

Background: Leptin inhibits insulin secretion by increasing β-cell KATP currents.
Results: Leptin causes a transient increase in surface KATP channel density. This increase is dependent on AMPK and PKA and actin depolymerization.
Conclusion: Leptin increases KATP currents by recruiting KATP channels to the β-cell membrane.
Significance: KATP channel trafficking is an important physiological mechanism to regulate channel activity.
Keywords: ABC Transporter, Actin, AMP-activated kinase (AMPK), Endocytosis, Potassium Channels, cAMP-dependent Protein Kinase (PKA), Signal Transduction
Abstract
Pancreatic β-cells secrete insulin in response to metabolic and hormonal signals to maintain glucose homeostasis. Insulin secretion is under the control of ATP-sensitive potassium (KATP) channels that play key roles in setting β-cell membrane potential. Leptin, a hormone secreted by adipocytes, inhibits insulin secretion by increasing KATP channel conductance in β-cells. We investigated the mechanism by which leptin increases KATP channel conductance. We show that leptin causes a transient increase in surface expression of KATP channels without affecting channel gating properties. This increase results primarily from increased channel trafficking to the plasma membrane rather than reduced endocytosis of surface channels. The effect of leptin on KATP channels is dependent on the protein kinases AMP-activated protein kinase (AMPK) and PKA. Activation of AMPK or PKA mimics and inhibition of AMPK or PKA abrogates the effect of leptin. Leptin activates AMPK directly by increasing AMPK phosphorylation at threonine 172. Activation of PKA leads to increased channel surface expression even in the presence of AMPK inhibitors, suggesting AMPK lies upstream of PKA in the leptin signaling pathway. Leptin signaling also leads to F-actin depolymerization. Stabilization of F-actin pharmacologically occludes, whereas destabilization of F-actin simulates, the effect of leptin on KATP channel trafficking, indicating that leptin-induced actin reorganization underlies enhanced channel trafficking to the plasma membrane. Our study uncovers the signaling and cellular mechanism by which leptin regulates KATP channel trafficking to modulate β-cell function and insulin secretion.
Introduction
Leptin, a peptide hormone predominantly secreted by white adipocytes, regulates energy homeostasis according to fat storage in the body. It is well established that leptin acts on leptin receptors in hypothalamic neurons to reduce appetite, and disruption of leptin signaling results in obesity. There is growing evidence that, independent of its action on the central nervous system, leptin also has a potent effect on glucose homeostasis by directly targeting pancreatic β-cells, which secrete insulin in response to elevated plasma glucose levels to promote glucose utilization and storage (1). It has been proposed that leptin and insulin form a dual hormonal feedback loop, termed the adipoinsular axis, to coordinate metabolic control (2). In this axis, insulin secreted from β-cells is adipogenic and stimulates leptin release; elevated leptin levels in turn suppress insulin secretion to limit a further increase in fat mass. Supporting this idea, β-cells express leptin receptors, and leptin reduces insulin secretion in isolated islets and intact animals (3). Dysregulation of the adipoinsular axis results in hyperleptinemia and hyperinsulinemia, leading to the eventual development of type 2 diabetes as seen in animals with β-cell deletion of leptin receptors (1, 4, 5).
Control of insulin secretion is critically dependent on ATP-sensitive potassium channels (KATP)3 present in the β-cell membrane (6–8). The β-cell KATP channel is an octameric complex of four pore-forming inwardly rectifying potassium channel Kir6.2 subunits and four sulfonylurea receptor 1 (SUR1) regulatory subunits (9–11). ATP inhibits channel activity by interacting with Kir6.2, whereas Mg-ATP/ADP stimulates channel activity by interacting with SUR1. As the intracellular ATP/ADP ratio is determined by glucose metabolism, this allows the channel to serve as a metabolic sensor and link glucose metabolism to membrane potential. Accordingly, when blood glucose levels are low, KATP channels are open, keeping β-cell membrane potential hyperpolarized to prevent insulin secretion. Glucose stimulation prompts closure of KATP channels, resulting in membrane depolarization, activation of voltage-gated Ca2+ channels, and insulin release. In addition to gating regulation by intracellular nucleotides, KATP conductance is also a function of channel abundance in the plasma membrane. An increase of channel density will increase the overall channel conductance and thus the threshold of glucose concentrations necessary to depolarize β-cell membrane and stimulate insulin secretion; by contrast, a decrease of channel density is expected to render membrane potential more easily depolarized at a given stimulatory glucose concentration (8). Compared with gating regulation, relatively little is known about how the number of KATP channels in the β-cell membrane is governed.
That leptin increases KATP conductance in β-cells was first documented more than 15 years ago (12, 13), and this effect has been proposed to underlie the inhibitory action of leptin on insulin secretion. Several studies have been published reporting the involvement of various signaling molecules and events (14–19); however, the precise mechanism by which leptin increases KATP channel conductance remains poorly understood. In this study, we show that leptin increases KATP channel conductance by recruiting channels to the plasma membrane. This regulation is mediated by a signaling mechanism involving the AMP-activated protein kinase (AMPK) and the cAMP-dependent protein kinase (PKA). We demonstrate that leptin signaling leads to F-actin depolymerization, which promotes channel trafficking to the plasma membrane. Our study identifies a physiological signaling mechanism that regulates KATP channel density at the β-cell membrane to control insulin secretion.
MATERIALS AND METHODS
Molecular Biology
Rat Kir6.2 cDNA is in the pCDNAI/Amp vector. A minimal α-BTX-binding (WRYYESSLEPYPD) peptide tag was placed at the N terminus of SUR1 (BTX tag-SUR1) in pECE. The BTX-tag SUR1 and Kir6.2 recombinant adenoviruses were constructed using a modified pShuttle plasmid (AdEasy kit, Stratagene) containing a tetracycline-inducible promoter as described previously (20). Recombinant viruses were amplified in HEK293 cells and purified according to the manufacturer's instructions.
Cell Culture and Viral Infection
INS-1 cells (clone 832/13) were cultured in RPMI 1640 medium with 11.1 mm d-glucose (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, 10 mm HEPES, 2 mm glutamine, 1 mm sodium pyruvate, and 50 μm β-mercaptoethanol (21). Cells at ∼70% confluency were washed once with phosphate-buffered saline (PBS) and then incubated for 1.5 h at 37 °C in OptiMEM and a mixture of viruses, including tetracycline-inhibited transactivator and a tetracycline-inhibited transactivator-regulated construct expressing BTX-SUR1 and Kir6.2. The multiplicity of infection for each virus was determined empirically. After 90 min, 2× growth medium was added, and the cells were incubated at 37 °C until reaching appropriate density for the various experiments. Transduction with adenoviruses carrying dominant-negative (AMPKα2-K45R) or constitutively active (AMPKγ1-H150R) AMPK subunits was performed as described previously (22).
Drug Treatments
All drugs in this section were purchased from Sigma. For stimulation with leptin, AICAR, forskolin, or 8-bromo-cAMP, INS-1 cells grown in 6-well plates were exposed to regular RPMI 1640 medium without serum for 30 min before treatment with leptin, AICAR, forskolin, or 8-bromo-cAMP for the indicated time or 30 min. Pharmacological inhibitors, including the AMPK inhibitor compound C (CC) or the PKA inhibitors, H89 or protein kinase A inhibitor fragment 14–22 (PKI), were added 30 min before leptin, AICAR, forskolin, or 8-bromo-cAMP treatment.
Electrophysiology
For measuring channel sensitivity to ATP and MgADP, the inside-out patch clamp recording configuration was used. Micropipettes had resistance typically ∼1–2 megohms. The bath (intracellular) and pipette (extracellular) solutions were K-INT. ATP was added as the potassium salt. The recording was performed at room temperature, and currents were measured at a membrane potential of −50 mV, and inward currents were shown as upward deflections.
Whole-cell patch clamp recording was used to measure KATP current density in INS-1 cells and β-cells dispersed from human islets (obtained through the Integrated Islets Distribution Program). To identify β-cells, dissociated human islet cells plated on coverslips were placed into the recording chamber and stained briefly (3–5 min) with 10 μg/ml dithizone solution (in PBS) followed by a few minutes of washout with Tyrode's solution. Previous studies have shown that at this low concentration and short incubation time, dithizone has no deleterious effect on β-cell function (23). Only dithizone-positive cells were recorded. Cells were held at −70 mV, and KATP currents were recorded at two voltage steps (−50 and −90 mV) applied every 2 s. Micropipettes were pulled from nonheparinized Kimble glass (Fisher) on a horizontal puller (Sutter Instrument, Novato, CA) and had typical resistance of 3–5 megohms when filled with K-INT solution (140 mm KCl, 10 mm K-HEPES, 1 mm K-EGTA, pH 7.3). Outside Tyrode's solution contained the following (in mm): NaCl, 137; KCl, 5.4; CaCl2, 1.8; MgCl2, 0.5; Na-HEPES, 5; NHCO3, 3; NaH2PO4, 0.16; pH 7.2. Diazoxide (200 μm) was applied to the bath solution immediately after break-in to maximally stimulate KATP channels. After the current had plateaued, 300 μm tolbutamide (a KATP channel antagonist) was applied to ensure the specificity of the KATP currents.
Surface Biotinylation
INS-1 cells were washed twice with cold PBS. Biotinylation of surface protein was carried out by incubating cells with 1 mg/ml of the membrane-impermeant, thiol-cleavable, amine-reactive biotinylation reagent, EZ-Link Sulfo-NHS-SS-Biotin (Pierce) in PBS for 30 min on ice. The reaction was terminated by incubating cells for 5 min with PBS containing 20 mm glycine, followed by three washes with cold PBS. Cells were then lysed in 300 μl of lysis buffer (50 mm Tris-HCl, 2 mm EDTA, 2 mm EGTA, 100 mm NaCl, 1% Triton X-100, pH 7.4, with complete protease inhibitor) for 30 min at 4 °C. Cell lysate was cleared by centrifugation at 21,000 × g for 10 min at 4 °C, and 500 μg of total lysate was incubated with 100 μl of ∼50% slurry of NeutrAvidin-agarose (Pierce) overnight at 4 °C. Biotinylated proteins were eluted with 2× protein loading buffer for 15 min at room temperature. Both eluent and input samples (50 μg of total cell lysate) were analyzed by immunoblotting using anti-SUR1 or anti-Kir6.2 antibodies as described previously (24, 25).
To monitor internalization of surface KATP channels (Fig. 5A), cells were subjected to surface biotinylation as described above and chased in RPMI 1640 medium containing vehicle or leptin for 15 or 30 min at 37 °C to allow internalization of biotinylated cell surface proteins. At the end of each chase, cells were treated with the membrane-impermeable reducing agent MENSA (50 mm in PBS) for 20 min at 4 °C to strip off the biotin label from proteins remaining at the cell surface. Internalized biotinylated SUR1 protein was then precipitated by NeutrAvidin-agarose beads and analyzed by immunoblotting.
FIGURE 5.
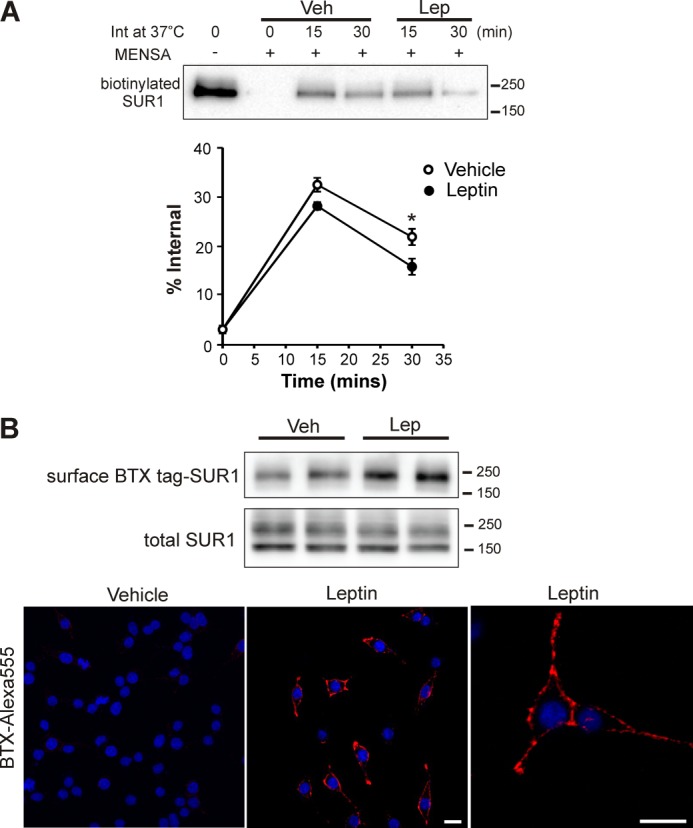
Leptin causes a mild reduction in internalization of surface KATP channels but a pronounced increase in KATP channel trafficking to the cell surface. A, INS-1 cells were subjected to surface biotinylation at 4 °C to label surface SUR1. Cells were then returned to 37 °C to allow protein trafficking to resume in the presence or absence of 10 nm leptin for 15 or 30 min. Residual surface biotin was stripped off using the reducing agent MENSA, and internalized biotinylated SUR1 was detected by precipitation using NeutrAvidin beads followed by immunoblotting with anti-SUR1 antibody. Top panel, a representative blot showing the amount of surface-biotinylated SUR1 prior to internalization, the amount of biotinylated SUR1 detected at time 0 of the 37 °C chase indicating the efficiency of surface biotin stripping by MENSA, and the amount of internalized biotinylated SUR1 at 15 or 30 min of chase in vehicle-treated or leptin-treated cells. Bottom panel, quantification of the amount of internalized surface SUR1 from the blot shown above expressed as % of total biotinylated surface SUR1 before internalization (n = 6 independent experiments; *, p < 0.01 comparing the leptin-treated group with control). B, INS-1 cells were transduced with adenoviruses carrying BTX-tag SUR1 and Kir 6.2. Cells were preincubated with unlabeled BTX for 60 min at 4 °C to bind pre-existing surface BTX-tag SUR1 and then returned to 37 °C in the presence of vehicle or 10 nm leptin for 30 min. BTX-tag SUR1 inserted into the plasma membrane during the 30-min 37 °C incubation was then either biotinylated using biotin-conjugated BTX and detected by Western blot (upper panel, duplicates are shown) or labeled with Alexa 555-conjugated BTX and viewed by confocal microscopy (lower panel). For the fluorescence images shown, red is Alexa 555, and blue is DAPI stain of nuclei; scale bar, 10 μm. Veh, vehicle; Lep, leptin.
For detecting channels trafficked to the plasma membrane during leptin treatment (Figs. 5B and 6B), INS-1 cells transduced with the BTX-tag SUR1 and Kir6.2 viruses were preincubated with 10 μg/ml unlabeled BTX (Molecular Probes) at 4 °C for 60 min, washed briefly, and then incubated in regular RPMI containing leptin, 8-bromo-cAMP, or AICAR in the absence or presence of PKA or AMPK inhibitors at 37 °C for 30 min. After 30 min, cells were washed with cold PBS, placed on ice, and incubated with 1 μg/ml BTX-conjugated biotin (BTX-biotin; Molecular Probes) for 1 h at 4 °C. Surface BTX-SUR1 labeled with BTX-biotin was then pulled down with NeutrAvidin beads and detected by immunoblotting.
FIGURE 6.

Leptin causes actin depolymerization and increased KATP channel trafficking to the cell surface in an AMPK- and PKA-dependent manner. A, images show surface staining of KATP channels (red) delivered to the plasma membrane during a 30-min time in response to leptin (10 nm), AICAR (250 μm), or 8-bromo-cAMP (8Br) (10 μm) in INS-1 cells expressing BTX-tag SUR1 and Kir 6.2 as described in Fig. 5B. Alexa 488-conjugated phalloidin was used to stain F-actin (green) as described under “Materials and Methods.” Inclusion of compound C during the incubation occluded the effect of leptin and AICAR but not 8-bromo-cAMP on actin depolymerization and KATP trafficking to the cell surface. Inclusion of PKI during the incubation blocked the effects of all three treatments, leptin (Lep), AICAR (AIC), and 8-bromo-cAMP (8-Br), on F-actin abundance and channel trafficking. Veh, vehicle. Scale bar, 10 μm. B, same as A except that BTX-tag SUR1 channels delivered to the cell surface were detected with BTX-biotin followed by precipitation with NeutrAvidin beads and immunoblotting with anti-SUR1 antibody. Upper panel, representative blots of surface and total SUR1. Lower panel, fold-increase in surface SUR1 relative to the upper band of total SUR1 was quantified from blots shown above. Values were normalized to that of vehicle control (n = 3; *, p < 0.05 by one-way ANOVA and Dunnett's post hoc test).
Immunoblotting
INS-1 cells were washed twice with ice-cold PBS and lysed in the lysis buffer described above at 4 °C with rotation for 30 min. Cell lysate was cleared by centrifugation at 21,000 × g for 10 min at 4 °C. Small aliquots of the lysates were used for protein determination by the Lowry method (Pierce) with bovine serum albumin as the standard. Proteins were separated by SDS-PAGE (7.5–12.5%) and transferred onto PVDF membranes (Millipore, Bedford, MA). Membranes were incubated overnight at 4 °C with a primary antibody diluted in the Tris-buffered saline plus 0.1% Tween 20 (TBST). The antibodies against SUR1 and Kir6.2 (1:500 dilutions) were made as described previously (24). The antibody against phosphoacetyl-CoA carboxylase at Ser-3 and phospho-AMPK at Thr-172 (1:1000 dilutions) was from Millipore. The antibody against IGF-1Rβ (1:1000) was from Santa Cruz Biotechnology, and α-AMPK (1:1000) was purchased from Cell Signaling. After three 10-min washes in TBST buffer, blots were incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibodies in TBST buffer as follows: 1:40,000 goat anti-rabbit IgG (GE Healthcare) for SUR1 and Kir6.2; 1:2000 goat anti-rabbit IgG for phospho-AMPK at Thr-172; 1:2000 horse anti-mouse IgG (GE Healthcare) for total AMPK. Finally, the blots were washed three times for 10 min in TBST and developed using the enhanced chemiluminescence detection kit (Super Signal West Femto, Pierce). The signals were imaged by AlphaView® (Cell Biosciences). Blots were stripped and re-probed with anti-tubulin as a loading control. The blots were quantified with ImageJ (National Institutes of Health) and normalized to the corresponding controls.
Fluorescence Microscopy
To visualize surface BTX-tag SUR1, INS-1 cells were infected with the BTX-tag SUR1 and Kir6.2 recombinant adenoviruses as described above and plated onto 18-mm, number 1.5 glass coverslips (Warner Instruments) 24 h post-infection. In images shown in Fig. 2D, cells were treated with leptin or vehicle control for 30 min and then surface BTX-tag SUR1-labeled by incubation with 1 μg/ml Alexa Fluor®555 α-bungarotoxin (Alexa 555-BTX; Molecular Probes) for 1 h at 4 °C. To visualize channels inserted into the plasma membrane during drug treatments (Figs. 5B and 6A), pre-existing surface BTX-tag SUR1 was blocked with unlabeled BTX as described above, and new BTX-tag SUR1 inserted into the membrane following various drug treatments was subsequently labeled with 1 μg/ml Alexa 555-BTX for 1 h at 4 °C. BTX florescence was imaged by confocal microscopy as described below.
FIGURE 2.

Leptin increases surface expression of KATP channels in β-cells. A, INS-1 cells were treated with 10 nm leptin for the times indicated and subjected to surface biotinylation. Left, representative Western blots show surface-biotinylated SUR1 pulled down with NeutrAvidin beads and total SUR1 in the cell lysate. Note only the upper band corresponding to the complex-glycosylated SUR1 and not the lower band corresponding to the ER-core glycosylated SUR1, was detected at the cell surface. Molecular mass markers in this and subsequent figures are in kDa. Right, bar graph shows the fold increase in surface SUR1 relative to the upper band of total SUR1 and normalized to time 0 (n = 4, *, p < 0.05 by one-way ANOVA and Dunnett's post hoc test). B, whole-cell recordings of dissociated human β-cells as described under “Materials and Methods.” Left, representative recordings from a human β-cell treated with vehicle (Veh) control and a cell treated with 10 nm leptin for 30 min. Diazoxide (Diaz) was applied to maximally stimulate channel activity, and tolbutamide (Tolb) was applied to ensure the current was from KATP channels. Right, averaged current density from cells treated with vehicle (n = 9) or 10 nm leptin (n = 11). *, p < 0.05 by unpaired Student's t test. Error bars are S.E. C, INS-1 cells were transduced with adenoviruses to express Kir6.2 and BTX-tag SUR1. Cells were treated with vehicle or 10 nm leptin for 30 min, and surface SUR1 was labeled with Alexa 555-BTX (red). Nuclei were stained with DAPI (blue). Scale bar, 10 μm. D, INS-1 cells were treated with 10 nm leptin for 15 or 30 min followed by surface biotinylation of membrane proteins. Biotinylated proteins were pulled down with the NeutrAvidin beads and blotted for IGF-1Rβ or SUR1. Total IGF-1Rβ and SUR1 present in the whole cell lysate are also shown for comparison.
For F-actin staining, INS-1 cells were fixed in 4% paraformaldehyde for 30 min. Then cells were washed in PBS and permeabilized in PBS, 0.5% Triton X-100 for 10 min, rinsed in PBS, blocked with 20% normal goat serum (Vector Laboratories) for 30 min, rinsed in PBS, and incubated with 2 units/ml Alexa 488-phalloidin (Invitrogen) for 90–120 min. All imaging experiments were performed on a Zeiss LSM710 three-channel spectral confocal microscope with a 63× 1.4 numerical aperture (NA) objective (Carl Zeiss) under identical conditions with randomly selected regions of each coverslip.
Statistical Analysis
All data were analyzed with the program GraphPad PrismTM. Results were expressed as mean ± S.E. Differences were tested using one-way analysis of variance (ANOVA) followed by the post hoc Dunnett's test for multiple comparisons. When only two groups were compared, unpaired Student's t test was used. The level of statistical significance was set at p < 0.05.
RESULTS
Leptin Increases Surface KATP Channel Expression
Leptin has been shown to increase KATP channel conductance in β-cells using cell-attached and whole-cell electrophysiological recordings (12, 13). However, it is not known whether this increase results from an effect on channel gating property or channel density. To address this question, we examined the effects of leptin using the rat insulinoma cells INS-1 that express endogenous KATP channels and leptin receptors (26, 27). INS-1 cells were treated with 10 nm leptin for 15 or 30 min, conditions previously shown to increase KATP conductance. Channel sensitivity to ATP and MgADP, two key physiological ligands that determine channel activity, were assessed by inside-out patch clamp recording. Leptin treatment did not alter channel sensitivity to ATP or MgADP, indicating that the increased conductance is unlikely due to altered channel gating (Fig. 1).
FIGURE 1.

Leptin treatment does not alter nucleotide sensitivities of KATP channels. A, representative inside-out patch clamp recordings from INS-1 cells treated with vehicle control or 10 nm leptin for 30 min. Recordings were made in symmetrical K-INT solution at −50 mV, and inward currents are shown as upward deflections. Patches containing KATP channels were exposed to bath (corresponding to intracellular) solutions containing varying concentrations of ATP and ADP as indicated by the bars above the recordings. B, quantification of currents observed in different solutions from recordings shown in A. Currents were expressed as % of those observed in K-INT solution with no ATP or ADP. No significant differences between control and leptin-treated groups (Student's t test) were observed under all solution conditions. Error bars are S.E.
Next, we tested whether leptin affects KATP channel abundance at the cell surface. Using surface protein biotinylation as described under “Materials and Methods,” a transient increase in surface SUR1 was observed in cells treated with 10 nm leptin for 15–180 min (Fig. 2A). At the peak effect of 30 min, surface SUR1 was ∼4-fold higher than that seen at time 0. Although Kir6.2 does not have extracellular lysine residues that could be biotinylated, it was co-purified with biotinylated SUR1 such that a corresponding increase in surface Kir6.2 was also observed. Note that because SUR1 is the subunit that is directly labeled by surface biotinylation, surface-biotinylated SUR1 was used to assess surface expression of KATP channels in all subsequent experiments. Interestingly, Western blots of total SUR1 and Kir6.2 proteins showed no difference between control and leptin-treated cells. These results indicate that leptin causes a rapid and transient increase in KATP channel density, likely through a post-translational trafficking event.
To confirm the effect of leptin in primary β-cells, we used whole-cell patch clamp recording to measure KATP current density in dissociated human β-cells treated with leptin or vehicle control. As shown in Fig. 2B, a clear increase in diazoxide-stimulated, tolbutamide-blocked currents characteristic of KATP channels was observed in leptin (10 nm)-treated cells compared with vehicle-treated cells. The averaged current density increase by leptin was 2.67-fold. A similar increase in KATP current density was also observed in INS-1 cells upon leptin treatment (∼2-fold; data not shown). Note the increase measured by whole-cell recording is not as great as that seen in surface biotinylation experiments likely because the time that it took to perform the recording did not allow for the capture of the peak effect in every cell.
To directly visualize KATP channels in the cell membrane, we transduced INS-1 cells with recombinant adenoviruses for Kir6.2 and a SUR1 tagged at the extracellular N terminus with a minimal bungarotoxin-binding motif (BTX tag-SUR1; see under “Materials and Methods”) (28, 29) and performed surface immunostaining using Alexa 555-BTX. As shown in Fig. 2C, there is a marked increase in the staining of BTX-tag SUR1 in leptin-treated cells compared with control cells, consistent with an increase in surface channel expression. This result also shows that like endogenous KATP channels, the exogenously expressed channels are subjected to leptin regulation.
To test if the effect of leptin is specific to KATP channels, we examined surface expression of another membrane protein, the insulin growth factor receptor 1 (IGF-1R), after 30 min of 10 nm leptin treatment. No difference in the abundance of surface-biotinylated IGF-1Rβ was seen between control and leptin-treated cells (Fig. 2D), indicating that leptin does not change global protein expression or membrane trafficking.
Effect of Leptin on Surface Expression of KATP Channels Is AMPK- and PKA-dependent
Leptin is known to activate a number of signaling pathways, including AMPK (30, 31). We found that in INS-1 cells, leptin transiently increased phosphorylation of AMPK at threonine 172 with a time course that corresponds to the increase in surface SUR1 (Fig. 3A). To determine whether the effect of leptin on channel expression is dependent on AMPK, we manipulated AMPK activity and tested the effect of leptin. As shown in Fig. 3B, treatment of INS-1 cells with AICAR (250 μm), an AMPK activator, for 30 min increased surface SUR1, mimicking the effect of leptin. Conversely, treatment with an AMPK inhibitor, compound C, precluded the effect of leptin. Moreover, we transduced INS-1 cells with adenoviruses carrying a constitutively active AMPK subunit (AMPKγ1-H150R) or a dominant-negative AMPK subunit (AMPKα2-K45R) to increase or reduce AMPK activity (22, 32), respectively, as confirmed by an increase or a decrease in the phosphorylation of Ser-79 in acetyl-CoA carboxylase, a downstream substrate of AMPK (Fig. 3, C and D) (30). Manipulations of AMPK activity by expressing the dominant-negative or constitutively active AMPK similarly occluded or mimicked the effect of leptin and AICAR on KATP channel surface expression (Fig. 3, C and D), whereas transduction of INS-1 cells with a control EGFP adenovirus (Ad-EGFP) was without effect. Taken together, the above results provide compelling evidence that the effect of leptin on KATP channels is mediated by AMPK.
FIGURE 3.

Leptin increases surface expression of KATP channels via activation of AMPK and PKA. A, upper panel, Western blots of AMPK phosphorylated at Thr-172 (pAMPK) and total AMPK in INS-1 cells treated with 10 nm leptin for the indicated time. Lower panel, quantification of pAMPK from blots like those shown in the upper panel. The pAMPK signal was normalized to the total AMPK signal and expressed as fold increase of the value seen at time 0. Each bar represents the mean ± S.E. of four separate experiments. (n = 4/group; *, p < 0.05 compared with time 0 by one-way ANOVA and Dunnett's post hoc test.) B, surface-biotinylated and total SUR1 in INS-1 cells pretreated with DMSO or 10 μm of the AMPK inhibitor compound C (CC) for 30 min before being treated with vehicle (Veh), 10 nm leptin (Lep), or 250 μm of the AMPK activator AICAR (AIC) for another 30 min. C, INS-1 cells were infected with control adenovirus carrying EGFP (Ad-EGFP) or adenovirus carrying a dominant-negative AMPK subunit (Ad-AMPK-DN) to reduce AMPK activity (multiplicity of infection (MOI) of 50 for 48 h). Cells were then treated with vehicle, leptin, or AICAR for 30 min and analyzed for surface and total SUR1. Cell lysate was also blotted for phosphoacetyl-coenzyme A carboxylase to monitor AMPK activity and for tubulin that serves as a loading control. D, AMPK activity was enhanced by infecting INS-1 cells with increasing amounts of adenovirus carrying a constitutively active AMPK subunit (Ad-AMPK-CA). Surface SUR1, total SUR1, and phosphoacetyl-CoA carboxylase were analyzed by immunoblotting. E, INS-1 cells were pretreated with DMSO, or the PKA inhibitor H89 (10 μm), or PKI (1 μm) for 30 min before adding vehicle or 10 nm leptin for another 30 min. Surface SUR1 and total SUR1 in the lysate were analyzed as described in the text. Both PKA inhibitors H89 and PKI significantly attenuated the effect of leptin on surface SUR1. F, stimulation of PKA by either forskolin (Fsk; 10 μm) or 8-bromo-cAMP (8-Br; 10 μm) for 30 min was sufficient to mimic the effect of leptin and increase surface SUR1 expression. As controls, co-administration of the PKA inhibitors H89 or PKI with forskolin or 8-bromo-cAMP abrogated the ability of forskolin or 8-bromo-cAMP to increase surface SUR1.
A previous study has reported that KATP channel surface expression can be up-regulated by high glucose exposure in a PKA-dependent manner (33). We sought to determine whether PKA has a role in mediating the effect of leptin on KATP channel surface expression. INS-1 cells preincubated with the PKA inhibitors H89 (10 μm) or PKI (1 μm) for 30 min before 10 nm leptin treatment failed to show increased KATP channel surface expression upon leptin treatment, as judged by the abundance of surface-biotinylated SUR1 (Fig. 3E). On the contrary, activation of PKA by treating cells with 10 μm forskolin or 8-bromo-cAMP for 30 min led to an increase in surface SUR1 (Fig. 3F), resembling that observed in cells treated with leptin. These results indicate that activation of both AMPK and PKA is required for leptin to exert its effect on KATP channel density in the plasma membrane.
AMPK Acts Upstream of PKA in the Leptin Signaling Cascade to Regulate Surface Expression of KATP Channels
Having demonstrated that both AMPK and PKA play a role in mediating the effect of leptin, we next investigated the relationship between AMPK and PKA in the leptin signaling cascade. If AMPK is acting upstream of PKA, it follows that inhibition of PKA activity should prevent the AMPK activator AICAR from mimicking the effect of leptin; moreover, activation of PKA should still increase KATP channel surface expression even when AMPK activity is blocked by compound C. However, if PKA acts upstream of AMPK, inhibition of AMPK activity should occlude the effect of PKA activation on KATP channels, and activating AMPK should bypass the need to activate PKA such that KATP surface expression still increases even when PKA activity is inhibited. Results of surface biotinylation experiments show that inhibition of PKA using PKI prevented the ability of the AMPK activator AICAR to increase surface KATP channels (Fig. 4A). However, inhibition of AMPK by compound C, although it abrogated the effect of leptin, failed to prevent two PKA activators, 8-bromo-cAMP and forskolin, from increasing surface channel expression (Fig. 4A). Because leptin increased the level of Thr(P)-172·AMPK (Fig. 3A), we further examined whether PKA activity affects AMPK phosphorylation. As shown in Fig. 4B, both leptin and AICAR increased Thr(P)-172·pAMPK signals after 30 min of treatment, but activation of PKA by 8-bromo-cAMP was without effect. Additionally, inhibition of PKA activity by PKI did not affect AMPK phosphorylation at Thr-172 induced by leptin or AICAR, although compound C diminished both leptin- and AICAR-induced AMPK phosphorylation (Fig. 4B). In summary, inhibition of PKA occluded the AMPK-mediated effect on surface SUR1 expression, but inhibition of AMPK did not alter the PKA-mediated effect. These results are consistent with AMPK acting upstream of PKA in the leptin signal transduction pathway that leads to increased KATP channel surface expression.
FIGURE 4.

AMPK acts upstream of PKA in the leptin signaling cascade that leads to increased surface KATP channel expression. A, INS-1 cells were pretreated with DMSO, 1 μm PKI, or 10 μm CC for 30 min before vehicle (Veh), 10 nm leptin (Lep), 10 μm 8-bromo-cAMP (8Br), or 250 μm AICAR (AIC) was applied for another 30 min. Surface SUR1 and total SUR1 were then analyzed as described under “Materials and Methods.” Inhibition of PKA activity by PKI significantly diminished the ability of leptin, AICAR, and 8-bromo-cAMP to increase surface SUR1. However, inhibition of AMPK activity with CC, although it abolished the effect of leptin or AICAR on surface SUR1 expression, did not block the ability of 8-bromo-cAMP to increase surface SUR1 levels. B, INS-1 cells treated as in A were lysed and subjected to immunoblotting analysis with phospho-AMPK and total AMPK antibodies. Top panel, representative blots show that phospho-AMPK signals were enhanced by leptin and AICAR but not by 8-bromo-cAMP. Pretreating cells with the PKA inhibitor PKI did not affect leptin or AICAR-induced AMPK phosphorylation; by contrast, pretreating cells with the AMPK inhibitor CC effectively abolished leptin or AICAR-induced AMPK phosphorylation. Bottom panel, bar graph shows the mean phospho-AMPK level relative to total AMPK normalized to that seen in vehicle (DMSO)-treated group (n = 4; *, p < 0.05 by one-way ANOVA and Dunnett's post hoc test).
Leptin Increases Surface KATP Channel Density by Promoting Channel Trafficking to the Plasma Membrane
The density of a membrane protein at the cell surface is a balance between its delivery to and removal from the plasma membrane. We asked whether the increased surface KATP channel expression upon leptin treatment is a consequence of reduced channel endocytosis or increased channel insertion into the plasma membrane. To test the former possibility, a pulse-chase protocol of surface-biotinylated SUR1 was employed. Surface SUR1 was biotinylated at 4 °C and chased for 15 or 30 min at 37 °C in the presence or absence of leptin. At the end of the chase, cells were treated with a reducing agent (MENSA; see under “Materials and Methods”) at 4 °C to remove the biotin label remaining at the cell surface. Internalized biotinylated SUR1 was then affinity- purified and detected by immunoblotting. At 15 min of chase, the amount of internalized biotinylated SUR1 in vehicle and leptin-treated cells was not significantly different (32.51 ± 1.41% versus 28.19 ± 0.79% of total biotinylated SUR1 at time 0). At 30 min, leptin-treated cells showed slightly reduced intracellular biotinylated SUR1 compared with control cells (15 ± 1.64% versus 21.91 ± 1.65% of total biotinylated SUR1 at time 0, p < 0.01, n = 6) (Fig. 5A). In parallel experiments where the residual surface biotin label was not stripped off at the end of each chase, there was no difference in total residual biotinylated SUR1 between leptin-treated and control cells (93.2 ± 6.83% for leptin versus 88.67 ± 11.23% for control, p > 0.05, n = 3), indicating leptin did not alter the degradation rate of surface KATP channels. These results suggest that reduced endocytic trafficking of KATP channels may contribute to the increased surface channel density observed in leptin-treated cells; however, the small decrease in the amount of endocytosed channels is insufficient to account for the increase in surface KATP channels engendered by leptin treatment (∼3–4-fold; Fig. 2, A and B).
We next examined whether leptin promotes channel delivery to the cell surface. To monitor channel delivery to the plasma membrane, we again took advantage of the BTX-tag SUR1/Kir6.2 variant, which we have shown to be subjected to leptin regulation like endogenous channels (Fig. 2C). INS-1 cells were transduced with BTX-tag SUR1 and Kir6.2 recombinant adenoviruses. Pre-existing surface BTX-tag SUR1 was first saturated with unlabeled BTX at 4 °C, and cells were then returned to 37 °C in a medium with or without leptin for 30 min. At the end of the incubation, newly inserted channels were labeled with either biotin-conjugated BTX at 4 °C and detected by immunoprecipitation followed by immunoblotting or with Alexa 555-conjugated BTX and detected by fluorescence microscopy. In both cases, a marked increase in newly inserted channels was observed in leptin-treated cells compared with control cells (Fig. 5B). Quantification of immunoblotting results showed that leptin induced a >2-fold increase in the amount of channels inserted during the 30-min time period (also see bar graph in Fig. 6). These results led us to conclude that leptin increases surface expression of KATP channels predominantly by promoting channel trafficking to the plasma membrane.
Leptin Induces F-actin Depolymerization via AMPK/PKA Signaling to Promote KATP Channel Trafficking to the Cell Surface
Cytoskeleton remodeling plays a role in membrane trafficking (34, 35). In previous studies, F-actin depolymerization has been found to coincide with leptin-induced increase of KATP channel conductance (18). We asked whether actin reorganization has a direct role in leptin-induced channel trafficking to the cell surface. Leptin treatment resulted in reduced F-actin staining by Alexa 488-conjugated phalloidin, indicating F-actin depolymerization (Fig. 6A). As the effect of leptin on surface KATP channel abundance is AMPK- and PKA-dependent, we examined whether the reduced F-actin staining observed upon leptin treatment shares the same signaling requirements. As shown in Fig. 6A, activation of AMPK by AICAR or PKA by 8-bromo-cAMP also led to reduced F-actin staining, whereas blocking AMPK with compound C or inhibition of PKA with PKI prevented leptin-induced F-actin remodeling. Consistent with the AMPK and PKA signaling relationship elucidated above, pretreating cells with compound C failed to block PKA-induced F-actin depolymerization. Using the experimental paradigm described above for Fig. 5B, we further determined whether F-actin depolymerization is correlated with increased channel insertion into the plasma membrane. These experiments show that treatments that led to F-actin depolymerization also led to increased channel insertion into the membrane; by contrast, treatments that prevented F-actin depolymerization also failed to yield enhanced channel trafficking to the cell surface (Fig. 6, A and B). These findings revealed a tight correlation between F-actin depolymerization and increased surface insertion of KATP channels evoked by the leptin/AMPK/PKA signaling cascade.
To determine whether F-actin depolymerization is required for KATP channel trafficking, we treated cells with drugs known to promote or block F-actin depolymerization and monitored cell surface expression of KATP channels in response to leptin signaling. In INS-1 cells pretreated for 10 min with 100 nm jasplakinolide, a cyclic peptide that binds and stabilizes filamentous actin (36), leptin failed to increase surface-biotinylated SUR1 (Fig. 7A). The same jasplakinolide treatment also blocked the ability of the AMPK activator AICAR as well as the PKA activators forskolin and 8-bromo-cAMP to increase surface SUR1 (Fig. 7B). These results support the notion that F-actin depolymerization is necessary for leptin, AMPK, and PKA to exert their effects on surface KATP channel expression. Conversely, treating cells with the F-actin depolymerizing drug latrunculin B (37) alone without leptin resulted in increased surface expression of KATP channels, indicating that F-actin depolymerization is sufficient to recruit channels to the plasma membrane.
FIGURE 7.

Actin depolymerization is required for increased KATP channel trafficking to the plasma membrane in response to leptin signaling. INS-1 cells were treated with 100 nm of the actin-stabilizing agent jasplakinolide (Jas) 10 min prior to treatment with 10 nm leptin (Lep), 250 μm AICAR, 10 μm forskolin (Fsk), or 8-bromo-cAMP (8Br) for 30 min. Veh, vehicle. Surface SUR1 and total SUR1 were analyzed as described in previous figures. Stabilization of F-actin by jasplakinolide prevented an increase in surface SUR1 caused by leptin (A), AICAR (B), or PKA activators (C). D, treating cells with the F-actin-destabilizing drug latrunculin B (100 nm) for 30 min resulted in increased surface SUR1 (triplicates are shown).
DISCUSSION
As the major determinant of the resting membrane potential of pancreatic β-cells, KATP channels serve as gatekeepers to control insulin secretion. In addition to the intricate gating regulation by ATP and ADP in response to glucose metabolism, variation in surface channel density in the plasma membrane is expected to alter total conductance to modulate membrane excitability. The study we present here demonstrates that surface expression of KATP channels is rapidly up-regulated by the hormone leptin. We show that leptin signals through AMPK and PKA to cause actin depolymerization and trafficking of KATP channels to the plasma membrane (Fig. 8). The study uncovers a signaling and cellular mechanism by which leptin regulates KATP channels and points to KATP channel trafficking regulation as an important physiological mechanism for controlling channel activity and thus insulin secretion by physiological signals such as leptin.
FIGURE 8.

Schematic diagram of leptin-induced trafficking of KATP channels to the plasma membrane in pancreatic β-cells. Binding of leptin to its receptor induces a signaling cascade that involves activation of AMPK and PKA. Activation of PKA leads to filamentous actin (F-actin) depolymerization to promote KATP channel trafficking to the plasma membrane.
How leptin increases β-cell KATP conductance has been under investigation ever since the phenomenon was first reported more than 15 years ago (12, 13). A number of signaling molecules, including PI3K and tyrosine kinases, were implicated in early studies (14, 16), although their roles were not further substantiated. Subsequent studies show that leptin causes inhibition of the lipid/protein phosphatase PTEN, accumulation of PIP3, and F-actin depolymerization, events that correlate with increased KATP conductance (15, 18, 19). Despite these efforts, the molecular and cellular events following leptin stimulation that lead to increased KATP activity remain incompletely understood. Because phosphoinositides phosphatidylinositol 4,5-bisphosphate and PIP3 are known to increase channel open probability and decrease channel sensitivity to ATP inhibition (38, 39), one possibility is that increased channel conductance results from increased PIP3 levels. Analysis of channel sensitivity to ATP inhibition and MgADP stimulation failed to show significant difference between control and leptin-treated cells (Fig. 1), disfavoring an effect of leptin signaling on channel gating property. These results steered us to focus on the alternative that leptin may increase KATP activity by increasing the number of channels present at the cell surface. The biochemical and imaging data we present here provide compelling evidence that leptin increases KATP conductance by promoting channel trafficking to the cell surface. Although we also observed a small but statistically significant reduction in the amount of endocytosed surface channels in leptin-treated cells, this mechanism is unlikely to be a major contributor when compared with the pronounced increase in the amount of channels delivered to the cell surface upon leptin stimulation. The findings that actin depolymerization is tightly coupled to the leptin-AMPK-PKA signaling axis and is both necessary and sufficient for increased KATP channel trafficking to the plasma membrane provide a mechanistic explanation for the link between actin reorganization and increased KATP conductance reported in previous studies (15, 18, 19). We note that a previous study by Lam et al. (17) showed that leptin reduces ATP concentrations in INS-1 cells by ∼30% by inhibiting glucose uptake. Although the current understanding of physiological regulation of KATP channels by intracellular ATP and ADP does not predict that a small decrease in ATP concentration will significantly increase channel activity (8), it remains a possibility that decreased ATP concentrations could potentially also contribute to the increased KATP conductance seen upon leptin treatment.
Actin remodeling has been shown to play an important role in vesicular transport associated with exocytosis (34, 35, 40, 41). In pancreatic β-cells, actin depolymerization precedes insulin granule exocytosis in response to glucose stimulation to facilitate SNARE protein interactions necessary for membrane fusion (42–44). This raises the question of how secretory vesicles that carry KATP channels are related to insulin granules. Interestingly, while early studies proposed that KATP channels reside predominantly in insulin granules (45, 46), a more recent study by Yang et al. (33) argues that KATP channels co-localize with chromogranin-positive but insulin-negative secretory granules. Clearly more work is needed to resolve this controversy. If KATP channel-carrying vesicles are distinct from insulin granules, it would be important to understand how actin depolymerization is temporally and spatially linked to specific signaling events to achieve regulated trafficking of distinct populations of vesicles in β-cells.
Leptin has been reported to activate AMPK in a number of cell systems (30, 47, 48). Our data show that leptin leads to increased phosphorylation of AMPK at Thr-172. Several kinases, including LKB1, CaMKK2, and TAK1, can phosphorylate AMPK (49). Although we did not investigate in this study how leptin leads to increased AMPK phosphorylation, a study just published by Park et al. (50), while this manuscript was under review, reported that leptin activates AMPK via CaMKK2, which is in turn activated by Ca2+ influx through TRPC4 channels to promote KATP channel trafficking to the cell surface. However, the study by Park et al. (50) did not explore the signaling and cellular events following AMPK activation that lead to increased channel trafficking to the cell surface. Interestingly, we found that both PKA inhibitors, H89 and PKI, abolished the ability of leptin and AMPK activators to induce actin depolymerization and KATP channel recruitment to the plasma membrane; moreover, PKA activators were sufficient to mimic the effect of leptin even in the presence of AMPK inhibitors. These results suggest that PKA acts downstream of AMPK in the leptin signaling pathway. To our knowledge, modulation of PKA activity by AMPK has not been previously documented, although a reverse regulation whereby phosphorylation of AMPK by PKA inhibits AMPK activity has been reported (51). If PKA is indeed activated by AMPK in the leptin signaling pathway, it would represent a novel downstream target of AMPK. However, until the complete signaling network is mapped out, we cannot exclude the possibility that PKA affects actin depolymerization and KATP channel trafficking through a parallel pathway that converges with the leptin-AMPK signaling cascade.
The molecular events that link AMPK and PKA activation to actin polymerization and KATP channel trafficking remain to be determined. Many proteins are involved in actin dynamics and play a role in vesicle trafficking in β-cells, including gelsolin, N-WASP, and cofilin (41, 44, 52). Some of these have been shown to be phosphorylated directly by PKA or AMPK, and some are regulated by kinases and phosphatases that are regulated by PKA or AMPK to promote actin depolymerization (53, 54). Actin depolymerization may then allow the mobilization of vesicles previously bound to the F-actin meshwork to permit membrane fusion (40, 44). AMPK and PKA could also phosphorylate KATP channels directly to affect channel trafficking. It is worth noting that a previous study has identified a PKA phosphorylation site in human SUR1 that appears to be involved in channel surface expression regulation (55). Phosphorylation of KATP channels by AMPK has also been proposed to modulate channel function (56, 57), although the details remain to be worked out. Whether channel phosphorylation is involved in the effect of leptin needs further investigation. In addition to proteins, phosphoinositides phosphatidylinositol 4,5-bisphosphate and PIP3 are well recognized for their roles in cytoskeleton remodeling and membrane trafficking in β-cells (58) and other cell types in general (59). As leptin signaling has also been reported to increase PIP3 levels (18, 19), an important question to address in the future is whether changes in phosphoinositides account for the observed F-actin depolymerization and KATP channel trafficking.
Leptin and insulin, secreted by adipocytes and β-cells, respectively, have been proposed to form a negative adipoinsular feedback loop to prevent hyperinsulinemia and hyperleptinemia. It is worth noting that in our experiments, the effect of leptin on AMPK phosphorylation and KATP channel surface expression peaked at ∼30 min and diminished with longer incubation (Fig. 2A). Intriguingly, circulating leptin levels in humans have been reported to follow a pulsatile pattern with ∼30 pulses every 24 h and each pulse lasting ∼30 min (60). The transient nature indicates that the leptin signaling pathway is tightly regulated. We speculate that the ability to turn off the leptin signal may prevent prolonged inhibition of insulin secretion when a subsequent rise in glucose calls for more insulin release.
In summary, we have uncovered a signaling pathway by which leptin modulates the abundance of KATP channels in the plasma membrane to regulate insulin secretion. Aside from β-cells, neurons in the central nervous system involved in a wide range of physiological and pathological processes, including energy homeostasis (18, 61–63), epilepsy (64, 65), and Parkinson disease (66), express KATP channels and are subjected to regulation by leptin and metabolic signals. Thus, the KATP channel regulatory mechanism identified here may extend well beyond pancreatic β-cells, with broad implications in metabolic regulation in health and disease.
Acknowledgments
We thank Dr. Christopher Newgard for the INS-1 cell clone 832/13 and Dr. Christopher Rhodes for providing the recombinant adenoviruses for the dominant-negative and constitutively active AMPK. We also thank Dr. Jeremy Bushman for help with the electrophysiology experiments on INS-1 cells, Erik Olson for technical assistance, and the staffs of the Advanced Light Microscopy Core at Jungers Center (Oregon Health and Science University, Portland, OR) for their expert help with image acquisition and analysis. Human islets were provided by the Integrated Islets Distribution Program through the Pilot Program for Human Islet Research.
Addendum
While this manuscript was being considered for publication, Park et al. (50) reported similar findings that leptin increases KATP channel trafficking to the plasma membrane.
This work was supported, in whole or in part, by National Institutes of Health Grant DK57699 (to S.-L. S.). This work was presented in abstract form at the 2013 American Society for Biochemistry and Molecular Biology (ASBMB) Annual Meeting.
This article was selected as a Paper of the Week.
- KATP channel
- ATP-sensitive potassium channel
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- AMPK
- AMP-activated protein kinase
- SUR1
- sulfonylurea receptor 1
- Kir6.2
- inwardly rectifying potassium channel Kir6.2
- ANOVA
- analysis of variance
- MENSA
- sodium 2-mercaptoethane sulfonate
- CC
- compound C
- BTX
- bungarotoxin
- PKI
- protein kinase A inhibitor
- PIP3
- phosphatidylinositol 3,4-bisphosphate
- EGFP
- enhanced GFP
- AICAR
- 5-aminoimidazole-4-carboxamide ribonucleotide
- K-INT
- internal potassium solution.
REFERENCES
- 1. Covey S. D., Wideman R. D., McDonald C., Unniappan S., Huynh F., Asadi A., Speck M., Webber T., Chua S. C., Kieffer T. J. (2006) The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 4, 291–302 [DOI] [PubMed] [Google Scholar]
- 2. Kieffer T. J., Habener J. F. (2000) The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 278, E1–E14 [DOI] [PubMed] [Google Scholar]
- 3. Kulkarni R. N., Wang Z. L., Wang R. M., Hurley J. D., Smith D. M., Ghatei M. A., Withers D. J., Gardiner J. V., Bailey C. J., Bloom S. R. (1997) Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Invest. 100, 2729–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morioka T., Asilmaz E., Hu J., Dishinger J. F., Kurpad A. J., Elias C. F., Li H., Elmquist J. K., Kennedy R. T., Kulkarni R. N. (2007) Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J. Clin. Invest. 117, 2860–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Niswender K. D., Magnuson M. A. (2007) Obesity and the beta cell: lessons from leptin. J. Clin. Invest. 117, 2753–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aguilar-Bryan L., Bryan J. (1999) Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20, 101–135 [DOI] [PubMed] [Google Scholar]
- 7. Ashcroft F. M. (2005) ATP-sensitive potassium channelopathies: focus on insulin secretion. J. Clin. Invest. 115, 2047–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476 [DOI] [PubMed] [Google Scholar]
- 9. Clement J. P., 4th, Kunjilwar K., Gonzalez G., Schwanstecher M., Panten U., Aguilar-Bryan L., Bryan J. (1997) Association and stoichiometry of KATP channel subunits. Neuron 18, 827–838 [DOI] [PubMed] [Google Scholar]
- 10. Inagaki N., Gonoi T., Seino S. (1997) Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 409, 232–236 [DOI] [PubMed] [Google Scholar]
- 11. Shyng S., Nichols C. G. (1997) Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110, 655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kieffer T. J., Heller R. S., Leech C. A., Holz G. G., Habener J. F. (1997) Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes 46, 1087–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harvey J., McKenna F., Herson P. S., Spanswick D., Ashford M. L. (1997) Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line, CRI-G1. J. Physiol. 504, 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harvey J., Ashford M. L. (1998) Role of tyrosine phosphorylation in leptin activation of ATP-sensitive K+ channels in the rat insulinoma cell line CRI-G1. J. Physiol. 510, 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harvey J., Hardy S. C., Irving A. J., Ashford M. L. (2000) Leptin activation of ATP-sensitive K+ (KATP) channels in rat CRI-G1 insulinoma cells involves disruption of the actin cytoskeleton [In Process Citation]. J. Physiol. 527, 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harvey J., McKay N. G., Walker K. S., Van der Kaay J., Downes C. P., Ashford M. L. (2000) Essential role of phosphoinositide 3-kinase in leptin-induced KATP channel activation in the rat CRI-G1 insulinoma cell line. J. Biol. Chem. 275, 4660–4669 [DOI] [PubMed] [Google Scholar]
- 17. Lam N. T., Cheung A. T., Riedel M. J., Light P. E., Cheeseman C. I., Kieffer T. J. (2004) Leptin reduces glucose transport and cellular ATP levels in INS-1 beta-cells. J. Mol. Endocrinol. 32, 415–424 [DOI] [PubMed] [Google Scholar]
- 18. Ning K., Miller L. C., Laidlaw H. A., Burgess L. A., Perera N. M., Downes C. P., Leslie N. R., Ashford M. L. (2006) A novel leptin signalling pathway via PTEN inhibition in hypothalamic cell lines and pancreatic beta-cells. EMBO J. 25, 2377–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ning K., Miller L. C., Laidlaw H. A., Watterson K. R., Gallagher J., Sutherland C., Ashford M. L. (2009) Leptin-dependent phosphorylation of PTEN mediates actin restructuring and activation of ATP-sensitive K+ channels. J. Biol. Chem. 284, 9331–9340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin Y. W., Bushman J. D., Yan F. F., Haidar S., MacMullen C., Ganguly A., Stanley C. A., Shyng S. L. (2008) Destabilization of ATP-sensitive potassium channel activity by novel KCNJ11 mutations identified in congenital hyperinsulinism. J. Biol. Chem. 283, 9146–9156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hohmeier H. E., BeltrandelRio H., Clark S. A., Henkel-Rieger R., Normington K., Newgard C. B. (1997) Regulation of insulin secretion from novel engineered insulinoma cell lines. Diabetes 46, 968–977 [DOI] [PubMed] [Google Scholar]
- 22. Meares G. P., Hughes K. J., Jaimes K. F., Salvatori A. S., Rhodes C. J., Corbett J. A. (2010) AMP-activated protein kinase attenuates nitric oxide-induced beta-cell death. J. Biol. Chem. 285, 3191–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Conget J. I., Sarri Y., González-Clemente J. M., Casamitjana R., Vives M., Gomis R. (1994) Deleterious effect of dithizone-DMSO staining on insulin secretion in rat and human pancreatic islets. Pancreas 9, 157–160 [DOI] [PubMed] [Google Scholar]
- 24. Yan F. F., Lin Y. W., MacMullen C., Ganguly A., Stanley C. A., Shyng S. L. (2007) Congenital hyperinsulinism-associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes 56, 2339–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin C. W., Lin Y. W., Yan F. F., Casey J., Kochhar M., Pratt E. B., Shyng S. L. (2006) Kir6.2 mutations associated with neonatal diabetes reduce expression of ATP-sensitive K+ channels: Implications in disease mechanism and sulfonylurea therapy. Diabetes 55, 1738–1746 [DOI] [PubMed] [Google Scholar]
- 26. Fehmann H. C., Peiser C., Bode H. P., Stamm M., Staats P., Hedetoft C., Lang R. E., Göke B. (1997) Leptin: a potent inhibitor of insulin secretion. Peptides 18, 1267–1273 [DOI] [PubMed] [Google Scholar]
- 27. Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., Newgard C. B. (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 [DOI] [PubMed] [Google Scholar]
- 28. Bruederle C. E., Gay J., Shyng S. L. (2011) A role of the sulfonylurea receptor 1 in endocytic trafficking of ATP-sensitive potassium channels. Traffic 12, 1242–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCann C. M., Bareyre F. M., Lichtman J. W., Sanes J. R. (2005) Peptide tags for labeling membrane proteins in live cells with multiple fluorophores. BioTechniques 38, 945–952 [DOI] [PubMed] [Google Scholar]
- 30. Minokoshi Y., Kim Y. B., Peroni O. D., Fryer L. G., Müller C., Carling D., Kahn B. B. (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343 [DOI] [PubMed] [Google Scholar]
- 31. Anderson K. A., Ribar T. J., Lin F., Noeldner P. K., Green M. F., Muehlbauer M. J., Witters L. A., Kemp B. E., Means A. R. (2008) Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 7, 377–388 [DOI] [PubMed] [Google Scholar]
- 32. Minokoshi Y., Alquier T., Furukawa N., Kim Y. B., Lee A., Xue B., Mu J., Foufelle F., Ferré P., Birnbaum M. J., Stuck B. J., Kahn B. B. (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428, 569–574 [DOI] [PubMed] [Google Scholar]
- 33. Yang S. N., Wenna N. D., Yu J., Yang G., Qiu H., Yu L., Juntti-Berggren L., Köhler M., Berggren P. O. (2007) Glucose recruits KATP channels via non-insulin-containing dense-core granules. Cell Metab. 6, 217–228 [DOI] [PubMed] [Google Scholar]
- 34. Muallem S., Kwiatkowska K., Xu X., Yin H. L. (1995) Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J. Cell Biol. 128, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson J. L., Monfregola J., Napolitano G., Kiosses W. B., Catz S. D. (2012) Vesicular trafficking through cortical actin during exocytosis is regulated by the Rab27a effector JFC1/Slp1 and the RhoA-GTPase-activating protein Gem-interacting protein. Mol. Biol. Cell 23, 1902–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holzinger A. (2009) Jasplakinolide: an actin-specific reagent that promotes actin polymerization. Methods Mol. Biol. 586, 71–87 [DOI] [PubMed] [Google Scholar]
- 37. Wakatsuki T., Schwab B., Thompson N. C., Elson E. L. (2001) Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J. Cell Sci. 114, 1025–1036 [DOI] [PubMed] [Google Scholar]
- 38. Baukrowitz T., Schulte U., Oliver D., Herlitze S., Krauter T., Tucker S. J., Ruppersberg J. P., Fakler B. (1998) PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282, 1141–1144 [DOI] [PubMed] [Google Scholar]
- 39. Shyng S. L., Nichols C. G. (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282, 1138–1141 [DOI] [PubMed] [Google Scholar]
- 40. Tobin V. A., Ludwig M. (2007) The actin filament and dendritic peptide release. Biochem. Soc. Trans. 35, 1243–1246 [DOI] [PubMed] [Google Scholar]
- 41. Tomas A., Yermen B., Min L., Pessin J. E., Halban P. A. (2006) Regulation of pancreatic beta-cell insulin secretion by actin cytoskeleton remodelling: role of gelsolin and cooperation with the MAPK signalling pathway. J. Cell Sci. 119, 2156–2167 [DOI] [PubMed] [Google Scholar]
- 42. Thurmond D. C., Gonelle-Gispert C., Furukawa M., Halban P. A., Pessin J. E. (2003) Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 17, 732–742 [DOI] [PubMed] [Google Scholar]
- 43. Jewell J. L., Luo W., Oh E., Wang Z., Thurmond D. C. (2008) Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J. Biol. Chem. 283, 10716–10726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Z., Thurmond D. C. (2009) Mechanisms of biphasic insulin-granule exocytosis-roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 122, 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Geng X., Li L., Watkins S., Robbins P. D., Drain P. (2003) The insulin secretory granule is the major site of KATP channels of the endocrine pancreas. Diabetes 52, 767–776 [DOI] [PubMed] [Google Scholar]
- 46. Varadi A., Grant A., McCormack M., Nicolson T., Magistri M., Mitchell K. J., Halestrap A. P., Yuan H., Schwappach B., Rutter G. A. (2006) Intracellular ATP-sensitive K+ channels in mouse pancreatic beta cells: against a role in organelle cation homeostasis. Diabetologia 49, 1567–1577 [DOI] [PubMed] [Google Scholar]
- 47. Gao S., Kinzig K. P., Aja S., Scott K. A., Keung W., Kelly S., Strynadka K., Chohnan S., Smith W. W., Tamashiro K. L., Ladenheim E. E., Ronnett G. V., Tu Y., Birnbaum M. J., Lopaschuk G. D., Moran T. H. (2007) Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc. Natl. Acad. Sci. U.S.A. 104, 17358–17363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Uotani S., Abe T., Yamaguchi Y. (2006) Leptin activates AMP-activated protein kinase in hepatic cells via a JAK2-dependent pathway. Biochem. Biophys. Res. Commun. 351, 171–175 [DOI] [PubMed] [Google Scholar]
- 49. Hardie D. G. (2004) The AMP-activated protein kinase pathway–new players upstream and downstream. J. Cell Sci. 117, 5479–5487 [DOI] [PubMed] [Google Scholar]
- 50. Park S. H., Ryu S. Y., Yu W. J., Han Y. E., Ji Y. S., Oh K., Sohn J. W., Lim A., Jeon J. P., Lee H., Lee K. H., Lee S. H., Berggren P. O., Jeon J. H., Ho W. K. (2013) Leptin promotes KATP channel trafficking by AMPK signaling in pancreatic beta-cells. Proc. Natl. Acad. Sci. U.S.A. 110, 12673–12678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Djouder N., Tuerk R. D., Suter M., Salvioni P., Thali R. F., Scholz R., Vaahtomeri K., Auchli Y., Rechsteiner H., Brunisholz R. A., Viollet B., Mäkelä T. P., Wallimann T., Neumann D., Krek W. (2010) PKA phosphorylates and inactivates AMPKα to promote efficient lipolysis. EMBO J. 29, 469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Uenishi E., Shibasaki T., Takahashi H., Seki C., Hamaguchi H., Yasuda T., Tatebe M., Oiso Y., Takenawa T., Seino S. (2013) Actin dynamics regulated by the balance of neuronal Wiskott-Aldrich syndrome protein (N-WASP) and cofilin activities determines the biphasic response of glucose-induced insulin secretion. J. Biol. Chem. 288, 25851–25864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee Y. M., Lee J. O., Jung J. H., Kim J. H., Park S. H., Park J. M., Kim E. K., Suh P. G., Kim H. S. (2008) Retinoic acid leads to cytoskeletal rearrangement through AMPK-Rac1 and stimulates glucose uptake through AMPK-p38 MAPK in skeletal muscle cells. J. Biol. Chem. 283, 33969–33974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bernstein B. W., Bamburg J. R. (2010) ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 20, 187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Béguin P., Nagashima K., Nishimura M., Gonoi T., Seino S. (1999) PKA-mediated phosphorylation of the human KATP channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 18, 4722–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang T. J., Chen W. P., Yang C., Lu P. H., Liang Y. C., Su M. J., Lee S. C., Chuang L. M. (2009) Serine-385 phosphorylation of inwardly rectifying K+ channel subunit (Kir6.2) by AMP-dependent protein kinase plays a key role in rosiglitazone-induced closure of the KATP channel and insulin secretion in rats. Diabetologia 52, 1112–1121 [DOI] [PubMed] [Google Scholar]
- 57. Yoshida H., Bao L., Kefaloyianni E., Taskin E., Okorie U., Hong M., Dhar-Chowdhury P., Kaneko M., Coetzee W. A. (2012) AMP-activated protein kinase connects cellular energy metabolism to KATP channel function. J. Mol. Cell. Cardiol. 52, 410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tomas A., Yermen B., Regazzi R., Pessin J. E., Halban P. A. (2010) Regulation of insulin secretion by phosphatidylinositol-4,5-bisphosphate. Traffic 11, 123–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Insall R. H., Weiner O. D. (2001) PIP3, PIP2, and cell movement–similar messages, different meanings? Dev. Cell 1, 743–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Licinio J., Mantzoros C., Negrão A. B., Cizza G., Wong M. L., Bongiorno P. B., Chrousos G. P., Karp B., Allen C., Flier J. S., Gold P. W. (1997) Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat. Med. 3, 575–579 [DOI] [PubMed] [Google Scholar]
- 61. Miki T., Liss B., Minami K., Shiuchi T., Saraya A., Kashima Y., Horiuchi M., Ashcroft F., Minokoshi Y., Roeper J., Seino S. (2001) ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat. Neurosci. 4, 507–512 [DOI] [PubMed] [Google Scholar]
- 62. Plum L., Ma X., Hampel B., Balthasar N., Coppari R., Münzberg H., Shanabrough M., Burdakov D., Rother E., Janoschek R., Alber J., Belgardt B. F., Koch L., Seibler J., Schwenk F., Fekete C., Suzuki A., Mak T. W., Krone W., Horvath T. L., Ashcroft F. M., Brüning J. C. (2006) Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J. Clin. Invest. 116, 1886–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang S. B., Tien A. C., Boddupalli G., Xu A. W., Jan Y. N., Jan L. Y. (2012) Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 75, 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Giménez-Cassina A., Martínez-François J. R., Fisher J. K., Szlyk B., Polak K., Wiwczar J., Tanner G. R., Lutas A., Yellen G., Danial N. N. (2012) BAD-dependent regulation of fuel metabolism and KATP channel activity confers resistance to epileptic seizures. Neuron 74, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liss B., Roeper J. (2001) A role for neuronal KATP channels in metabolic control of the seizure gate. Trends Pharmacol. Sci. 22, 599–601 [DOI] [PubMed] [Google Scholar]
- 66. Schiemann J., Schlaudraff F., Klose V., Bingmer M., Seino S., Magill P. J., Zaghloul K. A., Schneider G., Liss B., Roeper J. (2012) K-ATP channels in dopamine substantia nigra neurons control bursting and novelty-induced exploration. Nat. Neurosci. 15, 1272–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]