

Background: Clioquinol is a potent chelator of divalent metal ions including zinc.
Results: Clioquinol fits the zinc-centered active pocket of HDACs and inhibits HDAC activity, which results in cell cycle arrest and cancer cell apoptosis.
Conclusion: Clioquinol inhibits HDAC activity and induces blood cancer cell death.
Significance: This is the first report to demonstrate that clioquinol inhibits HDAC activity.
Keywords: Apoptosis, Computer Modeling, Enzyme Inhibitors, Histone Deacetylase, Multiple Myeloma, Clioquinol
Abstract
The antiparasitic clioquinol (CQ) represents a class of novel anticancer drugs by interfering with proteasome activity. In the present study, we found that CQ induced blood cancer cell apoptosis by inhibiting histone deacetylases (HDACs). CQ accumulated the acetylation levels of several key proteins including histone H3 (H3), p53, HSP90, and α-tubulin. In the mechanistic study, CQ was found to down-regulate HDAC1, -3, -4, and -5 in both myeloma and leukemia cells. Computer modeling analysis revealed that CQ was well docked into the active pocket of the enzyme, where the oxygen and nitrogen atoms in CQ formed stable coordinate bonds with the zinc ion, and the hydroxyl group from CQ formed an effective hydrogen bond with Asp-267. Moreover, co-treatment with CQ and zinc/copper chloride led to decreased Ac-H3. Furthermore, CQ inhibited the activity of Class I and IIa HDACs in the cell-free assays, demonstrating that CQ interfered with HDAC activity. By inhibiting HDAC activity, CQ induced expression of p21, p27, and p53, cell cycle arrest at G1 phase, and cell apoptosis. This study suggested that the HDAC enzymes are targets of CQ, which provided a novel insight into the molecular mechanism of CQ in the treatment of hematological malignancies.
Introduction
Acetylation is an important modification of lysine residues on histones and other proteins and plays a critical role in modulating gene expression as an epigenetic regulator (1–3). This acetylation/deacetylation process is controlled by two kinds of enzymes: histone acetyltransferases, which add acetyl groups to lysine residues, and histone deacetylases (HDACs),2 which remove the acetyl groups from substrate proteins. Acetylation neutralizes the positive charge in histone proteins by changing amines into amides, thus decreasing the ability of the histones to bind to DNA. This decreased binding allows chromatin expansion, providing access for transcription factors to bind to their target promoters and permitting genetic transcription to take place. In contrast, HDACs remove those acetyl groups, increasing the positive charge of histone tails and encouraging high affinity binding between histones and the DNA backbone. Thus HDACs modulate gene expression and cellular processes by deacetylating histones (1–4). Because deacetylation of histones condenses chromatin structure and represses gene transcription, it is hypothesized that by this mechanism, inhibition of HDACs slows down replication forks, activates dormant origins, and induces DNA damage and cancer cell apoptosis (5). HDACs are up-regulated in various cancers such as colorectal cancer, gastric cancer, and prostate cancer, and the depletion of HDACs could inhibit the growth of various human cancer cell lines (3, 6).
In recent years, accumulative evidence has suggested that inhibition of HDACs leads to the expression of tumor suppressor genes, such as p21 (7, 8), p27 (8, 9), and p53 (7, 10), therefore inducing cell cycle arrest and cell apoptosis (1, 9). HDAC inhibitors are emerging as a new class of anticancer agents for the treatment of solid and hematological malignancies (6). Several HDAC inhibitors such as suberoylanilide hydroxamic acid (6, 11) and the short chain fatty acid sodium butyrate have been under clinical evaluation for the treatment of blood cancers (6, 12).
HDACs could be divided into two groups based on their zinc dependence (4). Zinc-dependent HDACs include 11 members covering Classes I (HDAC1, -2, -3, and -8), II (HDAC4, -5, -6, -7, -9, and -10), and IV (HDAC11). The classic pharmacophore for HDAC inhibitors consists of three distinct structural motifs: the zinc-binding group, a hydrophobic linker, and a recognition cap group (13). The HDAC-specific zinc-binding motif adjacent to the active site is likely to participate in substrate recognition and protein-protein interaction and may provide a site for modulation of activity (14).
Clioquinol (CQ) is an antifungal and antiparasitic drug and is also a strong chelator of divalent metal ions such as zinc and copper (15). In the last decade, CQ (16–18) and its analogs including nitroxoline (19) and 5-amino-8-hydroxyl-quinoline (20) have been demonstrated to induce cancer cell death, including prostate cancer, bladder cancer, breast cancer, leukemia, and multiple myeloma (16, 18). Various mechanisms have been proposed in cell death by these agents. For example, nitroxoline kills bladder cancer by inhibiting type 2 methionine aminopeptidase protein and angiogenesis (19), although 5-amino-8-hydroxyl-quinoline is associated with proteasome inhibition (20). CQ is the most studied one. In the treatment of Alzheimer's disease, CQ acts as a chelator to bind to zinc and copper highly accumulated in the brain tissue (21). In the presence of zinc and copper, CQ displays great activity to inhibit proteasome activity and induces cancer cell death. CQ-induced cancer cell death is probably associated with lysosomal blockage (22) and reactive oxygen species induction (23). In the induction of cancer cell death, CQ is also found to inhibit NFκB and TNF signaling pathway, as well as modulation of zinc or copper transportation (24). These findings suggest that CQ probably acts in a broad panel of mechanisms. However, the capacity to chelate divalent metal ions is probably a key nature in the biological activity of CQ. Because divalent metal ions are essential for HDAC activity, we hypothesized that CQ may induce apoptosis by inhibiting HDAC activity via interfering with zinc in the active sites of these enzymes. In the present study, we analyzed the effects of CQ on HDAC activity by computer modeling and cell-free and cell-based assays and found that CQ-induced leukemia and myeloma cell death is associated with HDAC inhibition.
EXPERIMENTAL PROCEDURES
Cell Lines
Human multiple myeloma (MM) cell lines (RPMI-8226 and OPM2) and leukemia cell lines (U937, K562, and THP-1) were obtained from American Type Culture Collection, and MM cell lines (OCI-My5 and LP1) and leukemia cell line OCI-AML2 were generously provided by Dr. Aaron Schimmer (Ontario Cancer Institute, Toronto, Canada). All MM cell lines were maintained in Iscove's modified Dulbecco's medium (Invitrogen), and all leukemia cell lines were maintained in RPMI 1640 medium (Invitrogen). All media were supplemented with 10% fetal bovine serum (Invitrogen), 100 μg/ml of penicillin, and 100 units/ml of streptomycin (Hyclone).
Chemicals
Clioquinol was purchased from Calbiochem; ZnCl2 and CuCl2 were purchased from Sigma. Trichostatin A (TSA) was provided by Beyotime Institute of Biotechnology (Nantong, China).
Primary Chronic Myeloid Leukemia and MM Samples
Bone marrow from CML and MM patients was obtained from the First Affiliated Hospital of Soochow University with written permission from patients. The study was approved by the Ethical Review Board of Soochow University. Mononuclear cells were isolated from the samples by density centrifugation with Ficoll (Sigma). Primary cells were cultured at 37 °C in complete Iscove's modified Dulbecco's medium as described previously (8, 17).
Cell Cycle Analysis
Cells were treated with various concentrations of CQ for 24 h, harvested, washed with cold PBS, and then resuspended in 70% cold ethanol and incubated overnight at −20 °C. The cells were then treated with DNase-free RNase (Invitrogen) before staining with 50 μg/ml of propidium iodide. The detailed procedure and cell cycle analysis was performed as reported previously (25).
Western Blotting Analysis
Whole cell lysates were prepared in radioimmunoprecipitation assay buffer as described previously (26). Protein concentrations were determined by the BCA assay (Beyotime). Equal amounts (30 μg) of total proteins were subjected to fractionation by SDS-PAGE followed by transfer to PVDF membranes (Millipore). Anti-human antibodies against caspase-3, poly(ADP-ribose) polymerase, cyclin D2, cyclin D3, p21, p27, p53, HSP90, Histone H3, acetyl-histone 3 (Ac-H3K9), acetyl-p53 (Lys-379), acetyl-α-tubulin (Lys-40), and α-tubulin were purchased from Cell Signaling Technology. Anti-acetyl-HSP90 (Lys-294) was obtained from Rockland Immunochemicals. Antibodies against HDAC1, -2, -3, -4, and -5 were purchased from Biovision. Monoclonal antibodies against β-actin and GAPDH were purchased from Sigma. Secondary horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit IgG were purchased from Beyotime. Detection was performed by the Enhanced Chemical Luminescence method (Beyotime).
Molecular Docking and Molecular Simulation
The structure of CQ was constructed in Schrodinger (version 09). The structure of TSA complexed with HDAC8 (Protein Data Bank entry 1t64) (27) was used as the initial structure for the subsequent molecular docking studies. The Amber10 molecular simulation package was used to add hydrogen atoms and assign protonation states of residues in 1t64 (28). The atomic partial charges of CQ were derived by semiempirical AM1 geometry optimization and subsequent single-point Hartree-Fock/6-31G* calculations of the electrostatic potential, to which the charges were fitted using the RESP technique (29). The electrostatic potential was calculated using Gaussian09, and the partial charges and force field parameters of CQ were generated automatically using the antechamber program in AMBER10 (28). In simulations, the AMBER03 force field was used for proteins (30), and the general AMBER force field was used for ligands (31). The whole system was immersed in a rectangular box of TIP3P water molecules. The water box was extended 9 Å from solute atoms in all three dimensions. Then the complex was relaxed by 5,000 cycles of minimization procedure (500 cycles of steepest descent and 4,500 cycles of conjugate gradient minimization). The last structure from minimization was selected for the molecular docking studies. Then the inhibitor and the water molecules were removed from the minimized complex, and CQ was docked into the active site of HDAC8 using the Glide program in Schrodinger. The docking sites were restricted to a cube with a 15 Å radius around the zinc ion in the active site, and no additional constraints were defined. In molecular docking calculations, the precision level was set to extra precision.
Kinase Activity in Cell-free Assays
The effects of CQ on recombinant HDAC enzyme activities were performed by the Reaction Biology Corporation and determined with specific HDAC substrates. Briefly, recombinant HDACs and substrates were diluted in reaction buffer (50 mm Tris-HCl, pH 8.0, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2, 1 mg/ml BSA, 1% dimethyl sulfoxide). 5 nl of serial diluted CQ (in dimethyl sulfoxide) was added with the Echo 550 liquid handler (Labcyte Inc). According to the enzymatic specificity, a fluorogenic peptide from p53 residues 379–382 (RHKK(Ac)) was used as the substrate for Class I (HDAC1, -2, -3, and -5), Class IIb (HDAC6 and -10), and class IV HDACs (HDAC11), and a fluorogenic peptide from p53 residues 379–382 (RHK(Ac)K(Ac)) was used for HDAC8, whereas Boc-Lys(trifluoroacetyl)-AMC was used for Class IIA HDACs (HDAC4, -5, -7, and -9). The reaction was started by adding substrates into the reaction mixture and was stopped after 2 h of incubation at 30 °C. Substrate concentrations were analyzed at excitation and emission wavelengths of 360 and 460 nm.
Luciferase Activity Analysis
The structure of a luciferase reporter driven by cyclin D2 promoter was reported earlier (32). The reporter plasmid (5 μg) was transfected in murine NIH3T3 cell line for 24 h, followed by splitting and treatment with CQ for another 24 h. Luciferase activity in these cells were then measured with a Bright-GloTM luciferase assay system as reported previously (32).
RESULTS
Clioquinol Increases Acetylation of Histone H3 in Both Myeloma and Leukemia Cells
To determine the inhibition of CQ on HDAC, we first evaluated the effects of CQ on the acetylation status of histone H3, one of the main targets of HDACs and the biomarker of HDAC inhibition (1, 2). After incubation with 0, 5, 10, or 20 μm of CQ or 100 nm of TSA for 24 h, the Ac-H3 level was analyzed by Western blotting. The results showed that the pan-HDAC inhibitor TSA markedly increased the Ac-H3 level (Fig. 1). Similar to TSA, CQ increased Ac-H3 in both leukemia (THP-1) and myeloma (RPMI-8226) cells in a dose-dependent manner (Fig. 1a). To confirm this finding, we analyzed Ac-H3 expression in another leukemia (OCI-AML2) and MM cell line (LP1). Cells were treated with 10 μm of CQ for 12 -48 h and were found that Ac-H3 was induced by CQ in a time-dependent manner (Fig. 1b). In addition to Ac-H3, CQ also raised the acetylation level of p53, HSP90 (Fig. 1c), and α-tubulin (Fig. 1d), but p53 and HSP90 acetylation levels were seen at higher concentrations only, which was probably due to varied response sensitivity of individual proteins.
FIGURE 1.
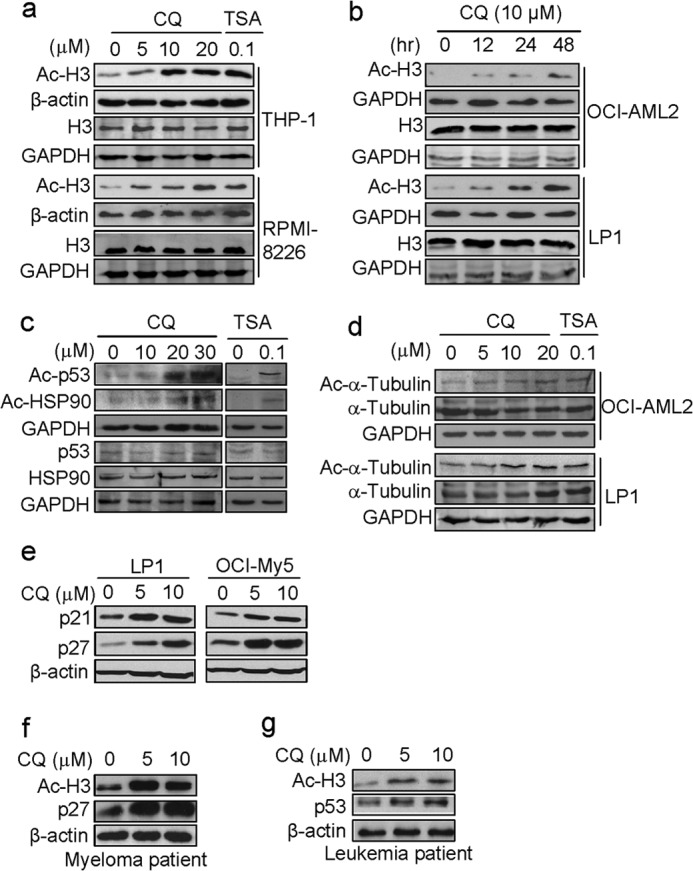
CQ increases protein acetylation. a, leukemic (THP-1) and myeloma (RPMI-8226) cells were treated with CQ (0, 5, 10, and 20 μm) or TSA for 24 h, followed by Ac-H3 and H3 analysis using Western blotting assay. b, leukemic (OCI-AML2) and myeloma (LP1) cells were treated with 10 μm of CQ for the indicated times, Ac-H3 and H3 was further evaluated by Western blotting. c, THP1 was treated with CQ (0, 10, 20, and 30 μm) for 24 h. Acetylated p53, acetylated HSP90, p53, and HSP90 levels were analyzed with specific antibodies. d, OCI-AML2 and LP1 cells was treated with CQ (0, 5, 10, and 20 μm) or TSA (0.1 μm) for 24 h. Cell lysates were applied for analysis of acetylated α-tubulin and α-tubulin. e, MM cells (LP1 and OCI-My5) were treated with CQ at indicated concentrations for 24 h. Proteins of p21 and p27 were evaluated using Western blotting against specific antibodies. f, mononuclear cells from the bone marrow of primary myeloma patients were treated with CQ (0, 5, and 10 μm) for 24 h. Cell lysates were then prepared and subjected to Western blotting analysis for Ac-H3 and p27. g, mononuclear cells from the bone marrow of primary leukemia patients were treated with CQ (0, 5, 10 μm) for 24 h. Cell lysates were then prepared and subjected to Western blotting analysis for Ac-H3 and p53.
Because p21 and p27 expression is closely modulated by histone hyperacetylation and inhibition of HDACs induces their expression (33, 34), we next examined the expression of p21 and p27 in MM cell lines LP1 and OCI-My5 using specific antibodies and found that both p21 and p27 were induced by CQ (Fig. 1e), which was consistent with previous studies (34). This effect was also observed in primary myeloma and leukemia cells. CQ raised the Ac-H3 level in primary cells, along with increased p27 (Fig. 1f) and p53 proteins (Fig. 1g).
Effects of Clioquinol on Expression of Histone Deacetylases
Increase of Ac-H3 may result from down-regulation of the expression of HDACs and/or the inhibition of HDAC activity (8). HDACs are expressed in most cancer cells, including leukemia (35, 36), lymphoma (36), and myelomas (8, 36), and these enzymes are partly associated with chemosensitivity and clinical outcome (37). To determine the effects of CQ on the expression of HDACs, we examined HDAC1, -2, -3, -4, and -5 in CQ-treated myeloma and leukemia cells. Western blotting analysis using specific anti-human HDAC enzymes indicated the effects of CQ on HDACs varied. HDAC2 expression was not significantly changed by CQ in all cell lines examined (Fig. 2, a and b), whereas HDAC1, -3, and -5 were decreased in all cell lines by CQ at 20 μm or higher concentrations (Fig. 2). Because CQ increased H3 acetylation level at 5 or 10 μm (Fig. 2a), other events were probably involved in CQ-induced HDAC inhibition, for example, direct interaction between CQ and HDAC molecules.
FIGURE 2.
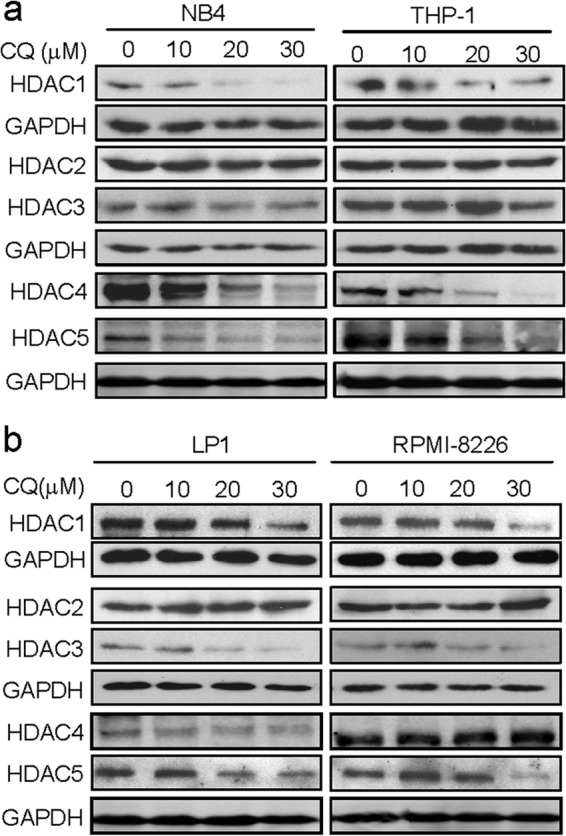
CQ suppresses HDAC expression. Leukemia (NB4 and THP-1) (a) and myeloma (LP1 and RPMI-8226) (b) cells were treated with CQ at indicated concentrations for 24 h. Cell lysates were then prepared and subjected to Western blotting analysis for the protein expression of representative HDAC enzymes (HDACs 1–5) using specific antibodies. GAPDH was used as a loading control.
Clioquinol Is Docked into the Active Pocket of HDAC8 and Inhibits HDAC Activity by Interfering with Zinc
To investigate whether CQ interacts with HDAC enzymes, we first predicted the interaction using the Glide program in Schrodinger as we did previously (8). HDAC8 has been well studied and thus was used as a prototype of HDACs (8, 38). The results suggested that CQ was well docked into the active pocket of HDAC8 (Fig. 3a), where it formed strong and stable coordinate bonds with zinc ion and interfered with the interaction of zinc and Asp-178 and of zinc and Asp-267 (Fig. 3b). Additionally, the hydroxyl group of CQ formed an effective hydrogen bond with Asp-267. Moreover, CQ formed extensive van der Waals interactions with three hydrophobic residues: Trp-141, Phe-152, and Tyr-306 (Fig. 3b). These results indicated that CQ strongly interacted with HDAC, which may contribute to the functional inhibition of HDAC.
FIGURE 3.

CQ inhibits HDAC activity by interacting with zinc and the active pocket of HDACs. a, HDAC8 was used as the prototype to analyze the interaction of CQ and HDAC using the Glide program in Schrodinger (version 09). CQ was well docked into the active pocket of HDAC8. b, CQ formed stable coordinate bonds with the zinc ion and interacted with residues around the active pocket of HDAC. c, zinc or copper did not increase Ac-H3 level. LP1 cells were treated ZnCl2, CuCl2, or TSA for 24 h. Cell lysates were then prepared for AC-H3 and H3 analysis. GAPDH was used as a loading control. d and e, addition of zinc or copper attenuated Ac-H3 accumulation raised by CQ. LP1 cells were co-treated with 15 μm of CQ and zinc (d) or copper (e) chloride for 24 h. Cell lysates were then prepared and subjected to Western blotting analysis for Ac-H3 and H3. GAPDH was used as a loading control.
To confirm our hypothesis that CQ inhibits HDAC by interfering with zinc ion, we performed an experiment by co-treating LP1 cells with CQ and increasing concentrations of ZnCl2 or CuCl2 followed by analysis of Ac-H3 level. As shown in Fig. 4c, zinc or cupper had no effects on H3 acetylation. However, the addition of zinc or copper attenuated the effects of CQ on accumulation of Ac-H3 in a concentration-dependent manner (Fig. 3, d and e). This result was possibly due to the formation of CQ-Zn or CQ-Cu complex, which prevented CQ from accessing to and interacting with HDACs.
FIGURE 4.

CQ inhibits HDAC activity. Recombinant Class IIa (HDAC4, -5, -7, and -9) enzymes were incubated with increased CQ and Boc-Lys (trifluoroacetyl)-AMC for 2 h at 30 °C followed by substrate measurement at excitation and emission wavelengths of 360 and 460 nm. a, HDAC4. b, HDAC5. c, HDAC7. d, HDAC9. Solid squares, Test 1 for CQ; open squares, Test 2 for CQ; solid triangles, TSA treatment.
Clioquinol Inhibits Class I and IIa but Not Class IIb or IV HDAC Activity
There are 18 HDACs in human cells, of which 11 are zinc-dependent. To find out the inhibition profile of CQ on these HDACs, we analyzed HDAC activities in a cell-free assay by incubating CQ with HDAC enzymes, followed by measuring the content of fluorogenic substrates as described in the section of “Experimental Procedures.” Compared with the pan-HDAC inhibitor TSA, CQ inhibited the activity of Class I (HDAC1, -2, and -8; Table 1) and Class IIa HDACs (HDAC4, -5, -7, and -9) (Fig. 4 and Table 1) but had no effects on the activity of Class IIb (HDAC6 and -10), Class IV (HDAC11), or HDAC3 (a member of Class I) (Table 1), suggesting that CQ selectively inhibited HDAC activity. Because CQ was the only factor to interfere with HDAC activity in the cell-free system, this experiment confirmed that CQ inhibited HDAC activity by interfering with the active pocket in HDAC, such as disrupting zinc function.
TABLE 1.
The IC50 values of CQ to zinc-dependent HDACs
Recombinant HDACs were incubated with CQ and individual substrates for 2 h at 30 °C before applying for substrate analysis. ND, not detectable; IC50, the concentration (mol/liter) that inhibits 50% of the HDAC activity.
| HDACs | CQ-TEST1 | CQ-TEST2 | Tricostatin A |
|---|---|---|---|
| Class I | |||
| HDAC1 | 1.06E-04 | 1.71E-04 | 5.22E-09 |
| HDAC2 | 1.16E-04 | 9.56E-05 | 1.15E-08 |
| HDAC3 | ND | ND | 5.58E-09 |
| HDAC8 | 2.07E-05 | 2.25E-05 | 7.01E-08 |
| Class 2A | |||
| HDAC4 | 3.11E-05 | 5.24E-05 | 3.23E-06 |
| HDAC5 | 3.07E-05 | 3.04E-05 | 1.48E-06 |
| HDAC7 | 9.68E-05 | 9.04E-05 | 2.95E-06 |
| HDAC9 | 5.70E-05 | 7.46E-05 | 4.04E-06 |
| Class IIB | |||
| HDAC6 | ND | ND | 1.22E-09 |
| HDAC10 | ND | ND | 1.40E-08 |
| Class IV | |||
| HDAC11 | ND | ND | 1.83E-08 |
Clioquinol Arrests Blood Cancer Cells at G0/G1 Phase
Because inductions of p21, p27, and p53 lead to cell cycle arrest in G0/G1 phase, which is also an important event, resulted in HDAC inhibition (6, 34), we next analyzed the cell cycle of the CQ-treated leukemia (OCI-AML2 and U937) and myeloma (LP1 and OCI-My5) cells. Cell cycle analysis by flow cytometry showed that all cells treated with 20 μm of CQ were arrested at G0/G1 phase (data not shown). Compared with the control, G0/G1 fractions were increased by 20–40%.
Clioquinol Induces Apoptosis in Both Myeloma and Leukemia Cell Lines
Inhibition of HDAC leads to cancer cell apoptosis, to investigate whether CQ can induce cell apoptosis, MM cell lines (LP1, OCI-My5) and leukemia cell lines (Jurkat, MDAY-D2) were treated with CQ for 24 h, followed by apoptosis analysis using Annexin V-FITC and PI double staining. All cells underwent apoptosis in the presence of CQ, and this effect was concentration-dependent (data not shown). CQ-induced apoptosis was further confirmed by the activation of executive apoptosis enzyme caspase-3, as well as the cleavage of poly(ADP-ribose) polymerase in both myeloma (RPMI-8226) and leukemia (NB4) cells (Fig. 5). Consistent with our hypothesis and previous studies, CQ induced leukemia and MM cell apoptosis.
FIGURE 5.
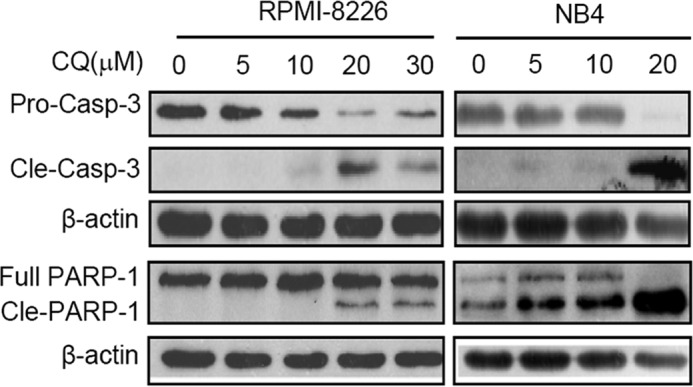
Clioquinol activates the apoptotic signals. MM cell line RPMI-8226 and leukemia cell line NB4 were used for apoptotic enzyme analysis. Cells were treated with indicated concentrations for 24 h, followed by total protein extraction and Western blotting analyses using specific antibodies against caspase-3 and poly(ADP-ribose) polymerase (PARP). β-Actin was used as a loading control.
Clioquinol Acts as an Inhibitor of Both HDACs and Proteasomes
The above study has shown that CQ was an inhibitor of HDACs and induced cell cycle arrest and apoptosis of both leukemia and myeloma cells. However, previous studies have well established that CQ is also an inhibitor of proteasomes (16–18). Cell cycle arrest and cell apoptosis can result from inhibition of both HDACs and proteasomes; to demonstrate that at least part of cell death or cell cycle arrest from CQ is via inhibition of HDAC, we examined the effect of CQ on cell cycle regulator D-cyclins. D-cyclin transcription is modulated by HDACs (39), whereas their degradation is processed via the proteasomes (40). Therefore, we first examined the expression of cyclin D3 (CCND3) in leukemia cell lines K562 and OCI-AML2 treated with CQ or bortezomib, a typical proteasome inhibitor. As shown in Fig. 6a, bortezomib treatment led to an increased CCND3 in both cell lines in a concentration-dependent manner. Notably, increase of CDND3 in K562 was not significant as that in OCI-AML2, which is probably due to overexpression of β5 subunit that renders K562 resistant to bortezomib (20). In contrast, CQ decreased CCND3 in both cell lines (Fig. 6b). To confirm this effect, we examined cyclin D2 (CCND2), another member in D-cyclins, in myeloma cells LP1 and OCI-My5 treated by CQ. Western blotting analysis revealed that CQ decreased CCND2 in both cell lines (Fig. 6c). Therefore, in terms of the effects on D-cyclins, CQ did not act as an inhibitor of proteasome. If CQ down-regulated D-cyclins as an inhibitor, it should suppress their transcription. To verify this hypothesis, we next made a luciferase construct driven by cyclin D2 promoter. This plasmid was transfected into NIH3T3 cells followed by CQ treatment for 24 h. Luciferase activity analysis revealed that cyclin D2 promoter-driven luciferase activity was decreased by CQ in a concentration-dependent manner (Fig. 6d). This finding thus suggested that CQ regulates CCND2 transcription by inhibiting its promoter activity. Therefore, taken together the present and previous studies, we could conclude that CQ induced blood cancer cell death via both HDAC and proteasome pathways.
FIGURE 6.
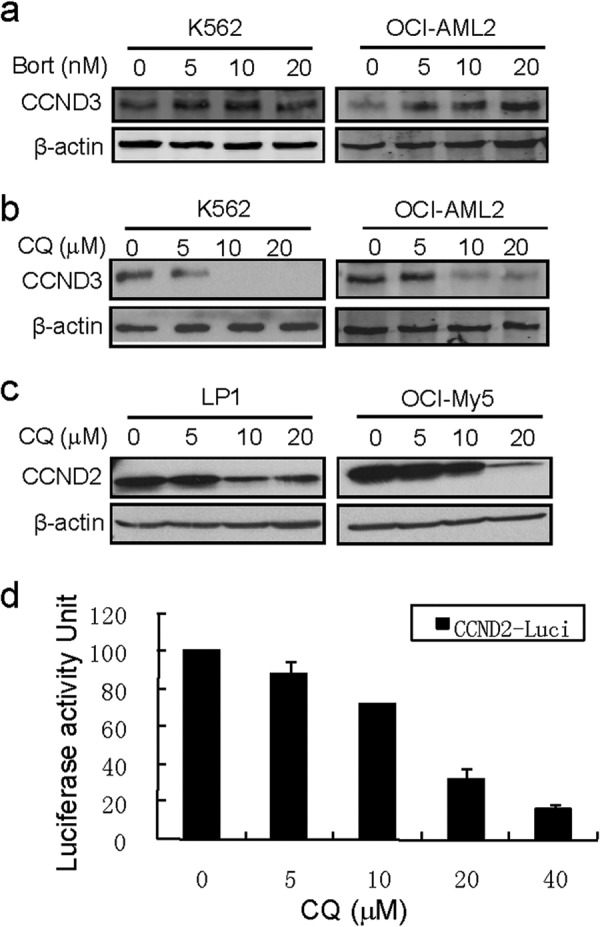
Clioquinol suppresses cyclin D transcription. a and b, K562 and OCI-AML2 cells were treated with bortezomib (Bort, a) or CQ (b) for 24 h at the indicated concentrations. CCND3 were evaluated by Western blotting. c, LP1 and OCI-My5 were treated with CQ for 24 h, and CCND2 expression was evaluated by Western blotting analysis. d, a luciferase construct driven by cyclin D2 promoter was transfected in NIH3T3 cells, 24 h later, cells were treated with CQ for another 24 h. Luciferase activity was measured by adding a luciferase substrate (Bright-GloTM; Promega) on a plate reader (Luminoskan; Fisher Thermo).
DISCUSSION
Through all above studies by computer modeling, cell-free and cell-based assays, we found that CQ inhibited HDAC activity probably by binding to and interfering with zinc in the active center of these enzymes. In the last decade, the antimicrobial agent CQ has been repurposed for the treatment for cancers (15), Alzheimer disease and Parkinson disease (15). In cancer treatment, CQ was found to bind to zinc or copper to form a novel class of proteasome inhibitors (24). CQ was also found to bind to zinc and to disable lysosomal activity (22). Another finding is that CQ binds to copper and zinc, thus modulating the transportation of these ions (41). In Alzheimer and Parkinson disease treatment, CQ binds to copper, thus decreasing copper concentration accumulated in brain tissues (21). CQ is also able to inhibit RNA-dependent DNA polymerase and inactivates Rous sarcoma virus and Herpes simplex virus by forming a complex with copper (42). By careful analysis, we can find that one of the key features in the physiochemical properties of CQ in the therapeutic activity is that CQ possesses the capacity to chelate divalent metal ions such as zinc and copper. Because zinc is essential for HDAC activity, CQ induces cancer cell death, probably by interfering with HDAC function.
In the family of HDACs, most members including Class I (HDAC1, -2, -3, and -8), Class II (HDAC4,-5, -6, 7-, 9, and -10), and Class IV (HDAC11) are zinc-dependent, and zinc is localized to the active center of these enzymes. One of important features of HDAC inhibitors is that they bear a strong Zn2+ binding group in the structure (13). For example, the classic HDAC inhibitors TSA and suberoylanilide hydroxamic acid are proposed to block the catalytic reaction by chelating the zinc ion in the active site pocket through its hydroxamic acid group (43) and to form close contact with the residues that make up the active site conserved across the HDAC family (43). CQ has adjacent nitrogen and hydroxyl groups that form the zinc-binding domain in its chemical structure. Computer modeling revealed that CQ forms strong and stable coordinates with HDACs. Specifically, CQ binds to the zinc atom at the active pocket of HDAC via its oxygen and nitrogen. In addition, CQ also formed strong van de Waals forces with three adjacent hydrophobic residues: Trp-141, Phe-152, and Tyr-306, and an effective hydrogen bond with Asp-267. Thus CQ interferes with HDAC active pocket, which probably leads to the change of the conformation of the enzyme or blocks the access of the substrate to the active pocket. This interaction of CQ with zinc pocket was confirmed by the addition of zinc, which resulted in attenuated H3 acetylation.
The inhibition of CQ on HDAC activity was further confirmed in the deacetylation assay by incubation of CQ and purified HDAC enzymes. In the cell-free enzyme system, CQ specifically inhibited Class I (except HDAC3) and IIa HDAC activities but had no effects on Class IIb or IV HDACs. This selectivity in HDAC inhibition is possibly due to the structural difference in individual enzymes. CQ also down-regulates the expression of HDACs including HDAC1, -3, -4, and -5, which solidifies the inhibition of HDAC activity, especially at high doses.
Acetylation is an important modification of proteins at the post-translational level and plays a critical role in regulation of gene transcription and protein stability (1, 2, 4). A direct outcome from HDAC inhibition is that hyperacetylation of histone enables the transcription of a small subset of tumor suppressor genes including p21 (1, 4). Additionally, HDAC1 can bind to the promoter of p21 and inhibits its expression (44). When HDAC is decreased, the inhibition will be lifted from p21 promoter and triggers p21 transcription (44). In our study, CQ led to increased expression of p21, p27, and p53, which could result from HDAC inhibition as reported previously (2). In contrast to the induction of p21 and p27, cyclin D was down-regulated by CQ, which is probably associated with hyperacetylation of NFκB because of the inhibition of HDACs. NFκB hyperacetylation status hinders the access of NFκB to the D-cyclin promoter (39). Thus, by binding to HDAC active pocket and decreasing expression of HDACs, CQ inhibits HDAC activity, which leads to condensed chromosomes, and selectively induces some key genes. Notably, CQ not only induced p53 expression but also increased its acetylation level. This is important for CQ-induced cell apoptosis because acetylation of p53 abrogates Mdm2-mediated repression by blocking the recruitment of Mdm2 to p53-responsive promoters, which leads to p53 activation independent of its phosphorylation status (45). Moreover, p53 acetylation can enhance its sequence-specific DNA binding thus regulated expression of some specific genes (46).
To be noted, CQ is also an inhibitor of proteasomes. As reported earlier, both CQ (16, 17, 47) and its analog 5-amino-8-hydroxyl-quinoline (20) inhibit proteasomal enzymatic activity both in vitro and in vivo. In this study, CQ also presents itself as an effective inhibitor of HDACs. Is a proteasomal inhibitor an HDAC inhibitor, or it is just a coincidence? A recent study may give the answer. Kikuchi et al. (48) reported that HDACs are critical targets of bortezomib, the typical proteasomal inhibitor, which specifically down-regulates the expression of class I HDACs (HDAC1, HDAC2, and HDAC3) in MM cell lines and primary MM cells at the transcriptional level, accompanied by histone hyperacetylation, as we showed in this study. Bortezomib down-regulates HDACs because it induces caspase-8-dependent degradation of Sp1 protein, the most potent transactivator of HDACs (48). The detailed mechanisms under the cross-talk between proteasome and HDAC signals in CQ-induced cell death should be further investigated.
Taken together, by computer modeling and in vitro and in vivo assays, we demonstrated that CQ inhibits HDAC activity by interacting and interfering with the residues and zinc in the active pocket of HDACs. Because it has also been demonstrated as a proteasome inhibitor, CQ could induce blood cancer cell apoptosis via inhibiting both HDAC and bortezomib pathways.
This work was supported by National Natural Science Foundation of China Grants 81272632, 81071935, 81101795, and 81320108023; Natural Science Foundation of Jiangsu Province Grants BK2011268 and BK2010218; National Basic Research Program of China Program 973 Grant 2011CB933501; and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
- HDAC
- histone deacetylase
- Ac-H3
- acetylated histone H3
- CQ
- clioquinol
- MM
- multiple myeloma
- TSA
- trichostatin A.
REFERENCES
- 1. Rodríguez-Paredes M., Esteller M. (2011) Cancer epigenetics reaches mainstream oncology. Nat. Med. 17, 330–339 [DOI] [PubMed] [Google Scholar]
- 2. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 3. Yang X. J., Seto E. (2007) HATs and HDACs. From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 [DOI] [PubMed] [Google Scholar]
- 4. Gallinari P., Di Marco S., Jones P., Pallaoro M., Steinkühler C. (2007) HDACs, histone deacetylation and gene transcription. From molecular biology to cancer therapeutics. Cell Res. 17, 195–211 [DOI] [PubMed] [Google Scholar]
- 5. Conti C., Leo E., Eichler G. S., Sordet O., Martin M. M., Fan A., Aladjem M. I., Pommier Y. (2010) Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 70, 4470–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lane A. A., Chabner B. A. (2009) Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 27, 5459–5468 [DOI] [PubMed] [Google Scholar]
- 7. Condorelli F., Gnemmi I., Vallario A., Genazzani A. A., Canonico P. L. (2008) Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br. J. Pharmacol. 153, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mao X., Hou T., Cao B., Wang W., Li Z., Chen S., Fei M., Hurren R., Gronda M., Wu D., Trudel S., Schimmer A. D. (2011) The tricyclic antidepressant amitriptyline inhibits D-cyclin transactivation and induces myeloma cell apoptosis by inhibiting histone deacetylases. In vitro and in silico evidence. Mol. Pharmacol. 79, 672–680 [DOI] [PubMed] [Google Scholar]
- 9. Chen J. S., Faller D. V. (2005) Histone deacetylase inhibition-mediated post-translational elevation of p27KIP1 protein levels is required for G1 arrest in fibroblasts. J. Cell. Physiol. 202, 87–99 [DOI] [PubMed] [Google Scholar]
- 10. Zhang F., Shi Y., Wang L., Sriram S. (2011) Role of HDAC3 on p53 expression and apoptosis in T cells of patients with multiple sclerosis. PLoS One 6, e16795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kirschbaum M., Frankel P., Popplewell L., Zain J., Delioukina M., Pullarkat V., Matsuoka D., Pulone B., Rotter A. J., Espinoza-Delgado I., Nademanee A., Forman S. J., Gandara D., Newman E. (2011) Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin's lymphoma and mantle cell lymphoma. J. Clin. Oncol. 29, 1198–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piekarz R., Bates S. (2004) A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr. Pharm. Des. 10, 2289–2298 [DOI] [PubMed] [Google Scholar]
- 13. Chen P. C., Patil V., Guerrant W., Green P., Oyelere A. K. (2008) Synthesis and structure-activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg. Med. Chem. 16, 4839–4853 [DOI] [PubMed] [Google Scholar]
- 14. Schuetz A., Min J., Allali-Hassani A., Schapira M., Shuen M., Loppnau P., Mazitschek R., Kwiatkowski N. P., Lewis T. A., Maglathin R. L., McLean T. H., Bochkarev A., Plotnikov A. N., Vedadi M., Arrowsmith C. H. (2008) Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 283, 11355–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mao X., Schimmer A. D. (2008) The toxicology of clioquinol. Toxicol. Lett. 182, 1–6 [DOI] [PubMed] [Google Scholar]
- 16. Daniel K. G., Chen D., Orlu S., Cui Q. C., Miller F. R., Dou Q. P. (2005) Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 7, R897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao X., Li X., Sprangers R., Wang X., Venugopal A., Wood T., Zhang Y., Kuntz D. A., Coe E., Trudel S., Rose D., Batey R. A., Kay L. E., Schimmer A. D. (2009) Clioquinol inhibits the proteasome and displays preclinical activity in leukemia and myeloma. Leukemia 23, 585–590 [DOI] [PubMed] [Google Scholar]
- 18. Chen D., Cui Q. C., Yang H., Barrea R. A., Sarkar F. H., Sheng S., Yan B., Reddy G. P., Dou Q. P. (2007) Clioquinol, a therapeutic agent for Alzheimer's disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 67, 1636–1644 [DOI] [PubMed] [Google Scholar]
- 19. Shim J. S., Matsui Y., Bhat S., Nacev B. A., Xu J., Bhang H. E., Dhara S., Han K. C., Chong C. R., Pomper M. G., So A., Liu J. O. (2010) Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J. Natl. Cancer Inst. 102, 1855–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li X., Wood T. E., Sprangers R., Jansen G., Franke N. E., Mao X., Wang X., Zhang Y., Verbrugge S. E., Adomat H., Li Z. H., Trudel S., Chen C., Religa T. L., Jamal N., Messner H., Cloos J., Rose D. R., Navon A., Guns E., Batey R. A., Kay L. E., Schimmer A. D. (2010) Effect of noncompetitive proteasome inhibition on bortezomib resistance. J. Natl. Cancer Inst. 102, 1069–1082 [DOI] [PubMed] [Google Scholar]
- 21. Bareggi S. R., Cornelli U. (2012) Clioquinol. Review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci. Ther. 18, 41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu H., Zhou Y., Lind S. E., Ding W. Q. (2009) Clioquinol targets zinc to lysosomes in human cancer cells. Biochem. J. 417, 133–139 [DOI] [PubMed] [Google Scholar]
- 23. Chen H. L., Chang C. Y., Lee H. T., Lin H. H., Lu P. J., Yang C. N., Shiau C. W., Shaw A. Y. (2009) Synthesis and pharmacological exploitation of clioquinol-derived copper-binding apoptosis inducers triggering reactive oxygen species generation and MAPK pathway activation. Bioorg. Med. Chem. 17, 7239–7247 [DOI] [PubMed] [Google Scholar]
- 24. Schimmer A. D. (2011) Clioquinol. A novel copper-dependent and independent proteasome inhibitor. Curr. Cancer Drug Targets 11, 325–331 [DOI] [PubMed] [Google Scholar]
- 25. Mao X., Liang S. B., Hurren R., Gronda M., Chow S., Xu G. W., Wang X., Beheshti Zavareh R., Jamal N., Messner H., Hedley D. W., Datti A., Wrana J. L., Zhu Y., Shi C. X., Lee K., Tiedemann R., Trudel S., Stewart A. K., Schimmer A. D. (2008) Cyproheptadine displays preclinical activity in myeloma and leukemia. Blood 112, 760–769 [DOI] [PubMed] [Google Scholar]
- 26. Mao X., Cao B., Wood T. E., Hurren R., Tong J., Wang X., Wang W., Li J., Jin Y., Sun W., Spagnuolo P. A., MacLean N., Moran M. F., Datti A., Wrana J., Batey R. A., Schimmer A. D. (2011) A small-molecule inhibitor of D-cyclin transactivation displays preclinical efficacy in myeloma and leukemia via phosphoinositide 3-kinase pathway. Blood 117, 1986–1997 [DOI] [PubMed] [Google Scholar]
- 27. Somoza J. R., Skene R. J., Katz B. A., Mol C., Ho J. D., Jennings A. J., Luong C., Arvai A., Buggy J. J., Chi E., Tang J., Sang B. C., Verner E., Wynands R., Leahy E. M., Dougan D. R., Snell G., Navre M., Knuth M. W., Swanson R. V., McRee D. E., Tari L. W. (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12, 1325–1334 [DOI] [PubMed] [Google Scholar]
- 28. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bayly C. I., Cieplak P., Cornell W. D., Kollman P. A. (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges. The Resp model. J. Phys. Chem. 97, 10269–10280 [Google Scholar]
- 30. Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., Kollman P. (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 [DOI] [PubMed] [Google Scholar]
- 31. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 32. Mao X., Stewart A. K., Hurren R., Datti A., Zhu X., Zhu Y., Shi C., Lee K., Tiedemann R., Eberhard Y., Trudel S., Liang S., Corey S. J., Gillis L. C., Barber D. L., Wrana J. L., Ezzat S., Schimmer A. D. (2007) A chemical biology screen identifies glucocorticoids that regulate c-maf expression by increasing its proteasomal degradation through up-regulation of ubiquitin. Blood 110, 4047–4054 [DOI] [PubMed] [Google Scholar]
- 33. Mercurio C., Minucci S., Pelicci P. G. (2010) Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacol. Res. 62, 18–34 [DOI] [PubMed] [Google Scholar]
- 34. Bolden J. E., Peart M. J., Johnstone R. W. (2006) Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug. Discov. 5, 769–784 [DOI] [PubMed] [Google Scholar]
- 35. Claus R., Lübbert M. (2003) Epigenetic targets in hematopoietic malignancies. Oncogene 22, 6489–6496 [DOI] [PubMed] [Google Scholar]
- 36. Melnick A., Licht J. D. (2002) Histone deacetylases as therapeutic targets in hematologic malignancies. Curr. Opin. Hematol. 9, 322–332 [DOI] [PubMed] [Google Scholar]
- 37. Marquard L., Gjerdrum L. M., Christensen I. J., Jensen P. B., Sehested M., Ralfkiaer E. (2008) Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 53, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wood T. E., Dalili S., Simpson C. D., Sukhai M. A., Hurren R., Anyiwe K., Mao X., Suarez Saiz F., Gronda M., Eberhard Y., MacLean N., Ketela T., Reed J. C., Moffat J., Minden M. D., Batey R. A., Schimmer A. D. (2010) Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol. Cancer Ther. 9, 246–256 [DOI] [PubMed] [Google Scholar]
- 39. Hu J., Colburn N. H. (2005) Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-κB/p65 DNA binding. Mol. Cancer Res. 3, 100–109 [DOI] [PubMed] [Google Scholar]
- 40. Hu X., Bryington M., Fisher A. B., Liang X., Zhang X., Cui D., Datta I., Zuckerman K. S. (2002) Ubiquitin/proteasome-dependent degradation of D-type cyclins is linked to tumor necrosis factor-induced cell cycle arrest. J. Biol. Chem. 277, 16528–16537 [DOI] [PubMed] [Google Scholar]
- 41. Zhai S., Yang L., Cui Q. C., Sun Y., Dou Q. P., Yan B. (2010) Tumor cellular proteasome inhibition and growth suppression by 8-hydroxyquinoline and clioquinol requires their capabilities to bind copper and transport copper into cells. J. Biol. Inorg Chem. 15, 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rohde W., Mikelens P., Jackson J., Blackman J., Whitcher J., Levinson W. (1976) Hydroxyquinolines inhibit ribonucleic acid-dependent deoxyribonucleic acid polymerase and inactivate Rous sarcoma virus and herpes simplex virus. Antimicrob. Agents. Chemother. 10, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Finnin M. S., Donigian J. R., Cohen A., Richon V. M., Rifkind R. A., Marks P. A., Breslow R., Pavletich N. P. (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193 [DOI] [PubMed] [Google Scholar]
- 44. Lagger G., Doetzlhofer A., Schuettengruber B., Haidweger E., Simboeck E., Tischler J., Chiocca S., Suske G., Rotheneder H., Wintersberger E., Seiser C. (2003) The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol. Cell. Biol. 23, 2669–2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. (2008) Acetylation is indispensable for p53 activation. Cell 133, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo J., Li M., Tang Y., Laszkowska M., Roeder R. G., Gu W. (2004) Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Du T., Filiz G., Caragounis A., Crouch P. J., White A. R. (2008) Clioquinol promotes cancer cell toxicity through tumor necrosis factor α release from macrophages. J. Pharmacol. Exp. Ther. 324, 360–367 [DOI] [PubMed] [Google Scholar]
- 48. Kikuchi J., Wada T., Shimizu R., Izumi T., Akutsu M., Mitsunaga K., Noborio-Hatano K., Nobuyoshi M., Ozawa K., Kano Y., Furukawa Y. (2010) Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 116, 406–417 [DOI] [PubMed] [Google Scholar]