

Background: Cell migration requires persistent PKC phosphorylation, which can be achieved through the RFFL-mTORC2 pathway.
Results: We show how Gα12 specifically activates ARAF to stimulate RFFL expression, which can also be stimulated by EGF, via CRAF, and activated BRAF.
Conclusion: Different signaling pathways, through different Raf proteins, stimulate RFFL expression to support cell migration.
Significance: The RFFL-PKC pathway has a broad significance in cell migration regulation.
Keywords: Cell Migration, Cell Signaling, G Proteins, Raf, Signal Transduction, Signaling
Abstract
We previously characterized a Gα12-specific signaling pathway that stimulates the transcription of the E3 ligase RFFL via the protein kinase ARAF and ERK. This pathway leads to persistent PKC activation and is important for sustaining fibroblast migration. However, questions remain regarding how Gα12 specifically activates ARAF, which transcription factor is involved in Gα12-mediated RFFL expression, and whether RFFL is important for cell migration stimulated by other signaling mechanisms that can activate ERK. In this study, we show that replacement of the Gα12 residue Arg-264 with Gln, which is the corresponding Gα13 residue, abrogates the ability of Gα12 to interact with or activate ARAF. We also show that Gα12 can no longer interact with and activate an ARAF mutant with its C-terminal sequence downstream of the kinase domain being replaced with the corresponding CRAF sequence. These results explain why Gα12, but not Gα13, specifically activates ARAF but not CRAF. Together with our finding that recombinant Gα12 is sufficient for stimulating the kinase activity of ARAF, this study reveals an ARAF activation mechanism that is different from that of CRAF. In addition, we show that this Gα12-ARAF-ERK pathway stimulates RFFL transcription through the transcription factor c-Myc. We further demonstrate that EGF, which signals through CRAF, and an activated BRAF mutant also activate PKC and stimulate cell migration through up-regulating RFFL expression. Thus, RFFL-mediated PKC activation has a broad significance in cell migration regulation.
Introduction
Heterotrimeric G proteins mediate signal transduction of a diverse range of biologically active molecules. Among the four families of Gα subunits, the Gα12/13 family, comprised of Gα12 and Gα13, is ubiquitously expressed. Although Gα12 and Gα13 share less than 70% amino acid sequence identity, mouse genetic studies have shown that these two Gα proteins are functionally redundant in regulating vascular smooth muscles, neuron migration, cardiac morphogenesis, T and B cell migration, and trafficking (1, 2). The redundant functions may be mediated by a group of Rho guanine nucleotide exchange factors that both Gα proteins activate (2). Nevertheless, evidence has emerged to suggest that these two proteins may also have additional functions that differ between the isoforms. Gα13 appears to regulate platelet activation (3, 4), PDGF-induced cell migration (5), and angiogenesis (6). On the other hand, G12 was shown to be involved in LPA-induced mitogenic activity (7), S1P-induced COX2 expression (8), and TCR-mediated IL-2 expression (9). Some of the specific in vivo functions of Gα13 may be mediated by its interaction with integrin αIIbβ3 and GEF115 (3, 10, 11).
We recently characterized a Gα12-specific signaling mechanism activated by lysophosphatidic acid (LPA)2 (12). LPA induces diverse cellular responses, including proliferation, adhesion, migration, morphogenesis, differentiation, and survival (13), and has an important role in pulmonary fibrosis (14). We found that LPA induced two phases of PKC hydrophobic motif (HM) phosphorylation. The late, sustained phase is mediated by G12 but not other G proteins, including its close homolog G13 (12). We also found that Gα12, but not Gα13, specifically interacted with ARAF, but not CRAF, to stimulate transcription of the E3 ubiquitin ligase RFFL via MEK and ERK. RFFL can poly-ubiquitinate and destabilize PRR5L, an mTORC2-associated protein that inhibits mTORC2-mediated HM phosphorylation of PKCδ but not AKT. Its elimination by RFFL resulted in PKCδ HM phosphorylation and activation. We further showed that this Gα12-mediated pathway is critically important for fibroblast migration and the development of pulmonary fibrosis (12).
The Raf family of protein kinases, consisting of ARAF, BRAF, and CRAF, plays important roles in signal transduction by regulating the MEK and ERK cascade. ARAF is the least studied among the three RAF isoforms (15, 16). It is poorly activated by growth factors, overexpressed tyrosine kinases, or activated Ras. Ras is considered the primary Raf upstream activator (17, 18). However, our recent study demonstrates that ARAF is primarily regulated by G protein-coupled receptors (GPCRs) via Gα12 independently of Ras (12). Thus, it would be important to understand why Gα12, but not Gα13, specifically interacts with and activates ARAF but not CRAF. Ras activates CRAF by binding to its N-terminal Ras-binding domain to relieve N terminus-mediated autoinhibition, so the question is whether Gα12 activates ARAF via a similar mechanism. Although the LPA-Gα12 pathway acted specifically through ARAF to activate ERK, leading to RFFL transcription activation, ERK can also be activated by CRAF and BRAF, which are activated by growth factors. Moreover, many cancer cells, particularly melanoma cancer cells, contain activated BRAF mutations, and activation of CRAF and BRAF by either growth factors or activated mutations is expected to activate ERK. The questions are whether these CRAF and BRAF activating mechanisms lead to RFFL expression and persistent PKC activation and whether RFFL-mediated persistent PKC activation is important in cell migration stimulated by growth factors and activated BRAF mutants.
In this report, we identified an important Gα12 amino acid required for its interaction with ARAF and the ARAF sequence required for its interaction with and activation by Gα12. We also demonstrated that EGF acted specifically through CRAF in mouse embryonic fibroblast (MEF) cells to up-regulate RFFL expression and stimulate cell migration. In addition, in two tumor cell lines that harbor the BRAF-activating V600E mutation, RFFL is involved in elevated PKC activation and plays an important role in cell migration.
EXPERIMENTAL PROCEDURES
Materials
Reagents were obtained from the following sources: protein A/G-PLUS-Sepharose from Santa Cruz Biotechnology, Inc.; antibodies to PKCδ, p44/42 MAPK, c-Myc, histone H3, phospho-p44/42 MAPK, phospho-PKCδ/θ(Ser-643/676), phospho-MARCKS(Ser-152/156), phospho-CRAF(Ser-338), phospho-ARAF(Ser-299), and phospho-MEK1/2(Ser-217/221) from Cell Signaling Technology, Inc.; antibodies to Gα12, Gα13, and Elk1 (H-160) from Santa Cruz Biotechnology, Inc.; Lipofectamine/Plus reagent and Lipofectamine RNAiMax from Invitrogen; L-α-lysophosphatidic acid from Sigma; a chromatin immunoprecipitation assay kit from Millipore; and His-MEK1(K97A) from SignalChem.
Cell Culture and Stimulation
HEK293T cells and MEFs were maintained in DMEM with 4.5 g/liter glucose supplemented with 10% FBS. WN266-4 cells were maintained in minimum Eagle's medium with 10% FBS. NCI-460 cells were maintained in RPMI 1640 with 10% FBS. Transient transfection was carried out using Lipofectamine and Plus (Invitrogen), and samples were collected 24 h after transfection. Synthetic siRNA oligos, which were obtained from Dharmacon (the sequences are listed in supplemental Table S1), were transfected using Lipofectamine RNAiMax, and samples were collected 48 h after transfection. Cells were starved overnight in DMEM without serum before being stimulated with LPA or EGF.
Protein Purification
GST-ARAF, ARAF mutants, or CRAF was expressed in Escherichia coli BL21 (DE3). After isopropyl-1-thio-β-d-galactopyranoside (100 μm) induction at 22 °C for 24 h, proteins were extracted with 0.5 mg/ml lysozyme in buffer A (20 mm Tris-HCl (pH 7.5), 50 mm NaCl, 0.5% Triton X-100, and 1 mm phenylmethylsulfonyl fluoride). The high-speed supernatant of the extract was loaded on glutathione-agarose and eluted with buffer A with 5 mm reduced glutathione. The recombinant protein was dialyzed into 20 mm Tris-HCl (pH 7.5), 100 mm NaCl, and 5% glycerol.
Immunoprecipitation
Cells were lysed with cell lysis buffer (20 mm Tris-HCl (pH 7.5), 100 mm NaCl, 0.7% Triton X-100, 1 mm EGTA, 5 mm MgCl2, protease inhibitor mixture, and phosphatase inhibitor mixture (Roche)) and AMF (50 μm AlCl3, 10 mm MgCl2, and 5 mm NaF). Immunoprecipitation was carried out using anti-Gα12 antibody.
In Vitro Binding Assay and Kinase Assay
Approximately 60 pmol of purified Gα12 or Gα13 was incubated with either 10 μm GTPγS for 2 h at 30 °C in buffer (50 mm HEPES (pH 8.0), 1 mm EDTA, 3 mm DTT, and 0.05% polyoxyethylene 10 lauryl ether containing 10 mm MgSO4). Reactions were divided and then incubated with glutathione-agarose-bound GST fusion proteins for 2 h at 4 °C. For the kinase assay, the above mixtures were incubated with 200 ng of His-MEK1 and 500 μm ATP for 30 min at 30 °C and stopped by SDS sample buffer. For the binding assay, the glutathione-agarose was pelleted, washed with buffer, and analyzed by Western blotting.
ChIP Assay
The ChIP assays were performed using the ChIP immunoprecipitation assay kit (Millipore, MA) and 107 HEK293T cells by following the instructions of the manufacturer.
Cell Migration Assays
For the scratch migration assay, cells were grown to confluency in a monolayer in six-well plates. A linear gap was generated by scratching the bottoms of the wells using a sterile 200-μl pipette tip. Phase-contrast microscopy images were acquired 0 and 16 h after the gaps were created. The distance migrated in 16 h by the control cells are taken as 1. The transwell assay was done using transwells coated with Matrigel (1 mg/ml). Cells were harvested by trypsin/EDTA, washed, and resuspended in medium containing 0.1% BSA at a density of 106 cells/ml. Cell suspensions (100 μl) were loaded onto the transwells and incubated at 37 °C for 6 h. The cells from the upper portion of the transwell filters were removed by cotton swab. Cells at the bottom of the filters were stained with crystal violet and counted under a light microscope.
RESULTS AND DISCUSSION
To understand how Gα12, but not its close homolog Gα13, specifically regulates ARAF, we generated a Gα12 chimeric mutant in which the Gα12 amino acid sequence from residues Val-225 to Leu-294 was substituted with the corresponding Gα13 sequence as depicted in Fig. 1A. The reason for selecting this stretch of amino acid sequence is that these regions of Gα subunits have been recognized for effector interactions (19–22). This Gα12 chimeric mutant also contains the constitutively active Q229L mutation and was designated as Gα12QL-Ch. We have shown previously that Gα12QL, but not Gα13QL, stimulated ERK phosphorylation in HEK293 cells and that Gα12QL-mediated ERK phosphorylation exclusively depends on ARAF in these cells (12). Thus, ERK phosphorylation was used as a surrogate marker to test Gα12 for its ability to activate ARAF. We found that expression of Gα12QL-Ch, unlike Gα12QL, failed to stimulate ERK phosphorylation in HEK293T cells (Fig. 1B), suggesting that the residues from Val-225 to Leu-294 may contain amino acids critical for ARAF activation.
FIGURE 1.
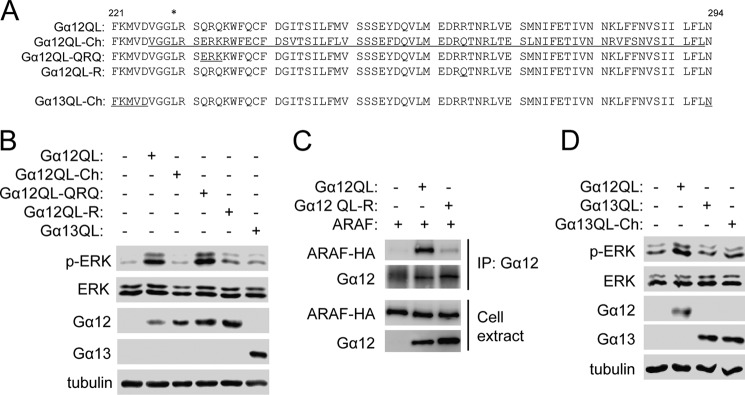
Residue Arg-264 is required for Gα12 interaction with and activation of ARAF. A, amino acid sequences of the effector interaction sites of Gα12 and Gα13 and their mutants. The asterisk denotes the Leu substitution for Gln of the activating Gα12 mutation. The amino acid sequence of Gα13 is underlined. B, the effect of Gα12QL and its mutants on ERK phosphorylation. HEK293T cells were transfected with the G proteins, and Western blot analyses were performed the next day. C, interaction of Gα12QL and its mutant with ARAF. HEK293T cells were transfected with the G proteins and HA-ARAF, and immunoprecipitation (IP) using anti-HA was carried out the next day, followed by Western blot analysis. D, the effect of Gα13QL and its mutant on ERK phosphorylation. HEK293T cells were transfected with the G proteins, and Western blot analyses were performed the next day.
Comparison of these Gα12 residues (Val-226 to Leu-293) with Gα13 revealed only a few residues that are not conserved, including Gln-232, Gln-234, and Arg-264 (Fig. 1A). We replaced Gln-232 and Gln-234 with corresponding Gα13 residues (Glu and Lys) to create Gα12QL-QRQ and replaced Arg-264 with Gln to create Gα12QL-R (Fig. 1A). Although Gα12QL-QRQ still retained its ability to stimulate ERK phosphorylation, Gα12QL-R showed a markedly reduced ability (Fig. 1B). These results suggest that the Gα12 residue Arg-264 is critically important for Gα12 to stimulate ERK phosphorylation. Consistent with the fact that Gα12 stimulates ERK phosphorylation via MEK1 (12), we also observed that expression of Gα12QL-R only weakly increased MEK-1 phosphorylation compared with that of Gα12QL (supplemental Fig. S1). To confirm that Gα12 Arg-264 is also important for Gα12 to bind to ARAF, we carried out the coimmunoprecipitation assay as we did previously (12) and found that the interaction of Gα12QL-R with ARAF was markedly reduced (Fig. 1C).
To determine whether the Gα12 residues Val-226 to Leu-293 are sufficient for activating ARAF and stimulating ERK phosphorylation, we generated a Gα13 chimera, designated as Gα13QL-Ch, by substituting these Gα12 residues for the corresponding Gα13 residues (Fig. 1A). Expression of Gα13QL-Ch had a marginal effect on ERK phosphorylation in HEK293T cells (Fig. 1D), suggesting that these Gα12 residues are largely insufficient to activate ARAF and stimulate ERK phosphorylation.
Next we investigated the basis for specific activation of ARAF, but not CRAF, by Gα12. There are three highly conserved regions (CR1–3) in the Raf family of protein kinases (Fig. 2A). However, sequences in regions other than these CRs are less conserved. We first generated two ARAF chimeric mutants with substitution of the CRAF sequences between CR1 and CR2 and between CR2 and CR3, respectively, for those in ARAF. When these chimeric mutants were tested in a cotransfection assay with Gα12, they showed similar synergistic effects with Gα12 compared with the wild-type ARAF, suggesting that these regions may not be important for Gα12 activation (data not shown). We then generated an ARAF chimera with substitution of the CRAF C terminus (starting at residues His-611) for ARAF C-terminal residues (starting at Arg-572), which was designated ARAF-C (Fig. 2A). In HEK293 cells, expression of ARAF alone induced little ERK phosphorylation. However, ERK phosphorylation was markedly greater in HEK293 cells coexpressing ARAF and Gα12QL than that in cells expressing Gα12QL alone. We used this synergistic effect of ARAF with Gα12QL to examine whether the ARAF mutants were able to be activated by Gα12QL. Unlike ARAF, ARAF-C and CRAF failed to show any synergistic effect with Gα12QL (Fig. 2B). In addition, ARAF-C appeared to reduce Gα12QL-induced ERK phosphorylation (Fig. 2B, compare lane 6 with lane 2), suggesting that it may act as a dominant negative mutant. These results together suggest that the C-terminal sequence downstream of the ARAF kinase domain is required for its activation by Gα12. To further confirm this conclusion, we examined the interaction of Gα12 with ARAF-C using a coimmunoprecipitation assay and found that ARAF-C acted like CRAF rather than ARAF and showed little interaction with Gα12 (Fig. 2C). Consistent with these coimmunoprecipitation results, recombinant GTPγS-bound Gα12 protein strongly interacted with the ARAF protein but not with ARAF-C or the CRAF protein (Fig. 2D). These results collectively indicate that the C-terminal region of ARAF is required for its interaction with and activation by Gα12.
FIGURE 2.

The ARAF C-terminal sequence is required for ARAF interaction with and activation by Gα12. A, schematic of the Raf family of kinases and the C-terminal amino acid sequences of ARAF and CRAF. The amino acid sequence of CRAF is underlined. B, the effect of ARAF C-terminal substitution on ERK phosphorylation stimulated by Gα12QL. HEK293T cells were transfected with Gα12QL and Rafs, and Western blotting was carried out the next day. C and D, the effect of ARAF C-terminal substitution on its interaction with Gα12. The interaction between Gα12 and Rafs was assessed by coimmunoprecipitation (IP) using HEK293 cells transfected with Gα12QL and Rafs (C) or by pull-down of recombinant GTPγS-loaded Gα12 or Gα13 and Raf proteins (D). E, the effect of CRAF C-terminal substitution on ERK phosphorylation stimulated by Gα12QL. HEK293 cells were transfected with Gα12QL and/or ARAF or CRAF-C, and Western blot analysis was performed 24 h after transfection.
To determine whether the C-terminal ARAF sequence is sufficient for activation by Gα12, we generated a CRAF chimeric mutant with substitution of the ARAF C-terminal sequence for the CRAF C-terminal sequence, designated as CRAF-C (Fig. 2A). CRAF-C showed no synergistic effect with Gα12 (Fig. 2E), thus suggesting that the ARAF C-terminal sequence is not sufficient for activation by Gα12. ARAF sequences beyond this C-terminal region might also be involved in the interaction with Gα12. This conclusion is consistent with the observation that ARAF-C acts as a dominant negative mutant (Fig. 2B).
Although we have shown that Gα12 directly interacts with and activates ARAF in transfected cells, it is important to also know whether Gα12 can activate ARAF kinase activity independently of other factors. This is particularly crucial because Gα12 binds to a region on ARAF that is different from the one to which the Ras proteins, the best characterized Raf activators, bind. Thus, we performed an in vitro kinase assay using recombinant Gα12 and Gα13 proteins that were loaded with GTPγS and recombinant ARAF, ARAF-C, or CRAF protein in the presence of ATP and kinase-dead MEK1 as substrates. The Raf kinase activity was detected by using an anti-phospho-MEK1 antibody. Gα12, but not Gα13, stimulated the phosphorylation of MEK1 only when ARAF, but not CRAF or ARAF-C, was present (Fig. 3). These results clearly demonstrate that Gα12 is sufficient for stimulating ARAF kinase activity.
FIGURE 3.
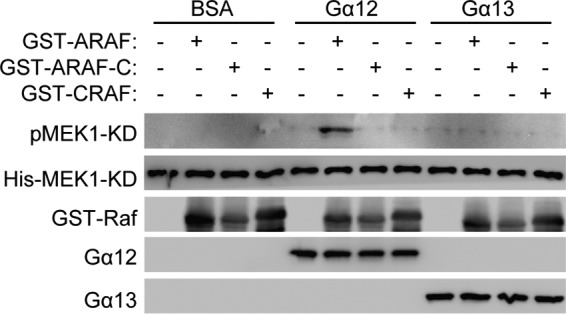
Direct stimulation of ARAF kinase activity by Gα12. Recombinant Gα12 or Gα13 protein was loaded with GTPγS and incubated with kinase-dead MEK1 protein and ARAF or CRAF protein in the presence of ATP. Phosphorylation of MEK1 was detected by Western blot analysis.
Because Gα12 and ARAF stimulate RFFL expression via ERK (12), we also tested if Gα12QL-R and ARAF-C could stimulate RFFL expression. Neither Gα12QL-R nor ARAF-C, when expressed in HEK293T cells, stimulated RFFL expression, whereas their wild-type proteins could (Fig. 4A). These results are consistent with their inability to stimulate ERK phosphorylation. We also investigated which transcription factors are involved in RFFL transcription regulation by this pathway. Elk and c-Myc are transcription factors known to be regulated by ERK. We examined their involvement by using a ChIP assay and found that LPA, which is upstream of Gα12 (12), stimulated the binding of c-Myc, but not Elk, to the RFFL promoter regions in a MEK-dependent manner (Fig. 4, B and C). In addition, expression of Gα12QL or ARAF, but not Gα12QL-R or ARAF-C, stimulated c-Myc binding to the RFFL promoter elements (Fig. 4D). To further characterize the importance of c-Myc in RFFL transcription regulation, we transfected the cells with c-Myc siRNAs, which silenced c-Myc expression (Fig. 4E), and inhibited LPA-stimulated expression of RFFL (F). Together, these data demonstrate that the LPA-Gα12-ARAF pathway up-regulates RFFL gene expression via c-Myc.
FIGURE 4.
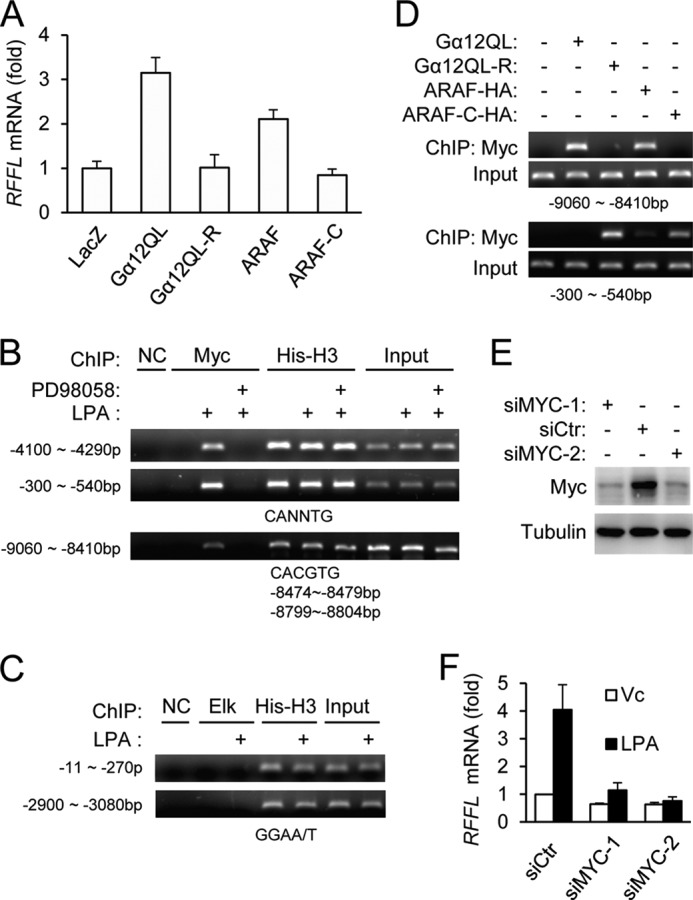
c-Myc is involved in RFFL transcription activation by the Gα12-ARAF pathway. A, expression of Gα12QL or ARAF, but not their mutants, was able to stimulate RFFL expression in HEK293T cells. B–D, stimulation of c-Myc binding to the RFFL promoter elements by LPA, Ga12QL, and ARAF. HEK293T cells were treated with LPA (400 nm) in the presence or absence of the MEK inhibitor PD98058 (1 μm) for 2 h at 37 °C (B and C) or transected with Gα12QL, ARAF, or their mutants (D). A chromosome immunoprecipitation assay was performed as described under “Experimental Procedures.” NC, negative control. E and F, the effect of c-Myc knockdown on RFFL expression. HEK293T cells were transfected with c-Myc siRNAs. Two days later, the cells were stimulated with LPA for 2 h, and their total RNAs and proteins were collected. The relative mRNA levels of RFFL were determined by quantitative RT-PCR (F), whereas c-Myc protein content was examined by Western blot analysis (E). siCTR, siRNA control; Vc, vehicle control.
Not only can ARAF activate the MEK-ERK pathway, but the ARAF homolog CRAF and BRAF are also known to activate the pathway, and they should up-regulate RFFL as well. In fact, expression of WT CRAF or mutant BRAF carrying an activating mutation, V600E, also led to up-regulation of RFFL expression in HEK293T cells (Fig. 5A). Interestingly, in MEFs, EGF-up-regulated RFFL expression primarily depended on CRAF, but not BRAF or ARAF, because knockdown of CRAF, but not BRAF or ARAF, inhibited EGF-induced RFFL expression (Fig. 5, B and C, and supplemental Fig. S2, A and B). Consistent with this observation, knockdown of CRAF, but not ARAF or BRAF, inhibited EGF-induced phosphorylation of PKCδ HM and MARCKS (Fig. 5D and supplemental Fig. S2C). MARCKS is a PKC substrate, and its phosphorylation indicates PKC activation (12). We have shown previously that RFFL mediated long-term PKCδ HM phosphorylation in MEFs, which is important for MEF migration (12). In agreement with these findings, RFFL knockdown primarily inhibited EGF-induced phosphorylation of PKCδ and MARCKS at 120 min rather than at 30 min (Fig. 5E and supplemental Fig. S2D) and impaired EGF-induced migration (Fig. 5F and supplemental Fig. S2E). We also investigated the role of RFFL in the migration of two tumor cell lines harboring the V600E BRAF mutation. They are WM266-4 (a melanoma cell line) and NCI-H460 (a non-small cell lung carcinoma cell line). Knockdown of either BRAF or RFFL inhibited migration of these cells (Fig. 5G and supplemental Fig. S2, A and F). Consistent with the effects on cell migration, BRAF or RFFL knockdown also decreased the phosphorylation of PKCδ and MARCKS (Fig. 5H and supplemental Fig. S2G).
FIGURE 5.
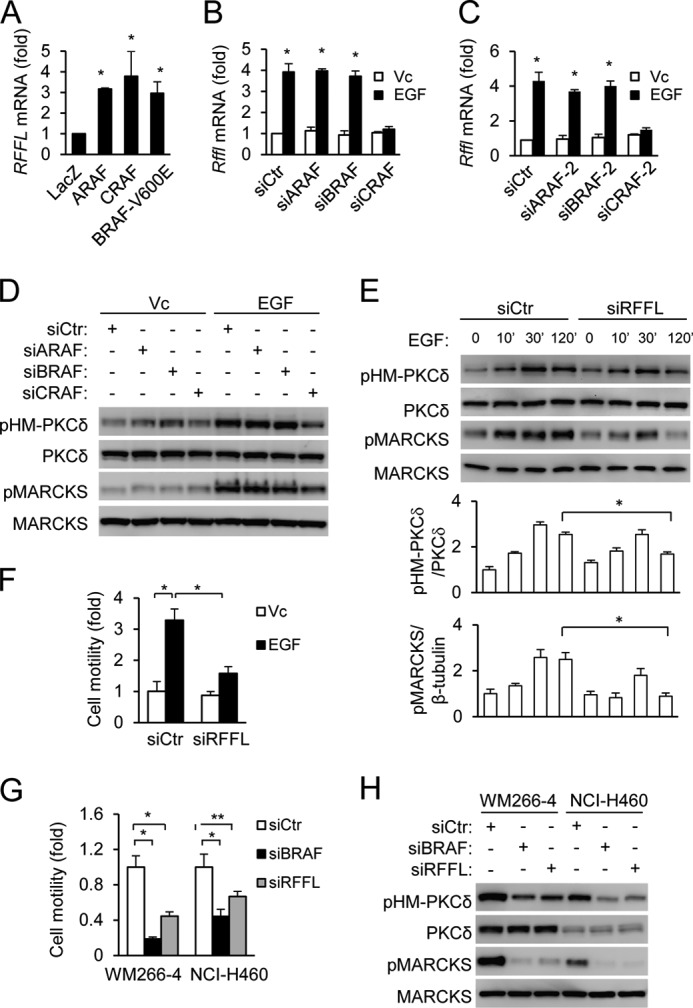
The importance of RFFL in EGF-CRAF and activated BRAF-stimulated sustained PKC HM phosphorylation and cell migration. A, elevation of RFFL mRNA levels by Raf expression. HEK293T cells were transfected with the Raf cDNAs, and the relative levels of RFFL mRNA were determined by quantitative RT-PCR the next day. Data are presented as mean ± S.D. *, p < 0.05 versus LacZ, Student's t test. B–D, CRAF is required for EGF-induced RFFL expression and phosphorylation of PKCδ and MARCKS. MEFs were transfected with the Raf siRNAs for 2 days and stimulated with EGF for 2 h before quantitative RT-PCT (B and C) or Western blot analysis (D). The relative mRNA levels of Rffl (B and C) were determined by quantitative RT-PCR. Data are presented as mean ± S.D. *, p < 0.05 versus Vc, Student's t test. siCTR, siRNA control; Vc, vehicle control. E and F, important roles of RFFL in EGF-induced PKCδ HM and MARCKS phosphorylation and cell migration. MEFs were transfected with RFFL siRNA for 2 days and analyzed by Western blotting (E) or scratch migration assay (F). The Western blot analysis was quantified from three blots. Quantitative data are presented as mean ± S.D. *, p < 0.05, Student's t test. G and H, important roles of RFFL in PKCδ HM and MARCKS phosphorylation and cell migration of tumor cells harboring activated BRAF. Cells were transfected with RFFL siRNA for 2 days and analyzed by a transwell migration assay (G) or Western blot analysis (H). The data in G are presented as mean ± S.D. *, p < 0.01; **, p < 0.05; Student's t test).
In this study, we provided insights into the molecular basis for the specific interactions between Gα12 and ARAF. In addition, we showed that although Gα12, unlike Ras, binds to the C terminus of ARAF, it may achieve a similar outcome in activation of ARAF, probably by relieving the autoinhibition by the N terminus. The fact that Gα12 alone is sufficient to activate ARAF kinase activity further confirms the above conclusion, although other cofactors, including KSR (kinase suppressor of RAS), Raf phosphorylation, and dimerization, may help to stabilize or further activate ARAF. Thus, the results in this study further solidify the conclusion that ARAF is a specific, direct effector of Gα12. In this study, we also demonstrated that EGF, acting through CRAF, and activated BRAF could also up-regulate RFFL expression to regulate PKC activity and cell migration. Thus, different signaling pathways, either activated by a GPCR ligand (such as LPA) via ARAF, a growth factor (such as EGF) via CRAF, or activated BRAF mutations, as depicted in Fig. 6, can achieve persistent PKC activation through the activation of ERK and RFFL transcription to support cell migration. This RFFL-mediated signaling pathway may have a much broader significance in cell migration regulation than illustrated in this and our previous study (12) because many tumor cells have amplified EGF signaling activity or mutated Ras and Raf genes that can also lead to RFFL expression and persistent PKC activation. Therefore, RFFL, mTORC2, and PKC may be potential targets for blocking tumor migration and, hence, metastasis, in addition to treating pulmonary fibrosis, as we indicated previously (12). This hypothesis needs to be tested with in vivo models, particularly the spontaneous genetic tumor models.
FIGURE 6.
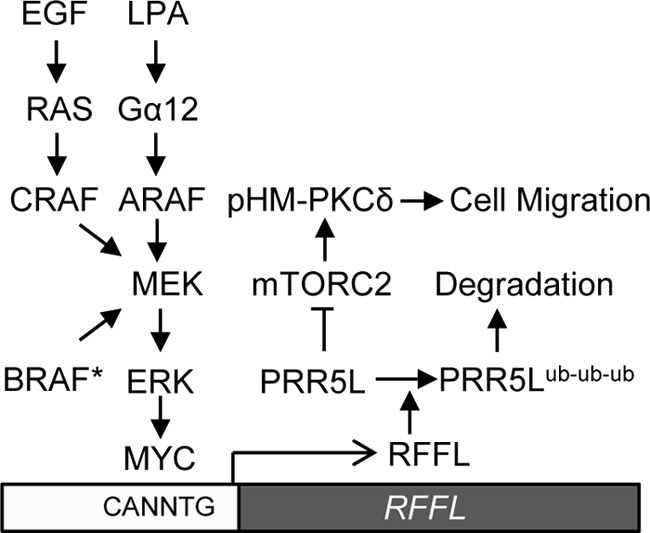
Schematic of a diverse range of signaling pathways that can lead to persistent PKC activation to support cell migration. Three Raf proteins activated through three different mechanisms can activate RFFL transcription via an ERK-MYC pathway, which leads to persistent mTORC2-mediated PKC HM phosphorylation and activation. Persistent PKC activation is important for sustaining the migration of fibroblasts and tumor cells. BRAF*, activated BRAF; ub, ubiquitin.
Questions also remain on how RFFL activates PKC. We have shown previously that RFFL induces PKC HM phosphorylation by polyubiquitinating and destabilizing PRR5L, a protein that inhibits mTORC2-mediated PKC HM phosphorylation. It is not known whether the phosphorylation at the HM site of PKCδ is sufficient for its activation. We have substituted a glutamine residue for the HM site serine residue but did not observe any change in the kinase activity (data not shown). Given the known caveat of phosphomimic mutations, additional studies are needed to resolve this question.
Acknowledgments
We thank Michelle Orsulak for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants HL070694 and HL080706 (to D. W.) and GM061454 (to T. K.).

This article contains supplemental Figs. S1 and S2 and Table S1.
- LPA
- lysophosphatidic acid
- HM
- hydrophobic motif
- MEF
- mouse embryonic fibroblast
- MARCKS
- myristoylated alanine-rich C kinase substrate
- CR
- conserved region.
REFERENCES
- 1. Herroeder S., Reichardt P., Sassmann A., Zimmermann B., Jaeneke D., Hoeckner J., Hollmann M. W., Fischer K. D., Vogt S., Grosse R., Hogg N., Gunzer M., Offermanns S., Wettschureck N. (2009) Guanine nucleotide-binding proteins of the G12 family shape immune functions by controlling CD4+ T cell adhesiveness and motility. Immunity 30, 708–720 [DOI] [PubMed] [Google Scholar]
- 2. Worzfeld T., Wettschureck N., Offermanns S. (2008) G(12)/G(13)-mediated signalling in mammalian physiology and disease. Trends Pharmacol. Sci. 29, 582–589 [DOI] [PubMed] [Google Scholar]
- 3. Gong H., Shen B., Flevaris P., Chow C., Lam S. C., Voyno-Yasenetskaya T. A., Kozasa T., Du X. (2010) G protein subunit Gα13 binds to integrin αIIbβ3 and mediates integrin “outside-in” signaling. Science 327, 340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moers A., Nieswandt B., Massberg S., Wettschureck N., Grüner S., Konrad I., Schulte V., Aktas B., Gratacap M. P., Simon M. I., Gawaz M., Offermanns S. (2003) G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 9, 1418–1422 [DOI] [PubMed] [Google Scholar]
- 5. Shan D., Chen L., Wang D., Tan Y. C., Gu J. L., Huang X. Y. (2006) The G protein G α(13) is required for growth factor-induced cell migration. Dev. Cell 10, 707–718 [DOI] [PubMed] [Google Scholar]
- 6. Sivaraj K. K., Takefuji M., Schmidt I., Adams R. H., Offermanns S., Wettschureck N. (2013) G13 controls angiogenesis through regulation of VEGFR-2 expression. Dev. Cell 25, 427–434 [DOI] [PubMed] [Google Scholar]
- 7. Radhika V., Hee Ha J., Jayaraman M., Tsim S. T., Dhanasekaran N. (2005) Mitogenic signaling by lysophosphatidic acid (LPA) involves Gα12. Oncogene 24, 4597–4603 [DOI] [PubMed] [Google Scholar]
- 8. Ki S. H., Choi M. J., Lee C. H., Kim S. G. (2007) Gα12 specifically regulates COX-2 induction by sphingosine 1-phosphate. Role for JNK-dependent ubiquitination and degradation of IκBα. J. Biol. Chem. 282, 1938–1947 [DOI] [PubMed] [Google Scholar]
- 9. Won H. Y., Min H. J., Lee W. H., Kim S. G., Hwang E. S. (2010) Gα12 is critical for TCR-induced IL-2 production and differentiation of T helper 2 and T helper 17 cells. Biochem. Biophys. Res. Commun. 394, 811–816 [DOI] [PubMed] [Google Scholar]
- 10. Hart M. J., Jiang X., Kozasa T., Roscoe W., Singer W. D., Gilman A. G., Sternweis P. C., Bollag G. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science 280, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 11. Kozasa T., Jiang X., Hart M. J., Sternweis P. M., Singer W. D., Gilman A. G., Bollag G., Sternweis P. C. (1998) p115 RhoGEF, a GTPase activating protein for Gα12 and Gα13. Science 280, 2109–2111 [DOI] [PubMed] [Google Scholar]
- 12. Gan X., Wang J., Wang C., Sommer E., Kozasa T., Srinivasula S., Alessi D., Offermanns S., Simon M. I., Wu D. (2012) PRR5L degradation promotes mTORC2-mediated PKC-δ phosphorylation and cell migration downstream of Gα(12). Nat. Cell Biol. 14, 686–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anliker B., Chun J. (2004) Cell surface receptors in lysophospholipid signaling. Semin. Cell Dev. Biol. 15, 457–465 [DOI] [PubMed] [Google Scholar]
- 14. Tager A. M., LaCamera P., Shea B. S., Campanella G. S., Selman M., Zhao Z., Polosukhin V., Wain J., Karimi-Shah B. A., Kim N. D., Hart W. K., Pardo A., Blackwell T. S., Xu Y., Chun J., Luster A. D. (2008) The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54 [DOI] [PubMed] [Google Scholar]
- 15. Roskoski R., Jr. (2010) RAF protein-serine/threonine kinases. Structure and regulation. Biochem. Biophys. Res. Commun. 399, 313–317 [DOI] [PubMed] [Google Scholar]
- 16. Udell C. M., Rajakulendran T., Sicheri F., Therrien M. (2011) Mechanistic principles of RAF kinase signaling. Cell. Mol. Life Sci. 68, 553–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marais R., Light Y., Paterson H. F., Mason C. S., Marshall C. J. (1997) Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J. Biol. Chem. 272, 4378–4383 [DOI] [PubMed] [Google Scholar]
- 18. Baljuls A., Mueller T., Drexler H. C., Hekman M., Rapp U. R. (2007) Unique N-region determines low basal activity and limited inducibility of A-RAF kinase. The role of N-region in the evolutionary divergence of RAF kinase function in vertebrates. J. Biol. Chem. 282, 26575–26590 [DOI] [PubMed] [Google Scholar]
- 19. Chen Z., Singer W. D., Sternweis P. C., Sprang S. R. (2005) Structure of the p115RhoGEF rgRGS domain-Gα13/i1 chimera complex suggests convergent evolution of a GTPase activator. Nat. Struct. Mol. Biol. 12, 191–197 [DOI] [PubMed] [Google Scholar]
- 20. Tesmer J. J., Sunahara R. K., Gilman A. G., Sprang S. R. (1997) Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα.GTPγS. Science 278, 1907–1916 [DOI] [PubMed] [Google Scholar]
- 21. Slep K. C., Kercher M. A., He W., Cowan C. W., Wensel T. G., Sigler P. B. (2001) Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature 409, 1071–1077 [DOI] [PubMed] [Google Scholar]
- 22. Kreutz B., Yau D. M., Nance M. R., Tanabe S., Tesmer J. J., Kozasa T. (2006) A new approach to producing functional G α subunits yields the activated and deactivated structures of G α(12/13) proteins. Biochem. 45, 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]