

Background: Apolipoproteins (apo) C-I and C-III regulate plasma triglyceride metabolism by inhibition of lipoprotein lipase (LPL) activity.
Results: ApoC-I or apoC-III prevents LPL from binding to lipid droplets. This results in inhibition of LPL.
Conclusion: Inhibition of LPL activity by apoC-I and apoC-III is due to displacement of LPL from lipid droplets.
Significance: Our proposed mechanism explains several effects of these apolipoproteins on lipoprotein metabolism.
Keywords: Apolipoproteins, Dyslipidemia, Lipolysis, Lipoprotein, Lipoprotein Metabolism, Triglyceride, Apolipoprotein C-I, Apolipoprotein C-III, Lipoprotein Lipase
Abstract
Apolipoproteins (apo) C-I and C-III are known to inhibit lipoprotein lipase (LPL) activity, but the molecular mechanisms for this remain obscure. We present evidence that either apoC-I or apoC-III, when bound to triglyceride-rich lipoproteins, prevent binding of LPL to the lipid/water interface. This results in decreased lipolytic activity of the enzyme. Site-directed mutagenesis revealed that hydrophobic amino acid residues centrally located in the apoC-III molecule are critical for attachment to lipid emulsion particles and consequently inhibition of LPL activity. Triglyceride-rich lipoproteins stabilize LPL and protect the enzyme from inactivating factors such as angiopoietin-like protein 4 (angptl4). The addition of either apoC-I or apoC-III to triglyceride-rich particles severely diminished their protective effect on LPL and rendered the enzyme more susceptible to inactivation by angptl4. These observations were seen using chylomicrons as well as the synthetic lipid emulsion Intralipid. In the presence of the LPL activator protein apoC-II, more of apoC-I or apoC-III was needed for displacement of LPL from the lipid/water interface. In conclusion, we show that apoC-I and apoC-III inhibit lipolysis by displacing LPL from lipid emulsion particles. We also propose a role for these apolipoproteins in the irreversible inactivation of LPL by factors such as angptl4.
Introduction
Lipoprotein lipase (LPL)2 has central functions in blood lipid metabolism (1, 2). The enzyme hydrolyzes triglycerides in chylomicrons and very low density lipoproteins (VLDL). The released fatty acids and monoglycerides are taken up in tissues for metabolic purposes or for storage. In addition, LPL is important for binding of lipoproteins to cell surfaces, and for receptor-mediated endocytosis of whole lipoprotein particles, by stimulating the interaction of lipoproteins with receptors (3). Loss-of-function mutations in the genes encoding LPL or its cofactor apolipoprotein C-II (apoC-II) lead to severe hypertriglyceridemia (4). In contrast, a premature stop mutation in apolipoprotein C-III (apoC-III) is associated with low levels of plasma triglycerides (5). This finding in humans is in line with studies demonstrating that apoC-III knock-out mice are hypotriglyceridemic (6), whereas overexpression of apoC-III in mice leads to hypertriglyceridemia (7–9). ApoC-III is known to inhibit LPL activity in vitro (10–12). Alaupovic and co-workers (13) concluded from studies of hypertriglyceridemic patients that apoC-III correlated positively with plasma triglyceride levels and that the inhibitory effect of human plasma on LPL activity was due to apoC-III. The general consensus is that apoC-III increases plasma triglycerides by affecting the function of LPL, but the mechanism for this inhibitory effect is not yet resolved. In addition, apoC-III has been reported to interfere with VLDL assembly and secretion (14), binding of lipoproteins to glycosaminoglycans (15), and lipoprotein remnant clearance by receptor-mediated endocytosis (16).
Apolipoprotein C-I (apoC-I) has been shown to inhibit LPL activity by studies in vitro (11, 17–19), and transgenic mice expressing human apoC-I display hypertriglyceridemia (20). Like apoC-III, apoC-I inhibits remnant clearance and lipoprotein-binding to receptors (16), whereas lipoprotein association to glycosaminoglycans was reported to be unaffected by apoC-I (21). The increased lipid levels in blood of apoC-I transgenic animals were concluded to be due to impaired uptake of VLDL by the liver rather than to an enhanced production or disturbed lipolysis of VLDL (22). This view has later been changed in favor of effects of apoC-I mainly on LPL and intravascular lipolysis (23, 24).
When compared with apolipoproteins of the C family, angiopoietin-like protein 4 (angptl4) is a more recently discovered modulator of LPL activity. Unlike the apolipoproteins, angptl4 causes irreversible inactivation of LPL by binding to the enzyme and converting active LPL dimers to inactive monomers (25). LPL is known to be stabilized by binding to detergents, emulsion particles, and lipoproteins. Recently, it was shown that inactivation of LPL by angptl4 is slower in the presence of lipoproteins (26). The expression of angptl4 is up-regulated by peroxisome proliferator-activated receptors, which in turn are activated by free fatty acids and other ligands (27, 28). Angptl4 is a strong candidate for the tissue-specific, post-translational regulation of LPL activity that is necessary for directing uptake of blood lipids according to the needs of different tissues of the body (2, 29) and for protecting tissues against deleterious effects of lipid overload (27, 28).
We have used a simple filtration technique for studies of the effects of apoC-I and apoC-III on binding of LPL to lipid emulsion particles. Our data indicate that a main factor in the inhibition of LPL by these apolipoproteins is that the enzyme is expelled from the substrate. As a consequence, LPL becomes more prone to inactivation, here demonstrated by increased sensitivity to angptl4.
EXPERIMENTAL PROCEDURES
Bovine LPL was purified from milk as described (30). 125I-labeled LPL was produced by the lactoperoxidase method and then purified by chromatography on heparin-Sepharose (31). Heparin was obtained from Leo Pharma AB. Bovine serum albumin (A-4503) was obtained from Sigma-Aldrich. Apolipoprotein A-I (10686-H07E) was obtained from Sino Biological Inc. Liposomes made from dimyristoylphosphatidylcholine (DMPC) were prepared as described (32).
Intralipid 10 and 20%, kindly donated by Fresenius Kabi (Stockholm, Sweden), was repeatedly washed to remove liposomes formed from excess phospholipids, and other potential smaller lipid particles, by floatation at 40,000 × g, 30,000 × g, and 20,000 × g, respectively, for 15 min at 15 °C. After each centrifugation, the infranatant was discarded, and buoyant lipid particles were dispersed in 50 mm Tris, pH 7.4. After the last centrifugation, the remaining lipid emulsion particles were dispersed to a final concentration of 10% triglycerides (w/v) in 50 mm Tris, pH 7.4, containing 2% glycerol (v/v). If not stated otherwise, 20% Intralipid was used as starting material. Rat intestinal lymph chylomicrons were isolated through cannulation of mesenteric lymph vessels as described (33).
ApoC-I and apoC-II were extracted from human plasma by absorption to Intralipid emulsion particles and then purified as described previously (34). The N-terminal domain of human angptl4 (amino acids 26–184) was cloned into pET29a. Human apoC-III (amino acids 21–99) was cloned into pET23b (35). Both proteins were expressed in the Escherichia coli strain BL21. Transfected bacteria were grown until A600 = 0.6, and expression was induced with 1 mm isopropyl-1-thio-β-d-galactopyranoside for 1 h for apoC-III, and for 4 h for angptl4, at 37 °C. Cultures were centrifuged at 3200 × g for 20 min at 4 °C and dissolved in 50 mm sodium phosphate buffer, pH 7.4, containing 0.3 m NaCl, 6 m guanidine:HCl, and 5 mm imidazole prior to freeze/thaw lysis at −80 °C. Angptl4 was purified using HisPur cobalt resin (Pierce) according to the manufacturer's protocol, under reducing conditions. The protein was eluted by 20 mm acetic acid, pH 4.0, and stored at −80 °C. ApoC-III was purified on a HisTrapTM FF crude column (GE Healthcare) using a gradient of increasing concentrations of imidazole (10 mm to 500 mm) in 50 mm sodium phosphate buffer, pH 7.4, containing 0.3 m NaCl and 6 m urea. Fractions eluting late in the gradient were dialyzed against 10 mm NH4HCO3 and later frozen at −20 °C prior to lyophilization. The dried product was dissolved in 10 mm Tris, pH 8.5, containing 5 m urea. Murine N-terminal angptl4 was expressed in HEK293 cells as described previously (25). Protein concentrations were determined by the bicinchoninic acid assay (Pierce).
LPL Activity Measurements
LPL activity was measured using emulsion particles isolated from Intralipid corresponding to 1–4 mg of triglyceride/ml in 0.15 m Tris, pH 8.5, containing 0.1 m NaCl, 6% BSA (w/v), and 16.7 units of heparin/ml. Human apoC-II was used in some experiments at the concentrations given in the figure legends. Incubations were made at room temperature for 30–45 min in 96-well microtiter plates (Nunc, catalog number 236108) containing a total volume of 150 μl/well. The plates were gently agitated on an orbital shaker (600 rpm). Different amounts of apolipoproteins were added from stock solutions containing 5 m urea. By dilutions in the same buffer, care was taken so that all wells contained the same volume of vehicle. Lipid hydrolysis was stopped by the addition of 50 μl of 10% Triton X-100 (v/v). Aliquots (3.5–10 μl) were transferred to another 96-well microtiter plate (Greiner Bio-One, catalog number 655101) containing 150 μl of NEFA-HR(2) R1 (Wako Chemicals) for determination of the amounts of fatty acids released (where NEFA indicates “non-esterified fatty acids”). The mixtures were incubated for 15 min at room temperature by gentle agitation on the orbital shaker (600 rpm). Then 75 μl of NEFA-HR(2) R2 (Wako Chemicals) was added, and the incubation was continued for an additional 10 min. Fatty acids were quantified by absorbance measurements on a SpectraMax 340 (Molecular Devices) using NEFA C standard (Wako Chemicals) as reference. LPL activity is expressed in units where 1 unit corresponds to the release of 1 μmol of fatty acid/min.
Preincubation Experiments
LPL was preincubated with emulsion particles isolated from Intralipid, corresponding to 2 mg of triglyceride/ml in 0.15 m Tris, pH 7.4, containing 0.1 m NaCl and 6% BSA (w/v). Preincubations were made in 96-well microtiter plates (Nunc catalog number 236108) at room temperature for 15 min on an orbital shaker (600 rpm) in the presence of different combinations of apoCs and angptl4 (N-terminal domain) in a final incubation volume of 150 μl. Aliquots of 5–10 μl were then transferred to incubation mixtures containing [9,10-3H]triolein-labeled Intralipid (courtesy of Pharmacia-Upjohn and Fresenius-Kabi) corresponding to 2 mg of triglyceride/ml in 0.15 m Tris, pH 8.5, containing 0.1 m NaCl, 6% BSA (w/v), 16.7 units of heparin/ml, and 5% (v/v) heat-inactivated rat serum as source of apoC-II. The contribution of lipid substrate from the first incubation did not influence activity measurements in the second incubation, which was carried out in a final volume of 200 μl at 25 °C for 30–60 min on an orbital shaker. Released fatty acids were extracted and quantified as described previously (36).
Binding Studies by Filtration
Trace amounts of 125I-labeled LPL were mixed with unlabeled LPL and added to the same incubation system (total volume 150 μl) as used for LPL activity measurements, but with washed, unlabeled Intralipid (see above). After 2 min at room temperature, 1 ml of ice-cold 0.15 m Tris, pH 8.5, was added, and the mixture was filtered through a 0.22-μm syringe filter (PALL PN4602). This filter prevented passage of large lipid emulsion particles. Radioactivity from 125I-labeled LPL was determined in the filtrate (flow-through) and was considered to represent unbound LPL. Error bars for these experiments were calculated using Gaussian approximation of mean error using Equation 1,
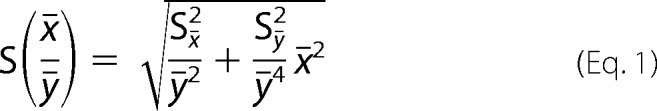 |
where x̄ and ȳ are mean values of the observed measurements. Sx̄ and Sȳ are mean errors for the observed measurements, respectively.
Folding Studies by CD Measurements
To assess interaction of apoC-III with lipids, solutions of apoC-III (0.2 mg/ml of wild type and mutants) in 20 mm potassium phosphate buffer, pH 7.4, were titrated with successive additions of liposomes made from DMPC. Circular dichroism spectra were recorded on a Jasco CD spectrophotometer using a 0.1-cm path length quartz cell at 20 °C. Spectra were obtained by averaging three scans in the wavelength range 190–260 nm. The signal obtained from buffer only was subtracted from the spectra. Estimation of protein secondary structure was made in CDPro (37) using the CDSSTR algorithm and SDP48 as reference set. Raw data in millidegrees were converted to per residue molar absorption units (Δϵ) using Equation 2,
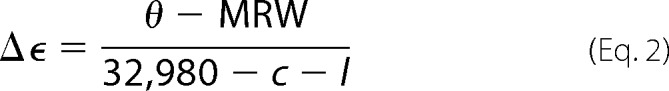 |
where θ is the observed raw measurement in millidegrees, MRW is the mean residual weight of the protein (molecular weight/total number of amino acid residues), c is the protein concentration in mg/ml, and l is the path length in cm.
Multiple Sequence Alignment and Mutagenesis of ApoC-III
The following sequences were obtained from the National Center for Biotechnology Information (NCBI): Homo sapiens, NP_000031.1; Mus musculus, NP_075603.1; Cavia porcellus, NP_001166386.1; Bos taurus, NP_001001175.1; Sus scrofa, NP_001002801.1; Canis lupus familiaris, NP_001003369.1; and Rattus norvegicus, NP_001257982.1. Alignments were made using ClustalW2 (38). Mutagenesis was made using QuikChange II (Stratagene) according to the supplier's instructions followed by DNA sequencing.
Heparin-Sepharose Chromatography
For separation of active LPL dimers from inactive, presumably monomeric, forms of LPL, we used affinity chromatography on heparin-Sepharose (25, 31, 39). Approximately 6 nm 125I-labeled LPL was added to 0.15 m Tris, pH 7.4, containing 0.1 m NaCl and 6% BSA (w/v) to a final volume of 300 μl. The samples were incubated in Eppendorf tubes at room temperature for 15 min on an orbital shaker (600 rpm), if not stated otherwise. Incubations were made in the absence and presence of 2 mg of triglyceride/ml from emulsion particles isolated from Intralipid containing either no apolipoproteins, 2 μm apoC-I, or 2 μm apoC-III. In addition, 73 nm angptl4 (N-terminal domain) was included for some experiments. After incubation, 900 μl of ice-cold 20 mm Tris, pH 7.4, containing 20% glycerol (v/v) was added, and mixtures were then filtered (PALL PN4602). A sample (500 μl) of the filtrate was loaded on a 1-ml HiTrap column (GE Healthcare) using the ÄKTA purifier system (GE Healthcare). A linear gradient from 0 to 2 m NaCl was applied (in 20 mm Tris, pH 7.4, 20% glycerol) to elute the bound LPL protein. Radioactivity of each fraction was determined using a Wallac Wizard 1480 (PerkinElmer).
RESULTS
This study was primarily designed to investigate the mechanism by which apoC-III inhibits LPL activity. The inhibition was found to be explained by the prevention of LPL from binding the lipid/water interface of lipid emulsion particles. ApoC-I was later included to investigate whether it affected LPL activity by a similar mechanism as apoC-III. For most of the studies, we used a commercial egg yolk phospholipid-stabilized emulsion of soy bean triglycerides (Intralipid) as substrate for LPL. This lipid emulsion is made to resemble triglyceride-rich lipoproteins and is used for parenteral nutrition of patients. The results with Intralipid were confirmed using rat lymph chylomicrons for some experiments. Crucial residues for lipid interaction were indentified in apoC-III by substitution of hydrophobic amino acid residues in amphipathic α-helical parts of apoC-III by alanines. Finally, the protective effect of triglyceride-rich emulsion particles on inactivation of LPL by angptl4 was investigated in the absence and presence of apoC-I and apoC-III.
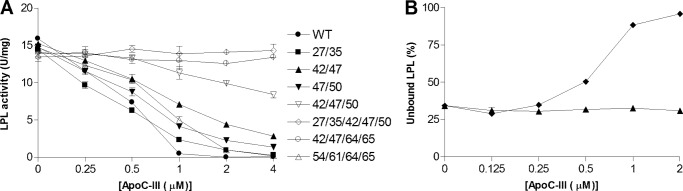
To investigate the mechanism for the inhibition of LPL activity by apoC-III, a filtration method was developed for rapid separation of the lipid emulsion particles from the rest of the incubation mixture. Using 125I-labeled LPL, we could demonstrate that apoC-III displaced LPL from the lipid emulsion particles in a dose-dependent manner (Fig. 1A). At a higher concentration of lipid emulsion, more apoC-III was required to observe the same displacement of LPL. A similar relationship was obtained for the inhibitory action of apoC-III on LPL activity, which also depended on the amount of lipid emulsion (Fig. 1B).
FIGURE 1.

ApoC-I and apoC-III prevent LPL from binding to lipid emulsion particles and inhibit triglyceride hydrolysis. A, unlabeled LPL (24 nm) and trace amounts of 125I-labeled LPL were added to emulsion particles isolated from Intralipid corresponding to 1 mg (●), 2 mg (▴) or 4 mg (♦) of triglyceride/ml with different amounts of either apoC-III or apoC-I. C, identical experiments were carried out using 2 mg of triglyceride/ml for apoC-I and apoA-I. E, the experiment with apoA-I was conducted using a different batch of isolated Intralipid particles. Therefore apoC-III was also used as a control in this experiment (dotted line). Solutions were filtered, and the amount of unbound LPL was determined in the filtrate as a percentage of LPL without Intralipid. B, D, and F, using identical conditions as in panels A, C, and E (but without 125I-labeled LPL), the solutions were incubated for 30 min at room temperature, and free fatty acids were then quantified to determine LPL activity. Data points represent mean values of duplicate samples ± Gaussian approximation of mean error (A, C, and E) or ± S.D. (B, D, and F).
We next questioned whether the inhibition of binding and activity of LPL was a feature unique for apoC-III by comparison with the effects of similar amounts of apoC-I and apoA-I. ApoC-I mimicked the effects of apoC-III by preventing LPL from binding to the large lipid emulsion particles and by inhibiting LPL activity (Fig. 1, C and D). ApoA-I had no effect on LPL displacement, nor did it inhibit LPL activity (Fig. 1, E and F).
Next we investigated whether our observations regarding binding of LPL to Intralipid were applicable to a natural substrate such as rat lymph chylomicrons. As in experiments with Intralipid, the addition of apoC-I or apoC-III resulted in reduced binding of LPL to the lipid particles (Fig. 2A) and inhibition of LPL activity (Fig. 2B). The amounts of apoC-I or apoC-III needed to displace LPL from the chylomicrons were considerably higher than those with Intralipid. This could be due to the presence of the LPL co-factor apoC-II on the chylomicrons (40), which is present at quantities sufficient for maximal LPL activation (41). The inhibition of LPL activity by apoC-III was previously shown to be related to the amount of bound apolipoprotein C-II on the lipid substrate (42). As expected, when Intralipid particles contained apoC-II, more apoC-I or apoC-III was required to displace LPL from the emulsion particles and to inhibit LPL activity (Fig. 2, C and D) when compared with emulsion particles without apoC-II. Analyses of the distribution of apoC-III in the incubation system by ELISA indicated that the presence of apoC-II did not prevent apoC-III from binding to the lipid particles. Approximately 10% of apoC-III was found in the filtrate both in the absence and in the presence of apoC-II (data not shown).
FIGURE 2.

Addition of human apoC-I or apoC-III to rat lymph chylomicrons prevents LPL binding and inhibits enzyme activity, but higher concentrations of the apolipoproteins were required than with emulsion particles isolated from Intralipid. A and C, unlabeled LPL (6 nm) and trace amounts of 125I-LPL were added to lipid particles corresponding to 2 mg of triglyceride/ml containing different amounts of either apoC-I (○) or apoC-III (▾). The solutions were then filtered to assess LPL binding to the lipid particles. The lipid particles were either rat lymph chylomicrons (A) or isolated lipid particles from Intralipid containing 0.5 μm apoC-II (C). B and D, using identical conditions as in panels A and C (but without 125I-labeled LPL), the solutions were incubated for 60 min at room temperature. Free fatty acids were then quantified to determine LPL activity for rat lymph chylomicrons (B) or Intralipid (D). Data points represent mean values of duplicate samples ± Gaussian approximation of mean error (A and C) or S.D. (B and D).
Our finding that the inhibitory action of apoC-III on LPL activity correlated negatively with the amount of lipid emulsion suggested that apoC-III competes with LPL for binding to the lipid emulsion particles, rather than acting directly on LPL. Starting from the amphipathic structure of apoC-III previously determined by solution NMR (43), we used site-directed mutagenesis to identify key residues for binding of apoC-III to the emulsified lipid. These sites were identified by comparing all available apoC-III sequences, and selection was made on basis of which hydrophobic residues were largely conserved across animal species (Fig. 3). The apoC-III variants with alanine replacements were first analyzed with regard to their ability to inhibit LPL activity. The double mutants L27A/V35A, W42A/F47A, and F47A/L50A all exhibited reduced ability to inhibit LPL activity when compared with wild-type apoC-III, with W42A/F47A being the least inhibitory (Fig. 4A). When this variant was further mutated to W42A/F47A/L50A, the inhibitory potential was greatly diminished. Replacement of two additional hydrophobic residues upstream toward the N terminus, generating the five-point mutant L27A/V35A/W42A/F47A/L50A, resulted in an apoC-III variant without any inhibitory effect on LPL activity. In contrast, replacements of four hydrophobic residues in the C-terminal half of apoC-III, W54A/F61A/F64A/W65A, resulted in a variant with moderately reduced effects on the inhibition of LPL activity when compared with wild-type apoC-III. Interestingly, the mutant W42A/F47A/F64A/W65A had lost all inhibitory effect on LPL activity. Binding studies with 125I-labeled LPL demonstrated that the most inactive mutant with regard to inhibition of LPL activity, L27A/V35A/W42A/F47A/L50A, lacked the ability to displace LPL from the emulsion particles (Fig. 4B).
FIGURE 3.

Multiple sequence alignment of apoC-III. The indicated positions in the (mature) human amino acid sequence of apoC-III were substituted by alanine residues in different combinations.
FIGURE 4.
Substitutions of hydrophobic residues in apoC-III with alanine resulted in variants with reduced ability to inhibit LPL activity due to inability of displacing the enzyme from lipid emulsion particles. A, LPL (24 nm) was added to emulsion particles isolated from Intralipid containing different amounts of mutated variants of apoC-III at 2 mg of triglyceride/ml. Incubations were carried out for 45 min at room temperature. Free fatty acids were then quantified to determine LPL activity. B, the ability to displace LPL from lipid emulsion particles was compared for wild-type apoC-III (♦) and the least inhibitory mutant apoC-III L27A/V35A/W42A/F47A/L50A (▴) using identical conditions as in panel A, but with the addition of a trace amount of 125I-LPL. Data points represent mean values of duplicate samples ± S.D. (A) or ± Gaussian approximation of mean error (B).
Interaction with lipids readily induces α-helical structure in apoC-III (44). Comparison of CD spectra from wild-type apoC-III with those of three of our mutated apoC-III variants revealed differences in secondary structure. As expected, the α-helical content increased in wild-type apoC-III with the addition of increasing amounts of liposomes made from DMPC (Fig. 5A). This response was also seen, but was less pronounced, with two of the double-point mutants where the CD spectra of F47A/L50A were more similar to those of wild-type apoC-III than those of W42A/F47A (Fig. 5, B and C). In comparison, mutant L27A/V35A/W42A/F47A/L50A displayed mainly unordered structure also in the presence of liposomes, indicating that this variant was unable to bind to lipid.
FIGURE 5.

Lack of induced secondary structure upon addition of liposomes parallels the inability of apoC-III variants to inhibit LPL activity against emulsified triglycerides. A–D, CD analysis of apoC-III variants in the absence of lipid (○) or with successive additions of liposomes made from DMPC to a final concentration of 0.25 mg (♦), 0.5 mg (▵), 0.75 mg (●), or 1 mg (▿) of DMPC/ml. ApoC-III variants were wild type (A), W42A/F47A (B), F47A/L50A (C), and L27A/V35A/W42A/F47A/L50A (D). Insets represent predicted α-helical content estimated by CDPro/CDSSTR. Each spectrum represents an average of three scans.
Previous studies had demonstrated that LPL is stabilized by interaction with detergents, with lipid emulsions, or with lipoproteins (45) and that the putative LPL-controlling protein angptl4 has reduced ability to inactivate LPL in the presence of triglyceride-rich lipoproteins (26). Given that apoC-I and apoC-III prevent LPL from binding to lipid emulsions and lipoproteins, we hypothesized that these apolipoproteins would make LPL more susceptible to inactivation by angptl4. Control experiments with incubation of LPL with emulsion particles isolated from Intralipid showed that neither apoC-I nor apoC-III caused accelerated LPL inactivation on their own (Fig. 6, A and C). As expected, angptl4 inactivated LPL in the absence of Intralipid but had limited effect when the enzyme was incubated in the presence of lipid emulsion particles (Fig. 6A). However, when apoC-I or apoC-III was added, inactivation of LPL was promoted (Fig. 6B). This effect was also confirmed with the N-terminal domain of mouse angptl4 expressed in HEK239 cells (data not shown). In the presence of apoC-II, more apoC-I or apoC-III (about 10-fold more) had to be added to expose LPL to inactivation by angptl4 (Fig. 6, C and D). The presence of chylomicrons gave similar results as those obtained in the presence of emulsion particles isolated from Intralipid with added apoC-II (Fig. 6, E–G).
FIGURE 6.

Triglyceride-rich lipid particles protect LPL from inactivation by angptl4, but addition of either apoC-I or apoC-III to the lipid particles renders LPL susceptible to inactivation. A, the first incubation (preincubation) contained emulsion particles isolated from Intralipid corresponding to 2 mg of triglyceride/ml, 73 nm angptl4, and the indicated amounts of apoC-I or apoC-III in different combinations. Solutions were preincubated with 24 nm LPL for 15 min at room temperature before the remaining LPL activity was measured by transfer of 5-μl aliquots to an assay system with radiolabeled Intralipid. B, using identical conditions as in panel A, the effects of LPL inactivation by increasing amounts of either apoC-I (▾) or apoC-III (♦) in combination with angptl4 were investigated. Experiments were repeated in the presence of 0.5 μm apoC-II during the preincubation. C and D, in this case, only 6 nm LPL was added, and aliquots of 10 μl were transferred to the assay system with radiolabeled Intralipid. E–G, next, experiments were made with rat lymph chylomicrons (Chylo, corresponding to 2 mg of triglyceride/ml) instead of isolated Intralipid particles. The amount of LPL was 24 nm, and the amount of angptl4 was 73 nm. Aliquots of 5 μl of the preincubated solutions were transferred to the assay with radiolabeled Intralipid for determination of remaining LPL activity. With these conditions, we examined the ability of chylomicrons to protect LPL from angptl4-dependent inactivation in the absence of exogenous apolipoproteins (E) or in the presence of apoC-I (F) or apoC-III (G). Data points represent mean values of duplicate samples ± S.D.
To investigate how apoC-I or apoC-III, either alone or in combination with angptl4, affected the activity status of LPL, we used heparin-Sepharose chromatography. This technique was previously shown to separate active LPL dimers with high affinity for heparin from inactive, monomeric forms of LPL with lower heparin affinity (25, 31, 39). 125I-labeled LPL was preincubated with or without angptl4, with or without apolipoproteins, and was thereafter applied on a heparin-Sepharose column and then eluted by a linear gradient of NaCl. Without preincubation, LPL eluted at about 1 m NaCl, as expected for active LPL dimers (39). Preincubation of LPL at 42 °C for 15 min resulted in a peak eluting already at 0.5 m NaCl, characteristic of LPL monomers (39). Preincubation of LPL with angptl4 resulted in a peak eluting at 0.5 m NaCl. Some radioactivity eluted at 1 m NaCl, presumably corresponding to remaining active LPL dimers. Forms of LPL eluting in between 0.5 and 1 m NaCl were also seen (Fig. 7A). In the corresponding experiment with preincubation of LPL alone for 15 min at room temperature, some radioactivity eluted already at 0.5 m NaCl. This demonstrated that LPL was not perfectly stable on its own, as expected, but that some degree of spontaneous inactivation occurred. The presence of lipid droplets from Intralipid containing either apoC-I or apoC-III during the preincubation resulted in almost identical elution patterns as when no Intralipid particles were present, supporting our findings that LPL was unable to bind to the emulsion particles under these conditions. As expected, the addition of angptl4 to the preincubation solution resulted in a substantial loss of LPL eluting at 1 m NaCl, corresponding to active dimers, and more LPL eluting at 0.5 m NaCl, as inactive monomers (Fig. 7, B and C).
FIGURE 7.
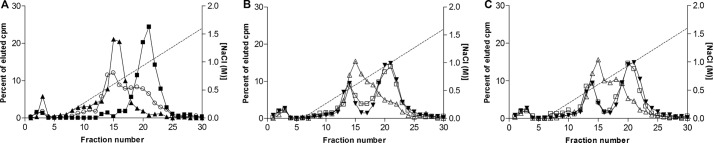
Angptl4 promotes conversion of LPL to forms with lower heparin affinity, and this is promoted in the presence of apolipoprotein-containing emulsion particles. A, 125I-labeled LPL (∼6 nm) was subjected to different conditions and was then applied to a heparin-Sepharose column and eluted by a gradient of NaCl to separate different molecular forms of the enzyme. LPL was applied directly on the column without any pretreatment (■), preincubated for 15 min at 42 °C (▴), or preincubated at room temperature for 15 min with 73 nm angptl4 (○). B, LPL was preincubated for 15 min in room temperature in the absence (▾) or presence of emulsion particles isolated from Intralipid with 2 μm apoC-I (□) or with 2 μm apoC-I and 73 nm angptl4 (▵). C, same as panel B but with apoC-III instead of apoC-I.
DISCUSSION
By a simple filtration technique, we were able to rapidly separate unbound LPL from an incubation system containing either large lipid emulsion particles isolated from Intralipid or chylomicrons and demonstrate that apoC-I and apoC-III prevented binding of LPL to both types of lipid particles. This correlated well with the observed reduction in LPL activity seen on the addition of apoC-I or apoC-III to these incubation systems. Higher concentrations of apoC-III were required for inhibition of activity and displacement of LPL when more lipid emulsion was used or when apoC-II was present. Similar results were obtained with apoC-I. To calculate the potential surface occupancy of the added apoCs, we used data from Wang et al. (46), who designed an apolipoprotein-derived peptide to examine the surface area that an amphipathic α-helical structure may occupy on a lipid/water interface. We assumed that each amino acid residue in apoC-III occupies a similar area as calculated for the peptide and that our isolated Intralipid particles range between 200 and 400 nm. By this, we could estimate that only 10–20% of the Intralipid particles were covered when 1 μm apoC-III was added to emulsion particles corresponding to 2 mg of triglyceride/ml. From these calculations, it seems unlikely that the apolipoproteins expel LPL from the lipid/water interface simply due to steric effects. Our findings rather suggest that apoC-I and apoC-III modify the surface properties of the lipid particles such that LPL is unable to bind to them. Alternatively, the apolipoproteins compete with LPL for binding to specific structures on the lipid particles. Interestingly, apoA-I did not decrease binding of LPL to Intralipid or cause inhibition of LPL activity, although this apolipoprotein was previously demonstrated to bind avidly to lipid particles isolated from Intralipid by floatation (47).
The filtration method has great advantages over other methods used to isolate lipid emulsion particles in that the filtration by syringe filters is simple and rapid so that separation can be obtained within seconds. There are minimal pressure effects on the system when compared with when centrifugation is used at high g forces (48). We found that the nonspecific binding of LPL to the filters varied with different types of syringe filters, but was efficiently reduced by the high concentration of BSA that is normally used as fatty acid acceptor during measurements of LPL activity. With the system we used, 93.8 ± 6.1% of the LPL protein was recovered after filtration in the absence of lipid emulsion particles. A possible drawback of the filter technique is that the lipid binding ability of LPL can be underestimated because some emulsion particles are small enough to pass through the filters. We observed differences in how much 125I-labeled LPL passed through the filter depending on the batch of lipid emulsion particles used. Intralipid differs in mean diameter size depending on the triglyceride/phospholipid ratio of the emulsion (49). We isolated the largest emulsion particles from Intralipid by floatation for our experiments, but still noted some variation between experiments. The presence of smaller sized particles can probably explain why as much as 60% of the enzyme passed through the filter when rat lymph chylomicrons were used because the chylomicrons were not washed by floatation.
Others have reported that apoC-III is able to remove phospholipids from monolayers (50). We found that the majority of the added apoC-III did not pass the syringe filters. It is therefore unlikely that apoC-III sequestered phospholipids from the emulsion particles to form smaller lipid particles that attracted LPL and carried the enzyme through the filter leading to an incorrect interpretation of our results.
The surface behavior of several apolipoproteins and their interaction with phospholipids spread at an air/water interface have been thoroughly investigated (51–53). It was demonstrated that apolipoproteins can penetrate a phospholipid monolayer, resulting in increased surface pressure as the interface becomes more crowded (46). If the surface pressure becomes too high, bound proteins are expelled from the interface. This particular pressure is called the exclusion pressure. Studies with surfaces more similar to lipoproteins such as the phospholipid/triolein/water interface showed that apoC-I remains bound even at high surface pressure, and the affinity of apoC-I for the lipid/water interface increases when the proportion of phospholipid over triolein is increased (54). It was suggested that the plasticity of the interaction of apoC-I with lipid could account for the finding that other more complex apolipoproteins such as apoA-I and apoE are displaced under conditions when apoC-I remains at the interface (54). Studies with LPL using lipid monolayers containing both phospholipid and triglycerides demonstrated decreased enzyme activity with increased surface pressure (55, 56). One could speculate that the observed decrease in enzymatic activity coincides with the exclusion pressure of LPL. Furthermore, the lipid binding properties of apoC-I, and possibly those of apoC-III, may resemble those of LPL. This would make these apolipoproteins specific in terms of their ability to prevent LPL from binding the lipid/water interface. In the presence of apoC-II, more of apoC-I or apoC-III was needed to obtain the same degree of displacement of LPL from the lipid particles and inhibition of LPL activity when compared with when apoC-II was absent. Similar data were previously reported from studies in other systems (40). Jackson et al. (42) showed that apoC-III displaced apoC-II from triglyceride-rich emulsion particles with a concomitant loss of LPL activity. Approximately 10 μm apoC-III was required to displace LPL and inhibit LPL activity in the presence of apoC-II at a concentration of emulsion particles from Intralipid corresponding to 2 mg of triglyceride/ml. We estimate that under these conditions, apoC-III could cover most of the lipid particles. The interaction between LPL and apoC-II is likely to create a higher affinity of the LPL-apoC-II complex for the lipid/water interface when compared with that for any of the individual proteins. This may explain the need for increased amounts of apoC-I or apoC-III to displace LPL in the presence of apoC-II. The exact mechanism by which apoC-II activates LPL is not fully understood. There are indications for a direct interaction between LPL and apoC-II (32), although it has not been possible to demonstrate the true mode of interaction between these proteins. We demonstrate here that the addition of exogenous apoC-I or apoC-III to rat chylomicrons displaced LPL and inhibited LPL activity. When compared with our model system, there were distinct differences that can in part be explained by the presence of apoC-II. However, other apolipoproteins on the chylomicrons could also affect LPL residency. Previous studies in vitro with chylomicrons from an apoC-II-deficient patient demonstrated that exogenous apoC-II was required for initiating LPL activity with this substrate in contrast to results with Intralipid (57). Hence, for the rare patients with missense mutations in the gene for apoC-II, a reduction in expression of apolipoproteins that prevents LPL binding could be helpful to control their hyperchylomicronemia.
Plasma concentrations of apoC-III in healthy individuals are ∼10 mg/dl (∼11 μm) of which close to 20% resides on triglyceride-rich lipoproteins (TRLs) (58). Hypertriglyceridemic subjects showed a 3.5-fold increase of apoC-III, whereas apoB levels were only increased 1.5-fold. Interestingly, these individuals also displayed an increased ratio of apoC-III per TRL/HDL, with ∼50% of apoC-III residing on TRLs (58). A similar observation was seen for apoC-I (59). The concentrations of apoC-I and apoC-III used in our model system thus fall within the physiological range.
Previous studies had shown that cleavage of apoC-III by thrombin generates two fragments (residues 1–40 and 41–78) (60, 61). No secondary structure was induced in the N-terminal fragment when added to DMPC liposomes. In contrast, the α-helical content was increased in the C-terminal fragment, indicating lipid binding (60). In that study, only the C-terminal fragment was capable of inhibiting LPL activity, but the fragment was somewhat less potent when compared with the full-length protein. Others had shown that the N-terminal fragment had some, yet limited, ability to inhibit LPL activity (61). Our results are in line with these findings. The C-terminal half of apoC-III appeared most important in terms of inhibition of LPL activity. Alanine substitutions of residues Leu-27 and Val-35 did not affect the inhibition of LPL activity by apoC-III, whereas substitutions of hydrophobic residues located more centrally in the apoC-III molecule (residues 42–50) resulted in marked reductions in the inhibitory potential. Previous studies reported that alanine substitutions of Phe-64 and Trp-65 decreased lipid binding of apoC-III, whereas they seemed to increase the inhibition of LPL activity (35). This was interpreted as proof that the main effect of apoC-III on LPL activity is due to a direct interaction with the enzyme. In our systems, we had limited effects when substituting residues in the C-terminal part of apoC-III. The mutant W54A/F61A/F64A/W65A was modestly less inhibitory than wild-type apoC-III. However, substitutions of the centrally located hydrophobic residues (Trp-42/Phe-47/Leu-50), in combination with either L27A and V35A or F64A and W65A, led to apoC-III variants where the potential for inhibition of LPL was abrogated. For apoC-III variants examined with measurements of CD, the inhibitory potential on LPL activity paralleled their ability to increase their content of α-helical structure in the presence of DMPC liposomes. Because apoC-III forms α-helices on binding to phospholipids (60), we assume that our apoC-III variants are limited in their lipid binding capability. However, we cannot rule out that the apoC-III variants were still residing on the lipid/water interface but without affecting LPL. Hydrophobic residues across the entire apoC-III protein are likely to be involved in the lipid binding (43), but those that are centrally located in the molecule seem to be the most important. It is possible that they are involved in the initial anchoring of apoC-III to the lipid/water interface, which in turn induces the necessary conformational changes to promote multisite interactions involving several of the six amphipathic α-helices (43). A similar mechanism can be anticipated for inhibition of LPL by apoC-I, but the molecular basis for this was not studied in detail.
Recently, Sundaram et al. (14) proposed that apoC-III plays a crucial role in VLDL assembly by incorporation of bulk triglycerides into precursor lipoproteins independently of microsomal transfer protein. In two following studies, the same group showed that two naturally occurring human mutations of apoC-III, A23T and K58E, were unable to promote triglyceride incorporation during VLDL assembly (73, 74). Plasma concentrations of triglycerides and apoC-III are lower in carriers of the A23T or K58E mutations (62, 63). Interestingly, recombinant A23T was reported to be as effective as wild-type apoC-III in inhibiting LPL activity (63). It remains to be examined whether K58E will affect the inhibitory potential on LPL activity. We have made single-point substitutions of other conserved charged residues (K21A, D25N, R40A, K51A, and D66N), and all of these mutants were as efficient as wild-type apoC-III in inhibiting LPL activity (data not shown). If A23T or K58E has an effect on intravascular lipolysis, we propose that this is due to decreased ratios between apoC-III/apoB on triglyceride-rich lipoproteins.
Angptl4 is a powerful player in controlling lipid metabolism (64). One of the direct effects of angptl4 is that it inactivates LPL by dissociating active LPL dimers to inactive monomers (25). This effect resides in the N-terminal domain of angptl4, which binds to LPL. We used the recombinant N-terminal domain of human angptl4 to study the effects of apoC-I and apoC-III on the inactivation of LPL in the presence of Intralipid or rat chylomicrons. We hypothesized that if inactivation of LPL by angptl4 occurs in the plasma compartment, the presence of elevated concentrations of apolipoproteins on the surface of lipoproteins should decrease binding of LPL to them and make the enzyme more prone to be inactivated by angptl4. This was found to be the case in vitro. It is not yet known where angptl4 acts on LPL in vivo, whether this occurs on the endothelium in contact with blood, or whether angptl4 acts on LPL along its transport route to the luminal side of the endothelium. Transgenic miniature pigs that overexpress human apoC-III have similar levels of LPL activity that can be released to blood by heparin when compared with their wild-type littermates, indicating that the luminal LPL activity is not affected (8). Effects on in the heparin-releasable LPL activity of one tissue could, however, be masked by release of LPL from other tissues as seen in heart-specific overexpression of angptl4 (65) and in mice deficient in LPL in adipose tissue (66). There are only few studies addressing the specific activity of LPL in plasma, i.e. activity/LPL mass. Interestingly, the levels of apoC-I were reported to correlate with low specific LPL activity (59). This could possibly be related to the enhanced action of angptl4 on LPL in the presence of higher concentrations of apoC-I (67). Angptl4 expression is up-regulated by fasting (68), and that is the condition known to decrease LPL activity in adipose tissue by post-translational mechanisms (39). One could therefore anticipate that LPL activity is suppressed by increased levels of apoC-I or apoC-III primarily in adipose tissue.
In summary, we suggest that apoC-I and apoC-III inhibit LPL activity by a similar mechanism. Both apolipoproteins can dose-dependently displace LPL from the lipid/water interface of lipoproteins and lipid emulsion particles. This results in decreased enzymatic activity, but not necessarily inactivation of the enzyme. However, the presence of increased levels of apoC-I or apoC-III may promote inactivation of LPL by factors such as angptl4, as demonstrated in vitro. Further studies are needed to corroborate this observation in vivo. Interestingly, patients with renal insufficiency have high plasma levels of both apoC-III and angptl4 (69, 70). These patients are known to have low levels of LPL activity in post-heparin plasma, elevated levels of plasma triglycerides, and increased risk for cardiovascular complications (71, 72). Our present results can possibly contribute to an explanation for the hypertriglyceridemia associated with kidney disease.
Acknowledgments
We are grateful for technical assistance, materials, and theoretical discussions from the following individuals: Dr. Marcus Wallgren, Umeå University (circular dichroism); Dr. Peter Anton, Umeå University (statistics); Dr. Valentina Sukonina, previously at Umeå University, now at University of Gothenburg (mouse Angptl4); and Dr. Stefan Nilsson (chylomicrons) and technician Solveig Nilsson (general laboratory work), both at Umeå University.
This work was supported by Swedish Science Council Grant 12203, The Swedish Heart and Lung Foundation, and British Heart Foundation Grant RG08/014.
- LPL
- lipoprotein lipase
- apo
- apolipoprotein
- angptl4
- angiopoietin-like protein 4
- DMPC
- dimyristoylphosphatidylcholine
- TRL
- triglyceride-rich lipoprotein.
REFERENCES
- 1. Wang H., Eckel R. H. (2009) Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab. 297, E271–E288 [DOI] [PubMed] [Google Scholar]
- 2. Olivecrona G., Olivecrona T. (2010) Triglyceride lipases and atherosclerosis. Curr. Opin. Lipidol. 21, 409–415 [DOI] [PubMed] [Google Scholar]
- 3. Beisiegel U., Weber W., Bengtsson-Olivecrona G. (1991) Lipoprotein lipase enhances the binding of chylomicrons to low density lipoprotein receptor-related protein. Proc. Natl. Acad. Sci. U.S.A. 88, 8342–8346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fojo S. S., Brewer H. B. (1992) Hypertriglyceridaemia due to genetic defects in lipoprotein lipase and apolipoprotein C-II. J. Intern. Med. 231, 669–677 [DOI] [PubMed] [Google Scholar]
- 5. Pollin T. I., Damcott C. M., Shen H., Ott S. H., Shelton J., Horenstein R. B., Post W., McLenithan J. C., Bielak L. F., Peyser P. A., Mitchell B. D., Miller M., O'Connell J. R., Shuldiner A. R. (2008) A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 322, 1702–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maeda N., Li H., Lee D., Oliver P., Quarfordt S. H., Osada J. (1994) Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 269, 23610–23616 [PubMed] [Google Scholar]
- 7. Ito Y., Azrolan N., O'Connell A., Walsh A., Breslow J. L. (1990) Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science 249, 790–793 [DOI] [PubMed] [Google Scholar]
- 8. Wei J., Ouyang H., Wang Y., Pang D., Cong N. X., Wang T., Leng B., Li D., Li X., Wu R., Ding Y., Gao F., Deng Y., Liu B., Li Z., Lai L., Feng H., Liu G., Deng X. (2012) Characterization of a hypertriglyceridemic transgenic miniature pig model expressing human apolipoprotein CIII. FEBS J. 279, 91–99 [DOI] [PubMed] [Google Scholar]
- 9. Ding Y., Wang Y., Zhu H., Fan J., Yu L., Liu G., Liu E. (2011) Hypertriglyceridemia and delayed clearance of fat load in transgenic rabbits expressing human apolipoprotein CIII. Transgenic Res. 20, 867–875 [DOI] [PubMed] [Google Scholar]
- 10. Brown W. V., Baginsky M. L. (1972) Inhibition of lipoprotein lipase by an apoprotein of human very low density lipoprotein. Biochem. Biophys. Res. Commun. 46, 375–382 [DOI] [PubMed] [Google Scholar]
- 11. Havel R. J., Kane J. P., Kashyap M. L. (1973) Interchange of apolipoproteins between chylomicrons and high density lipoproteins during alimentary lipemia in man. J. Clin. Invest. 52, 32–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krauss R. M., Herbert P. N., Levy R. I., Fredrickson D. S. (1973) Further observations on the activation and inhibition of lipoprotein lipase by apolipoproteins. Circ. Res. 33, 403–411 [DOI] [PubMed] [Google Scholar]
- 13. Wang C. S., McConathy W. J., Kloer H. U., Alaupovic P. (1985) Modulation of lipoprotein lipase activity by apolipoproteins. Effect of apolipoprotein C-III. J. Clin. Invest. 75, 384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sundaram M., Zhong S., Bou Khalil M., Links P. H., Zhao Y., Iqbal J., Hussain M. M., Parks R. J., Wang Y., Yao Z. (2010) Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J. Lipid Res. 51, 150–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ebara T., Ramakrishnan R., Steiner G., Shachter N. S. (1997) Chylomicronemia due to apolipoprotein CIII overexpression in apolipoprotein E-null mice. Apolipoprotein CIII-induced hypertriglyceridemia is not mediated by effects on apolipoprotein E. J. Clin. Invest. 99, 2672–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sehayek E., Eisenberg S. (1991) Mechanisms of inhibition by apolipoprotein C of apolipoprotein E-dependent cellular metabolism of human triglyceride-rich lipoproteins through the low density lipoprotein receptor pathway. J. Biol. Chem. 266, 18259–18267 [PubMed] [Google Scholar]
- 17. Bensadoun A., Ehnholm C., Steinberg D., Brown W. V. (1974) Purification and characterization of lipoprotein lipase from pig adipose tissue. J. Biol. Chem. 249, 2220–2227 [PubMed] [Google Scholar]
- 18. Ostlund-Lindqvist A. M., Iverius P. H. (1975) Activation of highly purified lipoprotein lipase from bovine milk. Biochem. Biophys. Res. Commun. 65, 1447–1455 [DOI] [PubMed] [Google Scholar]
- 19. Ekman R., Nilsson-Ehle P. (1975) Effects of apolipoproteins on lipoprotein lipase activity of human adipose tissue. Clin. Chim. Acta 63, 29–35 [DOI] [PubMed] [Google Scholar]
- 20. Simonet W. S., Bucay N., Pitas R. E., Lauer S. J., Taylor J. M. (1991) Multiple tissue-specific elements control the apolipoprotein E/C-I gene locus in transgenic mice. J. Biol. Chem. 266, 8651–8654 [PubMed] [Google Scholar]
- 21. Shachter N. S., Ebara T., Ramakrishnan R., Steiner G., Breslow J. L., Ginsberg H. N., Smith J. D. (1996) Combined hyperlipidemia in transgenic mice overexpressing human apolipoprotein Cl. J. Clin. Invest. 98, 846–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jong M. C., Hofker M. H., Havekes L. M. (1999) Role of ApoCs in lipoprotein metabolism: functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler. Thromb. Vasc. Biol. 19, 472–484 [DOI] [PubMed] [Google Scholar]
- 23. Berbée J. F., van der Hoogt C. C., Sundararaman D., Havekes L. M., Rensen P. C. (2005) Severe hypertriglyceridemia in human APOC1 transgenic mice is caused by apoC-I-induced inhibition of LPL. J. Lipid Res. 46, 297–306 [DOI] [PubMed] [Google Scholar]
- 24. van der Hoogt C. C., Berbée J. F. P., Espirito Santo S. M.., Gerritsen G., Krom Y. D., van der Zee A., Havekes L. M., van Dijk K. W., Rensen P. C. N. (2006) Apolipoprotein CI causes hypertriglyceridemia independent of the very-low-density lipoprotein receptor and apolipoprotein CIII in mice. Biochim. Biophys. Acta 1761, 213–220 [DOI] [PubMed] [Google Scholar]
- 25. Sukonina V., Lookene A., Olivecrona T., Olivecrona G. (2006) Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. U.S.A. 103, 17450–17455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nilsson S. K., Anderson F., Ericsson M., Larsson M., Makoveichuk E., Lookene A., Heeren J., Olivecrona G. (2012) Triacylglycerol-rich lipoproteins protect lipoprotein lipase from inactivation by ANGPTL3 and ANGPTL4. Biochim. Biophys. Acta 1821, 1370–1378 [DOI] [PubMed] [Google Scholar]
- 27. Lichtenstein L., Mattijssen F., de Wit N. J., Georgiadi A., Hooiveld G. J., van der Meer R., He Y., Qi L., Köster A., Tamsma J. T., Tan N. S., Müller M., Kersten S. (2010) Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab. 12, 580–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Georgiadi A., Lichtenstein L., Degenhardt T., Boekschoten M. V., van Bilsen M., Desvergne B., Müller M., Kersten S. (2010) Induction of cardiac Angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor β/δ and protects against fatty acid-induced oxidative stress. Circ. Res. 106, 1712–1721 [DOI] [PubMed] [Google Scholar]
- 29. Kroupa O., Vorrsjö E., Stienstra R., Mattijssen F., Nilsson S. K., Sukonina V., Kersten S., Olivecrona G., Olivecrona T. (2012) Linking nutritional regulation of Angptl4, Gpihbp1, and Lmf1 to lipoprotein lipase activity in rodent adipose tissue. BMC Physiol. 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bengtsson-Olivecrona G., Olivecrona T. (1991) Phospholipase activity of milk lipoprotein lipase. Methods Enzymol. 197, 345–356 [DOI] [PubMed] [Google Scholar]
- 31. Olivecrona G., Lookene A. (1997) Noncatalytic functions of lipoprotein lipase. Methods Enzymol. 286, 102–116 [DOI] [PubMed] [Google Scholar]
- 32. Shen Y., Lookene A., Nilsson S., Olivecrona G. (2002) Functional analyses of human apolipoprotein CII by site-directed mutagenesis: identification of residues important for activation of lipoprotein lipase. J. Biol. Chem. 277, 4334–4342 [DOI] [PubMed] [Google Scholar]
- 33. Hultin M., Savonen R., Olivecrona T. (1996) Chylomicron metabolism in rats: lipolysis, recirculation of triglyceride-derived fatty acids in plasma FFA, and fate of core lipids as analyzed by compartmental modelling. J. Lipid Res. 37, 1022–1036 [PubMed] [Google Scholar]
- 34. Astrup H. N., Bengtsson G. (1982) Activator proteins for lipoprotein lipase from bovine plasma: preparation by adsorption to Intralipid. Comp. Biochem. Physiol. B 72, 487–491 [DOI] [PubMed] [Google Scholar]
- 35. Liu H., Talmud P. J., Lins L., Brasseur R., Olivecrona G., Peelman F., Vandekerckhove J., Rosseneu M., Labeur C. (2000) Characterization of recombinant wild type and site-directed mutations of apolipoprotein C-III: lipid binding, displacement of ApoE, and inhibition of lipoprotein lipase. Biochemistry 39, 9201–9212 [DOI] [PubMed] [Google Scholar]
- 36. Bengtsson-Olivecrona G., Olivecrona T. (1992) Assay of lipoprotein lipase and hepatic lipase. in Lipoprotein Analysis: A Practical Approach (Converse C. A., Skinner E. R., eds), pp. 169–185, Oxford University Press, Cary, NC [Google Scholar]
- 37. Sreerama N., Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 38. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 39. Bergö M., Olivecrona G., Olivecrona T. (1996) Forms of lipoprotein lipase in rat tissues: in adipose tissue the proportion of inactive lipase increases on fasting. Biochem. J. 313, 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bengtsson G., Olivecrona T. (1977) Stimulation and inhibition of milk (lipoprotein) lipase by proteins from egg yolk lipoproteins. Eur. J. Biochem. 79, 225–231 [DOI] [PubMed] [Google Scholar]
- 41. Fielding C. J., Fielding P. E. (1976) Chylomicron protein content and the rate of lipoprotein lipase activity. J. Lipid Res. 17, 419–423 [PubMed] [Google Scholar]
- 42. Jackson R. L., Tajima S., Yamamura T., Yokoyama S., Yamamoto A. (1986) Comparison of apolipoprotein C-II-deficient triacylglycerol-rich lipoproteins and trioleoylglycerol/phosphatidylcholine-stabilized particles as substrates for lipoprotein lipase. Biochim. Biophys. Acta 875, 211–219 [DOI] [PubMed] [Google Scholar]
- 43. Gangabadage C. S., Zdunek J., Tessari M., Nilsson S., Olivecrona G., Wijmenga S. S. (2008) Structure and dynamics of human apolipoprotein CIII. J. Biol. Chem. 283, 17416–17427 [DOI] [PubMed] [Google Scholar]
- 44. Massey J. B., Gotto A. M., Jr., pownall H. J. (1979) Contribution of α helix formation in human plasma apolipoproteins to their enthalpy of association with phospholipids. J. Biol. Chem. 254, 9559–9561 [PubMed] [Google Scholar]
- 45. Bengtsson G., Olivecrona T. (1979) Binding of deoxycholate to lipoprotein lipase. Biochim. Biophys. Acta 575, 471–474 [DOI] [PubMed] [Google Scholar]
- 46. Wang L., Atkinson D., Small D. M. (2003) Interfacial properties of an amphipathic α-helix consensus peptide of exchangeable apolipoproteins at air/water and oil/water interfaces. J. Biol. Chem. 278, 37480–37491 [DOI] [PubMed] [Google Scholar]
- 47. Protter A. P., Vigne J.-L., Mallory J. B., Talmadge K. D., Kane J. P. (July 24, 1990) Mature apoai protein production under serum free culturing conditions. U. S. Patent 4,349,629
- 48. Formisano S., Brewer H. B., Jr., Osborne J. C., Jr. (1978) Effect of pressure and ionic strength on the self-association of Apo-A-I from the human high density lipoprotein complex. J. Biol. Chem. 253, 354–359 [PubMed] [Google Scholar]
- 49. Washington C., Davis S. S. (1988) The production of parenteral feeding emulsions by Microfluidizer. Int. J. Pharm. 44, 169–176 [Google Scholar]
- 50. Jackson R. L., Pattus F., Demel R. A. (1979) Interaction of plasma apolipoproteins with lipid monolayers. Biochim. Biophys. Acta 556, 369–387 [DOI] [PubMed] [Google Scholar]
- 51. Krebs K. E., Phillips M. C., Sparks C. E. (1983) A comparison of the surface activities of rat plasma apolipoproteins C-II, C-III-0, C-III-3. Biochim. Biophys. Acta 751, 470–473 [DOI] [PubMed] [Google Scholar]
- 52. Ibdah J. A., Krebs K. E., Phillips M. C. (1989) The surface properties of apolipoproteins A-I and A-II at the lipid/water interface. Biochim. Biophys. Acta 1004, 300–308 [DOI] [PubMed] [Google Scholar]
- 53. Weinberg R. B., Ibdah J. A., Phillips M. C. (1992) Adsorption of apolipoprotein A-IV to phospholipid monolayers spread at the air/water interface: a model for its labile binding to high density lipoproteins. J. Biol. Chem. 267, 8977–8983 [PubMed] [Google Scholar]
- 54. Meyers N. L., Wang L., Small D. M. (2012) Apolipoprotein C-I binds more strongly to phospholipid/triolein/water than triolein/water interfaces: a possible model for inhibiting cholesterol ester transfer protein activity and triacylglycerol-rich lipoprotein uptake. Biochemistry 51, 1238–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Demel R. A., Dings P. J., Jackson R. L. (1984) Effect of monolayer lipid structure and composition on the lipoprotein lipase-catalyzed hydrolysis of triacylglycerol. Biochim. Biophys. Acta 793, 399–407 [DOI] [PubMed] [Google Scholar]
- 56. Jackson R. L., Balasubramaniam A., Murphy R. F., Demel R. A. (1986) Interaction of synthetic peptides of apolipoprotein C-II and lipoprotein lipase at monomolecular lipid films. Biochim. Biophys. Acta 875, 203–210 [DOI] [PubMed] [Google Scholar]
- 57. Olivecrona G., Beisiegel U. (1997) Lipid binding of apolipoprotein CII is required for stimulation of lipoprotein lipase activity against apolipoprotein CII-deficient chylomicrons. Arterioscler. Thromb. Vasc. Biol. 17, 1545–1549 [DOI] [PubMed] [Google Scholar]
- 58. Fredenrich A., Giroux L. M., Tremblay M., Krimbou L., Davignon J., Cohn J. S. (1997) Plasma lipoprotein distribution of apoC-III in normolipidemic and hypertriglyceridemic subjects: comparison of the apoC-III to apoE ratio in different lipoprotein fractions. J. Lipid Res. 38, 1421–1432 [PubMed] [Google Scholar]
- 59. Cohn J. S., Tremblay M., Batal R., Jacques H., Veilleux L., Rodriguez C., Bernier L., Mamer O., Davignon J. (2002) Plasma kinetics of VLDL and HDL apoC-I in normolipidemic and hypertriglyceridemic subjects. J. Lipid Res. 43, 1680–1687 [DOI] [PubMed] [Google Scholar]
- 60. Sparrow J. T., Pownall H. J., Hsu F. J., Blumenthal L. D., Culwell A. R., Gotto A. M. (1977) Lipid binding by fragments of apolipoprotein C-III-1 obtained by thrombin cleavage. Biochemistry 16, 5427–5431 [DOI] [PubMed] [Google Scholar]
- 61. Lambert D. A., Catapano A. L., Smith L. C., Sparrow J. T., Gotto A. M., Jr. (1996) Effect of the apolipoprotein C-II/C-III1 ratio on the capacity of purified milk lipoprotein lipase to hydrolyse triglycerides in monolayer vesicles. Atherosclerosis 127, 205–212 [DOI] [PubMed] [Google Scholar]
- 62. von Eckardstein A., Holz H., Sandkamp M., Weng W., Funke H., Assmann G. (1991) Apolipoprotein C-III(Lys58 → Glu). Identification of an apolipoprotein C-III variant in a family with hyperalphalipoproteinemia. J. Clin. Invest. 87, 1724–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liu H., Labeur C., Xu C. F., Ferrell R., Lins L., Brasseur R., Rosseneu M., Weiss K. M., Humphries S. E., Talmud P. J. (2000) Characterization of the lipid-binding properties and lipoprotein lipase inhibition of a novel apolipoprotein C-III variant Ala23Thr. J. Lipid Res. 41, 1760–1771 [PubMed] [Google Scholar]
- 64. Mattijssen F., Kersten S. (2012) Regulation of triglyceride metabolism by Angiopoietin-like proteins. Biochim. Biophys. Acta 1821, 782–789 [DOI] [PubMed] [Google Scholar]
- 65. Yu X., Burgess S. C., Ge H., Wong K. K., Nassem R. H., Garry D. J., Sherry A. D., Malloy C. R., Berger J. P., Li C. (2005) Inhibition of cardiac lipoprotein utilization by transgenic overexpression of Angptl4 in the heart. Proc. Natl. Acad. Sci. U.S.A. 102, 1767–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Garcia-Arcos I., Hiyama Y., Drosatos K., Bharadwaj K. G., Hu Y., Son N. H., O'Byrne S. M., Chang C. L., Deckelbaum R. J., Takahashi M., Westerterp M., Obunike J. C., Jiang H., Yagyu H., Blaner W. S., Goldberg I. J. (2013) Adipose-specific lipoprotein lipase deficiency more profoundly affects brown than white fat biology. J. Biol. Chem. 288, 14046–14058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hansen J. B., Fernández J. A., Notø A. T., Deguchi H., Björkegren J., Mathiesen E. B. (2011) The apolipoprotein C-I content of very-low-density lipoproteins is associated with fasting triglycerides, postprandial lipemia, and carotid atherosclerosis. J. Lipids 2011, 271062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kersten S., Mandard S., Tan N. S., Escher P., Metzger D., Chambon P., Gonzalez F. J., Desvergne B., Wahli W. (2000) Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 275, 28488–28493 [DOI] [PubMed] [Google Scholar]
- 69. Attman P. O., Alaupovic P., Gustafson A. (1987) Serum apolipoprotein profile of patients with chronic renal failure. Kidney Int. 32, 368–375 [DOI] [PubMed] [Google Scholar]
- 70. Baranowski T., Kralisch S., Bachmann A., Lössner U., Kratzsch J., Blüher M., Stumvoll M., Fasshauer M. (2011) Serum levels of the adipokine fasting-induced adipose factor/angiopoietin-like protein 4 depend on renal function. Horm. Metab. Res. 43, 117–120 [DOI] [PubMed] [Google Scholar]
- 71. Crawford G. A., Savdie E., Stewart J. H. (1979) Heparin-released plasma lipases in chronic renal failure and after renal transplantation. Clin. Sci. (Lond.) 57, 155–165 [DOI] [PubMed] [Google Scholar]
- 72. Attman P. O., Samuelsson O. (2009) Dyslipidemia of kidney disease. Curr. Opin. Lipidol. 20, 293–299 [DOI] [PubMed] [Google Scholar]
- 73. Sundaram M., Zhong S., Bou Khalil M., Zhou H., Jiang Z. G., Zhao Y., Iqbal J., Hussain M. M., Figeys D., Wang Y., Yao Z. (2010) Functional analysis of the missense APOC3 mutation Ala23Thr associated with human hypotriglyceridemia. J. Lipid Res. 51, 1524–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Qin W., Sundaram M., Wang Y., Zhou H., Zhong S., Chang C. C., Manhas S., Yao E. F., Parks R. J., McFie P. J., Stone S. J., Jiang Z. H. G., Wang C., Figeys D., Jia W., Yao Z. (2011) Missense mutation in APOC3 within the C-terminal lipid binding domain of human apoC-III results in impaired assembly and secretion of triacylglycerol-rich very low density lipoproteins. Evidence that apoC-III plays a major role in the formation of lipid precursors within the microsomal lumen. J. Biol. Chem. 286, 27769–27780 [DOI] [PMC free article] [PubMed] [Google Scholar]