

Background: Preclinical evaluation of triptolide shows pancreatic tumor regression in animal models.
Results: Triptolide deregulates glycosylation of Sp1, leading to its decreased activity and causing pancreatic cancer cell death associated with down-regulating HSP70.
Conclusion: Triptolide down-regulation of HSP70 is associated with inhibition of Sp1 activity in pancreatic cancer.
Significance: This mechanism is of relevance, as its water-soluble prodrug, Minnelide, is currently under Phase 1 clinical trial.
Keywords: Heat Shock Protein, O-GlcNAc, Pancreas, Pancreatic Cancer, Sp1, Minnelide, Triptolide
Abstract
Pancreatic cancer, the fourth most prevalent cancer-related cause of death in the United States, is a disease with a dismal survival rate of 5% 5 years after diagnosis. One of the survival proteins responsible for its extraordinary ability to evade cell death is HSP70. A naturally derived compound, triptolide, and its water-soluble prodrug, Minnelide, down-regulate the expression of this protein in pancreatic cancer cells, thereby causing cell death. However, the mechanism of action of triptolide has not been elucidated. Our study shows that triptolide-induced down-regulation of HSP70 expression is associated with a decrease in glycosylation of the transcription factor Sp1. We further show that triptolide inhibits glycosylation of Sp1, inhibiting the hexosamine biosynthesis pathway, particularly the enzyme O-GlcNAc transferase. Inhibition of O-GlcNAc transferase prevents nuclear localization of Sp1 and affects its DNA binding activity. This in turn down-regulates prosurvival pathways like NF-κB, leading to inhibition of HSF1 and HSP70 and eventually to cell death. In this study, we evaluated the mechanism by which triptolide affects glycosylation of Sp1, which in turn affects downstream pathways controlling survival of pancreatic cancer cells.
Introduction
Pancreatic cancer is the fourth most prevalent cause of cancer-related death in the United Sates. The 5-year survival rate is a dismal 5% because of late diagnosis and lack of targeted therapy against this disease (1). Therefore, there is a need for a better understanding of the mechanisms that contribute to the progression of pancreatic cancer and for the design of more effective targeted treatment strategies.
One class of proteins responsible for the survival of pancreatic tumor cells is the heat shock proteins (HSPs).2 These proteins are overexpressed in a number of cancers, including pancreatic cancer, and play a leading role in cell survival mechanisms (2–4). HSP70 expression is activated by the hexosamine biosynthetic pathway (HBP) (5), a shunt pathway that has been implicated in cellular signaling cascades and regulation of transcription factors involved in cancer biology. Increased activation of the HBP in turn results in increased glycosylation by O-GlcNAc transferase (OGT), an enzyme that modifies many key cellular proteins and transcription factors (6). OGT is reported to be necessary for cell survival (7) and is overexpressed in a number of cancers (8–10). This pathway is thus an early cellular protective response to stress because this protein modification can occur rapidly (5). One of the transcription factors modified by the HBP is Sp1, a key transcription factor required for optimal HSP expression following stress (11, 12).
Sp1 is a zinc finger transcription factor with a GC-rich binding sequence in the gene promoter (13), and it regulates multiple aspects of tumor cell survival, growth, and angiogenesis. Thus, abnormal Sp1 expression and activation are thought to contribute to human cancer development and progression (14). Lines of evidence demonstrate that overactivation of Sp1 occurs frequently in a wide variety of human tumors, including pancreatic cancer, and that high Sp1 expression correlates with aggressive biology and poor clinical outcome (15, 16). Sp1 has also been shown to be overexpressed in pancreatic tumors (17, 18).
The transcriptional activity of Sp1 is regulated by a number of post-translational modifications. One of these is O-GlcNAcylation at Ser-484 catalyzed by the enzyme OGT, which facilitates its translocation to the nucleus and thus controls its activity (12). Expression of all heat shock-related proteins, such as HSF1, HSP70, and HSP27, relies on Sp1 binding to their promoter sequences for optimal transcriptional activity (19, 20).
Triptolide, a diterpene epoxide from the Chinese plant Tripterygium wilfordii, induces apoptotic cell death of pancreatic tumors via inhibition of HSP70 (21, 22). Triptolide is also effective against a number of additional malignancies, including cholangiocarcinoma (23), gastric cancer (24), osteosarcoma (25) and neuroblastoma (26, 27). Triptolide affects a number of prosurvival pathways in cancer cells. These include HSP70 and NF-κB in a number of cancers, including pancreatic cancer (28–31). Although triptolide is an effective anticancer compound in vitro, its use in animals has been restricted because of its limited solubility in water. Recently, a water-soluble prodrug of triptolide (Minnelide), synthesized by the University of Minnesota, has overcome this concern and shown considerable promise in preclinical studies (25, 28). Despite considerable research on triptolide, its exact mechanism of action has not been defined thus far.
In this study, we show that in pancreatic cancer, triptolide-induced down-regulation of HSP70, which leads to cell death, is mediated by impaired O-GlcNAc modification of Sp1. Triptolide decreases the expression and activity of OGT in these cells, resulting in reduced Sp1 translocation to the nucleus and reduced Sp1 activity. This in turn leads to lower expression of HSF1 and other HSPs, finally resulting in tumor cell death.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
The pancreatic cancer cell line MIA-PaCa2 (obtained from American Type Culture Collection) was grown and propagated in DMEM supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. The S2-013 and S2-VP10 cell lines (kind gift from Prof. D. Buschbaum, University of Alabama) were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. The AsPC1 cell line (obtained from American Type Culture Collection) was cultured in RPMI 1640 medium supplemented with 20% FBS. Human pancreatic ductal epithelial cells (obtained from American Type Culture Collection) were cultured in keratinocyte medium supplemented with bovine pituitary hormone and EGF. No authentication was done by the authors, but American Type Culture Collection authenticates using short tandem repeat profiling.
ON-TARGETplus SMARTpool human HSP70 siRNA (Dharmacon Inc., Lafayette, CO) and human OGT, human O-N-acetylglucosaminidase (OGA), human Sp1, and human glutamine:fructose aminotransferase (GFAT) siRNAs (Qiagen) were used to silence expression of the respective genes in the pancreatic cancer cell lines MIA-PaCa2 and S2-013. Transfections were done using HiPerFect (Qiagen) according to the manufacturer's instructions. A pool of four siRNAs was used for all of the above genes.
The activity of OGT was inhibited using the inhibitor ST045849 (TimTec Compound Libraries) at a concentration of 0.1–10 mm. The activity of GFAT was inhibited by 6-diazo-5-oxo-l-norleucine (DON). OGA activity was inhibited using O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino N-phenylcarbamate at a concentration of 0.15–150 μm. Triptolide and mithramycin were used at a concentrations of 25–200 nm for studying viability and proliferation. A rescue experiment was performed by the addition of 5 mm glucosamine to the cells in full medium.
Electric Cell Substrate Impedance Sensing
In this method, cells are grown on the surface of small and planar gold-film electrodes, and the AC impedance of the cell-covered electrode is measured continuously at a frequency of 64 kHz. Due to the insulating properties of cell membranes, the impedance increases with increasing coverage of the electrode until a confluent layer of cells is established. Arrays are inoculated at a low cell density, providing room for the dividing cell population. As the cell number increases, the amount of electrode area covered with the spread cells grows accordingly, causing the electrode resistance to rise. The change in resistance is thus a direct measure of proliferation of cells. In this study, 5 × 104 MIA-PaCa2 and S2-013 cells were plated onto 8W10E+ arrays and connected to the ECIS® instrument (Applied Biophysics). Wells were appropriately treated with either mithramycin or ST045849. The change in resistance was measured and plotted against untreated cells. Proliferation was measured in real time over 48 h.
Quantitative Real-time PCR
Quantitative real-time PCR for OGT, Sp1, HSF1, p50, and p65 was carried out using primers obtained from Qiagen (QuantiTect primer assay). Primers for HSP70 (forward Primer, ACCAAGCAGACGCAGATC; and reverse primer, CGCCCTCGTACACCTGGA) were synthesized by Life Technologies. RNA was isolated from the different cell lines and from the tumor samples according to the manufacturer's instructions using TRIzol (Invitrogen). Total RNA (1 μg) was used to perform real-time PCR with the QuantiTect SYBR Green PCR kit (Qiagen) according to the manufacturer's instructions and an Applied Biosystems 7300 real-time PCR system. All data were normalized to the housekeeping 18 S gene (18S QuantiTect primer assay, Qiagen).
Rescue Experiment with pCMV-Sp1 and pCMV-OGT
The nuclear localization signal (NLS) sequence aagaagcgtaaggta from plasmid pQC-NLS-CMV (Addgene) was cloned in the 5′-end of the Sp1 using the TOPO directional cloning kit with vector pcDNA3.1. The clones were confirmed by sequencing. Pancreatic cancer cells (MIA-PaCa2) were transfected with the Sp1-NLS plasmid for the rescue experiment with Sp1 with constitutive NLS or the pCMV-OGT plasmid (OriGene, Rockville, MD) for rescue with overexpressed OGT using Attractene (Qiagen) as a transfection reagent according to the manufacturer's instructions. Following 18 h of transfection, cells were treated with different doses of triptolide. Control sets of triptolide only and plasmid only were also kept in parallel. Cell viability in each set was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide-based viability assay. For studying activity, cells were processed for the dual-reporter assay (Qiagen) for Sp1, heat shock element (HSE), and NF-κB as detailed by the manufacturer.
Cell Viability Assay
Cell viability assays following siRNA or inhibitor treatment were performed by the trypan blue exclusion method or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium-based viability assay and expressed after normalizing to untreated cells. Proliferation of cells in response to inhibition of OGT was measured as a function of impedance using the ECIS® instrument.
Western Blotting and Hybridization
Protein from treated and untreated cell lysates was estimated using the BCA protein estimation assay (Thermo Scientific). For Western blotting, anti-OGT and anti-O-GlcNAc antibodies (Sigma), anti-HSP70 antibody (Stressgen), and anti-GFAT and anti-Sp1 antibodies (Cell Signaling) were used to check for levels of different proteins in the lysates.
Luciferase Reporter Assay for HSF1, NF-κB, and Sp1
Activity assays were performed using Cignal reporter assay kits for HSE, NF-κB, and Sp1 obtained from SABiosciences. Transfections were performed as specified in the manufacturer's instruction sheet. Cells were collected either 8 or 24 h post-treatment and analyzed using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's protocol.
Confocal Microscopy
Pancreatic cancer cells were grown on chamber slides and treated with 100 nm triptolide or 5 mm alloxan for 24 h, fixed with 4% paraformaldehyde for 15 min at room temperature, and permeabilized with 0.1% Triton X-100. Anti-Sp1 antibody was used at a dilution of 1:200 for 1 h at room temperature. After washing, cells were incubated with a 1:1200 dilution of Alexa Fluor 488-conjugated donkey anti-rabbit IgG secondary antibody (Molecular Probes) for 50 min at 4 °C. The slides were washed and mounted using Prolong Gold anti-fade agent containing DAPI (Molecular Probes). Immunofluorescence images were obtained on a Nikon Eclipse Ti confocal microscope using a 40× oil immersion objective. The nuclear localization of Sp1 was quantitated using ImageJ software.
Orthotopic Model for Pancreatic Cancer
Female nude mice (4–6 weeks old; Charles River Laboratories) were used for the pancreatic cancer mouse model. All procedures were carried out according to the guidelines of the University of Minnesota Institutional Animal Care and Use Committee. Briefly, 2 × 105 MIA-PaCa2 cells/10 μl of Matrigel were injected into the pancreases of 15 nude mice. After 10 days, treatment with mithramycin was started intraperitoneally on eight mice after randomization. Mithramycin (0.6 mg/kg of body weight) was injected every day into eight mice, whereas the remaining seven mice were injected with saline (vehicle). The treated and untreated mice were killed 30 days after introduction of MIA-PaCa2 cells. The tumor burden was evaluated by documenting the tumor volume and tumor weight following necropsy.
Tumor Xenograft Propagation in Mice
Pancreatic tumors resected during surgery were implanted in three SCID mice. Once the tumors had taken, they were re-implanted subcutaneously into fresh SCID mice. The implanted tumors retained stromal architecture from the parent tumor and were used for assessing Sp1 levels.
Immunohistochemistry
For immunohistochemistry, paraffin tissue sections were received mounted on charged slides. The slides were deparaffinized in xylene and hydrated through graded ethanols. Slides were steamed with a Reveal Decloaker (Biocare Medical, Concord, CA) to minimize background staining. Sniper Universal blocking sera (Biocare Medical) were used throughout the protocol. The slides were stained using a rabbit polyclonal antibody against Ki-67 (Thermo Scientific). A diaminobenzidine peroxidase substrate kit (Vector Laboratories) was used to reveal staining for Ki-67. The tissue sections were counterstained with Gill's hematoxylin (Vector Laboratories). The antibody was omitted for the negative omission controls.
Immunoprecipitation
Immunoprecipitation of Sp1 was done following an 18-h triptolide treatment. Treated and untreated lysates (500 μg) were incubated overnight with anti-Sp1 antibody. The immunoprecipitated complex was further captured using protein A-Sepharose beads (GE Healthcare). Western blotting was done with both anti-Sp1 and anti-O-GlcNAc antibodies to study glycosylation of this transcription factor.
Statistical Analysis
Values are expressed as the mean ± S.E. All in vitro experiments were performed at least three times. The significance of the difference between any two samples was analyzed by unpaired Student's t test; values of p < 0.05 were considered statistically significant.
RESULTS
Triptolide Down-regulates Pro-proliferative Pathways in Pancreatic Cancer
Pancreatic cancer cells have high expression of levels of HSPs and NF-κB activity, which protects them from cell death by regulating a number of prosurvival genes. Our laboratory has previously shown that triptolide induces tumor cell death by down-regulating HSPs (22, 28, 32). This down-regulation of HSP70 is mediated at the transcriptional level, and expression at both the mRNA and protein levels is reduced upon treatment with this compound. To see if triptolide affects the binding of HSF1 to the HSEs of its target genes, we performed a Dual-Luciferase assay for HSE binding. Our results show that triptolide indeed resulted in down-regulation of HSE binding by HSF1 (Fig. 1A). To see if this inhibition of HSF1 activity is due to repression of its expression, we also checked the expression of HSF1 at the transcriptional and translational levels. Triptolide caused a decrease in HSF1 RNA and protein (Fig. 1, B and C). A time course with triptolide further showed that HSF1 down-regulation corresponded with down-regulation of HSP70 in the pancreatic cancer cell line MIA-PaCa2. This indicated that HSP70 down-regulation by triptolide was mediated by events upstream of HSF1.
FIGURE 1.

Triptolide down-regulates prosurvival pathways. The pancreatic cancer cell line MIA-PaCa2 was used for this experiment. A, HSE binding activity of HSF1 after 100 nm triptolide treatment. B, HSF1 RNA expression after 100 nm triptolide treatment. C, expression levels of HSP70 and HSF1 after 100 nm triptolide treatment. D, NF-κB promoter binding activity. E and F, p50 and p65 mRNA expression after triptolide treatment, respectively. G and H, dose- and time-dependent Sp1 activity after triptolide treatment, respectively. I, Sp1 expression after triptolide treatment.
Triptolide is also reported to down-regulate NF-κB activity in a number of cancers, including pancreatic cancer (31). A Dual-Luciferase reporter assay to evaluate the effect of triptolide on the NF-κB pathway showed that in both MIA-PaCa2 and S2-013 cell lines, triptolide down-regulated NF-κB activity (Fig. 1D). Triptolide decreased the expression of p50/p65 subunits of the NF-κB transcription complex as well (Fig. 1E), indicating that the triptolide-induced reduction of NF-κB activity was a result of regulation of an upstream element.
Because Sp1 has been shown to be the transcription factor controlling transcription of NF-κB subunits p50 and p65 (33), we studied the effect of triptolide on Sp1 activity and expression. Although Sp1 mRNA expression remained unchanged after 24 h of triptolide treatment (Fig. 1I), Sp1 transcriptional activity was seen to be down-regulated by triptolide in a dose- and time-dependent manner (Fig. 1, G and H).
Transcription Factor Sp1 Is Overexpressed in Pancreatic Cancer Cells and Tumors
We studied the expression of Sp1 mRNA and protein in pancreatic cancer cell lines of varying aggressiveness and compared the -fold change in expression over normal human pancreatic ductal cells. Sp1 was found to be 4-fold overexpressed in the MIA-PaCa2 cells (derived from a primary tumor), 2–3-fold overexpressed in cell lines S2-013 and S2-VP10, and 3–4-fold overexpressed in the ascites-derived cell line AsPC1 compared with that in normal cells (Fig. 2A). Expression of Sp1 protein was also increased in these pancreatic cancer cell lines (Fig. 2B). Sp1 protein levels in the different pancreatic cancer cell lines corresponded with their HSP70 and HSF1 levels, indicating a correlation of expression levels of these proteins (Fig. 2B). Immunohistochemistry of the patient tumor xenograft propagated in SCID mice also showed increased expression of Sp1 compared with that in normal pancreas. These findings show that the transcription factor Sp1 was overexpressed in pancreatic tumors and tumor-derived cell lines (Fig. 2C).
FIGURE 2.
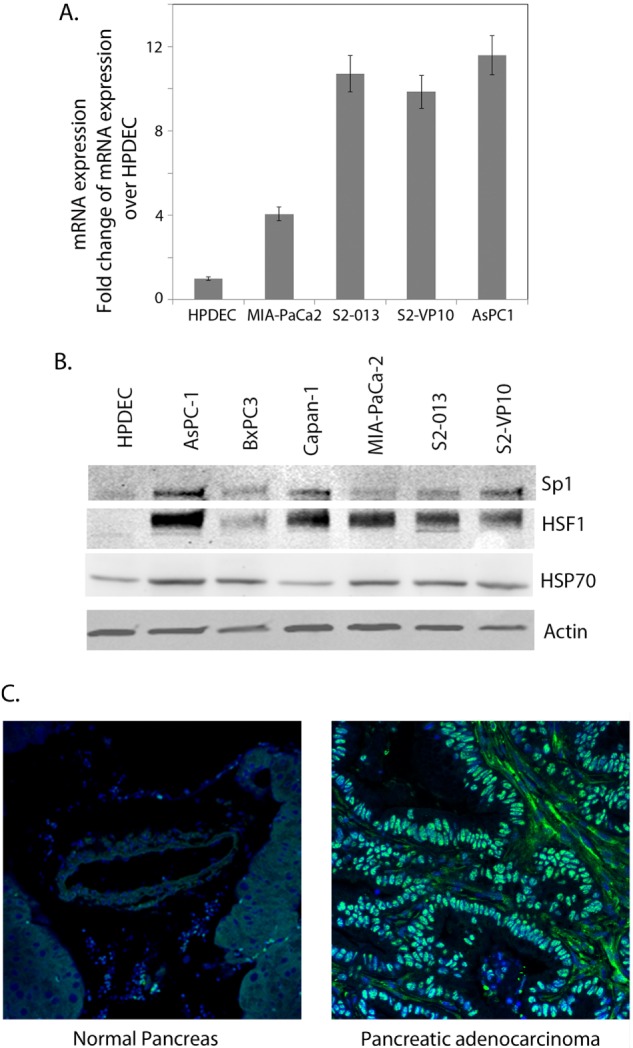
Sp1 is overexpressed in pancreatic cancer. A, Sp1 RNA expression in different pancreatic cancer cell lines compared with normal pancreatic ductal cells. B, Sp1 protein expression in pancreatic cancer cell lines compared with human pancreatic ductal cells. Expression of Sp1 was seen to correlate with HSF1 and HSP70 expression. C, immunohistochemical staining of Sp1 in a normal pancreas and overexpression of Sp1 versus a pancreatic tumor specimen.
Sp1 Expression Is Essential for Survival of Pancreatic Cancer Cells, and Its Down-regulation Leads to Loss of Viability and Proliferation both in Vitro and in Vivo
Inhibition of Sp1 expression by mithramycin, an inhibitor of Sp1 (34), decreased the viability of the pancreatic cancer cell lines MIA-PaCa2 and S2-013 (Fig. 3A). To further confirm this, we used siRNA to inhibit the expression of Sp1 (data reviewed but not shown) and monitored its viability and mechanism of cell death. As seen with mithramycin, Sp1 siRNA also resulted in a significant decrease in viability in a dose- and time-dependent manner in both cell lines (Fig. 3B). Cell death in both lines was apoptotic as measured by caspase-3/7 activity (data reviewed but not shown).
FIGURE 3.

Sp1 down-regulation results in loss of viability in cancer cells. Mithramycin treatment (A) or Sp1 inhibition by siRNA (B) resulted in MIA-PaCa2 and S2-013 cell death and decreased proliferation in MIA-PaCa2 (C) and S2-013 (D) cells. E, tumor regression in an orthotopic mouse model for pancreatic cancer after mithramycin treatment. F and G, tumor weight and tumor volume following mithramycin treatment, respectively. H and I, immunostaining for Ki-67 and quantitation of the same following mithramycin treatment, respectively. The asterisks indicate statistically significant change compared with the control.
To determine if inhibition of Sp1 results in loss of cell proliferation, we used electric cell-substrate impedance sensing to monitor cell proliferation after treatment with mithramycin, an Sp1 inhibitor (Fig. 3, C and D). Treatment with mithramycin resulted in decreased proliferation of both MIA-PaCa2 and S2-013 cells in this system. A decrease in proliferation was further confirmed by the trypan blue exclusion method (data not shown).
Mithramycin has been shown to be effective in tumor regression in pancreatic cancer in combination with other compounds. To confirm this, we injected MIA-PaCa2 pancreatic cancer cells orthotopically into the tails of the pancreases of 15 athymic nu/nu mice and treated eight of them with mithramycin and left seven untreated. The tumors in both groups were measured and documented at the end of experiment (Fig. 3E). Mithramycin was found to be effective in reducing tumor volume (Fig. 3F) and tumor weight (Fig. 3G). Ki-67 staining also confirmed fewer proliferating cells in the treated tumors (Fig. 3, H and I).
Sp1 Down-regulation Leads to Lower Expression of Pro-proliferative Genes
Because down-regulation of Sp1 resulted in cell death and decreased proliferation both in vitro and in vivo, we studied the effect of Sp1 down-regulation on prosurvival genes in pancreatic cancer cells. Down-regulation of Sp1 by both siRNA and mithramycin showed a decrease in expression of proteins belonging to the heat shock family, e.g. HSP70, HSP27, and HSF1 (Fig. 4, A and B). To see if the down-regulation in expression of HSPs occurs through inhibition of HSF1 binding to the HSEs in the promoter regions of HSPs, we performed a Dual-Luciferase reporter assay for HSE binding in the presence of mithramycin. As shown above with triptolide (Fig. 1), inhibition of Sp1 decreased HSE binding activity, demonstrating that Sp1 inhibition resulted in decreased promoter binding activity of HSF1 (Fig. 4C). This in turn resulted in decreased expression of HSP70 and HSP27. A similar decrease in HSE binding by HSF1 was observed in cells transfected with Sp1 siRNA (Fig. 4C).
FIGURE 4.

Sp1 down-regulation results in decreased expression of prosurvival genes. Mithramycin (A) and siRNA Sp1 (siSp1; B) down-regulation of HSP70, HSF1, and HSP27. Both HSF1 (C) and NF-κB (D) activities were reduced after Sp1 inhibition. Sp1 (E and F) and HSP70 (G and H) expression was reduced in tissue sections from the orthotopic mouse model treated with mithramycin (Mth). I, the tumors also showed decreased NF-κB activity. RLU, relative light units. The asterisks indicate statistically significant change compared to control.
To see if Sp1 inhibition results in decreased NF-κB promoter binding activity and reduced expression of prosurvival genes, we performed a Dual-Luciferase reporter assay for NF-κB binding. Once again, as shown in Fig. 1 with triptolide, both mithramycin-treated and Sp1 siRNA-transfected cells (Fig. 4D) showed significantly lower activity compared with untreated pancreatic cancer cells.
Tumor tissue from mithramycin-treated animals showed a decreased expression of Sp1 and HSP70 proteins (Fig. 4, F and H) compared with the untreated group (Fig. 4, E and G). An ELISA-based NF-κB activity assay showed that mithramycin-treated tumors had significantly less NF-κB activity compared with untreated controls (Fig. 4I). This indicated that just as triptolide decreased HSP70 expression and NF-κB activity, inhibition of Sp1 also resulted in similar down-regulation of HSP expression through inhibition of HSE binding by HSF1 along with down-regulation of NF-κB.
Sp1 Activity Is Controlled in Part by O-GlcNAc Modification
Because triptolide down-regulated the transcriptional activity of Sp1 but did not affect its transcription after 24 h of treatment, we studied the effect of triptolide on post-translational modifications of Sp1 that control its transcriptional activity. Glycosylation of Sp1 regulates its subcellular localization and thus affects its activity. The addition of a single N-acetylglucosamine at Ser-484 by the enzyme OGT enables its nuclear translocation, whereas removal of this sugar by OGA promotes phosphorylation at these sites and subsequent DNA binding by Sp1 (35). To see if triptolide affects O-GlcNAc modification of Sp1, MIA-PaCa2 cells were treated with 100 nm triptolide for 18 h, and 500 μg of cell lysate from both groups was immunoprecipitated with anti-Sp1 antibody. The immunoprecipitated samples were immunoblotted with anti-O-GlcNAc and anti-Sp1 antibodies. Although the Sp1 level remained unchanged in both the control and treated samples, the O-GlcNAc levels of Sp1 were decreased in the treated samples (Fig. 5A). This indicated that in pancreatic cancer cells, Sp1 was modified by O-GlcNAc addition and that triptolide altered the O-GlcNAc levels of Sp1 protein by inhibiting its glycosylation.
FIGURE 5.

Triptolide inhibits O-GlcNAc modification of Sp1. A, immunoprecipitation (Ip) showing that Sp1 was glycosylated with O-GlcNAc modification and decreased after treatment with triptolide. S, supernatant; Ip, immunoprecipitated pellet. B, inhibition of O-GlcNAcylation down-regulated Sp1 activity. Triptolide decreased expression of OGT (C) and GFAT (D). Protein expression of OGT (E) and GFAT (F) was also reduced after triptolide treatment. G, quantitation of the immunofluorescence of nuclear and cytosolic Sp1 following 100 nm triptolide treatment. H, immunofluorescence showed the cytosolic accumulation of Sp1 in triptolide-treated cells. The asterisks indicate statistically significant differences compared with the control. IB, immunoblot.
Because Sp1 activity is regulated by inhibition of O-GlcNAc modification, we performed an Sp1 activity assay following treatment of MIA-PaCa2 and S2-013 cells with inhibitors for O-GlcNAc modification. ST045849 (1 mm) was used as an inhibitor for OGT, and the deglycosylase OGA was inhibited using O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino N-phenylcarbamate (40 μm). Inhibition of both OGT and OGA resulted in decreased Sp1 activity (Fig. 5B).
To rule out nonspecific effects of pharmacological inhibitors, we also used siRNAs against OGT and OGA to inhibit these enzymes and tested for Sp1 activity. As with the inhibitors, Sp1 activity was decreased with OGT and OGA siRNAs (Fig. 5B), as observed above with triptolide (Fig. 1).
To see if triptolide inhibits Sp1 O-GlcNAcylation by altering the substrate pool of UDP-GlcNAc, we studied Sp1 promoter binding activity in the presence of DON, a pharmacological inhibitor of GFAT, as well as after transfecting cells with GFAT siRNA. GFAT is a rate-limiting enzyme of the HBP and is responsible for synthesis of UDP-GlcNAc, which is the substrate for OGT. In both cases, Sp1 activity was found to be decreased in both MIA-PaCa2 and S2-013 cells (Fig. 5B).
Because Sp1 activity was affected by alteration of O-GlcNAc modification, we studied the effect of triptolide on the protein and RNA expression of both OGT and GFAT. Expression of OGT and GFAT was down-regulated following triptolide treatment at both the transcriptional (Fig. 5, C and D) and translational (Fig. 5, E and F) levels. This confirmed that triptolide affected Sp1 activity by altering its O-GlcNAc modification via inhibition of the enzymes responsible for this modification.
To confirm that inhibition of OGT interferes with the subcellular localization of Sp1, MIA-PaCa2 pancreatic cancer cells were plated onto chamber slides and treated with triptolide for 18 h. Sp1 localization was then visualized by immunofluorescence. Nuclei were stained with DAPI. Although the control showed an almost equal distribution of cells with Sp1 in the nucleus and cytosol, triptolide-treated slides had mostly cells with Sp1 in the cytosol (Fig. 5, G and H). This indicated that triptolide treatment inhibited the nuclear translocation of Sp1.
Inhibition of both OGA and OGT is known to affect Sp1 activity. Triptolide decreased O-GlcNAc modification of Sp1 and prevented its nuclear translocation. This indicated that the predominant action of triptolide was on the glycosylating enzyme, not the deglycosylating one.
Inhibition of OGT Leads to Cell Death, Decreased Proliferation, and Inhibition of Prosurvival Pathways
Because triptolide was seen to down-regulate the O-GlcNAc modification of Sp1, we studied the effect of OGT inhibition on pancreatic cancer cells. The viability of MIA-PaCa2 (Fig. 6A) and S2-013 (Fig. 6B) pancreatic cancer cells was studied following inhibition of OGT by siRNA or ST045849. Cell viability decreased to 40–60% of controls following inhibition of OGT. Because OGT enzyme activity is driven by its substrate concentration, we inhibited the rate-limiting enzyme GFAT1 of the HBP. Inhibition of this enzyme by either a pharmacological inhibitor (DON) or GFAT1 siRNA resulted in a significant loss of viability of both MIA-PaCa2 (Fig. 6C) and S2-013 (Fig. 6D) cells. Inhibition of both GFAT and OGT also resulted in decreased proliferation as measured by electric cell-substrate impedance sensing (Fig. 6E). Furthermore, down-regulation of GFAT and OGT also resulted in down-regulation of NF-κB activity (Fig. 6F), HSE promoter binding activity of HSF1 (Fig. 6G), and decreased expression of HSP70 (Fig. 6H).
FIGURE 6.

Inhibition of O-GlcNAc modification results in loss of viability and proliferation. Inhibition of OGT with either ST045849 (an OGT inhibitor) or siRNA resulted in loss of viability of MIA-PaCa2 (A) and S2-013 (B) cells. Inhibition of GFAT with DON and GFAT siRNA (siGFAT) also decreased viability of MIA-PaCa2 (C) and S2-013 (D) cells. E, both ST045849 and DON also inhibited proliferation of pancreatic cancer cells. NF-κB (F) and HSF1 (G) activities were reduced on inhibition of O-GlcNAc modification. H, HSP70 expression was decreased when O-GlcNAcylation was inhibited. The asterisks indicate statistically significant differences compared with the control.
Overexpressing Sp1 Results in Rescue from Cell Death and Reverses the Inhibitory Effect of Triptolide on Pro-proliferative Pathways
To confirm the triptolide action is indeed mediated through inhibition of the nuclear localization of Sp1, we cloned a NLS signal into the 5′-end of Sp1 and expressed these Sp1-NLS constructs (clones Sp1-NLS1 and Sp1-NLS2) in pancreatic cancer cells. To determine whether OGT was the key enzyme through which triptolide was acting, we used glucosamine as a rescue compound. Because glucosamine is one of the key components that fuel the HBP in a cell and is instrumental in the synthesis of UDP-GlcNAc, the primary substrate for glycosylation by OGT, we hypothesized that if triptolide acts by inhibiting OGT activity, replenishing its substrate pool by adding glucosamine in the presence of triptolide should rescue cells from triptolide-mediated cell death. We further confirmed this by overexpressing OGT (pCMV-OGT) in MIA-PaCa2 cells. We hypothesized that if triptolide acts by inhibiting OGT activity and thus preventing Sp1 from localizing in the nucleus, replenishing OGT or constitutively targeting Sp1 to the nucleus in the presence of triptolide should rescue cells from triptolide-mediated cell death. Cell viability was assessed after overexpressing Sp1-NLS1, and Sp1-NLS2 and then treating cells with 100 nm triptolide. Overexpression of Sp1-NLS rescued pancreatic cancer cells from triptolide-induced cell death. Similarly, treatment with glucosamine and overexpression of OGT followed by treatment with triptolide also rescued cells from death (Fig. 7A).
FIGURE 7.

Sp1 overexpression rescues pancreatic cells from triptolide-induced death. A, clones Sp1-NLS1 and Sp1-NLS2 (expressing the NLS), Sp1 overexpression (without NLS), OGT overexpression, and stimulation of cells with 5 mm glucosamine rescued pancreatic cancer cells from triptolide-induced cell death. Both Sp1-NLS overexpression and OGT overexpression also rescued cells from triptolide-induced loss of Sp1 (B) and NF-κB (C) activities. D, overexpression of Sp1 and OGT also rescued cells from inhibition of HSP70. The asterisks indicate statistically significant differences compared with the control. E, Western blot showing overexpression of OGT and increased O-GlcNAcylation of proteins in MIA-PaCa2 pancreatic cancer cells. F, Western blot confirming overexpression of Sp1 in Sp1-NLS-overexpressing cells. The asterisks indicate statistically significant change compared with the control.
Furthermore, overexpression of Sp1 and OGT rescued pancreatic cancer cells from triptolide-induced down-regulation of Sp1 activity (Fig. 7B). Overexpression of both Sp1 and OGT also restored the NF-κB activity inhibited by triptolide (Fig. 7C). Similarly, overexpression of both Sp1 and OGT in triptolide-treated pancreatic cancer cells also resulted in reversing the down-regulation of HSP70 expression after triptolide treatment (Fig. 7D).
DISCUSSION
Triptolide has been used as an anticancer compound for over a decade. However, its mechanisms of action have remained elusive. Studies using non-physiological doses of 10 μm have indicated that this compound inhibits the XPB subunit of RNA polymerase IIB and shuts down global transcription (36). However, for most cancer cells, the LD50 for triptolide has been found to be 100 nmol/liter (37), at which XPB remains unaffected. The use of a water-soluble derivative of triptolide (Minnelide) in an animal model for over 300 days without any adverse effect on the normal physiology of the animals further indicates that this compound does not result in a global transcriptional shutdown and that an alternative mechanism of action exists for this compound at physiological doses (28). Triptolide is known to affect a number of prosurvival pathways in a cancer cell. These include down-regulation of HSP70 and NF-κB, which in turn control cell survival and proliferation. Whether these pathways work in unison or in a hierarchical manner has not been reported previously. In this study, we have shown that triptolide may affect all of these pro-proliferative pathways by regulating modification of the transcription factor Sp1.
Recently, Sp1 was reported to be responsible for the transcription of NF-κB subunits p50 and p65 (33). In the context of that study, we observed that down-regulation of Sp1 by mithramycin, siRNA, or triptolide resulted in decreased NF-κB activity in both cell lines and orthotopic animal models. In contrast, inhibition of NF-κB by BAY11-7085 did not affect Sp1 activity (Fig. 8A), clearly establishing the hierarchy between these two pathways. Furthermore, down-regulation of Sp1 activity also resulted in reduced HSP70 expression and decreased binding of HSF1 to HSEs in their promoter regions. Similarly, silencing HSF1 did not have any effect on Sp1 promoter binding activity (Fig. 8B). This observation indicates that Sp1 is a master regulator for HSP expression and NF-κB activation in pancreatic cancer cells (Fig. 8C). Thus, in the presence of triptolide, Sp1 promoter binding activity is inhibited, resulting in decreased NF-κB and HSF1 activities and leading to reduced HSP expression culminating in cell death. This is further strengthened when Sp1 overexpression in triptolide-treated cells is able to rescue them from drug-induced cell death and also to revert to NF-κB and HSF1 promoter binding activity.
FIGURE 8.
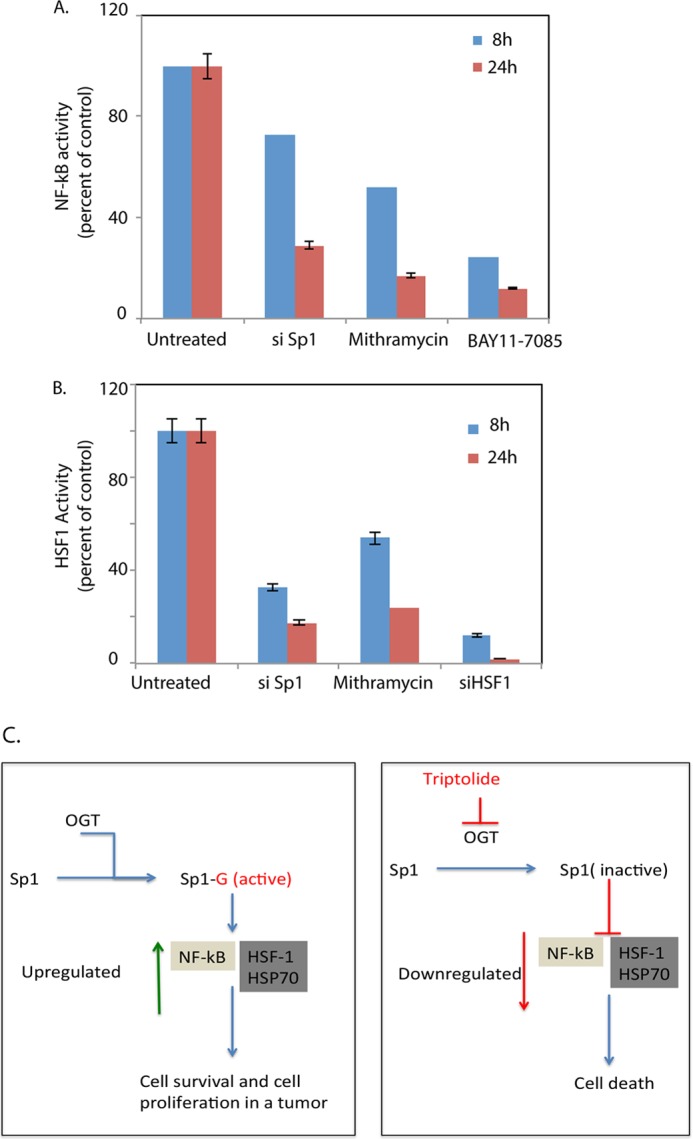
Sp1 inhibition down-regulates NF-κB and HSF1 promoter binding activity. A, inhibition of Sp1 by siRNA (si Sp1) or mithramycin resulted in down-regulation of NF-κB activity. BAY11-7085, a NF-κB pathway inhibitor, was used as a control. B, inhibition of Sp1 decreased HSF1 activity. HSF1 siRNA was used as a control. C, schematic flowchart demonstrating the possible mechanism of action of triptolide in pancreatic cancer cells.
Interestingly, although triptolide treatment resulted in a decrease in Sp1 activity and subsequent reduction in HSP expression, reduced HSE binding, and decreased NF-κB activity, it did not decrease Sp1 transcript levels after 24 h. This indicated that regulation of Sp1 activity by triptolide was mediated through its post-translational modifications. Sp1 protein is modified by the addition of a single O-linked N-acetylglucosamine at Ser-484 (35). This enables the nuclear translocation of Sp1 for further transactivation and DNA binding. Our study showed that inhibition of OGT by siRNA or its inhibitor ST045849 (38) resulted in the expected cytosolic accumulation of Sp1 and decreased Sp1 activity. Similarly, treatment with triptolide also resulted in a cytosolic accumulation of Sp1 and a subsequent decrease in activity (Fig. 5). Triptolide treatment was also found to inhibit OGT transcription and to decrease total O-GlcNAc modification in pancreatic cancer cells.
It has been reported that O-GlcNAc modification is dynamic and that both the addition and removal of O-GlcNAc from a modified transcription factor can influence its activity (39). Thus, in Sp1, the sites glycosylated by OGT need to be deglycosylated by OGA to be phosphorylated, bind to the DNA, and induce transcription of downstream elements. Consistent with this, our results showed that inhibition of OGA with either siRNA or GlcNAcstatin (data not shown) also inhibited Sp1 activity.
OGT is the last enzyme in the HBP, which orchestrates the modification of several cellular proteins. Its activity is dependent on the substrate UDP-GlcNAc in the cells. To completely inhibit OGT activity, we depleted the UDP-GlcNAc pool in pancreatic cancer cells by inhibiting an earlier enzyme in the HBP, GFAT. GFAT is a key regulator of the HBP, catalyzing the conversion of glutamine and fructose 6-phosphate to synthesize glucosamine 6-phosphate, which in turn is metabolized to synthesize UDP-GlcNAc in several steps. Inhibition of GFAT resulted in cell death, indicating that depletion of UDP-GlcNAc led to reduced OGT activity and decreased downstream events (Fig. 6) culminating in death. GFAT can be bypassed in the HBP by adding glucosamine to cells. This results in replenishing UDP-GlcNAc levels in cells and triggering downstream pathways that are dependent on O-GlcNAc modification. In our study, the addition of glucosamine, as well as overexpression of OGT, rescued cells from triptolide-induced death (Fig. 7), similar to that seen when Sp1 with a constitutive NLS was overexpressed in these cells. This confirmed that both OGT and Sp1 were targeted by triptolide in these cells. Because the nuclear localization of Sp1 was dependent on O-GlcNAc modification and thus OGT activity, it can be concluded that triptolide induces cell death by interfering with O-GlcNAc modification of Sp1.
It can thus be summarized that triptolide affects O-GlcNAc modification of Sp1, preventing its translocation to the nucleus. This results in decreased promoter binding activity of Sp1 for its target genes, such as NF-κB (p50/p65) and HSF1. The cumulative effect of reduced NF-κB and HSF1 activities leads to lowered expression of prosurvival genes in pancreatic cancer cells, eventually leading to cell death.
CONCLUSION
Triptolide or its prodrug (Minnelide) has shown immense promise in preclinical models of pancreatic cancer. However, its mechanism of action has remained elusive. Our study shows for the first time that triptolide-induced cell death in pancreatic cancer is mediated by alteration of O-GlcNAc modification of Sp1. Understanding the mode of action of this compound will be of great usefulness in delineating its biological function, as Minnelide is currently undergoing Phase 1 clinical trials.
This work was supported, in whole or in part, by National Institutes of Health UAB/UMN SPORE in Pancreatic Cancer Grant P50 CA101955 from NCI (to S. M. V.), Training Grant T32CA132715 (to R. C.), and Grants R01 CA170946 and CA124723 (to A. K. S.). University of Minnesota has filed a patent for Minnelide, which has been licensed to Minneamrita Therapeutics, LLC. A. K. S. and S. M. V. have financial interests in this company. A. K. S., S. M. V., and R. C. are inventors on this patent.
- HSP
- heat shock protein
- HBP
- hexosamine biosynthesis pathway
- OGT
- O-GlcNAc transferase
- OGA
- O-N-acetylglucosaminidase
- GFAT
- glutamine:fructose aminotransferase
- DON
- 6-diazo-5-oxo-l-norleucine
- NLS
- nuclear localization signal
- HSE
- heat shock element.
REFERENCES
- 1. Jemal A., Siegel R., Xu J., Ward E. (2010) Cancer statistics, 2010. CA Cancer J. Clin. 60, 277–300 [DOI] [PubMed] [Google Scholar]
- 2. Dudeja V., Mujumdar N., Phillips P., Chugh R., Borja-Cacho D., Dawra R. K., Vickers S. M., Saluja A. K. (2009) Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology 136, 1772–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aghdassi A., Phillips P., Dudeja V., Dhaulakhandi D., Sharif R., Dawra R., Lerch M. M., Saluja A. (2007) Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 67, 616–625 [DOI] [PubMed] [Google Scholar]
- 4. Banerjee S., Mujumdar N., Dudeja V., Mackenzie T., Krosch T. K., Sangwan V., Vickers S. M., Saluja A. K. (2012) MUC1c regulates cell survival in pancreatic cancer by preventing lysosomal permeabilization. PLoS ONE 7, e43020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zachara N. E., O'Donnell N., Cheung W. D., Mercer J. J., Marth J. D., Hart G. W. (2004) Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 279, 30133–30142 [DOI] [PubMed] [Google Scholar]
- 6. Zachara N. E., Hart G. W. (2006) Cell signaling, the essential role of O-GlcNAc! Biochim. Biophys. Acta 1761, 599–617 [DOI] [PubMed] [Google Scholar]
- 7. Zhu Q., Zhou L., Yang Z., Lai M., Xie H., Wu L., Xing C., Zhang F., Zheng S. (2012) O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med. Oncol. 29, 985–993 [DOI] [PubMed] [Google Scholar]
- 8. Krzeslak A., Forma E., Bernaciak M., Romanowicz H., Brys M. (2012) Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin. Exp. Med. 12, 61–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lynch T. P., Ferrer C. M., Jackson S. R., Shahriari K. S., Vosseller K., Reginato M. J. (2012) Critical role of O-linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J. Biol. Chem. 287, 11070–11081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mi W., Gu Y., Han C., Liu H., Fan Q., Zhang X., Cong Q., Yu W. (2011) O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim. Biophys. Acta 1812, 514–519 [DOI] [PubMed] [Google Scholar]
- 11. Kudlow J. E. (2006) Post-translational modification by O-GlcNAc: another way to change protein function. J. Cell. Biochem. 98, 1062–1075 [DOI] [PubMed] [Google Scholar]
- 12. Majumdar G., Wright J., Markowitz P., Martinez-Hernandez A., Raghow R., Solomon S. S. (2004) Insulin stimulates and diabetes inhibits O-linked N-acetylglucosamine transferase and O-glycosylation of Sp1. Diabetes 53, 3184–3192 [DOI] [PubMed] [Google Scholar]
- 13. Suske G., Bruford E., Philipsen S. (2005) Mammalian SP/KLF transcription factors: bring in the family. Genomics 85, 551–556 [DOI] [PubMed] [Google Scholar]
- 14. Safe S., Abdelrahim M. (2005) Sp transcription factor family and its role in cancer. Eur. J. Cancer 41, 2438–2448 [DOI] [PubMed] [Google Scholar]
- 15. Shi Q., Le X., Abbruzzese J. L., Peng Z., Qian C. N., Tang H., Xiong Q., Wang B., Li X. C., Xie K. (2001) Constitutive Sp1 activity is essential for differential constitutive expression of vascular endothelial growth factor in human pancreatic adenocarcinoma. Cancer Res. 61, 4143–4154 [PubMed] [Google Scholar]
- 16. Wang L., Wei D., Huang S., Peng Z., Le X., Wu T. T., Yao J., Ajani J., Xie K. (2003) Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin. Cancer Res. 9, 6371–6380 [PubMed] [Google Scholar]
- 17. Blasco F., S., Cascall M., Hernndez J. L., Alemany C., Masa M., J., Soler M., Nicols M., Prez-Torras S., Gmez A., Tarrasn G., V., Mazo A., Ciudad C. J., Piulats J. (2004) Expression profiles of a human pancreatic cancer cell line upon induction of apoptosis search for modulators in cancer therapy. Oncology 67, 277–290 [DOI] [PubMed] [Google Scholar]
- 18. Jiang N. Y., Woda B. A., Banner B. F., Whalen G. F., Dresser K. A., Lu D. (2008) Sp1, a new biomarker that identifies a subset of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 17, 1648–1652 [DOI] [PubMed] [Google Scholar]
- 19. Bevilacqua A., Fiorenza M. T., Mangia F. (2000) A developmentally regulated GAGA box-binding factor and Sp1 are required for transcription of the hsp70.1 gene at the onset of mouse zygotic genome activation. Development 127, 1541–1551 [DOI] [PubMed] [Google Scholar]
- 20. Porter W., Wang F., Wang W., Duan R., Safe S. (1996) Role of estrogen receptor/Sp1 complexes in estrogen-induced heat shock protein 27 gene expression. Mol. Endocrinol. 10, 1371–1378 [DOI] [PubMed] [Google Scholar]
- 21. Westerheide S. D., Kawahara T. L., Orton K., Morimoto R. I. (2006) Triptolide, an inhibitor of the human heat shock response that enhances stress-induced cell death. J. Biol. Chem. 281, 9616–9622 [DOI] [PubMed] [Google Scholar]
- 22. Phillips P. A., Dudeja V., McCarroll J. A., Borja-Cacho D., Dawra R. K., Grizzle W. E., Vickers S. M., Saluja A. K. (2007) Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 67, 9407–9416 [DOI] [PubMed] [Google Scholar]
- 23. Tengchaisri T., Chawengkirttikul R., Rachaphaew N., Reutrakul V., Sangsuwan R., Sirisinha S. (1998) Antitumor activity of triptolide against cholangiocarcinoma growth in vitro and in hamsters. Cancer Lett. 133, 169–175 [DOI] [PubMed] [Google Scholar]
- 24. Yang S., Chen J., Guo Z., Xu X. M., Wang L., Pei X. F., Yang J., Underhill C. B., Zhang L. (2003) Triptolide inhibits the growth and metastasis of solid tumors. Mol. Cancer Ther. 2, 65–72 [PubMed] [Google Scholar]
- 25. Banerjee S., Thayanithy V., Sangwan V., Mackenzie T. N., Saluja A. K., Subramanian S. (2013) Minnelide reduces tumor burden in preclinical models of osteosarcoma. Cancer Lett. 335, 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Antonoff M. B., Chugh R., Borja-Cacho D., Dudeja V., Clawson K. A., Skube S. J., Sorenson B. S., Saltzman D. A., Vickers S. M., Saluja A. K. (2009) Triptolide therapy for neuroblastoma decreases cell viability in vitro and inhibits tumor growth in vivo. Surgery 146, 282–290 [DOI] [PubMed] [Google Scholar]
- 27. Krosch T. C., Sangwan V., Banerjee S., Mujumdar N., Dudeja V., Saluja A. K., Vickers S. M. (2013) Triptolide-mediated cell death in neuroblastoma occurs by both apoptosis and autophagy pathways and results in inhibition of nuclear factor-κB activity. Am. J. Surg. 205, 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chugh R., Sangwan V., Patil S. P., Dudeja V., Dawra R. K., Banerjee S., Schumacher R. J., Blazar B. R., Georg G. I., Vickers S. M., Saluja A. K. (2012) A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci. Transl. Med. 4, 156ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang M., Huang J., Pan H. Z., Jin J. (2008) Triptolide overcomes dexamethasone resistance and enhanced PS-341-induced apoptosis via PI3K/Akt/NF-κB pathways in human multiple myeloma cells. Int. J. Mol. Med. 22, 489–496 [PubMed] [Google Scholar]
- 30. Qiu D., Kao P. N. (2003) Immunosuppressive and anti-inflammatory mechanisms of triptolide, the principal active diterpenoid from the Chinese medicinal herb Tripterygium wilfordii Hook. f. Drugs R D 4, 1–18 [DOI] [PubMed] [Google Scholar]
- 31. Sclabas G. M., Uwagawa T., Schmidt C., Hess K. R., Evans D. B., Abbruzzese J. L., Chiao P. J. (2005) Nuclear factor κB activation is a potential target for preventing pancreatic carcinoma by aspirin. Cancer 103, 2485–2490 [DOI] [PubMed] [Google Scholar]
- 32. Mujumdar N., Mackenzie T. N., Dudeja V., Chugh R., Antonoff M. B., Borja-Cacho D., Sangwan V., Dawra R., Vickers S. M., Saluja A. K. (2010) Triptolide induces cell death in pancreatic cancer cells by apoptotic and autophagic pathways. Gastroenterology 139, 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jutooru I., Chadalapaka G., Lei P., Safe S. (2010) Inhibition of NFκB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. J. Biol. Chem. 285, 25332–25344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blume S. W., Snyder R. C., Ray R., Thomas S., Koller C. A., Miller D. M. (1991) Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J. Clin. Invest. 88, 1613–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang X., Su K., Roos M. D., Chang Q., Paterson A. J., Kudlow J. E. (2001) O-Linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc. Natl. Acad. Sci. U.S.A. 98, 6611–6616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Titov D. V., Gilman B., He Q. L., Bhat S., Low W. K., Dang Y., Smeaton M., Demain A. L., Miller P. S., Kugel J. F., Goodrich J. A., Liu J. O. (2011) XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 7, 182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Visp S., DeVries L., Crancier L., Besse J., Brand S., Hobson D. J., Svejstrup J. Q., Annereau J. P., Cussac D., Dumontet C., Guilbaud N., Barret J. M., Bailly C. (2009) Triptolide is an inhibitor of RNA polymerase I- and II-dependent transcription leading predominantly to down-regulation of short-lived mRNA. Mol. Cancer Ther. 8, 2780–2790 [DOI] [PubMed] [Google Scholar]
- 38. Gross B. J., Kraybill B. C., Walker S. (2005) Discovery of O-GlcNAc transferase inhibitors. J. Am. Chem. Soc. 127, 14588–14589 [DOI] [PubMed] [Google Scholar]
- 39. Ozcan S., Andrali S. S., Cantrell J. E. (2010) Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 1799, 353–364 [DOI] [PMC free article] [PubMed] [Google Scholar]