

Abstract
The exact causes of inflammatory bowel disease (IBD) are not yet fully defined. From a vast body of literature, we know that the immune response has long been involved in the pathogenesis of IBD, including both ulcerative colitis and Crohn’s disease. A variety of specific alterations can lead to immune activation and inflammation directed to the colon, as revealed by some animal models. Current research has focused on the role of antibodies in downstream events and mechanisms of autoimmunity and inflammation. It is not well known whether the production of antibodies is a serologic consequence of IBD, or if it is a result of barrier dysfunction induced by inflammation. Here, we present a new hypothesis to distinguish the complex links between genetic susceptibility, barrier dysfunction, commensal and pathologic microbial factors and inflammatory response (especially autoantibodies) in the pathogenesis of IBD. To ascertain the hypothesis, we developed a pilot model with the concept of the presence of antibodies against enteric bacterial antigens in IBD. Results confirmed our hypothesis. Our hypothesis suggests the possibility of subcutaneous vaccination of animals with administration of all or specific enteric bacterial antigens.
Keywords: Inflammatory bowel disease rat model, Pathogenesis, Barrier dysfunction, Microbial factor
Core tip: We present a new hypothesis to distinguish the complex links between genetic susceptibility, barrier dysfunction, commensal and pathologic microbial factors and inflammatory response (especially autoantibodies) in the pathogenesis of inflammatory bowel disease (IBD). In our hypothesis, we suggest that prior activation of adaptive immunity against microbial flora antigens could initiate an IBD-like chronic inflammation if something like ethanol disturbs barrier function. If this hypothesis is supported with further experiments, it would illustrate unknown aspects of IBD pathogenesis. On this basis, we have developed a new immune-based model of IBD with the presence of antibodies against enteric bacterial antigens.
INTRODUCTION
Etiology
Investigations have demonstrated that the pathophysiology of inflammatory bowel disease (IBD) is multifactorial, but briefly host (e.g., genetics, intestinal barrier and immune system function) and exogenous factors (e.g., normal luminal flora) are two basic themes[1]. The normal intestine contains a large number of immune cells in a chronic state of so-called physiologic inflammation to control the gut and to prepare it for any immunologic response. Lack of immune responsiveness to lumen antigens may be a result of oral tolerance[2]. Multiple mechanisms are involved in the induction of oral tolerance. For instance, deletion or anergy of antigen-reactive T cells or activation of regulatory CD4 T cells suppresses gut inflammation through secretion of inhibitory cytokines such as interleukin (IL)-10 and transforming growth factor β (TGF-β). In addition, a selectively permeable barrier prevents unwanted solutes, microorganisms, and luminal antigens from confronting the immune system in the internal mucosa[2-4]. In IBD, this tolerance is altered and leads to an uncontrolled inflammation; thus, IBD is considered as a breakdown in the regulatory constraints on mucosal immune response to the microbial flora or their products within the intestine. Most of this process is mediated through components of the autoimmune response to self-antigens[5].
A variety of specific alterations can lead to immune activation and inflammation directed to the colon, as revealed in animal models demonstrating murine genetic models (transgenic models). These models showed us that deleting loci of specific cytokines (e.g., IL-2, IL-10, TGF-β) or their receptors or T cell antigen recognition molecules (e.g., T cell antigen receptors) or interfering with intestinal barrier integrity (e.g., mucus glycoprotein, deleting N-cadherin or nuclear factor κB) leads to inflammation[4].
It has been suggested that the continuous penetration of luminal antigens and unremitting stimulation of the mucosal immune system due to an increased permeability of the intestine epithelial cells may be the primary defect in patients suffering from IBD[3]. Therefore, if we consider increased epithelial permeability as the trigger, the tragedy of IBD initiates after a disruption occurring in the mucosal integrity. Then lots of macromolecule antigens in the lumen penetrate into internal compartments of the mucosa and submucosa, and subsequently become recognized by the gut immune system. Later, interstitial macrophages and dendritic cells are locally activated and release cytokines to recruit more macrophages and monocytes from the systemic circulation[6]. In normal subjects, this acute response is subsided after controlling the invasion. In genetically susceptible subjects [defects in innate immunity response (e.g., NOD2, α-defensins mutations)] or in the long-term exposure to penetrated antigens in the situation of persistent integrity perturbations, or if the invader was a specific uncontrollable pathogen (e.g., Salmonella sp., Shigella sp., Campylobacter sp., Clostridium difficile), antigen presenting cells (APCs) secrete cytokines, which leads to induction of differentiation in various T-cells. In this way, the adaptive immune response is turned on[7-11]. Activation of both TH1 or TH2 leads to an inflammatory response[12]. This ignition can be turned off by the regulatory systems. Generally, recovery is achieved after repair of the first alteration in intestinal permeability.
Microbial factors
Microorganisms are a likely factor in the initiation of inflammation in IBD[13]. However, the unanswered question in this area is whether microorganisms involved in the pathogenesis of IBD are commensal flora or invasive microbial pathogens?
Normal intestinal microflora may contribute to the development of IBD in susceptible individuals. This finding has been demonstrated repeatedly in murine models of IBD[14,15]. As an example, animals which are genetically altered (e.g., deficient in IL-2 and IL-10) to be susceptible to IBD do not develop the disease when raised under germ-free conditions[13]. Also, intestinal lesions in IBD typically predominate in areas of the highest bacterial exposure (e.g., in distal ileum and colon with 1012 organisms/g).
On the other hand, a number of studies have evaluated the possible role of specific infectious agents in the pathogenesis of IBD. This role has been evaluated in two ways: the relation between specific microorganisms and IBD (e.g., presence of specific antibodies in serologic findings of IBD patients[16]), and the association between some acute gastroenteritis and IBD[17].
Pathogens that could be directly responsible for initiating IBD are those that the mucosal immune system may fail to control in terms of the inflammatory response (e.g., Salmonella sp., Shigella sp.). These bacteria are rich in peptides having chemotactic properties (e.g., formyl-methionyl-leucyl-phenylalanine). The super-antigens capable of global T-lymphocyte stimulation and subsequent inflammatory response, and those producing toxins (necrotoxins, hemolysins, and enterotoxins), cause mucosal damage[8,9,16,18]. In summary, an acute infection with specific pathogens leads to a permanent uncontrollable perturbation in intestinal integrity, even though after the acute phase there is perhaps mediation of some cytokines (e.g., IFN-γ), and permeability changes across the epithelium are induced. This results in continuous exposure and stimulation of the mucosal immune system with commensal flora antigens[3,19-21].
IMMUNE REGULATION AND INFLAMMATORY CASCADE DEFECTS IN IBD
As discussed later, the mucosal immune system is normally nonresponsive to luminal contents due to oral tolerance. Once inflammation is initiated, the immune inflammatory response is propagated by T cell activation in the lamina propria. CD4 T cells are of three major types: TH1, TH2, and TH17 cells. The TH1 cells secrete predominantly IFN-γ, TNF-α, IL-2, and IL-12, which activate cell-mediated immunity by CD8 T cells (cytotoxic) resulting in transmural granulomatous inflammation resembling CD. Meanwhile, the TH2 cells can induce B-cell differentiation and humoral immunity by secreting predominantly IL-4, IL-5, and IL-13 with superficial mucosal inflammation features resembling UC[22]. TH17 cells secrete predominantly IL-17, IL-6, and granulocyte colony-stimulating factor and seem responsible for neutrophilic recruitment[5,8,23]. After activation of these cells, they produce specific cytokines and, consequently, the epithelial barrier permeability (e.g., IFN-γ) is increased. Some of these cytokines have destructive and apoptotic effects on mucosal cells, which eventually allow more antigens to pass and produce more agitation of immune cells amplifying the inflammatory cascade[3,8]. In normal situations, an activated response is subsided with regulatory T cells, including designated TH3, Tr1, and CD4, and CD25 cells[23]. Their function is blocking or down-regulating the response of TH1 and TH2 either by producing specific cytokines (IL-10 and TGF-β) or via cell-cell contact. There is evidence which demonstrates some defects in this regulatory system in IBD-susceptible subjects[24,25].
INTESTINAL BARRIER DYSFUNCTION
The intestine is covered by a monolayer of simple columnar and non-ciliated epithelial cells that are a type of brush border cells. These are joined together by intercellular and circumferential tight junctions to form a selectively permeable membrane. This barrier prevents unwanted solutes, microorganisms, and luminal antigens from entering the internal parts. They are also part of the immune system, acting as a first-line pathogen-recognition system because they present antigens similar to classical APC. They also express toll-like receptor (TLR) 4 and, furthermore, secrete antimicrobial peptides (e.g., cryptidins and defensins)[2-4,26]. However, the epithelial barrier has some guards of the innate immune system to ensure permanent immune responsiveness (e.g., DC, interstitial macrophages)[27]. If anything alters the barrier function, lots of luminal antigens could pass through the submucosal layer resulting in recruitment of neutrophils and macrophages. If these cells can control the invasion, it is not necessary to call adaptive immunity components, but if the invasion takes long then adaptive immune response component will be activated. In this process, if the regulatory systems are not able to overcome the inflammatory cascade, the secreted cytokines will deteriorate and amplify the first defect in the epithelial barrier by inducing apoptosis and necrosis in the epithelial cells. In addition, a number of studies have shown that inflammatory cytokines like TNF-κ and IFN-γ may have a role in increasing intestinal barrier permeability[3,4,8]. Some animal models of IBD have shown alterations in barrier function as the first trigger contributing to pathogenesis of IBD. Furthermore, abundant evidence indicates an increased intestinal permeability in IBD patients suggesting the permanent stimulation of the mucosal immune system as the primary defect in the pathogenesis of IBD[3,28,29].
STEPS OF AUTOIMMUNITY IN IBD
The pathogenesis of IBD and most of its extra-intestinal manifestations is immunologically mediated and appears to be mainly due to an autoimmune-related process[30,31]. As discussed, after a permanent alteration in barrier function, various antigens pass through the interstitial space which finally activates T cells. In normal subjects, the response is directed definitively against the specific epitope of antigens, but commensal organisms in the lumen have adhesive antigens (e.g., flagellar antigens) which adhere to the surface proteins of mucosal cells. If there are some predisposing factors, then there is a chance for APCs to process epitopes of these antigens, with parts of the mucosal surface proteins, which activate T lymphocytes against mucosal cell surface protein[8,31]. Another scenario happens when the response to the specific epitopes of antigens is cross-reactive to auto-antigens. There is evidence demonstrating relations between precise human leukocyte antigen (HLA) molecules and cross-reactive cellular antigens[32,33]. However, in the TH2-mediated immune response in UC, it is thought that perhaps development of self-reactive B cells, which are triggered to produce mucosal IgG autoantibodies, results in an inflammatory response. Meanwhile, TH1 cell-mediated immunity and auto-reactive T cells (CD4 or CD8) may be primed by microbial antigens that are cross-reactive to autoantigens[34].
A long series of studies demonstrated that IBD patients possess autoantibodies, some of which became serologic biomarkers to diagnose or distinguish subtypes of this disease, such as anti-lymphocyte, anti-goblet cell, pancreatic autoantibodies, the autoantibody against tropomyosin isoform 5 (a cytoskeletal protein found in colon epithelial cells), and antibodies against red blood cell membrane antigens that cross-react with enteropathogens such as Campylobacter sp.[31,34,35].
We will now discuss some of the known autoantibodies in IBD pathogenesis. There is a form of perinuclear antineutrophil cytoplasmic antibody (pANCA) which is non-reactive to myeloperoxidase. It is well defined that 60%-70% of UC patients and 5%-15% of their first-degree relatives are pANCA-positive, whereas this applies to only 2%-3% of the general population. There is a relation between positive pANCA antibody status and severity of UC disease and other complications. Interestingly, pANCA in CD is associated with colonic disease that resembles UC[31,34,35]. The definite antigens to which these antibodies are directed have not been identified, but they have cross-reactions with enteric bacterial antigens.
Other studies demonstrated the presence of another autoantibody, which is specific to patients with UC; it is an IgG autoantibody bound to a subtype of tropomyosin of colonic epithelial cell antigen. The capability of this antibody to initiate extracellular signal-regulated kinase (ERK) 1/2 signaling and up-regulating of the TLR and production of cytokines, and also the correlation between the titers of this antibody and the severity of colitis, suggest the possibility that such a protein could represent autoantigen- or complement-mediated responses[13,31].
Although the presence of antibodies directed against microbial antigens has been illustrated in the serum of CD patients, a shared epitope among the host antigens is not clearly defined. For example, 55% of CD patients have antibodies against outer membrane porin C of Escherichia coli, and 50% have immunoglobulins that are reactive to a homologue of the bacterial transcription-factor families from a Pseudomonas fluorescens-associated sequence (I2). Around 50% of CD patients have serum reactivity to Cbir1, an immunodominant antigen of the enteric microbial flora. This antigen can strongly induce B cells and CD4+ T cell responses. Transferring of Cbir1-specific CD4+ TH1 T cells to C3H/SCID mice generates a severe colitis dependent on exogenous expression of Cbir1 flagellin in the colon. In 60%-70% of CD patients, anti-Saccharomyces cerevisiae antibodies have been found. A mannose sequence in the cell wall of this commensal flora has been defined[35,36].
HYPOTHESIS
Although the above-mentioned studies support the concept of the presence of antibodies against enteric bacterial antigens in IBD, we propose a model to investigate whether the production of antibodies is a result of barrier dysfunction induced by inflammation or a serologic finding secondary to IBD. The hypothesis would result in a reliable model of IBD studies in animals. Our hypothesis suggests the possibility of subcutaneous vaccination of animals with administration of all or specific enteric bacterial antigens. In this way, production of immunoglobulin against these antigens would prevent intestinal inflammation. Anything that alters the function of this barrier and increases barrier permeability would result in inflammatory responses. To test this hypothesis, we have designed a pilot study and examined the model in male Wistar rats, which were immunized with anaerobic and aerobic enteric bacteria with and without an adjuvant. After assessing the IgG titers in the rats’ plasma, well-immunized rats were anesthetized and then chitosan and ethanol were instilled intrarectally as a tight junction opener and a barrier breaker, respectively. This protocol induced a chronic inflammatory response with inflammatory features in the ethanol group with persistent lesions. We propose that this model of chronic intestinal inflammation would be a reliable model of human IBD. Of course, further studies would need to prove immunization with specific bacteria (Figures 1 and 2).
Figure 1.
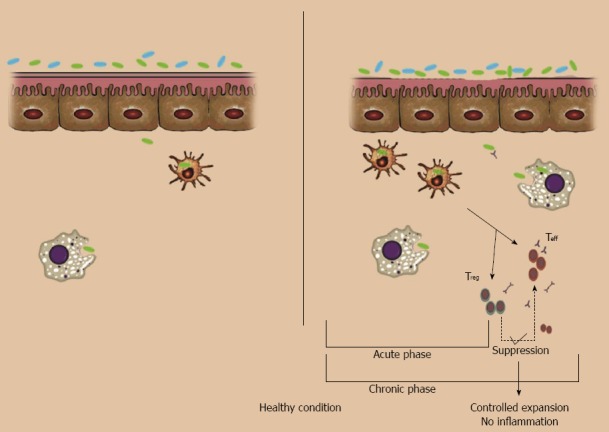
intestinal barrier dysfunction. Left (normal conditions): no or few commensal bacteria can pass the normal epithelial barrier and those that pass will be swallowed by interstitial macrophages and dendritic cells; it is not necessary to call for adaptive immune cells. Right: in normal situations, if something breaks the barrier (e.g. pathogens and barrier breaker chemicals like ethanol 30%) lots of commensal bacteria in the lumen will pass through the epithelial layer. This acute invasion will be controlled with recruiting of neutrophils and lymphocytes. Even after activation of B cells or T cells, if the defect in barrier function is resolved, apoptotic pathways will control the activated colonies of lymphocytes.
Figure 2.
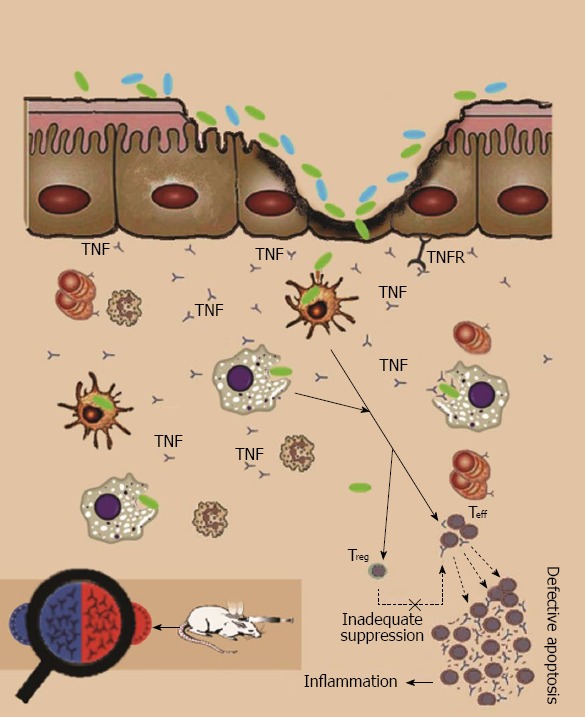
Steps of autoimmunity. If anything alters the barrier function, lots of luminal antigens can pass to the submucosal layer. If these cells can control the invasion, it is not necessary to call adaptive immunity components, but if the invasion take longer (e.g., in altered tight junction structure) or there are some defects in innate immunity response (e.g., mutations in Toll-like receptors), T cells are activated. If the regulatory system cannot overcome the inflammatory systems, cytokines and reactive destructive mediators further deteriorate the first defect via inducing apoptosis and necrosis in epithelial cells. Activation of T helper type 2 cells leads to a humoral response; also administration of a vaccine of commensal bacteria leads to a humoral immune response to their antigens, so after a disruption in barrier integrity with ethanol, the inflammatory cascade will turn on and induce inflammatory bowel disease in the animal as shown by histopathological findings. TNF: Tumor necrosis factor; TNFR: TNF receptor.
CONCLUSION
In this article, we addressed some known immune derangements involved in the initiation and pathogenesis of IBD. The following general principles are highlighted for better understanding of the possible mechanisms involved in the IBD pathogenesis.
Increased barrier permeability secondary to a genetic susceptibility, a specific infectious pathogen or their toxins and activation of T cells create a positive feedback to amplify the first barrier dysfunction and initiate an inflammatory cascade.
Two common features of autoimmunity processes may differ in activation of autoreactive T or B cells, involving a variety of imbalances in cytokine production and the development of autoantibodies. In IBD, these antibodies are directed against shared enteric flora antigens and epithelial cell-surface proteins.
In this study, we focused on autoantibodies. It is not well defined whether various autoantibodies found in the serologic assessment of IBD patients are destructive or involved in pathogenesis of the disease, or whether they are produced after tissue damage due to releasing of sequestered antigens[31]. We suggest that antibodies which are secreted in UC are catastrophic and are involved in the inflammatory response, but antibodies which are produced in CD are not involved in the pathogenesis and are secreted post-release of sequestered antigens. However, antibodies in both UC and CD patients are involved in extraintestinal complications, while there are various overlaps between these two subtypes.
In our hypothesis, we suggest that prior activation of adaptive immunity against microbial flora antigens in the way described could initiate an IBD-like chronic inflammation (especially in UC). Further experiments are essential to test various aspects of the method and unknown points of IBD pathogenesis.
Empirical data
After developing the hypothesis, we designed a pilot study. Six groups of male rats containing three rats in each group were considered. An extemporaneous vaccine was prepared with a mixture of heat-treated colonic commensal bacteria, which were obtained from cultured samples, and complete Freund’s adjuvant. This vaccine was injected subcutaneously into nine rats on days 0 and day 14. On day 28, a blood sample was taken from each rat to assess immunoglobulin titers. All of the test animals showed an elevated titer. Then these rats were divided into three groups; intra-colonic ethanol 30% was instilled in two groups, and in the third group, normal saline was instilled instead of ethanol and this group was assigned as the vaccine group. The two groups which received ethanol were: the model group that received no treatment and an infliximab-treated group that received 5 mg/kg per day of infliximab for 10 consecutive days after ethanol instillation. The first of the other three groups consisted of an ethanol group that received intracolonic ethanol 30% with no pre- or post-treatment. An established colitis model was induced with instillation of 10 mg of trinitrobenzene sulfonic acid dissolved in 30% ethanol as the vehicle in another group. And the last group was normal rats (sham group), which received normal saline intracolonically. The animals were sacrificed, and colon samples were removed for histopathological assays. Details of microscopic assessments are described in Figure 3.
Figure 3.
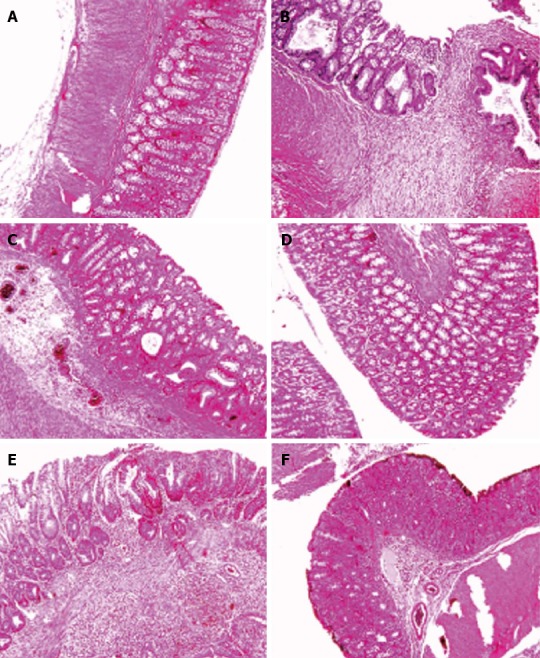
Histological images of colon tissues obtained from different groups. Microscopic evaluation of the trinitrobenzene sulfonic acid (TNBS) group shows villus atrophy, extensive severe transmural inflammation, granuloma with necrosis and crypt destruction, whereas features in the Sham group were normal. Histological examination of the Ethanol group showed a mild crypt distortion and some crypt abscess, whereas features in the Vaccine group were normal. Microscopic evaluation of the Model group showed mucosal inflammation and crypt distortion, branching and some ulceration with moderate to severe crypt destruction in ulcerated regions. Mild focal inflammation, minimal inflammatory cell infiltration and slight crypt branching were observed in the Infliximab group. A: Sham; B: TNBS; C: Ethanol; D: Vaccine; E: Model; F: Infliximab.
ACKNOWLEDGMENTS
The authors thank Tehran University of Medical Sciences for partial assistance.
Footnotes
P- Reviewers: Bonaz B, Blanco Luz P, Day AS, da Silva Figueredo C, Louwen R, Ingle SB, Nayci A, Yang PC S- Editor: Song XX L- Editor: Logan S E- Editor: Zhang DN
References
- 1.Rezaie A, Parker RD, Abdollahi M. Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig Dis Sci. 2007;52:2015–2021. doi: 10.1007/s10620-006-9622-2. [DOI] [PubMed] [Google Scholar]
- 2.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 3.Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2008;14:401–407. doi: 10.3748/wjg.14.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber CR, Turner JR. Inflammatory bowel disease: is it really just another break in the wall? Gut. 2007;56:6–8. doi: 10.1136/gut.2006.104182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esmaily H, Hosseini-Tabatabaei A, Rahimian R, Khorasani R, Baeeri M, Barazesh-Morgani A, Yasa N, Yassaman Khademi, Mohammad Abdollahi. On the benefits of silymarin in murine colitis by improving balance of destructive cytokines and reduction of toxic stress in the bowel cells. Cent Eur J Biol. 2009;4:204–213. [Google Scholar]
- 6.Hosseini-Tabatabaei A, Esmaily H, Rahimian R, Khorasani R, Baeeri M, Barazesh-Morgani A, Sari-Aslani F, Abdollahi M. Benefit of nicorandil on an immunologic murine model of experimental colitis. Cent Eur J Biol. 2009;4:74–85. [Google Scholar]
- 7.Jurjus AR, Khoury NN, Reimund JM. Animal models of inflammatory bowel disease. J Pharmacol Toxicol Methods. 2004;50:81–92. doi: 10.1016/j.vascn.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Esmaily H, Vaziri-Bami A, Miroliaee AE, Baeeri M, Abdollahi M. The correlation between NF-κB inhibition and disease activity by coadministration of silibinin and ursodeoxycholic acid in experimental colitis. Fundam Clin Pharmacol. 2011;25:723–733. doi: 10.1111/j.1472-8206.2010.00893.x. [DOI] [PubMed] [Google Scholar]
- 9.Gradel KO, Nielsen HL, Schønheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology. 2009;137:495–501. doi: 10.1053/j.gastro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Issa M, Vijayapal A, Graham MB, Beaulieu DB, Otterson MF, Lundeen S, Skaros S, Weber LR, Komorowski RA, Knox JF, et al. Impact of Clostridium difficile on inflammatory bowel disease. Clin Gastroenterol Hepatol. 2007;5:345–351. doi: 10.1016/j.cgh.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 11.Wen Z, Fiocchi C. Inflammatory bowel disease: autoimmune or immune-mediated pathogenesis? Clin Dev Immunol. 2004;11:195–204. doi: 10.1080/17402520400004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521–533. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 13.Abdolghaffari AH, Nikfar S, Rahimi HR, Abdollahi M. A comprehensive review of antibiotics in clinical trials for inflammatory bowel disease. Int J Pharmacol. 2012;8:596–613. [Google Scholar]
- 14.Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, Balish E, Hammer RE. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180:2359–2364. doi: 10.1084/jem.180.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Contractor NV, Bassiri H, Reya T, Park AY, Baumgart DC, Wasik MA, Emerson SG, Carding SR. Lymphoid hyperplasia, autoimmunity, and compromised intestinal intraepithelial lymphocyte development in colitis-free gnotobiotic IL-2-deficient mice. J Immunol. 1998;160:385–394. [PubMed] [Google Scholar]
- 16.Peyrin-Biroulet L, Standaert-Vitse A, Branche J, Chamaillard M. IBD serological panels: facts and perspectives. Inflamm Bowel Dis. 2007;13:1561–1566. doi: 10.1002/ibd.20226. [DOI] [PubMed] [Google Scholar]
- 17.Porter CK, Tribble DR, Aliaga PA, Halvorson HA, Riddle MS. Infectious gastroenteritis and risk of developing inflammatory bowel disease. Gastroenterology. 2008;135:781–786. doi: 10.1053/j.gastro.2008.05.081. [DOI] [PubMed] [Google Scholar]
- 18.Nikfar S, Mirfazaelian H, Abdollahi M. Efficacy and tolerability of immunoregulators and antibiotics in fistulizing Crohn’s disease: a systematic review and meta-analysis of placebo-controlled trials. Curr Pharm Des. 2010;16:3684–3698. doi: 10.2174/138161210794079236. [DOI] [PubMed] [Google Scholar]
- 19.Farrell RJ, LaMont JT. Microbial factors in inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31:41–62. doi: 10.1016/s0889-8553(01)00004-8. [DOI] [PubMed] [Google Scholar]
- 20.Fasano A, Baudry B, Pumplin DW, Wasserman SS, Tall BD, Ketley JM, Kaper JB. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl Acad Sci USA. 1991;88:5242–5246. doi: 10.1073/pnas.88.12.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berkes J, Viswanathan VK, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdolghaffari AH, Baghaei A, Moayer F, Esmaily H, Baeeri M, Monsef-Esfahani HR, Hajiaghaee R, Abdollahi M. On the benefit of Teucrium in murine colitis through improvement of toxic inflammatory mediators. Hum Exp Toxicol. 2010;29:287–295. doi: 10.1177/0960327110361754. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–4288. doi: 10.3748/wjg.14.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rezaie A, Khalaj S, Shabihkhani M, Nikfar S, Zamani MJ, Mohammadirad A, Daryani NE, Abdollahi M. Study on the correlations among disease activity index and salivary transforming growth factor-beta 1 and nitric oxide in ulcerative colitis patients. Ann N Y Acad Sci. 2007;1095:305–314. doi: 10.1196/annals.1397.034. [DOI] [PubMed] [Google Scholar]
- 25.Jahanshahi G, Motavasel V, Rezaie A, Hashtroudi AA, Daryani NE, Abdollahi M. Alterations in antioxidant power and levels of epidermal growth factor and nitric oxide in saliva of patients with inflammatory bowel diseases. Dig Dis Sci. 2004;49:1752–1757. doi: 10.1007/s10620-004-9564-5. [DOI] [PubMed] [Google Scholar]
- 26.Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:416–422. doi: 10.1038/ncpgasthep0259. [DOI] [PubMed] [Google Scholar]
- 27.Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. doi: 10.1053/gast.2002.30294. [DOI] [PubMed] [Google Scholar]
- 28.Blumberg RS, Saubermann LJ, Strober W. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr Opin Immunol. 1999;11:648–656. doi: 10.1016/s0952-7915(99)00032-1. [DOI] [PubMed] [Google Scholar]
- 29.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 30.Das KM. Relationship of extraintestinal involvements in inflammatory bowel disease: new insights into autoimmune pathogenesis. Dig Dis Sci. 1999;44:1–13. doi: 10.1023/a:1026629528233. [DOI] [PubMed] [Google Scholar]
- 31.Sandborn WJ. Serologic markers in inflammatory bowel disease: state of the art. Rev Gastroenterol Disord. 2004;4:167–174. [PubMed] [Google Scholar]
- 32.Turkcapar N, Toruner M, Soykan I, Aydintug OT, Cetinkaya H, Duzgun N, Ozden A, Duman M. The prevalence of extraintestinal manifestations and HLA association in patients with inflammatory bowel disease. Rheumatol Int. 2006;26:663–668. doi: 10.1007/s00296-005-0044-9. [DOI] [PubMed] [Google Scholar]
- 33.Orchard TR, Chua CN, Ahmad T, Cheng H, Welsh KI, Jewell DP. Uveitis and erythema nodosum in inflammatory bowel disease: clinical features and the role of HLA genes. Gastroenterology. 2002;123:714–718. doi: 10.1053/gast.2002.35396. [DOI] [PubMed] [Google Scholar]
- 34.Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol. 2006;6:244–251. doi: 10.1038/nri1784. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura RM, Matsutani M, Barry M. Advances in clinical laboratory tests for inflammatory bowel disease. Clin Chim Acta. 2003;335:9–20. doi: 10.1016/s0009-8981(03)00286-9. [DOI] [PubMed] [Google Scholar]
- 36.Ferrante M, Henckaerts L, Joossens M, Pierik M, Joossens S, Dotan N, Norman GL, Altstock RT, Van Steen K, Rutgeerts P, et al. New serological markers in inflammatory bowel disease are associated with complicated disease behaviour. Gut. 2007;56:1394–1403. doi: 10.1136/gut.2006.108043. [DOI] [PMC free article] [PubMed] [Google Scholar]