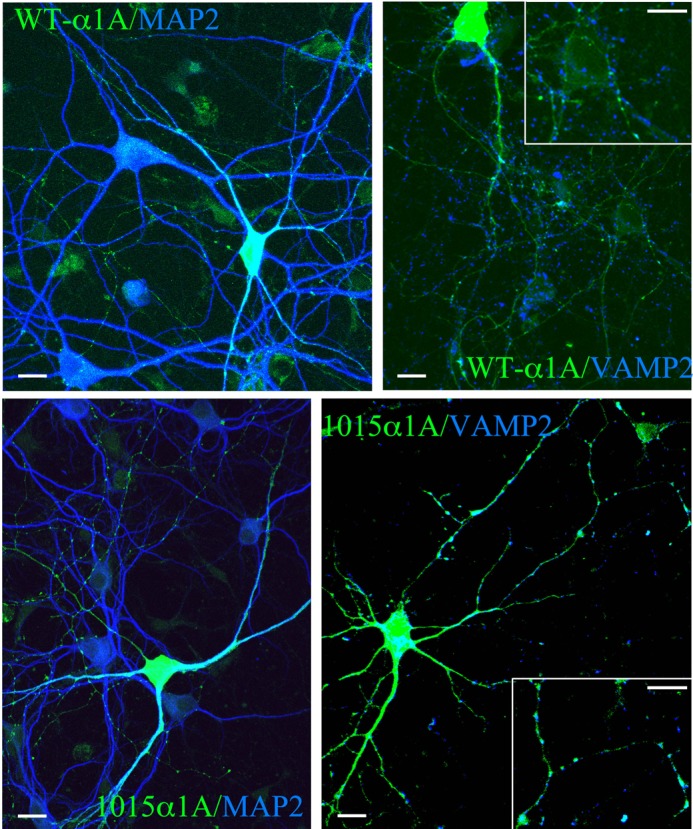
E1015K CaV2. 1 is delivered to somato-dendritic and presynaptic compartments. Hippocampal neurons (6 days in vitro) transfected with WT or E1015K α1A were fixed after 5 days and probed with antibodies against MAP-2 or synaptobrevin-2. Note the distribution of both EGFP-α1AWT EGFP-α1AE1015K in the soma and dendrites immunolabeled for MAP-2. In addition, co-localization with synaptobrevin is observed in punctate-like structures demonstrating localization of both WT and the E1015K α1A in the presynaptic terminals. Scale bars, 10 μm.
Increased Function in E1015K Channels Relative to WT
To investigate the effect that the E1015K variant had on CaV2.1 channel function, whole cell Ba2+ and Ca2+ currents were recorded in CaV2.1-transfected HEK293 cells. Fig. 5A compares representative I-V traces from WT and E1015K-transfected cells showing that Ba2+ currents were larger from E1015K channels. This increased current density was apparent across a broad voltage range but was not associated with any voltage shift in the I-V relationship (Fig. 5B). Migraine mutations in CaV2.1 channels have also been shown to impact on calcium-dependent gating properties (27). To determine whether the E1015K variant influenced Ca2+ currents, whole cell recordings were performed with Ca2+ as the charge carrier. Although a reduced current density overall and a 10-mV depolarizing shift in the peak current density were observed as expected using the less permeant cation, Ca2+ currents were significantly elevated over a broad voltage range for E1015K CaV2.1 similar to results obtained with Ba2+ (Fig. 5, C and D). These results indicate that the E1015K variant results in a significantly increased current density that is independent of the permeant cation.
FIGURE 5.
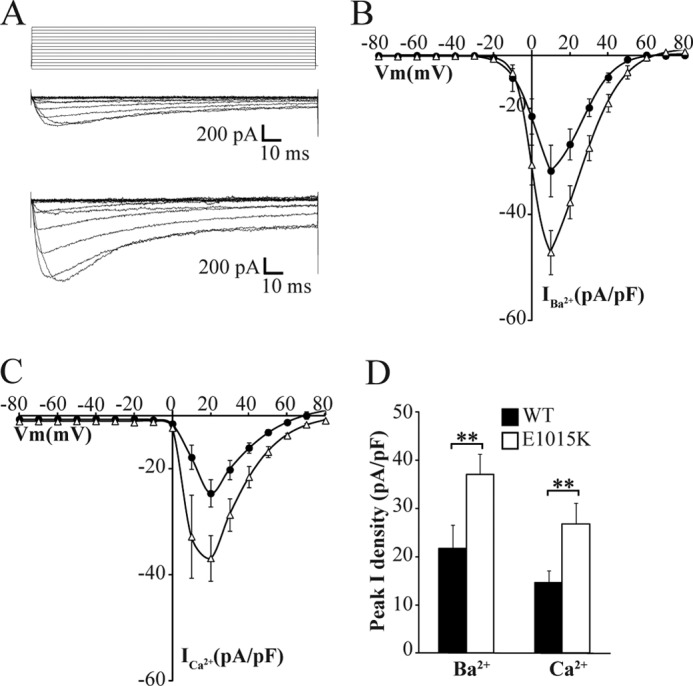
Increased current density in E1015K CaV2.1 compared with WT. A, representative whole cell inward Ba2+ currents (IBa) in response to the indicated voltage protocol (top) recorded from HEK293 cells transfected with WT (middle) and E1015K (bottom) CaV2.1 channel subunits. B and C, mean I–V relationships of peak IBa (B) (WT; n = 11 and E1015K; n = 12) and ICa (C) (WT; n = 12 and E1015K; n = 9) current density in CaV2.1-transfected cells. D, mean peak IBa and ICa current density in WT (n = 11–12) and E1015K-transfected (n = 9–12) cells measured at holding potentials of 10 and 20 mV respectively where ** indicates p < 0.01. Error bars, S.E.
To further characterize the functional effects of the E1015K mutation, activation and inactivation properties were measured. No significant difference was detected in either the voltage dependence of activation or the activation time constant between WT and E1015K CaV2.1 (data not shown). However, various inactivation parameters of E1015K CaV2.1 were altered. Open state inactivation measured via a 1-s depolarization to 0 mV induced diverging current responses between WT and E1015K (Fig. 6A). Whereas no effect was observed in the fast component of the inactivation rate (Fig. 6B), the rate of the slow component was significantly slower for E1015K CaV2.1 compared with WT (Fig. 6C). Similar results were also obtained with depolarizations to 10 and 20 mV (data not shown). In steady-state inactivation experiments, the voltage dependence of inactivation was shifted to more depolarized potentials for E1015K (Fig. 6D). This resulted in a significant difference in the half-inactivation voltage (Vhalf) of 36.4 ± 1.6 mV for WT versus 29.1 ± 1.4 mV for E1015K. This demonstrates a greater availability of channels for opening at a given voltage for E1015K compared with WT. The time course of recovery from inactivation also revealed substantial differences between the two channel types. E1015K channels were quicker to recover from inactivation at a recovery potential of −90 mV than WT (Fig. 6E) where a single exponential fit of the time constant produced recovery rates of 398.1 ± 56.0 ms and 715.6 ± 76.1 ms, respectively. Taken together, these results indicate that the E1015K variant causes profound differences in channel properties, particularly in how the channel inactivates.
FIGURE 6.

Inactivation properties of E1015K CaV2.1 channels are significantly altered. A, representative IBa current traces in response to the depolarization step shown (top) were normalized and aligned to compare the open-state inactivation of WT and E1015K CaV2.1 currents. Fast (τfast) (B) and slow (τslow) (C) inactivation rate constants were obtained from a double-exponential fit of the current decay shown in A from cells transfected with WT (n = 8) and E1015K (n = 9) CaV2.1 where ** indicates p < 0.01. D, voltage dependence of steady-state inactivation curves were recorded from WT (n = 15) or E1015K (n = 15) CaV2.1-transfected cells. E, time course of recovery from inactivation following a 4-s depolarization step to 20 mV for WT (n = 10) and E1015K (n = 9) CaV2.1 currents. Data represent the mean fractional current amplitude at different recovery times and were fit with a single exponential function. Error bars, S.E.
SNARE Protein Regulation of E1015K Channels
SNAREs modulate CaV2.1 channel function by binding to the synprint site, therefore an amino acid substitution in this region could perturb this important mode of channel regulation. To test this hypothesis, we co-transfected SNAP-25 or syntaxin 1A together with WT or E1015K CaV2.1. First, we analyzed the efficiency of protein co-expression by immunofluorescence (data not shown) and Western blotting (Fig. 7A). The results revealed that SNAREs were expressed with CaV2.1 channels in the majority of the cells and that the level of syntaxin 1A or SNAP-25 detected in transfected cells was comparable when expressed with the WT or mutated channels.
FIGURE 7.

SNARE proteins do not modulate E1015K CaV2.1 channel function. A, representative Western blots of cell lysates (30 μg of total proteins) obtained from HEK cells co-expressing syntaxin 1A or SNAP-25 with either WT or E1015K CaV2.1 and probed with antibodies against human α1A, β3, syntaxin 1A, or SNAP-25. B, representative current traces from cells expressing WT or E1015K CaV2.1 channels in the presence or absence of syntaxin 1A. C and D, effect of syntaxin 1A or SNAP-25 expression on steady-state inactivation curves recorded from WT (C) or E1015K (D) CaV2.1-expressing cells (n = 7–9). E, syntaxin 1A and SNAP-25 significantly decreased the Vhalf of inactivation of WT channels (where ** indicates p < 0.01) but had no effect on E1015K channels. Error bars, S.E.
Because SNAREs have been shown to regulate inactivation properties (9, 10, 28) and because these differ significantly in the E1015K channel, steady-state voltage-dependent inactivation was examined in the presence of SNAP-25 or syntaxin 1A. Fig. 7B shows that syntaxin 1A co-expression reduced the relative amount of WT CaV2.1 current after long depolarizing prepulses as described previously (10, 14). However, syntaxin 1A had comparatively little effect on E1015K channel currents where the proportion of current remaining was similar with or without syntaxin 1A co-expression (Fig. 7C). Similar results were obtained for SNAP-25 (data not shown). Accordingly, both syntaxin 1A and SNAP-25 shifted voltage-dependent inactivation curves of WT CaV2.1 channels to more hyperpolarized potentials (Fig. 7D). This resulted in a significant decrease of the Vhalf of inactivation (Fig. 7E) in agreement with prior studies (10, 14). In contrast, neither syntaxin 1A nor SNAP-25 had any effect on the voltage-dependent inactivation curve (Fig. 7D) or Vhalf of inactivation of E1015K CaV2.1 channels (Fig. 7E).
Next, we investigated whether the interactions between CaV2.1 channels and t-SNAREs were affected by the E1015K variant using co-immunoprecipitation experiments. As shown in Fig. 8, the proportion of syntaxin 1A co-immunoprecipitated relative to the total amount of immunoprecipitated α1A was similar between the WT and E1015K α1A subunits of CaV2.1 (data were from five experiments and are expressed as mean ± S.E.: syntaxin 1A/EGFP-α1AWT = 0.76 ± 0.088, syntaxin 1A/EGFP-α1AE1015K = 0.81 ± 0.21). Similar results were obtained with SNAP-25 (data were from two experiments; SNAP-25/EGFP-α1AWT = 0.46 ± 0.17, SNAP-25/EGFP-α1AE1015K = 0.48 ± 0.11), indicating that, at least by co-immunoprecipitation, no major difference exists in the binding of t-SNAREs to the synprint site of WT or E1015K channels.
FIGURE 8.

Binding of syntaxin 1A and SNAP-25 to E1015K CaV2.1 channel is not altered. A, representative Western blots of cell lysates (30 μg of protein) from HEK293 cells expressing CaV2.1 or EGFP (input) and of immunoisolated proteins (IP) are shown using μMACS anti-GFP columns. B, co-immunoisolation of EGFPα1AWT or EGFP-α1AE1015K with syntaxin 1A (CoIP). In A and B, blots were immunolabeled with antibodies against human α1A, GFP, β3, and syntaxin 1A (Syn1A). C, the density of EGFPα1AWT, EGFP-α1AE1015K, and syntaxin 1A or SNAP-25 bands was measured in autoradiograms from five or two independent experiments. The data are expressed as the ratio of syntaxin 1A or SNAP-25 co-immunoisolated with either EGFPα1AWT or EGFP-α1AE1015K (Syn1A/α1A n = 5; paired t test: p = 0.72: SNAP-25/α1A n = 2). Error bars, S.E.
DISCUSSION
Our study describes for the first time how a variant associated with migraine in the synprint site of the CaV2.1 channel impacts on channel function and regulation. Whereas both the biosynthesis and membrane trafficking of E1015K channels were unaltered compared with WT channels, significant differences in channel activity and regulation by SNARE proteins were observed. These results show that a novel variant in the synprint domain associated with migraine causes a gain-of-function via effects on both channel gating and regulation.
The E1015K Variant and Migraine
Association of the E1015K variant to migraine was observed in five patients from three unrelated Italian families. The variant is reported in the 1000 genomes database as a rare variant with a frequency of 0.002. The three families displaying the variant belong to a cohort of 226 migraine patients referred to the diagnostic laboratory, thus the E1015K variant seems to be more frequent among the group of patients (3 of 226; 0.013) than in the general population.
Segregation of the variant with the affected phenotype was demonstrated only for Family 1, where it was present in affected siblings (II.1, II.2) and maternally transmitted (I.2). In the two siblings, a pure HM phenotype was observed, whereas their mother (I.2) experienced only one hemiplegic episode thus not fully satisfying diagnostic IHS criteria. In Family 2, the proband (II.1) experienced several attacks, without cerebellar symptoms. In Family 3, the proband suffered from MA. In this family we were unable to obtain more detailed clinical information for the proband and other family members, so we can only speculate about the association of the E1015K variant with MA without hemiplegia. Several studies have investigated the role of the FHM1 locus in common migraine with conflicting results (29). An hypothesis was that milder mutations may cause common migraine, whereas our evidence indicates that the same variant may be associated to both HM and MA. This evidence could be explained by the existence of modifier genes, able to change or ameliorate the functional effect of the HM variant. An alternative explanation could be the presence of an environmental factor, able to influence the phenotype.
Although it may be possible that the E1015K amino acid substitution does not represent the disease-causing mutation, the following considerations support a role for this variant in migraine: (i) segregation analysis is consistent with the proposed hypothesis, because the variant co-segregates with the migraine phenotype; (ii) the variant was not found in a sample (380 chromosomes) of the general population drawn from the same area of origin of the patients; (iii) there is a high degree of conservation of Glu-1015 between species; (iv) the multiple sequence alignment “Align-GVGD” software predicted for Lys-1015 a likely interference with function with a C55 score (30); (v) no mutations have been found in the other known FHM genes, ATP1A2 and SCN1A.
Cellular Processing and Targeting of the E1015K Channel
The thoroughly investigated FHM mutations T666M, V714A, and I1815L have been shown to decrease channel density at the membrane in both HEK293 cells (2) and cultured neurons (31). In the present study, total protein expression of the E1015K variant was found to be comparable with WT. Furthermore, membrane delivery of the E1015K channel was not affected in either HEK293 cells or hippocampal neurons as measured by cell surface biotinylation and immunofluorescence. These results confirm that the variant is unlikely to cause dramatic changes in protein folding or affect cellular quality control processes that influence its degradation. Similarly, the targeting of the channel to the membrane is not altered by the E1015K substitution, suggesting that the protein-protein interactions that control membrane targeting of the CaV2.1 channel are not compromised. Interestingly, this process is critically dependent on membrane localized t-SNAREs binding to the synprint site of the channel (reviewed in Ref. 32). Therefore, despite an amino acid substitution in the synprint site, the E1015K channel must still be able to interact with SNAREs to localize to somatodendritic and presynaptic membrane compartments. This conclusion is supported by our co-immunoprecipitation results which show that the E1015K channel physically interacts with t-SNAREs to an extent similar to the WT channel. Interestingly, a recent study characterized a CACNA1A variant in the cytoplasmic loop connecting domain I and II (p. A454T-rs41276886) that modifies the functional interaction between SNAREs and CaV2.1, resulting in channels that are less efficiently coupled to secretion (21). Whether this variant also affects SNARE-dependent membrane targeting of the CaV2.1 variant was not investigated, however.
Impact of the E1015K Variant on Channel Function and Regulation
In addition to affecting channel density, many FHM1 mutations that occur in functionally important regions of the α1A subunit such as the voltage sensor S4 segments or the pore region (S5, S6, and membrane-associated loops) cause a gain-of-function phenotype (2, 33–35). Our electrophysiology experiments show that mutations in the synprint site also result in a gain-of-function reflected by an increased current density. Because total membrane expression was similar between WT and E1015K channels, the increase in current density is likely to be caused by alterations in channel activity rather than total channel numbers.
The observation that similar increases in function were detected with either Ba2+ or Ca2+ as the charge carrier indicates that modifications to channel gating rather than permeability underlie the increases in channel activity. This is reinforced by our finding that all of the channel inactivation parameters we studied were found to differ significantly between WT and E1015K channels. Indeed, we conclude that the major functional effect of the E1015K variant is to perturb CaV2.1 inactivation. In contrast, many FHM1 mutations cause a gain-of-function by affecting CaV2.1 activation properties (2, 31). Although the gain-of-function mechanism differs, the overall effect of the E1015K variant would be similar to FHM1 channels; i.e. an increased Ca2+ influx at depolarized potentials compared with WT. Given that neurotransmitter release is critically dependent on synaptic Ca2+ concentrations (36), it is possible that synapses with E1015K channels have enhanced neurotransmitter release contributing to the pathophysiology of migraine. However, additional studies using a knock-in mouse model as elegantly performed using the R192Q knock-in (37) are required to verify the downstream effects of the E1015K variant on neurotransmission.
Combined with defects in basic channel function, E1015K channels are also compromised by a lack of regulation by SNARE proteins, one of the major mechanisms for down-regulating CaV channel activity (8–10). This appears to be due to how SNARES influence inactivation as our biochemical results indicate that SNARE binding to the synprint site is not affected. It is possible that the E1015K substitution affects structural features of the synprint site required for normal inactivation gating which is supported by our E1015K channel inactivation data. Indeed, substitution of the negatively charged glutamate residue to the positively charged lysine would be expected to produce an allosteric change in the protein which may represent the mechanism behind the functional changes we have described. In this altered conformation, SNARE binding to the synprint site may no longer exert the same forces that are required for inactivation of the channel. Similar studies have also shown that migraine mutations in CaV2.1 affect the influence of regulatory proteins including SNAREs (21), G proteins (38, 39), and calmodulin-mediated facilitation (27).
The physiological consequences of this absence of SNARE-dependent down-regulation of CaV2.1 channel activity would exacerbate the already augmented function of the E1015K channel. Therefore, under conditions at which WT presynaptic CaV2.1 channels are normally inhibited by interactions with syntaxin or SNAP-25, E1015K channels would not be subjected to this inhibition and would exhibit the enhanced activity that we have described here. This would lead to increased Ca2+ influx before association with primed vesicles which could potentially impact on both spontaneous and evoked transmitter release to contribute to the downstream manifestation of migraine.
In conclusion, although future studies are required to explain how the changes in function contribute to the disease pathogenesis, our study has characterized how a genetic variant in the CaV2.1 channel associated with migraine leads to increases in channel function and disrupts key regulatory processes required for limiting Ca2+ influx. Future investigations into other CaV2.1 variants and how changes in function contribute to the disease pathogenesis will further advance our understanding of the diversity of defects and mechanisms that underlie both common and inherited forms of migraine. This may lead to the development of targeted treatment options for individuals with different migraine phenotypes.
Acknowledgments
We thank Dr. Jörg Striessing (Institute of Pharmacy, Pharmacology and Toxicology, Innsbruck, Austria) for the wild type human α1A, β3, and α2δ constructs; Dr. Renato Longhi (Consiglio Nazionale delle Ricerche Institute of Chemistry and Molecular Recognition, Milan, Italy) for peptide synthesis and their conjugation to keyhole limpet hemocyanin; Peter Reid and Celine Duynstee (University of Otago, New Zealand) for excellent technical assistance; and the Monzino Foundation, Milan, Italy, for the confocal microscopes and for the support to A. F.
This work was supported by a University of Otago research grant and the Department of Physiology, University of Otago (to S. B. C.), Fondazione Cariplo Grants 2004.1600 (to P. R. and P. C.) and 2012.0546, and Regione Lombardia Project NUTEC 30263049 (to P. R. and M. P.)
- CaV
- voltage-gated calcium
- EGFP
- enhanced green fluorescent protein
- FHM
- familial hemiplegic migraine
- HM
- hemiplegic migraine
- MA
- migraine with aura
- SNAP-25
- synaptosomal-associated protein of 25 kDa
- SNARE
- soluble N-ethylmaleimide-sensitive factor attachment protein receptor.
REFERENCES
- 1. Ophoff R. A., Terwindt G. M., Vergouwe M. N., van Eijk R., Oefner P. J., Hoffman S. M., Lamerdin J. E., Mohrenweiser H. W., Bulman D. E., Ferrari M., Haan J., Lindhout D., van Ommen G. J., Hofker M. H., Ferrari M. D., Frants R. R. (1996) Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552 [DOI] [PubMed] [Google Scholar]
- 2. Hans M., Luvisetto S., Williams M. E., Spagnolo M., Urrutia A., Tottene A., Brust P. F., Johnson E. C., Harpold M. M., Stauderman K. A., Pietrobon D. (1999) Functional consequences of mutations in the human α1A calcium channel subunit linked to familial hemiplegic migraine. J. Neurosci. 19, 1610–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van den Maagdenberg A. M., Kors E. E., Brunt E. R., van Paesschen W., Pascual J., Ravine D., Keeling S., Vanmolkot K. R., Vermeulen F. L., Terwindt G. M., Haan J., Frants R. R., Ferrari M. D. (2002) Episodic ataxia type 2: three novel truncating mutations and one novel missense mutation in the CACNA1A gene. J. Neurol. 249, 1515–1519 [DOI] [PubMed] [Google Scholar]
- 4. Wappl E., Koschak A., Poteser M., Sinnegger M. J., Walter D., Eberhart A., Groschner K., Glossmann H., Kraus R. L., Grabner M., Striessnig J. (2002) Functional consequences of P/Q-type Ca2+ channel CaV2.1 missense mutations associated with episodic ataxia type 2 and progressive ataxia. J. Biol. Chem. 277, 6960–6966 [DOI] [PubMed] [Google Scholar]
- 5. Zhuchenko O., Bailey J., Bonnen P., Ashizawa T., Stockton D. W., Amos C., Dobyns W. B., Subramony S. H., Zoghbi H. Y., Lee C. C. (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nat. Genet. 15, 62–69 [DOI] [PubMed] [Google Scholar]
- 6. Catterall W. A., Leal K., Nanou E. (2013) Calcium channels and short term synaptic plasticity. J. Biol. Chem. 288, 10742–10749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zamponi G. W., Bourinet E., Nelson D., Nargeot J., Snutch T. P. (1997) Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature 385, 442–446 [DOI] [PubMed] [Google Scholar]
- 8. Yokoyama C. T., Myers S. J., Fu J., Mockus S. M., Scheuer T., Catterall W. A. (2005) Mechanism of SNARE protein binding and regulation of CaV2 channels by phosphorylation of the synaptic protein interaction site. Mol. Cell. Neurosci. 28, 1–17 [DOI] [PubMed] [Google Scholar]
- 9. Zhong H., Yokoyama C. T., Scheuer T., Catterall W. A. (1999) Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin, and synaptotagmin. Nat. Neurosci. 2, 939–941 [DOI] [PubMed] [Google Scholar]
- 10. Bezprozvanny I., Scheller R. H., Tsien R. W. (1995) Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature 378, 623–626 [DOI] [PubMed] [Google Scholar]
- 11. Rettig J., Sheng Z. H., Kim D. K., Hodson C. D., Snutch T. P., Catterall W. A. (1996) Isoform-specific interaction of the α1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc. Natl. Acad. Sci. U.S.A. 93, 7363–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sheng Z. H., Rettig J., Cook T., Catterall W. A. (1996) Calcium-dependent interaction of N-type calcium channels with the synaptic core complex. Nature 379, 451–454 [DOI] [PubMed] [Google Scholar]
- 13. Cohen-Kutner M., Nachmanni D., Atlas D. (2010) CaV2.1 (P/Q channel) interaction with synaptic proteins is essential for depolarization-evoked release. Channels 4, 266–277 [DOI] [PubMed] [Google Scholar]
- 14. Sutton K. G., McRory J. E., Guthrie H., Murphy T. H., Snutch T. P. (1999) P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature 401, 800–804 [DOI] [PubMed] [Google Scholar]
- 15. De Fusco M., Marconi R., Silvestri L., Atorino L., Rampoldi L., Morgante L., Ballabio A., Aridon P., Casari G. (2003) Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump α2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 33, 192–196 [DOI] [PubMed] [Google Scholar]
- 16. Dichgans M., Freilinger T., Eckstein G., Babini E., Lorenz-Depiereux B., Biskup S., Ferrari M. D., Herzog J., van den Maagdenberg A. M., Pusch M., Strom T. M. (2005) Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet 366, 371–377 [DOI] [PubMed] [Google Scholar]
- 17. Linetti A., Fratangeli A., Taverna E., Valnegri P., Francolini M., Cappello V., Matteoli M., Passafaro M., Rosa P. (2010) Cholesterol reduction impairs exocytosis of synaptic vesicles. J. Cell Sci. 123, 595–605 [DOI] [PubMed] [Google Scholar]
- 18. Condliffe S. B., Corradini I., Pozzi D., Verderio C., Matteoli M. (2010) Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons. J. Biol. Chem. 285, 24968–24976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taverna E., Saba E., Linetti A., Longhi R., Jeromin A., Righi M., Clementi F., Rosa P. (2007) Localization of synaptic proteins involved in neurosecretion in different membrane microdomains. J. Neurochem. 100, 664–677 [DOI] [PubMed] [Google Scholar]
- 20. Müllner C., Broos L. A., van den Maagdenberg A. M., Striessnig J. (2004) Familial hemiplegic migraine type 1 mutations K1336E, W1684R, and V1696I alter CaV2.1 Ca2+ channel gating: evidence for β-subunit isoform-specific effects. J. Biol. Chem. 279, 51844–51850 [DOI] [PubMed] [Google Scholar]
- 21. Serra S. A., Cuenca-León E., Llobet A., Rubio-Moscardo F., Plata C., Carreño O., Fernàndez-Castillo N., Corominas R., Valverde M. A., Macaya A., Cormand B., Fernández-Fernández J. M. (2010) A mutation in the first intracellular loop of CACNA1A prevents P/Q channel modulation by SNARE proteins and lowers exocytosis. Proc. Natl. Acad. Sci. U.S.A. 107, 1672–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Witcher D. R., De Waard M., Liu H., Pragnell M., Campbell K. P. (1995) Association of native Ca2+ channel β subunits with the α1 subunit interaction domain. J. Biol. Chem. 270, 18088–18093 [DOI] [PubMed] [Google Scholar]
- 23. Liu H., De Waard M., Scott V. E., Gurnett C. A., Lennon V. A., Campbell K. P. (1996) Identification of three subunits of the high affinity ω-conotoxin MVIIC-sensitive Ca2+ channel. J. Biol. Chem. 271, 13804–13810 [PubMed] [Google Scholar]
- 24. Pichler M., Cassidy T. N., Reimer D., Haase H., Kraus R., Ostler D., Striessnig J. (1997) β-Subunit heterogeneity in neuronal L-type Ca2+ channels. J. Biol. Chem. 272, 13877–13882 [DOI] [PubMed] [Google Scholar]
- 25. Cao Y. Q., Piedras-Rentería E. S., Smith G. B., Chen G., Harata N. C., Tsien R. W. (2004) Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron 43, 387–400 [DOI] [PubMed] [Google Scholar]
- 26. Mochida S., Westenbroek R. E., Yokoyama C. T., Zhong H., Myers S. J., Scheuer T., Itoh K., Catterall W. A. (2003) Requirement for the synaptic protein interaction site for reconstitution of synaptic transmission by P/Q-type calcium channels. Proc. Natl. Acad. Sci. U.S.A. 100, 2819–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Adams P. J., Rungta R. L., Garcia E., van den Maagdenberg A. M., MacVicar B. A., Snutch T. P. (2010) Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc. Natl. Acad. Sci. U.S.A. 107, 18694–18699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Condliffe S. B., Matteoli M. (2011) Inactivation kinetics of voltage-gated calcium channels in glutamatergic neurons are influenced by SNAP-25. Channels 5, 304–307 [DOI] [PubMed] [Google Scholar]
- 29. de Vries B., Frants R. R., Ferrari M. D., van den Maagdenberg A. M. (2009) Molecular genetics of migraine. Hum. Genet. 126, 115–132 [DOI] [PubMed] [Google Scholar]
- 30. Tavtigian S. V., Deffenbaugh A. M., Yin L., Judkins T., Scholl T., Samollow P. B., de Silva D., Zharkikh A., Thomas A. (2006) Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 43, 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tottene A., Fellin T., Pagnutti S., Luvisetto S., Striessnig J., Fletcher C., Pietrobon D. (2002) Familial hemiplegic migraine mutations increase Ca2+ influx through single human CaV2.1 channels and decrease maximal CaV2.1 current density in neurons. Proc. Natl. Acad. Sci. U.S.A. 99, 13284–13289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jarvis S. E., Zamponi G. W. (2007) Trafficking and regulation of neuronal voltage-gated calcium channels. Curr. Opin. Cell Biol. 19, 474–482 [DOI] [PubMed] [Google Scholar]
- 33. Kraus R. L., Sinnegger M. J., Glossmann H., Hering S., Striessnig J. (1998) Familial hemiplegic migraine mutations change α1A Ca2+ channel kinetics. J. Biol. Chem. 273, 5586–5590 [DOI] [PubMed] [Google Scholar]
- 34. Tottene A., Pivotto F., Fellin T., Cesetti T., van den Maagdenberg A. M., Pietrobon D. (2005) Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J. Biol. Chem. 280, 17678–17686 [DOI] [PubMed] [Google Scholar]
- 35. Kraus R. L., Sinnegger M. J., Koschak A., Glossmann H., Stenirri S., Carrera P., Striessnig J. (2000) Three new familial hemiplegic migraine mutants affect P/Q-type Ca2+ channel kinetics. J. Biol. Chem. 275, 9239–9243 [DOI] [PubMed] [Google Scholar]
- 36. Neher E., Sakaba T. (2008) Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron 59, 861–872 [DOI] [PubMed] [Google Scholar]
- 37. Tottene A., Conti R., Fabbro A., Vecchia D., Shapovalova M., Santello M., van den Maagdenberg A. M., Ferrari M. D., Pietrobon D. (2009) Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in CaV2.1 knock-in migraine mice. Neuron 61, 762–773 [DOI] [PubMed] [Google Scholar]
- 38. Melliti K., Grabner M., Seabrook G. R. (2003) The familial hemiplegic migraine mutation R192Q reduces G-protein-mediated inhibition of P/Q-type (CaV2.1) calcium channels expressed in human embryonic kidney cells. J. Physiol. 546, 337–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garza-López E., Sandoval A., González-Ramirez R., Gandini M. A., Van den Maagdenberg A., De Waard M., Felix R. (2012) Familial hemiplegic migraine type 1 mutations W1684R and V1696I alter G protein-mediated regulation of CaV2.1 voltage-gated calcium channels. Biochim. Biophys. Acta 1822, 1238–1246 [DOI] [PubMed] [Google Scholar]