

Background: Nucleotide-binding domains (NBDs) of ABC proteins bind two ATPs, but it is unknown whether dissociation follows hydrolysis of one or both ATPs.
Results: NBD dimers with one or two sites capable of ATP hydrolysis dissociate at the same rate.
Conclusion: NBD dimers dissociate following a single ATP hydrolysis event.
Significance: Understanding ABC proteins will help develop strategies to modify their function in disease.
Keywords: ABC Transporter, ATPases, CFTR, Fluorescence, Kinetics, Multidrug Transporters, LRET, Luminescence Resonance Energy Transfer, Thermophile
Abstract
The functional unit of ATP-binding cassette (ABC) transporters consists of two transmembrane domains and two nucleotide-binding domains (NBDs). ATP binding elicits association of the two NBDs, forming a dimer in a head-to-tail arrangement, with two nucleotides “sandwiched” at the dimer interface. Each of the two nucleotide-binding sites is formed by residues from the two NBDs. We recently found that the prototypical NBD MJ0796 from Methanocaldococcus jannaschii dimerizes in response to ATP binding and dissociates completely following ATP hydrolysis. However, it is still unknown whether dissociation of NBD dimers follows ATP hydrolysis at one or both nucleotide-binding sites. Here, we used luminescence resonance energy transfer to study heterodimers formed by one active (donor-labeled) and one catalytically defective (acceptor-labeled) NBD. Rapid mixing experiments in a stop-flow chamber showed that NBD heterodimers with one functional and one inactive site dissociated at a rate indistinguishable from that of dimers with two hydrolysis-competent sites. Comparison of the rates of NBD dimer dissociation and ATP hydrolysis indicated that dissociation followed hydrolysis of one ATP. We conclude that ATP hydrolysis at one nucleotide-binding site drives NBD dimer dissociation.
Introduction
Typical ATP-binding cassette (ABC)2 transporters are membrane proteins that couple the energy from ATP hydrolysis to the transport of a variety of substrates (1, 2); most them import or export solutes, but some are ion channels and/or regulate ion channels (1–3). The basic ABC protein functional unit consists of two transmembrane domains and two nucleotide-binding domains (NBDs) (1, 2). ABC proteins exhibit considerable variability in primary sequence, number of transmembrane domains, quaternary structure, and functions; yet, the structure of the NBDs is highly conserved (1, 2). NBDs are responsible for nucleotide binding and hydrolysis and contain several conserved sequences involved in interaction with the nucleotides (2). The Walker A and B motifs are common to many ATPases and are involved in binding of nucleotide phosphates and coordination of Mg2+ and water at the catalytic site, respectively (see Fig. 1A); the Walker B motif contains a catalytic glutamate. The NBDs also contain the ABC signature motif (motif C), involved in ATP binding, which is unique to ABC proteins. The two ATP-bound NBDs dimerize forming an ATP “sandwich” in a head-to-tail arrangement (Fig. 1A), where each of the two nucleotide-binding sites (NBSs) is formed by contribution of the Walker A and Walker B motifs of one NBD and the signature motif of the other NBD (2). In many ABC proteins, including the multidrug-resistance protein P-glycoprotein (MDR1, ABCB1), both NBSs are capable of ATP hydrolysis; whereas in others, one of the NBS is hydrolysis-defective due to substitutions in residues critical for catalysis (1, 2). These ABC proteins include the TAP1/TAP2 heterodimer (ABCB2/ABCB3) and several proteins of the ABCC subfamily such as the cystic fibrosis transmembrane conductance regulator (CFTR, ABCC7) and multidrug-resistance proteins such as MRP1 (ABCC1).
FIGURE 1.
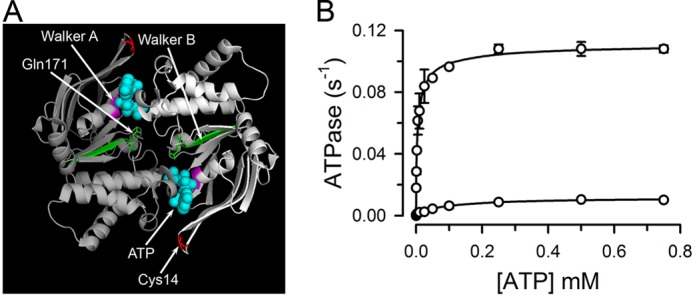
Structure and function of the NBD dimer. A, structure of the NBD dimer. Each monomer is represented in a different tone of gray. Walker motifs A and B are labeled and shown in purple and green, respectively. The two ATPs (cyan, spheres), Cys-14 (red, sticks), and Gln-171 (green, sticks) are also labeled. The figure displays the structure of the nucleotide-bound MJI based on PDB 1L2T. Glu replaces Gln-171 in MJ. B, dependence of the ATPase activity on ATP concentration. Data are means ± S.E. of two independent measurements. The line is a fit of the Hill equation to the data.
We have recently shown dynamic association and dissociation of NBDs during the ATP catalytic cycle in both isolated NBDs (MJ0796) and NBDs in the full-length transporter MsbA. MJ0796 is homolog to the NBD of the Gram-negative bacterial Lol complex that transports lipoprotein from the inner to the outer membrane, whereas MsbA is a bacterial lipid flippase homolog to P-glycoprotein (4, 5). Our kinetic studies support monomer/dimer modes of operation (6–14), as opposed to models where the NBDs remain in contact during the catalytic cycle (15–18). In monomer/dimer models ATP hydrolysis is followed by dissociation of the NBDs, and the energy that drives the conformational changes that lead to substrate transport (power stroke) is provided by NBD association/dissociation. In these models, NBD association/dissociation controls the alternate accessibility of the drug-binding pocket to each side of the membrane. One important unresolved issue is whether dissociation of the ATP-bound NBD dimers follows hydrolysis of one or both bound ATP molecules. Here, we addressed this issue using luminescence resonance energy transfer (LRET), a spectroscopic technique that allows for measurements of conformational changes in real time with Angstrom resolution.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Mutants of Methanocaldococcus jannaschii MJ0796 were expressed in Escherichia coli and purified by anion-exchange and gel-filtration chromatography as described (5, 19). The properties of the Cys-less-MJ0796-G174W (MJ-CL: Cys-53 and Cys-128 replaced with Gly and Ile, respectively) and the two single-Cys mutants MJ0796-G174W-G14C (MJ) and MJ0796-G174W-E171Q-G14C (MJI) have been published (5, 19). MJ is catalytically active, whereas MJI is catalytically deficient due to the replacement of the catalytic carboxylate Glu-171 with Gln (Fig. 1, A and B). MJI has essentially no ATPase activity but still binds ATP with high affinity (5, 19). All of the mutants employed have an introduced Trp at position 174 (G174W, a good probe to assess dimerization by Trp quenching), which was used to test the quality of the preparations. The Trp-Trp interaction seems to improve the stability of the dimer, which allowed us to detect for the first time the association/dissociation of a hydrolytically active NBD by size-exclusion chromatography (19). The G174W mutation does not affect ATPase activity, but it does increase the affinity for NaATP (5, 7, 14, 19).
LRET Studies
Single-Cys mutants in 1 mm EDTA, 50 mm Tris-HCl, pH 7.6, were labeled as described (5), with the thiol-reactive Tb3+ chelate DTPA-cs124-EMPH or fluorescein maleimide (Invitrogen) for 2 h at room temperature, at a 2:1 molar excess. Unreacted probes were removed by gel filtration (Zeba columns; Thermo Fisher Scientific), and donor-labeled and acceptor-labeled NBDs were mixed at a 1:1 molar ratio prior to LRET experiments. Emission was measured on an Optical Building Blocks phosphorescence lifetime photometer (EasyLife L; Optical Building Blocks, Birmingham, NJ), with a 200-μs delay from the beginning of the excitation pulse (gated mode). Excitation light from a xenon lamp was directed through a narrow-band 335-nm filter, and emission was collected through a 520-nm narrow-band filter. With this gated protocol, the long lifetime fluorescein emission (sensitized emission) arises from energy transfer from the Tb3+ donor. Data were typically collected at 100 Hz, at 20 °C, using 1 μm monomeric protein in 200 mm NaCl, 1 mm EDTA, 10% glycerol, 1 mm Tris-HCl, pH 7.6. Additional details, including a discussion of the advantages of recording LRET in a gated mode, have been published recently (4, 5).
Stop-flow LRET Studies
The kinetics of the dissociation of ATP-bound NBD dimers following ATP hydrolysis was monitored using a stop-flow mixing accessory (RX2000; Applied Photophysics, Surrey, UK) with a 20-μl cell, placed into the sample compartment of the OBB LRET system. The stop-flow device was driven by pressurized N2 and was set to mix equal volumes of two syringes in <10 ms. The first syringe contained 2 μm labeled proteins, dimerized by preincubation with 0.5 mm ATP in the LRET buffer described above. The second syringe contained LRET buffer with 0.5 mm ATP and 20 mm MgCl2. Final concentrations after mixing were 1 μm protein, 0.5 mm ATP, and 10 mm MgCl2. In most experiments, a 10-fold excess (20 μm) of the unlabeled, catalytically active MJ was also present in the second syringe (see “Results”). Changes in sensitized fluorescein emission were recorded as described for the standard LRET experiments, except for an increase in the data acquisition rate to 250 Hz.
ATPase Measurements
The rate of ATP hydrolysis was determined by a variant of the linked-enzyme assay (20), in the same buffer used for LRET experiments, with the addition of 1 μm protein, 10 mm MgCl2, and an ATP-regenerating system. The latter consisted of 3 mm phosphoenolpyruvate, 50 μg/ml pyruvate kinase, 25 μg/ml lactase dehydrogenase, and 0.7 mm NADH. The hydrolysis of ATP was determined from the decrease in NADH (absorbance measured at 340 nm) at room temperature. In some experiments, the rate of ATP hydrolysis was determined in the absence of an ATP-regenerating system, using [γ-32P]ATP (21), under conditions equivalent to those used during the LRET experiments. The proteins, at a concentration of 1 μm in LRET buffer, were incubated at room temperature and saturating ATP (0.5 mm). The reaction was initiated by the addition of 10 mm MgCl2 and allowed to proceed for 30 min.
Data Presentation and Statistics
Data are shown as means ± S.E. Statistical comparisons were performed by the Student's t test for paired or unpaired data, as appropriate. p < 0.05 in a two-tail analysis was considered significant. The number of experiments, n, corresponds to independent measurements from at least two different protein preparations. The goodness of fit for the analysis of stop-flow LRET decays was determined from the residual analysis scatter plots, which showed random distribution, and χ2 values near unity.
RESULTS
ATP Hydrolysis Activity of MJ and MJI
The rate of ATP hydrolysis by the single-Cys MJ (5) is similar to that of wild-type MJ0796 (14), whereas the mutation of the catalytic carboxylate Glu-171 to Gln essentially abolishes the ATPase activity of MJ0796 (14). Fig. 1B confirmed the effect of the E171Q mutation on the single-Cys mutant MJI used for the LRET studies presented here (see “Protein Expression and Purification” for details on the mutant). Under our experimental conditions, at a saturating [ATP] of 0.5 mm and room temperature, MJ hydrolysis rate was 0.11 ± 0.01 ATP/s (n = 12), with a calculated Kd of 4.6 μm, whereas MJI showed basically undetectable activity (0.003 ± 0.001 ATP/s, n = 12). The ATPase activity measured in the absence of an ATP-regenerating system (similar to the LRET experiments presented below) was not different (0.15 ± 0.04 ATP/s, n = 14) from that in the presence of the system (see above). During the time course of the LRET stop-flow experiments (60 s) the decrease in [ATP] is expected to be relatively minor. For a hydrolysis rate of 0.11 ATP s−1, [ATP] will decrease by ∼73 μm; 1.21 μm ATP/s by 11 μm MJ (1 μm labeled plus 10 μm unlabeled), and [ATP] is expected to decrease from 500 to ∼430 μm, remaining almost 100-fold above the Kd for dimerization (∼5 μm for MgATP) (19). In addition, because the measured ATPase activities in the presence and absence of an ATP-regenerating system were the same, no inhibitory effect of ADP on MJ was detected under our experimental conditions.
Dynamic MJ Monomer/Dimer Equilibrium during ATP Hydrolysis
A mix of Tb3+-labeled MJ (MJ-Tb3+) and fluorescein-labeled MJ (MJ-F) displays a slow increase in fluorescence (in the order of minutes) in response to the addition of a saturating [ATP] (Fig. 2A). We have shown that the increase in fluorescence is the result of ATP-induced dimerization that occurs with an apparent Kd for NaATP of approximately 50 μm (5). In the absence of ATP the monomers are far apart, and essentially no energy transfer occurs between donor/acceptor pairs; ATP addition promotes NBD dimerization that brings the donor and acceptor closer, to a distance of ∼50 Å, which can be followed from the increase in energy transfer between Tb3+ and fluorescein (5). The observed fluorescence increase originates from donor/acceptor dimers (expected to be ∼50% in a randomly distributed mix); donor/donor (∼25%) or acceptor/acceptor (∼25%) dimers do not display LRET. The addition of Mg to ATP-bound dimers produces a rapid decrease in fluorescence due to the dissociation of the dimers that results from ATP hydrolysis (Fig. 2A). The fluorescence in the presence of MgATP reached a level intermediate between those in the absence of ATP (nucleotide-free) and in the presence of ATP alone (ATP-bound dimers). This intermediate LRET value represents a dynamic equilibrium between the monomeric and dimeric NBDs: dimers dissociate after ATP hydrolysis, and the monomers bind ATP and reassociate (5, 19). We have previously shown that when Mg is present (MgATP versus NaATP) ATP-induced dimerization of MJ is ∼30-fold faster, and the apparent Kd for ATP is at least 10 times lower (<5 μm) (19). Under continuous ATP hydrolysis, in the presence of saturating [MgATP], neither association nor dissociation is favored, and approximately half of the NBDs are monomers and half-form dimers (5, 19). Further evidence of such dynamic equilibrium is provided by the complete decrease in fluorescence observed after addition of a 10-fold excess of unlabeled MJ (Fig. 2A). In the presence of excess unlabeled MJ, the labeled NBDs that dissociate from dimers after ATP hydrolysis have a greater probability of associating with unlabeled NBD, and the newly formed labeled/unlabeled dimers do not display LRET. This experiment confirms our previous findings that there is complete dissociation of NBD dimers during the catalytic cycle and a dynamic monomer/dimer equilibrium during continuous hydrolysis (5, 19). A similar experiment performed with the hydrolysis-deficient MJI shows ATP-induced dimerization but no significant effects of addition of Mg or excess unlabeled MJ (Fig. 2B). This result confirms that ATP-bound catalytically deficient MJI dimers are stable and do not dissociate during the time course of our studies (5, 7, 19).
FIGURE 2.
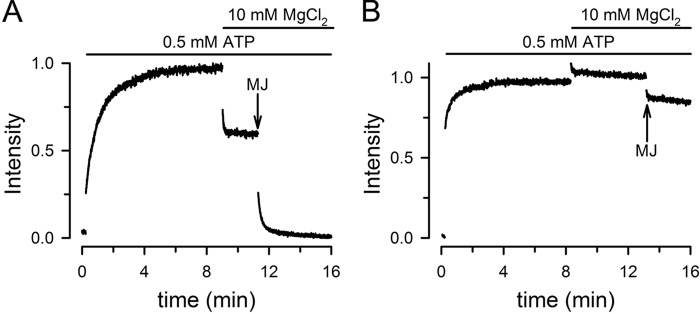
Association/dissociation of MJ and MJI homodimers followed by changes in LRET. A, MJ. MJ concentration was 1 μm. Half of the MJ was labeled with Tb3+ and the other half with fluorescein. Effects of successive addition of 0.5 mm ATP, 10 mm MgCl2, and 10 μm unlabeled MJ (arrow) are shown. The signal was normalized to the ATP-induced change. B, MJI. The experiment was similar to that in A, but used 1 μm MJI.
Dissociation of Dimers with Only One Hydrolysis-competent NBS
The MJ dimer contains two NBSs capable of hydrolysis, but it is still unresolved whether one or two ATP molecules are hydrolyzed during the monomer/dimer cycle. To address this issue we studied heterodimers formed by one active (MJ) and one hydrolysis-deficient (MJI) NBD. These heterodimers have two NBSs, but only one can hydrolyze ATP effectively because the second site has a mutation in the catalytic glutamate (E171Q). MJ labeled with donor (MJ-Tb3+) and MJI labeled with acceptor (MJI-fluorescein) were used to follow heterodimer association and dissociation. Ideally, an equimolar mix of MJ and MJI will produce ∼25% of MJ/MJ homodimers, 50% MJ/MJI heterodimers, and 25% of MJI/MJI homodimers. However, only MJ/MJI dimers containing donor/acceptor pairs are visible by LRET, allowing for selective measurements from normal/mutant dimers within the complex population. When ATP was added to a 1-to-1 mix of MJ-Tb3+ and MJI-fluorescein the LRET signal increased in an [ATP]-dependent fashion (black, pink, and red traces, Fig. 3A). The Kd for ATP-induced dimerization of the heterodimers, calculated from the changes in LRET intensity, was ∼25 μm (Fig. 3B). This value falls between those of 5 and 50 μm estimated for MJI and MJ homodimers, respectively (5, 19). The observed dimerization rate (Fig. 3A) for the heterodimers at saturating [ATP] (red) also showed an intermediate value between those of MJ (blue) and MJI (green) homodimers. We have previously shown that in absence of Mg, ATP-induced dimerization of MJI is ∼2 times faster than that of MJ (19). The higher affinity of MJI for ATP and its faster dimerization are likely related to the changes in electrostatics at the dimer interface in the E171Q mutant (7, 19). The heterodimers display characteristics intermediate between those of MJ and MJI (Fig. 3A), where one E171Q mutation per dimer produces smaller increases in the affinity for ATP and dimerization rate than the two E171Q mutations in MJI homodimers. It is important to note that the E171Q mutation does not decrease the dimerization capability of MJI. Size-exclusion chromatography analysis has demonstrated that monomeric MJ or MJI is completely converted to dimers in the presence of saturating [ATP] (19). The comparable maximal LRET intensities at 0.5 mm NaATP in our experiments (green, red, and blue traces in Fig. 3A) indicate that the extent of MJ/MJI heterodimer formation is similar to those of MJ and MJI homodimers.
FIGURE 3.
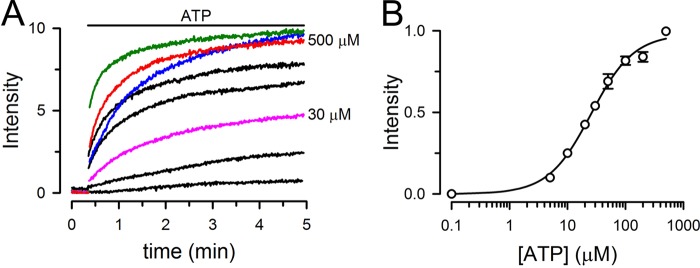
Dependence of MJ/MJI heterodimer formation on ATP concentration. A, typical LRET intensity changes in response to different ATP concentrations in the absence of hydrolysis (nominally divalent cation-free plus 1 mm EDTA). The sensitized fluorescein emission from MJ/MJI heterodimers was measured at ATP concentrations ranging from 0 to 500 μm (red trace labeled as 500 μm). Intermediate ATP concentrations were 5, 10, 30 (pink trace labeled as 30 μm), 50, and 100 μm. The time courses of MJ (blue) and MJI (green) homodimer formation in response to 500 μm ATP are shown for comparison. For the heterodimer formation experiments, Tb3+-labeled MJ and fluorescein-labeled MJI were mixed at a 1:1 molar ratio. For the homodimer formation experiments, Tb3+-labeled and fluorescein-labeled MJ or MJI was employed (1:1 molar ratio). Intensities are presented in arbitrary fluorescence units, without normalization. The total concentration of NBDs was 1 μm. B, summary of the dependence of MJ/MJI heterodimer formation on ATP concentration. Data are means ± S.E. (n = 3). S.E. values smaller than the symbols are not shown. The Kd and Hill coefficient calculated from the Hill equation were 27 ± 3 μm and 1.2 ± 0.1, respectively.
Fig. 4A shows MJ/MJI heterodimer formation initiated by the addition of saturating ATP and the subsequent dimer dissociation after addition of Mg. This MJ/MJI heterodimer dissociation demonstrates that one ATP hydrolysis is sufficient to dissociate the dimers. Contrary to the partial reversal of the ATP-induced increase in LRET signal by Mg in MJ homodimers (Fig. 2A), the reversal for the MJ/MJI heterodimers was almost complete. We attribute this behavior to the diminished availability of MJI for reassociation with MJ. Before Mg addition, approximately 25% of MJI is expected to form homodimers. Because MJI homodimers do not dissociate after adding Mg (Fig. 2B), that ∼25% of MJI protein is “trapped” as stable homodimers. In the presence of MgATP, newly dissociated MJI from MJ/MJI heterodimers forms additional MJI homodimers that increase the MJIs trapped as homodimers, unavailable for reassociation with MJ. Therefore the probability of heterodimer formation decreases after each catalytic cycle. Consistent with this interpretation, addition of MJ labeled with fluorescein in the presence of MgATP increases the LRET signal due to formation of MJ-Tb3+/MJ-fluorescein dimers (Fig. 4A). From these results we conclude that dimers with one catalytically active and one catalytically deficient NBS can undergo complete dissociation during the NBD hydrolysis cycle.
FIGURE 4.
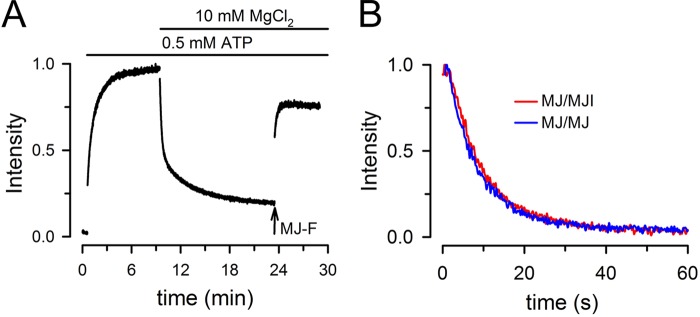
Association/dissociation of MJ/MJI heterodimers. A, changes in sensitized fluorescein emission from MJ/MJI heterodimers during the hydrolysis cycle. Tb3+-labeled MJ and fluorescein-labeled MJI were mixed at a 1:1 molar ratio (0.5 μm each), and the changes in LRET were measured in response to ATP and then in MgATP (i.e. during continuous ATP hydrolysis). MJ-F denotes the addition of 0.5 μm MJ labeled with fluorescein, which formed homodimers with the Tb3+-labeled MJ already present. The trace was normalized to the peak sensitized-emission in 0.5 mm ATP. Before addition of ATP the solution was nominally divalent cation-free and had 1 mm EDTA. B, time course of dissociation of MJ homodimers and MJ/MJI heterodimers. Tb3+-labeled MJ and fluorescein-labeled MJI were mixed at a 1:1 molar ratio for the heterodimer experiments, whereas Tb3+-labeled and fluorescein-labeled MJ were mixed at the same ratio for the homodimer experiments. ATP-bound dimers were preformed by >10-min incubation in the presence of 0.5 mm ATP (in a nominally divalent cation-free solution with 1 mm EDTA). Data acquisition followed the rapid mixing in the stop-flow cell of the labeled ATP-bound dimers with an equal volume of an identical buffer except for the presence of MgCl2 and unlabeled MJ. The final concentrations of labeled NBDs, ATP, MgCl2, and unlabeled MJ were 1 μm, 0.5 mm, 10 mm, and 10 μm, respectively.
The Dissociation Rates of Dimers with One and Two Hydrolysis-competent NBSs Are the Same
The experiments above showed that ATP hydrolysis results in dissociation of MJ/MJI heterodimers that have only one hydrolysis-competent NBS (Fig. 4B). However, it is possible that the rate of dimer dissociation following ATP hydrolysis is slower than that of MJ homodimers that have two hydrolysis-competent NBSs. To address this possibility, we compared the hydrolysis-induced dissociation rates of MJ homodimers and MJ/MJI heterodimers. Dimers were preformed by incubation of the labeled NBDs with saturating [ATP], and rates were measured under conditions that minimized the contribution of NBD redimerization, which therefore represent true dissociation rates. The ATP-bound dimers were mixed in a stop-flow cell with magnesium and a 10-fold excess of unlabeled MJ. Under these conditions, labeled/unlabeled dimers that do not produce LRET are more likely to be formed after the first dissociation, minimizing the contribution of reassociation of labeled MJ/MJI dimers. The results in Fig. 4B illustrate such experiments. There was an essentially complete decrease in sensitized fluorescein emission in the samples of MJ homodimers and MJ/MJI heterodimers. The decreases in sensitized fluorescein emission were well fitted by a single-exponential decay function with indistinguishable rates for the MJ homodimers (0.15 ± 0.03 s−1; n = 8) and the MJ/MJI heterodimers (0.13 ± 0.02 s−1; n = 8). These rates indicate that dimers with one or two catalytically active NBSs dissociate at the same speed. Although the E171Q mutation affects the dimerization rate (19), our results indicate that once the ATP-bound dimers are formed, their dissociation rate is not affected by the mutation.
The dimer dissociation rates presented above were measured in the presence of a reporter Trp introduced at the dimer interface (G174W) (5, 19). As mentioned under “Experimental Procedures” (“Protein Expression and Purification”) the effects of the Trp-Trp interaction are mild, producing only a minor increase in dimer stability (19). Independently of the possibility of small effects of the G174W mutation on the absolute dissociation rate, our results indicate that dimers with two catalytic carboxylates (MJ/MJ) and those with one E171Q mutation (MJ/MJI) dissociate at the same speed.
The absence of a difference between the dissociation rates of MJ homodimers and MJ/MJI heterodimers cannot rule out the possibility that both ATPs in the MJ homodimers are hydrolyzed before dissociation. The hydrolysis rate of the MJ/MJI mix (0.05 ± 0.01 ATP s−1) was approximately half of the hydrolysis rate of MJ/MJ homodimers when the total [NBDs] were the same. Assuming a random association pattern of the MJ/MJI mix, ∼25% of the NBDs will be MJ homodimers and 50% will be MJ/MJI heterodimers. Then, a 50% activity would be compatible with MJ homodimers with twice the activity of the heterodimers, as suggested in other studies (22, 23). However, under hydrolysis conditions (saturating [MgATP]), the lifetime of the MJ/MJI heterodimer is brief (<20 s, Fig. 4B), whereas MJI homodimers are very stable. Therefore, MJI available to form heterodimers decreases with each hydrolysis cycle (see explanation of Fig. 2B). The result is that under steady-state conditions the measured activity originates from the population of 50% of the NBDs that form MJ homodimers. Most importantly, the similarity between the ATP hydrolysis rate of MJ homodimers (averaging 0.13 ± 0.02 ATP s−1 from all measurements) and their dissociation rate (0.15 ± 0.03 s−1) strongly suggests that MJ homodimers dissociate after one ATP hydrolysis event. If both bound ATPs were hydrolyzed, the rate of ATP hydrolysis would be twice the dissociation rate (∼0.3 ATP s−1).
DISCUSSION
We have recently shown the usefulness of LRET for the study of NBD dimerization during the ATP hydrolysis cycle (4, 5, 19). In LRET, the rare elements Tb3+ and Eu3+ are used as donors. LRET has many advantages for our experiments versus traditional fluorescence resonance energy transfer. These advantages depend on the atomic-like and long-lifetime emission of the lanthanides and result in a very low background, high signal-to-noise ratio and independence of the sensitized emission lifetime from labeling stoichiometry (24). The LRET probes (donor or acceptor) are attached to a single-Cys residue in the protein, and alterations in the distance that separate the probes can be followed by changes in the efficiency of energy transfer and the resulting changes in sensitized-emission intensity. The residue at position 14 of MJ0796 (G14C mutant) was chosen as labeling target because it is exposed to the aqueous solvent (facilitating labeling), it is not part of the active site or dimer interface (Fig. 1A), has no effect on the ATPase activity, and the distance between the two Cys-14 residues in the ATP-bound dimer is ∼50 Å (5). This distance is nearly ideal to follow dimerization by the sensitized fluorescein emission using the Tb3+/fluorescein LRET pair, which has a Förster distance of ∼46 Å. In addition, the steep dependence of LRET on the distance between the probes minimizes the intermolecular signal between monomers in solution. In the present studies, we found that in the presence of MgATP (during continuous hydrolysis), the LRET signal that arises from ATP-bound dimers disappears rapidly following addition of an excess of catalytically active unlabeled NBDs. We interpret this as the result of formation of dimers consisting of labeled and unlabeled NBDs, which do not produce LRET; the ATP-bound dimers dissociate following ATP hydrolysis, with reassociation of the monomers with unlabeled NBDs. This is consistent with our previous observations (5, 19) and strongly supports the notion that NBD dimers dissociate completely during the hydrolysis cycle.
It is still unknown whether dissociation of ATP-bound NBD dimers follows hydrolysis of one or both of the bound ATP molecules. To address this question we used LRET on single-Cys NBDs labeled with either LRET donor or acceptor probes. We studied mixtures of “normal” donor-labeled NBDs and “hydrolysis-defective” acceptor-labeled NBDs. Because only the dimers containing donor/acceptor pairs are visible by LRET (homodimers formed by normal or hydrolysis-deficient NBDs are invisible to LRET), we selectively measured the behavior of normal/mutant dimers within the complex population. Our data show not only heterodimer dissociation under hydrolysis conditions (in the presence of MgATP) of ATP-bound NBD dimers with only one hydrolysis-competent NBS, but also that dissociation occurs at the same rate as in dimers with two hydrolysis-competent NBSs. Because the rate of dimer dissociation of the latter equals the rate of ATP hydrolysis under the same conditions, our results support a model where one ATP hydrolysis event occurs during the hydrolysis cycle. We conclude that hydrolysis at one of the NBSs dissociates the ATP-bound dimers and that this is true even in the case of identical NBDs.
Mutation of either of the equivalent catalytic Glu residues (to Gln) in each of the P-glycoprotein NBDs produces a dramatic decrease in ATPase activity (25). The severe decrease in ATPase activity when only one NBS is mutated suggests that hydrolysis by both NBDs is required for a normal cycle. Therefore, it is possible that P-glycoprotein and other ABC transporters behave differently from the simpler isolated NBDs studied here. However, the mechanism of the mutations in P-glycoprotein is not entirely clear. Because the P-glycoprotein mutants were able to reach the transition state (measured as vanadate-induced ADP trapping) and went through multiple turnover cycles, it was suggested that the reduced ATPase activity could be the result of slower NBD dimer formation and/or distortion of the dimer interface (25). In agreement with this interpretation, we have shown slower dimer formation in response to MgATP and decreased magnesium selectivity (versus sodium) in MJI (19), which can be explained by changes in the electrostatics at the dimer interface (19). Molecular dynamic simulations are consistent with our results showing that hydrolysis of one ATP is enough to destabilize the dimer interface and promote NBD dimer dissociation. Some molecular dynamic simulations suggest that ATP hydrolysis at one site is sufficient to induce opening of the dimer (26) and can even induce loosening of the nucleotide binding in the second site (27). In this context, vanadate-induced nucleotide-trapping experiments showed a maximal stoichiometry of 1 mol/mol of P-glycoprotein, suggesting that after ATP binding the NBD dimer moves to an asymmetric state where only one NBS binds ATP tightly, is committed to hydrolysis, and can be trapped (3, 28, 29). Such NBD asymmetry has been observed in other ABC transporters, including the homodimeric MsbA (31). Although NBD asymmetry may require the presence of the transmembrane domains, we found that only one ATP is hydrolyzed before MJ dimer dissociation. Therefore, asymmetry in isolated NBD dimers cannot be ruled out. In summary, the observations above are consistent with the model proposed by Tombline et al., where only one ATP is committed to hydrolysis (28), and our data suggest that the power stroke requires hydrolysis of only one ATP.
Another controversial question about the molecular mechanism ABC transporters is whether NBDs dissociate completely after ATP hydrolysis (monomer/dimer models) or whether they remain in close proximity during the catalytic cycle (constant-contact models). By measuring distances between LRET probes attached to selected residues in MJ0796 we have shown that isolated NBDs dimerize upon ATP binding and dissociate completely following ATP hydrolysis, with the establishment of a dynamic equilibrium where approximately half of the NBDs are monomers and half-dimers (5, 19, this paper). Furthermore, studies on the ABC exporter MsbA showed that the behavior of this full-length protein was basically similar to that of the isolated NBDs. However, instead of observing two conformations (dimer and monomer), there were three dominant conformations in each state during the hydrolysis cycle: open-state conformation (distance of ∼53 Å, dominant in the nucleotide-free state, equivalent to monomeric NBDs), and closed and partially open NBD dimers (∼30 Å and ∼ 36 Å, dominant in the ATP-bound and vanadate-inhibited states) (4). The proportion of MsbA molecules in the three conformational states changed among the apo, ATP-bound, continuous-hydrolysis, and vanadate-inhibited states; i.e. specific conformations were populated, without distinct conformations specific to one state (4). Additional support for large distance changes between MsbA NBDs during the hydrolysis cycle has come from double electron-electron resonance experiments (32). However, cross-linking experiments in P-glycoprotein suggest that hydrolysis can proceed without large separation between the NBDs. Cross-linking between a pair of Cys in the C-terminal ends of the NBDs does not abolish drug-stimulated ATPase activity, suggesting that these parts of the NBDs do not require a major separation for hydrolysis (33). Moreover, cross-linking of two introduced Cys that lie close to the signature motif of NBD1 (P517C) and the Walker A motif of NBD2 (I1050C) with an 8-Å bifunctional cross-linker increased the ATPase activity, like binding of drug substrates, suggesting that the increase in drug-stimulated ATPase activity occurs with the NBDs closely associated (30). It seems that the NBDs have the ability to dissociate completely during the ATP hydrolysis cycle, but regulatory factors like information transmitted from the transmembrane domains and presence of substrate drugs can restrict NBD separation, facilitating reassociation and increasing ATPase rates.
In summary, our results indicate that hydrolysis of one ATP promotes dissociation of the MJ0796 homodimer. Therefore, it is possible that ABC transporters with two hydrolysis-competent NBSs (like P-glycoprotein) and those with only one hydrolysis-competent NBS (like CFTR) have a comparable hydrolysis cycle mechanism. Even in transporters where both NBSs are identical, hydrolysis of one ATP would be sufficient to destabilize the dimer. Additional studies will be needed to determine whether the conformational change elicited by hydrolysis of one ATP powers substrate transport.
Acknowledgments
We thank Drs. Luis Reuss and Ina Urbatsch for comments.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM79629 and 3R01GM079629-03S1. This work was also supported by Cancer Prevention and Research Institute of Texas Grant RP101073.
- ABC
- ATP-binding cassette
- LRET
- luminescence resonance energy transfer
- MJ-CL, Cys-less
- single-Trp MJ0796 mutant (MJ0796-C53G/C128I/G174W)
- MJ
- single-Cys mutant (G14C) on the MJ-CL background
- MJI-C14
- catalytically deficient mutant (E171Q) on the MJ background
- NBD
- nucleotide-binding domain
- NBS
- nucleotide-binding site.
REFERENCES
- 1. Al-Shawi M. K. (2011) Catalytic and transport cycles of ABC exporters. Essays Biochem. 50, 63–83 [DOI] [PubMed] [Google Scholar]
- 2. Bouige P., Laurent D., Piloyan L., Dassa E. (2002) Phylogenetic and functional classification of ATP-binding cassette (ABC) systems. Curr. Protein Pept. Sci. 3, 541–559 [DOI] [PubMed] [Google Scholar]
- 3. Sharom F. J. (2008) ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 9, 105–127 [DOI] [PubMed] [Google Scholar]
- 4. Cooper R. S., Altenberg G. A. (2013) Association/dissociation of the nucleotide-binding domains of the ATP-binding cassette protein MsbA measured during continuous hydrolysis. J. Biol. Chem. 288, 20785–20796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zoghbi M. E., Krishnan S., Altenberg G. A. (2012) Dissociation of ATP-binding cassette nucleotide-binding domain dimers into monomers during the hydrolysis cycle. J. Biol. Chem. 287, 14994–15000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hopfner K. P., Karcher A., Shin D. S., Craig L., Arthur L. M., Carney J. P., Tainer J. A. (2000) Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell 101, 789–800 [DOI] [PubMed] [Google Scholar]
- 7. Smith P. C., Karpowich N., Millen L., Moody J. E., Rosen J., Thomas P. J., Hunt J. F. (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 10, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J., Lu G., Lin J., Davidson A. L., Quiocho F. A. (2003) A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell 12, 651–661 [DOI] [PubMed] [Google Scholar]
- 9. Higgins C. F. (2007) Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749–757 [DOI] [PubMed] [Google Scholar]
- 10. Janas E., Hofacker M., Chen M., Gompf S., van der Does C., Tampé R. (2003) The ATP hydrolysis cycle of the nucleotide-binding domain of the mitochondrial ATP-binding cassette transporter Mdl1p. J. Biol. Chem. 278, 26862–26869 [DOI] [PubMed] [Google Scholar]
- 11. Oswald C., Holland I. B., Schmitt L. (2006) The motor domains of ABC transporters. What can structures tell us? Naunyn Schmiedebergs Arch. Pharmacol. 372, 385–399 [DOI] [PubMed] [Google Scholar]
- 12. Vergani P., Lockless S. W., Nairn A. C., Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones P. M., O'Mara M. L., George A. M. (2009) ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem. Sci. 34, 520–531 [DOI] [PubMed] [Google Scholar]
- 14. Moody J. E., Millen L., Binns D., Hunt J. F., Thomas P. J. (2002) Cooperative, ATP-dependent association of the nucleotide-binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 277, 21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones P. M., George A. M. (2009) Opening of the ADP-bound active site in the ABC transporter ATPase dimer: evidence for a constant contact, alternating sites model for the catalytic cycle. Proteins 75, 387–396 [DOI] [PubMed] [Google Scholar]
- 16. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 17. Jones P. M., George A. M. (2007) Nucleotide-dependent allostery within the ABC transporter ATP-binding cassette: a computational study of the MJ0796 dimer. J. Biol. Chem. 282, 22793–22803 [DOI] [PubMed] [Google Scholar]
- 18. Jones P. M., George A. M. (2011) Molecular-dynamics simulations of the ATP/apo state of a multidrug ATP-binding cassette transporter provide a structural and mechanistic basis for the asymmetric occluded state. Biophys. J. 100, 3025–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zoghbi M. E., Fuson K. L., Sutton R. B., Altenberg G. A. (2012) Kinetics of the association/dissociation cycle of an ATP-binding cassette nucleotide-binding domain. J. Biol. Chem. 287, 4157–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Urbatsch I. L., Sankaran B., Weber J., Senior A. E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270, 19383–19390 [DOI] [PubMed] [Google Scholar]
- 21. De La Cruz E. M., Ostap E. M. (2009) Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 455, 157–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nikaido K., Ames G. F. (1999) One intact ATP-binding subunit is sufficient to support ATP hydrolysis and translocation in an ABC transporter, the histidine permease. J. Biol. Chem. 274, 26727–26735 [DOI] [PubMed] [Google Scholar]
- 23. Zaitseva J., Jenewein S., Wiedenmann A., Benabdelhak H., Holland I. B., Schmitt L. (2005) Functional characterization and ATP-induced dimerization of the isolated ABC-domain of the haemolysin B transporter. Biochemistry 44, 9680–9690 [DOI] [PubMed] [Google Scholar]
- 24. Selvin P. R. (2002) Principles and biophysical applications of lanthanide-based probes. Annu. Rev. Biophys. Biomol. Struct. 31, 275–302 [DOI] [PubMed] [Google Scholar]
- 25. Tombline G., Bartholomew L. A., Tyndall G. A., Gimi K., Urbatsch I. L., Senior A. E. (2004) Properties of P-glycoprotein with mutations in the “catalytic carboxylate” glutamate residues. J. Biol. Chem. 279, 46518–46526 [DOI] [PubMed] [Google Scholar]
- 26. Wen P. C., Tajkhorshid E. (2008) Dimer opening of the nucleotide binding domains of ABC transporters after ATP hydrolysis. Biophys. J. 95, 5100–5110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gyimesi G., Ramachandran S., Kota P., Dokholyan N. V., Sarkadi B., Hegedus T. (2011) ATP hydrolysis at one of the two sites in ABC transporters initiates transport related conformational transitions. Biochim. Biophys. Acta 1808, 2954–2964 [DOI] [PubMed] [Google Scholar]
- 28. Tombline G., Muharemagić A., White L. B., Senior A. E. (2005) Involvement of the “occluded nucleotide conformation” of P-glycoprotein in the catalytic pathway. Biochemistry 44, 12879–12886 [DOI] [PubMed] [Google Scholar]
- 29. Carrier I., Gros P. (2008) Investigating the role of the invariant carboxylate residues E552 and E1197 in the catalytic activity of Abcb1a (mouse Mdr3). FEBS J. 275, 3312–3324 [DOI] [PubMed] [Google Scholar]
- 30. Loo T. W., Bartlett M. C., Detty M. R., Clarke D. M. (2012) The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J. Biol. Chem. 287, 26806–26816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mittal A., Böhm S., Grütter M. G., Bordignon E., Seeger M. A. (2012) Asymmetry in the homodimeric ABC transporter MsbA recognized by a DARPin. J. Biol. Chem. 287, 20395–20406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zou P., Bortolus M., McHaourab H. S. (2009) Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J. Mol. Biol. 393, 586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Verhalen B., Wilkens S. (2011) P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J. Biol. Chem. 286, 10476–10482 [DOI] [PMC free article] [PubMed] [Google Scholar]