

Background: RhoG is a ubiquitously expressed member of the Rho family of GTPases.
Results: RhoG-deficient platelets display severely impaired GPVI-dependent platelet activation.
Conclusion: RhoG plays an important role in GPVI-FcRγ complex-mediated platelet activation and thrombus formation.
Significance: Our study enhances the understanding of the molecular mechanisms of GPVI-FcRγ complex activation.
Keywords: Collagen, Fc Receptors, Platelets, Rho GTPases, Thrombosis, Glycoprotein VI
Abstract
We investigated the mechanism of activation and functional role of a hitherto uncharacterized signaling molecule, RhoG, in platelets. We demonstrate for the first time the expression and activation of RhoG in platelets. Platelet aggregation, integrin αIIbβ3 activation, and α-granule and dense granule secretion in response to the glycoprotein VI (GPVI) agonists collagen-related peptide (CRP) and convulxin were significantly inhibited in RhoG-deficient platelets. In contrast, 2-MeSADP- and AYPGKF-induced platelet aggregation and secretion were minimally affected in RhoG-deficient platelets, indicating that the function of RhoG in platelets is GPVI-specific. CRP-induced phosphorylation of Syk, Akt, and ERK, but not SFK (Src family kinase), was significantly reduced in RhoG-deficient platelets. CRP-induced RhoG activation was consistently abolished by a pan-SFK inhibitor but not by Syk or PI3K inhibitors. Interestingly, unlike CRP, platelet aggregation and Syk phosphorylation induced by fucoidan, a CLEC-2 agonist, were unaffected in RhoG-deficient platelets. Finally, RhoG−/− mice had a significant delay in time to thrombotic occlusion in cremaster arterioles compared with wild-type littermates, indicating the important in vivo functional role of RhoG in platelets. Our data demonstrate that RhoG is expressed and activated in platelets, plays an important role in GPVI-Fc receptor γ-chain complex-mediated platelet activation, and is critical for thrombus formation in vivo.
Introduction
Platelets play a key role in hemostasis and thrombosis. Contact of circulating platelets with exposed collagen leads to platelet activation, resulting in platelet aggregation. Adhesion of platelets to collagen is mediated mainly by integrin α2β1, whereas activation is mediated by the signal-transducing glycoprotein VI (GPVI)3 (1, 2). GPVI is a platelet collagen receptor that is constitutively associated with the Fc receptor γ-chain (FcRγ) (3, 4). FcRγ is phosphorylated by SFK (Src family kinase) at the tyrosine residue in its immunoreceptor tyrosine-based activation motif (ITAM) upon GPVI-collagen binding, and the tyrosine kinase Syk (spleen tyrosine kinase) binds to the ITAM and becomes autophosphorylated (5–8). Activation of Syk leads to phosphorylation of a protein complex signalosome, such as LAT (linker for T cell activation) and SLP76 (Src homology 2-containing leukocyte protein 76); recruitment of Btk (Bruton's tyrosine kinase); and activation of PI3K (9–12). These adaptor proteins interact with several other signaling molecules to regulate phospholipase Cγ2 (PLCγ2) (13), the major effector of the GPVI signaling cascade, which leads to intracellular calcium mobilization and PKC activation.
Rho GTPases are members of the Ras superfamily and play crucial role in the regulation of many cellular functions, including cell adhesion, motility, proliferation, and differentiation (14). RhoA, Rac, and Cdc42 are the most characterized members, and their major function is to regulate the assembly and organization of the actin cytoskeleton (15). RhoA, Rac, and Cdc42 have been shown to control the assembly of focal adhesion complexes associated with actin-myosin filaments, lamellipodia, and filopodia, respectively (16). RhoG, a ubiquitously expressed member of the Rho family of GTPases (17–19), is most similar to Rac1 and Cdc42 in sequence identity and function (20). RhoG−/− mice have elevated levels of IgG1 and IgG2b antibodies. There is a mild hyper-reactivity defect in RhoG-deficient B2 cells or T cells (18). RhoG is thought to induce actin rearrangements and membrane protrusion by Rac activation through the ELMO-DOCK180 pathway. Binding to ELMO allows RhoG to link to Rac activation by the Rac guanine nucleotide exchange factor DOCK180, resulting in the formation of lamellipodial protrusions (21–24). RhoG may also influence the microtubule system through a specific interaction with the protein kinectin (25). In endothelial cells, constitutively active RhoG binds to the PI3K regulatory subunit p85α and induces PI3K-dependent phosphorylation of Akt (26). Although RhoG is involved in the regulation of actin cytoskeletal rearrangements, nothing has been studied regarding the role of RhoG in platelet activation.
In this study, we investigated the role of RhoG in platelet functional responses and the mechanism of RhoG activation in platelets. Our studies indicate that the small GTPase RhoG is present in both human and mouse platelets. RhoG is activated by the GPVI-specific agonist collagen-related peptide (CRP) in platelets. We also show that RhoG regulates GPVI-FcRγ complex-mediated signaling and is involved in the regulation of Syk, Akt, and ERK downstream of GPVI. Moreover, the role of RhoG in GPVI-FcRγ complex signaling appears to contribute to platelet function in vivo because RhoG−/− mice are protected from thrombotic injury. We provide evidence that RhoG plays an important role in ITAM-coupled GPVI-FcRγ complex signaling in platelets.
EXPERIMENTAL PROCEDURES
Materials
2-MeSADP, sodium citrate, fucoidan, acetylsalicylic acid, apyrase (type VII), and bovine serum albumin (fraction V) were purchased from Sigma. AYPGKF was custom-synthesized by Invitrogen. CRP was purchased from Dr. Richard Farndale at the University of Cambridge. Convulxin was purified as described (27). Wortmannin and PP2 were from Enzo Life Sciences (Plymouth Meeting, PA). OXSI-2 was from Calbiochem. Phycoerythrin-conjugated antibody JON/A, FITC-conjugated anti-P-selectin antibody, and FITC-conjugated anti-GPVI antibody were from Emfret Analytics (Wurzburg, Germany). Anti-phospho-Syk (Tyr525/Tyr526), anti-phospho-Akt (Ser473), anti-phospho-Src (Tyr416), anti-phospho-ERK (Thr202/Tyr204), and anti-β-actin antibodies were purchased from Cell Signaling Technology (Beverly, MA). HRP-labeled secondary antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). All other reagents were reagent-grade, and deionized water was used throughout.
Animals
RhoG-deficient mice (28) were obtained from Dr. Len Stephens (The Babraham Institute, Cambridge, United Kingdom).
Preparation of Human and Mouse Platelets
Human blood was obtained from a pool of healthy volunteers in a 0.16 volume of acid/citrate/dextrose. Platelet-rich plasma was prepared by centrifugation of citrated blood at 230 × g for 20 min at room temperature. Acetylsalicylic acid was added to platelet-rich plasma to a final concentration of 1 mm, and the preparation was incubated for 45 min at 37 °C, followed by centrifugation at 980 × g for 10 min at room temperature.
Mouse blood was collected from anesthetized mice into syringes containing 0.1 blood volume of 3.8% sodium citrate as anticoagulant. Red blood cells were removed by centrifugation at 100 × g for 10 min at room temperature. Platelet-rich plasma was recovered, and platelets were pelleted at 400 × g for 10 min at room temperature. The platelet pellet was resuspended in Tyrode's buffer (pH 7.4) containing 0.05 units/ml apyrase to a density of 1.5 × 108 cells/ml.
Platelet Aggregation and Secretion
Platelet aggregation was measured using a lumi-aggregometer (Chrono-log Corp., Havertown, PA) at 37 °C under stirring conditions. A 0.5-ml sample of washed platelets was stimulated with different agonists, and the change in light transmission was measured.
Platelet secretion was determined by measuring the release of ATP by adding luciferin/luciferase reagent. Platelet ATP release and aggregation were performed simultaneously in a lumi-aggregometer at 37 °C.
Flow Cytometry
Washed murine platelets were used to measure the agonist-dependent level of activated αIIbβ3 receptors with phycoerythrin-conjugated antibody JON/A and the surface exposure of P-selectin with FITC-conjugated anti-P-selectin antibody as described previously (29). All determinations were performed on a FACSCalibur flow cytometer (BD Biosciences).
Western Blotting
Platelets were stimulated with agonists for the appropriate time, and phosphorylation events were measured as described previously (30).
Measurement of RhoG Activation
Full-length ELMO (a gift from Dr. Kodi Ravichandran, University of Virginia) was expressed in Escherichia coli as a fusion protein with GST and purified on glutathione-Sepharose beads. For measurement of RhoG activity, washed platelets (5 × 108 cells) were activated with CRP as described in the figure legends. Cells were then lysed with an equal volume of 2× ice-cold cell lysis buffer (50 mm HEPES (pH 7.5), 100 mm NaCl, 20 mm MgCl2, 2% Nonidet P-40, 4% glycerol, 1 mm PMSF, 10 μg/ml leupeptin, and 20 mm GTP) and quickly frozen in a methanol/dry ice bath until the experiment was completed. Samples were defrosted on ice and centrifuged at 10,000 × g for 15 min at 4 °C, and the supernatants were incubated with GST-ELMO-Sepharose beads for 60 min at 4 °C. The beads were washed three times with lysis buffer, and bound proteins were analyzed by SDS-PAGE and immunoblotting. Relative RhoG activity was determined by the amount of RhoG bound to GST-ELMO normalized to the amount of RhoG in cell lysates.
Platelet Spreading and Clot Retraction
Washed platelets (1 × 107 platelets/ml) were plated on fibrinogen-coated coverslips and incubated at 37 °C in a CO2 incubator. Adherent cells were fixed with 3.7% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 3 min. Rhodamine-phalloidin (Invitrogen) was used to stain F-actin. For clot retraction, platelets (5 × 108 platelets/ml) were added to a glass cuvette and mixed with 1 mm CaCl2 and 0.2 units/ml thrombin. Exogenous fibrinogen was added to a final concentration of 0.1 mg/ml. Platelets were allowed to retract and were then photographed at the indicated time points.
Photo Dye-induced in Vivo Thrombosis Model
The photo dye-induced thrombosis model was modified from a previously reported model (31). Briefly, mice were anesthetized, and the cremaster muscle was exteriorized. 5% FITC-dextran (10 ml/kg) was slowly injected retro-orbitally and allowed to circulate for 10 min prior to photoactivation of the vessel. Photoactivation was then initiated by exposing 100 μm of vessel length to epi-illumination. Epi-illumination was applied continuously, and the progress of platelet adhesion and thrombus formation was monitored. Microvessel data were obtained using an upright Olympus BX51 microscope with a water immersion objective, and the image was recorded using a Photometrics QuantEM digital camera. Data were further analyzed using MetaMorph software. The occlusion time was determined at the time that the vessel lumen was occluded by the thrombus.
Statistical Analysis
All statistical tests were carried out using Prism software (version 3.0). Data are presented as means ± S.E. Statistical significance was determined by Student's t test and analysis of variance. p < 0.05 was considered statistically significant.
RESULTS
Expression of the Small GTPase RhoG in Platelets
While we were evaluating the proteins in the platelets using proteomic approaches, we identified RhoG in platelets. It has not been known previously whether RhoG is expressed in platelets or not. Hence, we first evaluated the expression levels of RhoG in human platelets. Western analysis revealed that RhoG is expressed in human platelets (Fig. 1A). We obtained mice lacking RhoG for our study and then determined the RhoG expression in these mice. As shown in Fig. 1B, wild-type littermate platelets contained RhoG, whereas RhoG was not detected in RhoG−/− platelets, confirming the genotype of these mice. These results confirm that RhoG is expressed in both human and mouse platelets.
FIGURE 1.
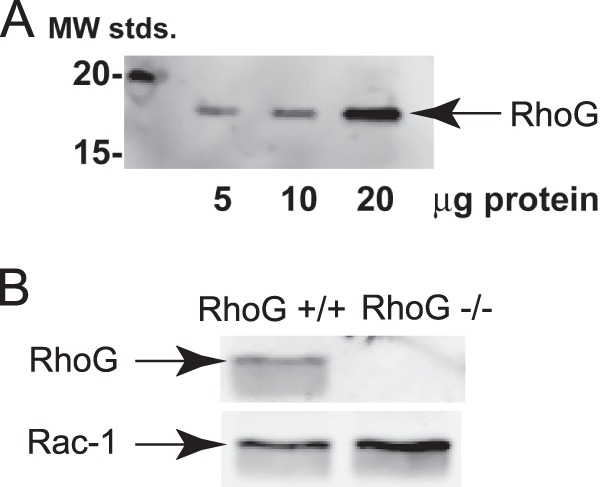
Expression of RhoG in human and mouse platelets. A, increasing amounts of human platelet lysate (in μg) were separated by SDS-PAGE, Western-blotted, and probed with anti-RhoG antibody. MW stds., molecular weight standards. B, RhoG expression in platelet lysates from RhoG-null mice (RhoG−/−) and wild-type littermates (RhoG+/+) was probed by Western blotting RhoG. Rac1 was probed to verify protein loading. The blot shown is representative of three independent experiments.
Impaired Platelet Functional Responses Downstream of GPVI Pathways in the Absence of RhoG
There are no known pharmacological inhibitors of RhoG. Thus, to evaluate the functional implications of RhoG in platelets, we compared the various agonist-induced platelet responses in wild-type and RhoG-deficient platelets. As shown in Fig. 2A, we found that platelet aggregation and dense granule secretion induced by the GPVI-specific agonists CRP and convulxin were significantly inhibited in RhoG-deficient platelets compared with wild-type littermate platelets. However, platelet aggregation and secretion in response to the PAR4-activating peptide AYPGKF or 2-MeSADP were not affected in RhoG-deficient platelets (Fig. 2B), indicating that G protein-coupled receptor agonists induce normal platelet aggregation and secretion in the absence of RhoG. Thus, these results suggest that RhoG selectively regulates platelet functional responses downstream of GPVI pathways.
FIGURE 2.

Agonist-induced platelet aggregation and dense granule secretion in RhoG-deficient platelets. Washed platelets from RhoG−/− mice and RhoG+/+ littermates were stimulated with the GPVI agonists CRP (2.5 μg/ml) and convulxin (CVX; 100 ng/ml) (A) and the G protein-coupled receptor agonists 2-MeSADP (30 nm) and AYPGKF (100 μm) (B) for 3.5 min under stirring conditions. Platelet aggregation and ATP secretion were measured by aggregometry. Tracings are representative of three independent experiments.
A similar approach was taken to analyze the activation of integrin αIIbβ3 by measuring binding of the conformation-dependent antibody JON/A and α-granule secretion by analysis of P-selectin expression using flow cytometry. Consistent with the results shown in Fig. 2, we found that both JON/A binding and P-selectin expression induced by different concentrations of CRP were significantly reduced in RhoG-deficient platelets compared with wild-type platelets (Fig. 3A). However, at both low and high concentrations, AYPGKF-induced JON/A binding and P-selectin expression were not affected in RhoG-deficient platelets (Fig. 3B), confirming that G protein-coupled receptor agonists cause integrin αIIbβ3 activation and α-granule secretion independently of RhoG.
FIGURE 3.

Integrin αIIbβ3 activation and α-granule secretion in RhoG-deficient platelets. Washed platelets from RhoG−/− mice and RhoG+/+ littermates were stimulated with the indicated doses of CRP (A) and AYPGKF (B) for 15 min in the presence of phycoerythrin-labeled antibody JON/A to determine integrin αIIbβ3 activation or FITC-labeled anti-P-selectin (CD62) antibody to evaluate α-granule secretion by flow cytometry. C, surface expression of the GPVI receptor in RhoG+/+ (gray) and RhoG−/− (black line) mice determined by flow cytometry. The data shown are representative of three experiments. Data are means ± S.E. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.005 compared with wild-type mice. MFI, mean fluorescence intensity.
To determine whether the role of RhoG in GPVI pathways is due to down-regulation of GPVI receptor expression, we measured the level of platelet surface expression of GPVI in RhoG−/− platelets by flow cytometry. We found that the level of GPVI receptor expression in RhoG−/− platelets was similar to that in wild-type platelets (Fig. 3C), indicating that the expression of GPVI is not affected by the absence of RhoG.
GPVI-mediated Signaling Events Are Inhibited in RhoG-deficient Platelets
To investigate the signaling events regulated by RhoG, platelets from RhoG-null and wild-type littermates were stimulated with CRP and AYPGKF, and the phosphorylation of downstream signaling molecules was evaluated by immunoblotting with phosphospecific antibodies. Fig. 4A shows that CRP-induced Syk, Akt, and ERK phosphorylation was significantly inhibited in RhoG-deficient platelets. Interestingly, tyrosine phosphorylation of SFK (upstream of Syk) was not altered. When platelets from RhoG−/− mice were stimulated with AYPGKF, phosphorylation of Akt and ERK was not affected by the absence of RhoG. These results indicate that RhoG plays an important role downstream of GPVI and upstream of Syk, possibly at the FcRγ level.
FIGURE 4.
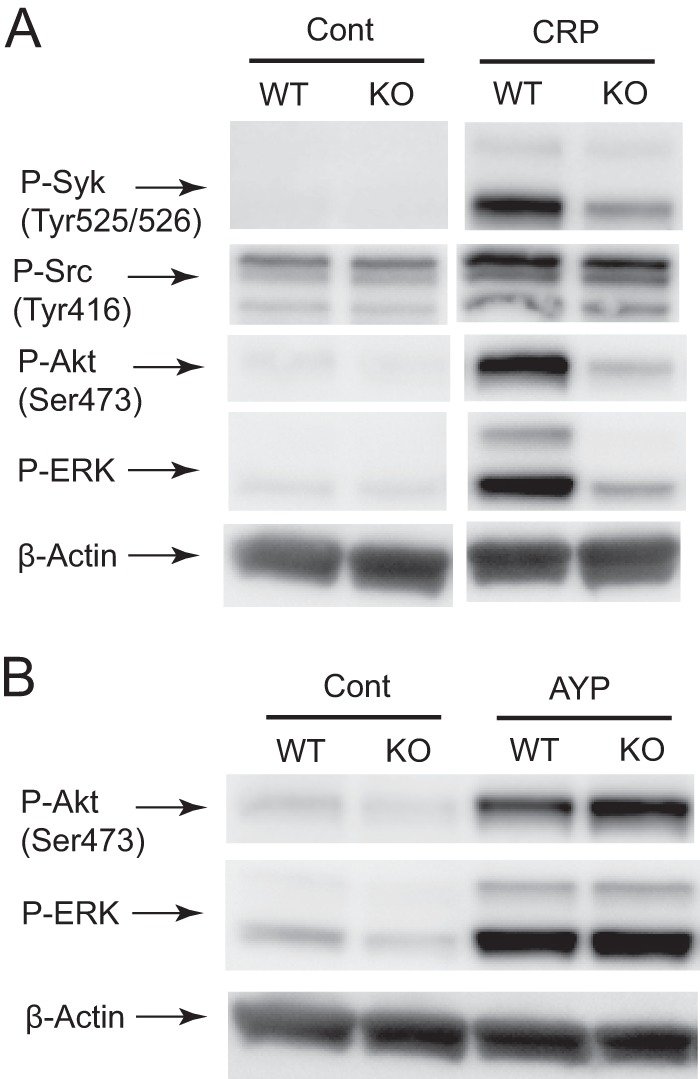
RhoG deficiency inhibits CRP-induced Syk, Akt, and ERK phosphorylation, but not Src phosphorylation. Washed platelets from RhoG−/− mice and RhoG+/+ littermates were stimulated with 2.5 μg/ml CRP (A) and 100 μm AYPGKF (B) at 37 °C for 2 min and probed with anti-phospho (P)-Syk (Tyr525/Tyr526), anti-phospho-Src (Tyr416), anti-phospho-Akt (Ser473), anti-phospho-ERK, and anti-β-actin (lane loading control) antibodies by Western blotting. The Western analysis shown is representative of three experiments. Cont, control; KO, knock-out.
RhoG Activation and Its Regulation Induced by CRP
We next determined whether RhoG is activated downstream of GPVI pathways. As shown in Fig. 5A, RhoG was activated by CRP in a time-dependent manner, where active GTP-bound RhoG was detected as early as 30 s after stimulation. To determine the signaling mechanism underlying the regulation of RhoG activation downstream of GPVI, we first evaluated the role of SFK in RhoG activation. Consistent with the results shown in Fig. 4A, where SFK phosphorylation was not affected by the absence of RhoG, CRP-induced RhoG activation was completely blocked by the pan-SFK inhibitor PP2 (Fig. 5B), suggesting that GPVI-mediated RhoG activation is regulated by SFK. However, CRP-induced RhoG activation was not inhibited by the Syk inhibitor OXSI-2 or the PI3K inhibitor wortmannin (Fig. 5B), indicating that Syk and PI3K are not required for RhoG activation.
FIGURE 5.
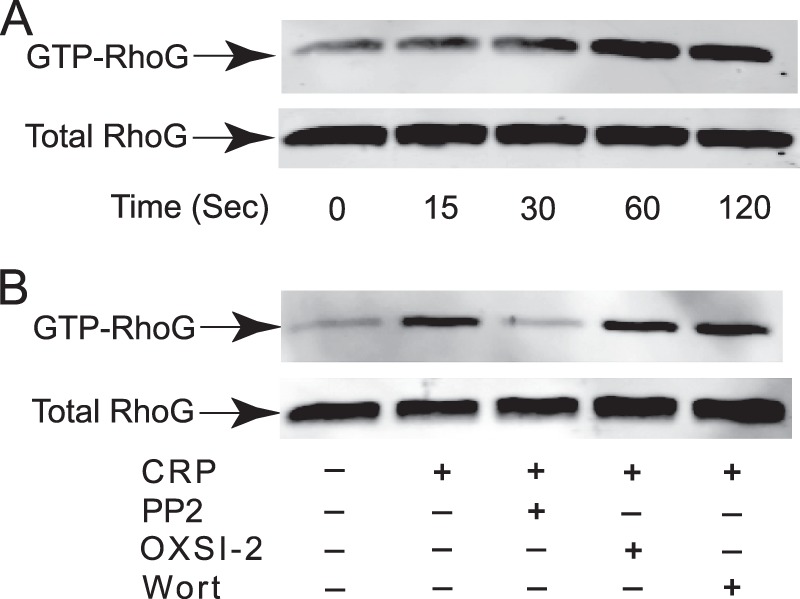
CRP-induced RhoG activation and its inhibition by PP2. A, RhoG activation was measured upon stimulation of washed human platelets with 5 μg/ml CRP for various times. Washed platelets were lysed, and active GTP-bound RhoG was determined by pulldown analysis using bacterially expressed GST-ELMO. B, RhoG activation induced by 5 μg/ml CRP for 60 s was evaluated in the presence and absence of 10 μm PP2, 2 μm OXSI-2, or 100 nm wortmannin (Wort). The blot shown is representative of three independent experiments.
CLEC-2 Agonist Fucoidan-induced Platelet Aggregation and Phosphorylation Events in RhoG-deficient Platelets
Our data indicate that RhoG is upstream of Syk in ITAM signaling. Of the ITAM receptors, CLEC-2 and GPVI cause similar signaling pathways leading to activation of Syk and PLCγ2, but they differ in the ITAM component (32, 33). GPVI utilizes FcRγ, whereas CLEC-2 does not. To evaluate the role of FcRγ in GPVI-mediated RhoG activation, we activated RhoG−/− platelets with fucoidan, a recently identified agonist for CLEC-2 (34). Fucoidan activates GPVI downstream signaling responses but bypasses the GPVI-FcRγ complex. Interestingly, we found that fucoidan (unlike CRP)-induced platelet aggregation was normal in RhoG−/− mice (Fig. 6A). In addition, fucoidan-induced Syk and Akt phosphorylation was not inhibited in RhoG−/− platelets. These results indicate that RhoG is not important for platelet aggregation or phosphorylation of Syk and Akt downstream of the CLEC-2 receptor and suggest that RhoG associates with the GPVI-FcRγ complex to propagate signaling leading to platelet activation.
FIGURE 6.
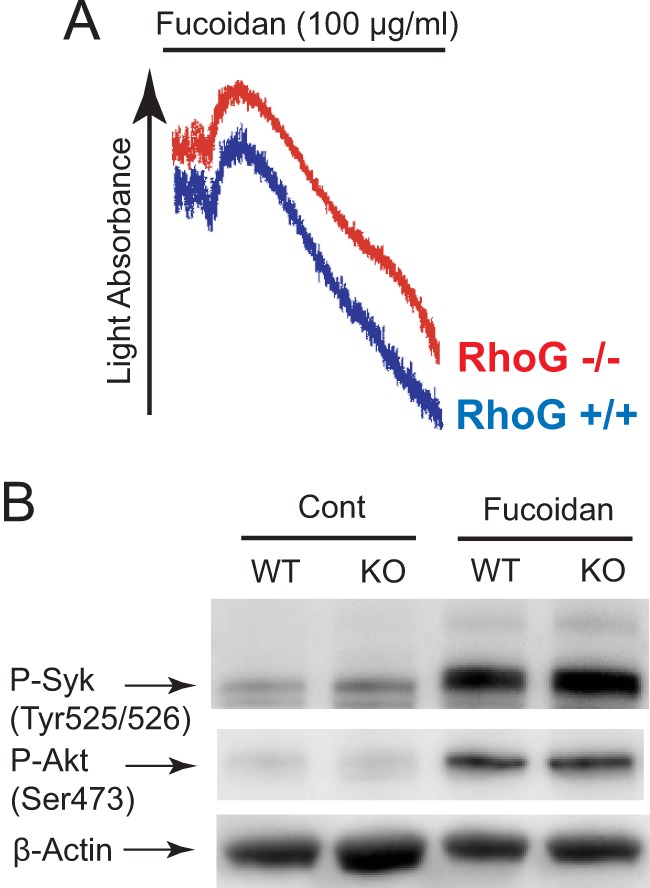
Fucoidan-induced platelet responses are not affected by RhoG deficiency. A, washed platelets from RhoG−/− mice and RhoG+/+ littermates were stimulated with 100 μg/ml fucoidan (a selective CLEC-2 agonist), and platelet aggregation was measured by aggregometry. B, wild-type and RhoG-deficient platelets were stimulated with 100 μg/ml fucoidan and probed with anti-phospho (P)-Syk (Tyr525/Tyr526), anti-phospho-Akt (Ser473), and anti-β-actin (lane loading control) antibodies by Western blotting. Data are representative of three independent experiments. Cont, control; KO, knock-out.
Regulation of in Vivo Thrombosis, Platelet Spreading, and Clot Retraction by RhoG
To evaluate the contribution of RhoG to the function of platelets in vivo, we used the microvascular thrombosis model with light/dye-induced injury as described previously (31). As shown in Fig. 7A, we found that blood flow in cremaster arterioles stopped within 30 min (29.5 ± 1.58 min, n = 5) in WT mice. However, RhoG knock-out mice had a significant delay in time to thrombotic occlusion in cremaster arterioles compared with wild-type littermate controls (38.5 ± 1.95 min, n = 5; p < 0.01). Images taken 30 min after the injury showed that RhoG+/+ mice had complete arteriolar occlusion, whereas thrombus growth was dramatically inhibited in RhoG−/− mice (Fig. 7B). These results indicate that RhoG plays an important role in thrombus formation in vivo.
FIGURE 7.

Analysis of thrombus formation, platelet spreading, and clot retraction in RhoG−/− mice. A, the time required for occlusion of cremaster arterioles in RhoG+/+ and RhoG−/− mice was measured using the microvascular thrombosis model with light/dye-induced injury as described under “Experimental Procedures.” Five mice of each genotype were used, and statistical analysis revealed a significant difference between the two genotypes of mice. *, p < 0.01. B, representative images of cremaster arterioles were taken from RhoG+/+ and RhoG−/− mice 30 min after injury. As seen by the outline (arrows) of the thrombus formed, thrombus formation was inhibited in RhoG−/− mice. C, washed platelets (1 × 107 platelets/ml) from RhoG+/+ and RhoG−/− mice were plated on fibrinogen-coated coverslips. After washing three times with PBS, adherent cells were fixed with 3.7% paraformaldehyde, stained with rhodamine-phalloidin, and analyzed by fluorescence microscopy. D, clot retraction analysis of platelets from RhoG+/+ and RhoG−/− mice. Images are representative of three independent experiments.
We tested the role of RhoG in outside-in signaling by examining the platelet adhesion and spreading on immobilized fibrinogen and clot retraction. We found that the extent of platelet spreading on immobilized fibrinogen (Fig. 7C) and clot retraction (Fig. 7D) were normal in RhoG-deficient platelets, indicating that RhoG does not play a role in αIIbβ3-mediated outside-in signaling in platelets.
DISCUSSION
In this study, we investigated the mechanism of activation and functional role of a hitherto uncharacterized signaling molecule, RhoG, in platelets. We have demonstrated for the first time the expression and activation of RhoG in platelets. Platelets deficient in RhoG show a marked impairment in platelet aggregation and secretion and integrin αIIbβ3 activation induced by the GPVI agonists CRP and convulxin, whereas the G protein-coupled receptor agonists AYPGKF and 2-MeSADP and the CLEC-2 agonist fucoidan normally activate RhoG-deficient platelets. Our results reveal the involvement of RhoG in the regulation of Syk in GPVI signaling because CRP-induced Syk (but not Src) phosphorylation is significantly reduced in RhoG-deficient platelets. Finally, our results show that RhoG plays an important role in thrombus formation in vivo. Because RhoG is ubiquitously expressed, our in vivo thrombus assay using a global RhoG knock-out mouse cannot rule out the effect of RhoG loss in other cell types on thrombus formation.
It has been shown that CLEC-2 mediates a signaling cascade that is similar to that of the collagen receptor GPVI (35). GPVI signaling depends on FcRγ activation leading to the downstream tyrosine phosphorylation signaling cascade, including Syk and PLCγ2. However, the CLEC-2 agonist rhodocytin activates platelets deficient in the GPVI-FcRγ complex, indicating that CLEC-2 does not utilize FcRγ. We have recently shown that fucoidan is a novel agonist for CLEC-2 (34), and our studies show that fucoidan can induce normal platelet aggregation and Syk phosphorylation in RhoG-deficient platelets. Because fucoidan activates ITAM and Syk phosphorylation signaling independently of the GPVI-FcRγ complex, our findings demonstrate the involvement of RhoG in the signaling pathways selective for the GPVI-FcRγ complex. They also raise the possibility that FcRγ might directly regulate RhoG recruitment and activation downstream of GPVI. Consistent with our results obtained using RhoG−/− mice, it has been shown that FcRγ−/− mice have impaired occlusive platelet microthrombi and that Syk phosphorylation is inhibited, but no difference was found in bleeding time between WT and knock-out mice (36). However, the mechanism through which FcRγ regulates RhoG recruitment and activation downstream of GPVI is unclear.
The CLEC-2 agonist rhodocytin and CRP have been shown to stimulate tyrosine phosphorylation of Syk and many other signaling proteins in GPVI pathways, including the GTP exchange factors Vav1 and Vav3 (35, 37, 38). Vav family proteins are GTP exchange factors for the Rho family of small G proteins. Vav proteins have been shown to be tyrosine-phosphorylated by Src and Syk and interact with several proximal molecules in the ITAM-dependent signaling cascade. Vav1 has been shown to selectively activate Rac1, Rac2, RhoG, and, to a lesser extent, RhoA, whereas Vav2 and Vav3 activate RhoA and RhoG but show less activity toward Rac1 (39, 40). Platelets deficient in both Vav1 and Vav3 exhibit a marked impairment in functional responses to GPVI, which is associated with loss of phosphorylation of PLCγ2 (37). Although the platelet functional defect in RhoG−/− and Vav1/Vav3−/− mice is very similar, there is an interesting difference in the regulation of Syk phosphorylation in these mice. CRP-induced Syk phosphorylation is inhibited in RhoG-deficient platelets, indicating a role for RhoG in Syk activation, whereas Syk phosphorylation in response to CRP is not altered in platelets deficient in Vav1/Vav3. Thus, it is unlikely that Vav1 and Vav3 are involved in the activation of RhoG in platelets. A more recent study demonstrated that another Rho guanine nucleotide exchange factor, Trio, is able to exchange GTP on RhoG and that it regulates lamellipodial formation to mediate cell spreading and migration in other cells (41). Trio is ubiquitously expressed, but the role of Trio in platelet function has not been established. In vitro, Trio has been shown to be phosphorylated by the Src kinase Fyn (42). Our study has shown that RhoG activation is regulated by SFK, and the role of Fyn in the regulation of GPVI-mediated platelet activation is well known. Thus, in future studies, it will be noteworthy to evaluate the role of Fyn and Trio in the regulation of RhoG activation to define the molecular mechanism governing RhoG activity in platelets.
It has been shown that coexpression of Vav1 in A5 cells stably transfected with integrin αIIbβ3 is essential for Rac activation and lamellipodial formation by fibrinogen (43, 44). In other cell systems, RhoG is known to activate Rac via interaction with ELMO and DOCK180 (21–23). Previous studies have shown that Rac1 is activated in platelets by a number of agonists (45–47) and plays an important role in aggregation, secretion, and clot retraction. Interestingly, Rac1 is known to be essential for the regulation of PLCγ2 in platelets downstream of GPVI or CLEC-2 (48, 49). In the presence of pharmacological inhibitors of Rac, as well as in Rac1-deficient murine platelets, agonist-induced dense granule secretion is inhibited (50). It has also been shown that formylmethionylleucylphenylalanine-stimulated activation of Rac1 and Rac2 is diminished in RhoG−/− neutrophils at very early times. In contrast to the requirement of Rac1 in integrin αIIbβ3-mediated outside-in signaling, we found that platelet spreading on immobilized fibrinogen and clot retraction were normal in RhoG-deficient platelets, suggesting that RhoG is not involved in the regulation of Rac1 activation in platelets. This finding is consistent with the observation that GPVI-mediated Rac1 activation is not affected in RhoG−/− platelets (51). Our studies clearly show the difference in the role of RhoG in regulating the signaling cascade by ITAM-coupled receptors in platelets compared with other cell types.
In conclusion, we used RhoG−/− mice to delineate the role of RhoG and the differential signaling events in which RhoG plays an important role in GPVI-FcRγ-mediated platelet activation and thrombus formation. Our studies enhance our understanding of the molecular mechanisms of GPVI-FcRγ complex activation and the signaling events downstream of GPVI stimulation.
This work was supported by National Institutes of Health Grants HL93231 and HL118593 (to S. P. K.). This work was also supported by American Heart Association Grant 12SDG8980013 (to S. K.).
- GPVI
- glycoprotein VI
- FcRγ
- Fc receptor γ-chain
- ITAM
- immunoreceptor tyrosine-based activation motif
- PLCγ2
- phospholipase Cγ2
- CRP
- collagen-related peptide.
REFERENCES
- 1. Watson S. P., Auger J. M., McCarty O. J., Pearce A. C. (2005) GPVI and integrin αIIbβ3 signaling in platelets. J. Thromb. Haemost. 3, 1752–1762 [DOI] [PubMed] [Google Scholar]
- 2. Watson S. P., Gibbins J. (1998) Collagen receptor signalling in platelets: extending the role of the ITAM. Immunol. Today 19, 260–264 [DOI] [PubMed] [Google Scholar]
- 3. Gibbins J. M., Okuma M., Farndale R., Barnes M., Watson S. P. (1997) Glycoprotein VI is the collagen receptor in platelets which underlies tyrosine phosphorylation of the Fc receptor γ-chain. FEBS Lett. 413, 255–259 [DOI] [PubMed] [Google Scholar]
- 4. Nieswandt B., Bergmeier W., Schulte V., Rackebrandt K., Gessner J. E., Zirngibl H. (2000) Expression and function of the mouse collagen receptor glycoprotein VI is strictly dependent on its association with the FcRγ chain. J. Biol. Chem. 275, 23998–24002 [DOI] [PubMed] [Google Scholar]
- 5. Ezumi Y., Shindoh K., Tsuji M., Takayama H. (1998) Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor γ chain complex on human platelets. J. Exp. Med. 188, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ichinohe T., Takayama H., Ezumi Y., Arai M., Yamamoto N., Takahashi H., Okuma M. (1997) Collagen-stimulated activation of Syk but not c-Src is severely compromised in human platelets lacking membrane glycoprotein VI. J. Biol. Chem. 272, 63–68 [DOI] [PubMed] [Google Scholar]
- 7. Poole A., Gibbins J. M., Turner M., van Vugt M. J., van de Winkel J. G., Saito T., Tybulewicz V. L., Watson S. P. (1997) The Fc receptor γ-chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. EMBO J. 16, 2333–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quek L. S., Pasquet J. M., Hers I., Cornall R., Knight G., Barnes M., Hibbs M. L., Dunn A. R., Lowell C. A., Watson S. P. (2000) Fyn and Lyn phosphorylate the Fc receptor γ chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood 96, 4246–4253 [PubMed] [Google Scholar]
- 9. Asazuma N., Wilde J. I., Berlanga O., Leduc M., Leo A., Schweighoffer E., Tybulewicz V., Bon C., Liu S. K., McGlade C. J., Schraven B., Watson S. P. (2000) Interaction of linker for activation of T cells with multiple adapter proteins in platelets activated by the glycoprotein VI-selective ligand, convulxin. J. Biol. Chem. 275, 33427–33434 [DOI] [PubMed] [Google Scholar]
- 10. Gibbins J. M., Briddon S., Shutes A., van Vugt M. J., van de Winkel J. G., Saito T., Watson S. P. (1998) The p85 subunit of phosphatidylinositol 3-kinase associates with the Fc receptor γ-chain and linker for activitor of T cells (LAT) in platelets stimulated by collagen and convulxin. J. Biol. Chem. 273, 34437–34443 [DOI] [PubMed] [Google Scholar]
- 11. Lagrue A. H., Francischetti I. M., Guimarães J. A., Jandrot-Perrus M. (1999) Phosphatidylinositol 3′-kinase and tyrosine-phosphatase activation positively modulate convulxin-induced platelet activation. Comparison with collagen. FEBS Lett. 448, 95–100 [DOI] [PubMed] [Google Scholar]
- 12. Quek L. S., Bolen J., Watson S. P. (1998) A role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Curr. Biol. 8, 1137–1140 [DOI] [PubMed] [Google Scholar]
- 13. Daniel J. L., Dangelmaier C., Smith J. B. (1994) Evidence for a role for tyrosine phosphorylation of phospholipase Cγ2 in collagen-induced platelet cytosolic calcium mobilization. Biochem. J. 302, 617–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bishop A. L., Hall A. (2000) Rho GTPases and their effector proteins. Biochem. J. 348, 241–255 [PMC free article] [PubMed] [Google Scholar]
- 15. Spiering D., Hodgson L. (2011) Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh. Migr. 5, 170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nobes C. D., Hall A. (1995) Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 17. O'Kane E. M., Stone T. W., Morris B. J. (2003) Distribution of Rho family GTPases in the adult rat hippocampus and cerebellum. Brain Res. Mol. Brain Res. 114, 1–8 [DOI] [PubMed] [Google Scholar]
- 18. Vigorito E., Bell S., Hebeis B. J., Reynolds H., McAdam S., Emson P. C., McKenzie A., Turner M. (2004) Immunological function in mice lacking the Rac-related GTPase RhoG. Mol. Cell. Biol. 24, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vincent S., Jeanteur P., Fort P. (1992) Growth-regulated expression of rhoG, a new member of the ras homolog gene family. Mol. Cell. Biol. 12, 3138–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gauthier-Rouvière C., Vignal E., Mériane M., Roux P., Montcourier P., Fort P. (1998) RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol. Biol. Cell 9, 1379–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. deBakker C. D., Haney L. B., Kinchen J. M., Grimsley C., Lu M., Klingele D., Hsu P. K., Chou B. K., Cheng L. C., Blangy A., Sondek J., Hengartner M. O., Wu Y. C., Ravichandran K. S. (2004) Phagocytosis of apoptotic cells is regulated by a UNC-73/TRIO-MIG-2/RhoG signaling module and armadillo repeats of CED-12/ELMO. Curr. Biol. 14, 2208–2216 [DOI] [PubMed] [Google Scholar]
- 22. Katoh H., Hiramoto K., Negishi M. (2006) Activation of Rac1 by RhoG regulates cell migration. J. Cell Sci. 119, 56–65 [DOI] [PubMed] [Google Scholar]
- 23. Katoh H., Negishi M. (2003) RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature 424, 461–464 [DOI] [PubMed] [Google Scholar]
- 24. Ravichandran K. S., Lorenz U. (2007) Engulfment of apoptotic cells: signals for a good meal. Nat. Rev. Immunol. 7, 964–974 [DOI] [PubMed] [Google Scholar]
- 25. Vignal E., Blangy A., Martin M., Gauthier-Rouvière C., Fort P. (2001) Kinectin is a key effector of RhoG microtubule-dependent cellular activity. Mol. Cell. Biol. 21, 8022–8034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamaki N., Negishi M., Katoh H. (2007) RhoG regulates anoikis through a phosphatidylinositol 3-kinase-dependent mechanism. Exp. Cell Res. 313, 2821–2832 [DOI] [PubMed] [Google Scholar]
- 27. Francischetti I. M., Saliou B., Leduc M., Carlini C. R., Hatmi M., Randon J., Faili A., Bon C. (1997) Convulxin, a potent platelet-aggregating protein from Crotalus durissus terrificus venom, specifically binds to platelets. Toxicon 35, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 28. Vigorito E., Billadeu D. D., Savoy D., McAdam S., Doody G., Fort P., Turner M. (2003) RhoG regulates gene expression and the actin cytoskeleton in lymphocytes. Oncogene 22, 330–342 [DOI] [PubMed] [Google Scholar]
- 29. Daniel J. L., Dangelmaier C. A., Mada S., Buitrago L., Jin J., Langdon W. Y., Tsygankov A. Y., Kunapuli S. P., Sanjay A. (2010) Cbl-b is a novel physiologic regulator of glycoprotein VI-dependent platelet activation. J. Biol. Chem. 285, 17282–17291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim S., Jin J., Kunapuli S. P. (2006) Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood 107, 947–954 [DOI] [PubMed] [Google Scholar]
- 31. Patel K. N., Soubra S. H., Bellera R. V., Dong J. F., McMullen C. A., Burns A. R., Rumbaut R. E. (2008) Differential role of von Willebrand factor and P-selectin on microvascular thrombosis in endotoxemia. Arterioscler. Thromb. Vasc. Biol. 28, 2225–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Séverin S., Pollitt A. Y., Navarro-Nuñez L., Nash C. A., Mourão-Sá D., Eble J. A., Senis Y. A., Watson S. P. (2011) Syk-dependent phosphorylation of CLEC-2: a novel mechanism of hem-immunoreceptor tyrosine-based activation motif signaling. J. Biol. Chem. 286, 4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watson S. P., Herbert J. M., Pollitt A. Y. (2010) GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 8, 1456–1467 [DOI] [PubMed] [Google Scholar]
- 34. Manne B. K., Getz T. M., Hughes C. E., Alshehri O., Dangelmaier C., Naik U. P., Watson S. P., Kunapuli S. P. (2013) Fucoidan is a novel platelet agonist for the C-type lectin-like receptor 2 (CLEC-2). J. Biol. Chem. 288, 7717–7726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Suzuki-Inoue K., Fuller G. L., García A., Eble J. A., Pöhlmann S., Inoue O., Gartner T. K., Hughan S. C., Pearce A. C., Laing G. D., Theakston R. D., Schweighoffer E., Zitzmann N., Morita T., Tybulewicz V. L., Ozaki Y., Watson S. P. (2006) A novel Syk-dependent mechanism of platelet activation by the C-type lectin receptor CLEC-2. Blood 107, 542–549 [DOI] [PubMed] [Google Scholar]
- 36. Takaya N., Katoh Y., Iwabuchi K., Hayashi I., Konishi H., Itoh S., Okumura K., Ra C., Nagaoka I., Daida H. (2005) Platelets activated by collagen through the immunoreceptor tyrosine-based activation motif in the Fc receptor γ-chain play a pivotal role in the development of myocardial ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 39, 856–864 [DOI] [PubMed] [Google Scholar]
- 37. Pearce A. C., Senis Y. A., Billadeau D. D., Turner M., Watson S. P., Vigorito E. (2004) Vav1 and Vav3 have critical but redundant roles in mediating platelet activation by collagen. J. Biol. Chem. 279, 53955–53962 [DOI] [PubMed] [Google Scholar]
- 38. Pearce A. C., Wilde J. I., Doody G. M., Best D., Inoue O., Vigorito E., Tybulewicz V. L., Turner M., Watson S. P. (2002) Vav1, but not Vav2, contributes to platelet aggregation by CRP and thrombin, but neither is required for regulation of phospholipase C. Blood 100, 3561–3569 [DOI] [PubMed] [Google Scholar]
- 39. Movilla N., Bustelo X. R. (1999) Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol. Cell. Biol. 19, 7870–7885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schuebel K. E., Movilla N., Rosa J. L., Bustelo X. R. (1998) Phosphorylation-dependent and constitutive activation of Rho proteins by wild-type and oncogenic Vav-2. EMBO J. 17, 6608–6621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Rijssel J., van Buul J. D. (2012) The many faces of the guanine-nucleotide exchange factor Trio. Cell Adh. Migr. 6, 482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DeGeer J., Boudeau J., Schmidt S., Bedford F., Lamarche-Vane N., Debant A. (2013) Tyrosine phosphorylation of the Rho guanine nucleotide exchange factor Trio regulates netrin-1/DCC-mediated cortical axon outgrowth. Mol. Cell. Biol. 33, 739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miranti C. K., Leng L., Maschberger P., Brugge J. S., Shattil S. J. (1998) Identification of a novel integrin signaling pathway involving the kinase Syk and the guanine nucleotide exchange factor Vav1. Curr. Biol. 8, 1289–1299 [DOI] [PubMed] [Google Scholar]
- 44. Obergfell A., Judd B. A., del Pozo M. A., Schwartz M. A., Koretzky G. A., Shattil S. J. (2001) The molecular adapter SLP-76 relays signals from platelet integrin αIIbβ3 to the actin cytoskeleton. J. Biol. Chem. 276, 5916–5923 [DOI] [PubMed] [Google Scholar]
- 45. Azim A. C., Barkalow K., Chou J., Hartwig J. H. (2000) Activation of the small GTPases, Rac and Cdc42, after ligation of the platelet PAR-1 receptor. Blood 95, 959–964 [PubMed] [Google Scholar]
- 46. Gratacap M. P., Payrastre B., Nieswandt B., Offermanns S. (2001) Differential regulation of Rho and Rac through heterotrimeric G-proteins and cyclic nucleotides. J. Biol. Chem. 276, 47906–47913 [DOI] [PubMed] [Google Scholar]
- 47. Soulet C., Gendreau S., Missy K., Benard V., Plantavid M., Payrastre B. (2001) Characterisation of Rac activation in thrombin- and collagen-stimulated human blood platelets. FEBS Lett. 507, 253–258 [DOI] [PubMed] [Google Scholar]
- 48. Pleines I., Elvers M., Strehl A., Pozgajova M., Varga-Szabo D., May F., Chrostek-Grashoff A., Brakebusch C., Nieswandt B. (2009) Rac1 is essential for phospholipase Cγ2 activation in platelets. Pflugers Arch. 457, 1173–1185 [DOI] [PubMed] [Google Scholar]
- 49. Pollitt A. Y., Grygielska B., Leblond B., Désiré L., Eble J. A., Watson S. P. (2010) Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood 115, 2938–2946 [DOI] [PubMed] [Google Scholar]
- 50. Akbar H., Kim J., Funk K., Cancelas J. A., Shang X., Chen L., Johnson J. F., Williams D. A., Zheng Y. (2007) Genetic and pharmacologic evidence that Rac1 GTPase is involved in regulation of platelet secretion and aggregation. J. Thromb. Haemost. 5, 1747–1755 [DOI] [PubMed] [Google Scholar]
- 51. Goggs R., Harper M. T., Pope R. J., Savage J. S., Williams C. M., Mundell S. J., Heesom K. J., Bass M., Mellor H., Poole A. W. (2013) RhoG protein regulates platelet granule secretion and thrombus formation in mice. J. Biol. Chem. 288, 34217–34229 [DOI] [PMC free article] [PubMed] [Google Scholar]