

Abstract
Alzheimer’s disease, Familial British dementia, Familial Danish dementia, Type 2 diabetes mellitus, plus Creutzfeldt-Jakob disease are associated with amyloid fibril deposition and oxidative stress. The antioxidant enzyme catalase is a neuroprotective amyloid binding protein. Herein the effects of catalase overexpression in SH-SY5Y neuronal cells on the toxicity of amyloid-β (Aβ), amyloid-Bri (ABri), amyloid-Dan (ADan), amylin (IAPP), and prion protein (PrP) peptides were determined. Results showed catalase overexpression was neuroprotective against Aβ, ABri, ADan, IAPP, and PrP peptides. The catalase inhibitor 3-amino-1,2,4-triazole (3-AT) and catalase-amyloid interaction inhibitor benzothiazole aniline tetra(ethylene glycol) (BTA-EG4) significantly enhanced neurotoxicity of amyloid peptides in catalase overexpressing neuronal cells. This suggests catalase neuroprotection involves breakdown of hydrogen peroxide (H2O2) plus a direct binding interaction between catalase and the Aβ, ABri, ADan, IAPP, and PrP peptides. Kisspeptin 45–50 had additive neuroprotective actions against the Aβ peptide in catalase overexpressing cells. The effects of 3-AT had an intracellular site of action, while catalase-amyloid interactions had an extracellular component. These results suggest that the 3-AT and BTA-EG4 compounds may be able to inhibit endogenous catalase mediated neuroprotection. Use of BTA-EG4, or compounds that inhibit catalase binding to amyloid peptides, as potential therapeutics for Neurodegenerative diseases may therefore result in unwanted effects.
Keywords: Catalase overexpression; benzothiazole aniline tetra(ethylene glycol); 3-amino-1,2,4-triazole; kisspeptin; amyloid peptide; SH-SY5Y neuronal cells
Catalase is a well-characterized antioxidant enzyme and amyloid binding protein.1,2 Overexpression of the enzyme has been linked to longevity, reduced processing of the amyloid-precursor protein plus amyloid-β peptide (Aβ) generation, and also memory function, suggesting a potential role for catalase in Alzheimer’s disease (AD).3−5 The blood levels of catalase protein are unchanged in AD; however, there is an observed decrease in the activity levels.6,7 In AD patients, the catalase activity is increased in the hippocampus, suggesting oxidative stress.8 In mouse models of AD, changes in antioxidants, including catalase, are seen.9 Specific changes in catalase mRNA plus enzyme activity are seen in the early stages before the onset of symptomatic changes in the Tg2576 mouse model of AD, with increases in the neocortex but decreases in the hippocampus, suggesting that oxidative stress may be an early event in the disease progression.10
The original discovery that catalase specifically bound the Aβ peptide also demonstrated that the interaction resulted in specific inhibition of the hydrogen peroxide (H2O2) degrading activity of the enzyme.2 The catalase activity in human erythrocytes can be inhibited by Aβ, suggesting a possible mechanism for the decrease in erythrocyte catalase activity in AD.6,11 Catalase binding to Aβ involves an interaction between the 25–35 region of Aβ and a specific domain in the wrapping loop of the catalase protein.2,12,13 The catalase binding is also seen with fibrillar Aβ forms containing residues 29–32 and can be inhibited by the Aβ 31–35 fragment suggesting an important role for the Ile residues at 31 and 32 of the Aβ peptide.14 The Aβ 31–35 fragment is the smallest fragment that inhibits the H2O2 degrading activity of catalase.2 The binding of catalase to Aβ deposits has been observed in AD brains and oxidative stress involving H2O2 is a component of the disease.8,15−17 As such it was suggested that compounds that disrupt catalase-Aβ interactions may be potential therapeutic agents for AD.2,18−20 Subsequent studies demonstrated that catalase also specifically bound both the islet amyloid polypeptide (IAPP) and prion protein (PrP), suggesting that catalase-amyloid interactions may play a role in amyloid diseases including AD, Creutzfeldt-Jakob Disease (CJD), and Type 2 diabetes mellitus (T2DM).21
A range of compounds have been identified that specifically disrupt catalase–Aβ interactions, including benzothiazole aniline tetra(ethylene glycol) (BTA-EG4) and benzothiazole aniline hexa(ethylene glycol) (BTA-EG6), which are derivatives of thioflavin-T.18,20 The BTA-EG4 binds to multiple regions of Aβ and is able to block not only binding of catalase but also other enzymes including the amyloid-binding alcohol dehydrogenase (ABAD).20 The BTA-EG4 compound has recently been shown to be active in vivo in an animal model of AD where it has beneficial actions and has been suggested as a therapeutic agent for AD.22,23 Peptides that specifically target the catalase binding domain of Aβ, including the endogenous kisspeptin (KP) peptides, have been shown to be neuroprotective; likewise, peptides targeting the ABAD binding domain of Aβ are also neuroprotective.13,24−26 Inhibition of catalase with either 3-amino-1,2,4-triazole (3-AT) or homocysteine significantly enhances the neurotoxicity of the Aβ peptide and suggests a role for the enzyme activity in endogenous neuroprotection against Aβ.27,28 Catalase overexpression in combination with glutathione peroxidase is neuroprotective against the Aβ peptide, which specifically activates H2O2 production in vitro.29−31
Herein we have used stable overexpression of the human catalase gene in human SH-SY5Y neuronal cultures to determine the mechanisms of catalase neuroprotection against the amyloid peptides Aβ, amyloid-Bri (ABri), amyloid-Dan (ADan), IAPP, and PrP. The catalase overexpression model has been characterized and the effects of the specific catalase inhibitor 3-AT plus the BTA-EG4 inhibitor of catalase-amyloid interactions on catalase neuroprotection have been determined.
Results and Discussion
Characterization of Catalase Overexpressing SH-SY5Y Neuronal Cells
The overexpression of the human catalase gene in the PCat SH-SY5Y neuronal cells, stably transfected with the pcDNA4/TO/myc–His expression vector containing the human catalase gene, was confirmed using immunocytochemistry (Figure 1A), which showed that the CAT 505 monoclonal anti-catalase staining was found within the cytoplasm. The staining of PVect control cells, stably transfected with the pcDNA4/TO/myc–His expression vector, showed considerably less CAT 505 monoclonal anti-catalase staining (Figure 1B). Western blotting using CAT 505 monoclonal anti-catalase staining showed specific staining of a 60 kDa band and increased levels in the PCat cell extracts compared to PVect cell extracts. To confirm that the transfected catalase gene was expressed, cells were analyzed by RT-PCR. Results showed a high level of catalase mRNA in the PCat SH-SY5Y neuronal cells compared to that found in PVect SH-SY5Y neuronal cells (Figure 1C). The activity of catalase, determined in extracts from PVect SH-SY5Y neuronal cells and PCat SH-SY5Y neuronal cells, showed a 6-fold higher activity in the PCat SH-SY5Y neuronal cells compared to the level of 18.3 ± 1.9 U/mg (mean ± SEM) in PVect SH-SY5Y neuronal cells (Figure 1D). The activity of PCat SH-SY5Y neuronal extracts was reduced to below the detection limit of the activity assay (<2 U/mg) by treatment with the catalase inhibitor 3-AT.
Figure 1.
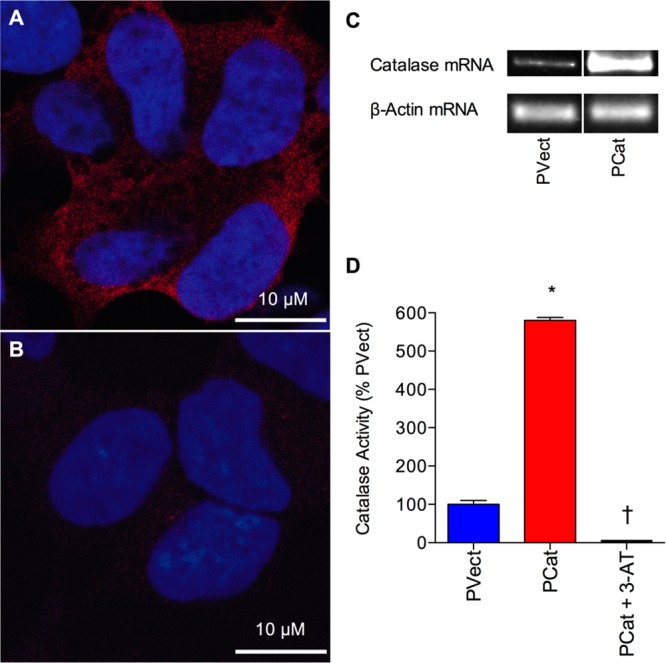
Characterization of catalase gene overexpression in SH-SY5Y neuronal cells. (A) Immunocytochemistry of human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) showing localization of catalase in the cytoplasm. (B) Immunocytochemistry of human SH-SY5Y neuronal cells stably expressing the pcDNA4/TO/myc–His expression vector (PVect) showing low level localization of catalase above background. Catalase appears red (CAT 505 monoclonal anti-catalase staining), and the nucleus appears blue (TO-PRO-3 iodide staining). Bars = 10 μm. (C) RT-PCR analysis of catalase and β-actin mRNA in human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect). (D) Catalase activity in extracts from Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect), plus the effect of the catalase inhibitor 50 mM 3-AT on PCat cell extract catalase activity. Results are expressed as % PVect catalase activity (mean ± SEM). *P < 0.05 vs PVect extracts; †P < 0.05 vs PCat extracts; one-way ANOVA.
Effects of Hydrogen Peroxide on Catalase Overexpressing SH-SY5Y Neuronal Cells
The increase in catalase activity (Figure 1D) in the PCat SH-SY5Y neuronal cells suggests that they should be resistant to H2O2 toxicity. The effects of 2 h treatment with a range of H2O2 concentrations (0–1000 μM) on PVect and PCat SH-SY5Y neuronal viability were determined. Results showed that the PCat SH-SY5Y neuronal cells were significantly resistant to H2O2 toxicity at doses up to 1000 μM (Figure 2A). To confirm that the resistance to H2O2 toxicity was due to the catalase activity, and not other endogenous peroxidases, the PVect and PCat SH-SY5Y neuronal cells were treated with 500 μM H2O2 plus a range of concentrations of 3-AT (0–50 mM).32 Results showed that 3-AT significantly reduced the H2O2 toxicity (Figure 2B).
Figure 2.
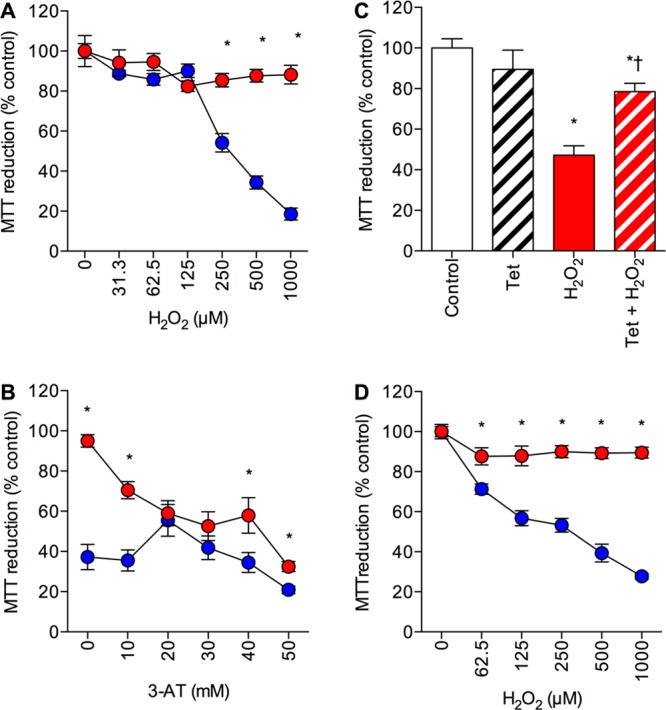
Effects of hydrogen peroxide on catalase overexpressing SH-SY5Y neuronal cells. (A) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat; closed red circles) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect; closed blue circles) were exposed to 0–1000 μM H2O2 and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs PVect; one-way ANOVA. (B) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat; closed red circles) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect; closed blue circles) were pretreated with 0–50 mM 3-AT prior to exposure to 500 μM H2O2 and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs PVect; one-way ANOVA. (C) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector plus the pcDNA6/TR vector (PCatIND) were exposed to 1 μg/mL tetracycline (Tet; open hatched column), 500 μM H2O2 (closed red column) or 1 μg/mL Tet plus 500 μM H2O2 (hatched red column) and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs control; †P < 0.05 vs H2O2 alone; one-way ANOVA. (D) Naïve (untransfected) SH-SY5Y neuronal cells were exposed to 500 μM H2O2 plus conditioned medium from PVect SH-SY5Y neuronal cells (closed blue circles) or PCat SH-SY5Y neuronal cells (closed red circles). Results are mean ± SEM; *P < 0.05 vs PVect; one-way ANOVA.
The pcDNA4/TO/myc–His expression vector contains tetracycline (Tet) operator sequences that allow suppression of the expression with a Tet repressor (TR) protein. PCat SH-SY5Y neuronal cells were stably transfected with the pcDNA6/TR vector containing the TR protein to create an inducible catalase expression line (PCatIND). Treatment of the PCatIND SH-SY5Y neuronal cells with 1000 μM H2O2 caused significant neurotoxicity that was reduced by pretreatment of the cells with 1 μg/mL Tet (Figure 2C) confirming that the overexpression of catalase was responsible for the neuroprotection against H2O2.
Catalase is known to be released from some cell types and the deposits of catalase in the AD brain are clearly extracellular.15,16 Overexpression systems often create cell lines that release excessive levels of the overexpressed proteins into the medium, raising the possibility that extracellular catalase could be responsible for the neuroprotection against H2O2 toxicity in the PCat SH-SY5Y neuronal cells. Conditioned medium from PVect and PCat SH-SY5Y neuronal cultures was harvested after 24 h incubation with fresh medium. The effects of 2 h treatment with a range of H2O2 concentrations (0–1000 μM) on naïve (untransfected) SH-SY5Y neuronal cells in the presence of conditioned medium from PVect or PCat SH-SY5Y neuronal cells was determined. Results showed that the conditioned medium from PCat SH-SY5Y neuronal cells was significantly protective against H2O2 toxicity (Figure 2D).
These results confirm that the increased catalase expression in the PCat SH-SY5Y neuronal cells is neuroprotective against H2O2 toxicity in agreement with previous studies using neuronal catalase overexpression.33 This suggests that the PCat SH-SY5Y neuronal model may be suitable for investigating the role of catalase in neuroprotection.
Effects of Cobalt Chloride on Catalase Overexpressing SH-SY5Y Neuronal Cells
The toxicity of a range of neurotoxins can be inhibited by catalase overexpression; however, cobalt chloride induced oxidative stress appears to be unaffected by catalase.29,34 The effects of 24 h treatment with a range of cobalt chloride concentrations (0–500 μM) on PVect and PCat SH-SY5Y neuronal viability were determined. Results showed that both PCat and PVect SH-SY5Y neuronal cells were susceptible to cobalt chloride toxicity at doses up to 500 μM (Figure 3). The dose response curves showed a similar pattern and there were no significant differences between the toxicity observed in PCat and PVect SH-SY5Y neuronal cells. This provides a positive control toxin that both PCat and PVect SH-SY5Y neuronal cells are equally susceptible to and has been used in subsequent experiments. The results confirm the suggestion that cobalt chloride neurotoxicity is not mediated via actions on H2O2 and demonstrate the specificity of the neuronal catalase overexpression model.34
Figure 3.
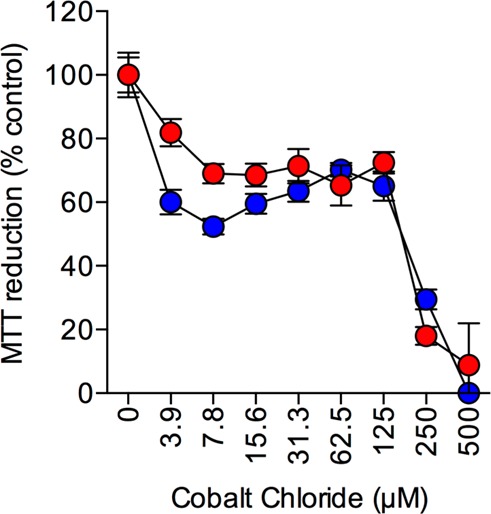
Effects of cobalt chloride on catalase overexpressing SH-SY5Y neuronal cells. Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat; closed red circles) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect; closed blue circles) were exposed to 0–500 μM cobalt chloride and cell viability determined by MTT reduction. Results are mean ± SEM.
Effects of Amyloid Peptides on Catalase Overexpressing SH-SY5Y Neuronal Cells
The resistance of PCat SH-SY5Y neuronal cells to H2O2 toxicity (Figure 2) suggests that the cell line may be suitable to determine the role of catalase in amyloid peptide toxicity. The Aβ, ABri, ADan, IAPP, and PrP peptides play a key role in the pathology of AD, Familial British dementia, Familial Danish dementia, T2DM, and CJD, respectively.35−40 With the exception of IAPP, all of the amyloid peptides are produced in the brain and all are neurotoxic.35−40 In the case of IAPP, a recent study has demonstrated IAPP deposits in the brains of T2DM patients, suggesting a need for neuroprotection in the treatment of T2DM.41 The diseases associated with amyloid peptides all have an oxidative stress component.38 Catalase has previously been shown to bind Aβ, IAPP plus PrP fibrils and is neuroprotective against Aβ and the rat IAPP in SH-SY5Y neuronal cells.2,11−14,21,26 The catalase binding region of the Aβ peptide is found within the 25–35 sequence, which shows similarity to regions of IAPP, PrP, ABri, and ADan peptides (Figure 4A).2,14 The sequence similarities between the amyloid peptides and their similar mechanisms of neurotoxicity which can be counteracted by amyloid binding compounds suggest that catalase overexpression may be protective against the Aβ, ABri, ADan, IAPP, and PrP peptides.25,29,42 The effects of 24 h treatment with 25 μM of the amyloid peptides, Aβ 25–35, ABri 1–34, ADan 1–34, IAPP 20–29, and PrP 106–126, on PVect and PCat SH-SY5Y neuronal viability were determined. Results showed that the PCat SH-SY5Y neuronal cells were significantly resistant to Aβ, ABri, ADan, IAPP, and PrP toxicity (Figure 4B). This is the first demonstration that catalase overexpression is neuroprotective against the ABri, ADan, IAPP, and PrP peptides, suggesting that catalase neuroprotection is not specific to a given amyloid peptide. This is in agreement with the observations that Aβ, IAPP, and PrP share a similar mechanism of toxicity and suggests that ABri plus ADan can be added to this group of amyloid neurotoxins acting via similar mechanisms.42 The dose–response curve for Aβ 25–35 neurotoxicity over the range 0–50 μM showed that the PCat SH-SY5Y neuronal cells were resistant to Aβ 25–35 doses up to 50 μM (Figure 4C) in agreement with previous studies that suggest a major role for catalase in neuroprotection against the Aβ peptide.29
Figure 4.

Effects of amyloid peptides on catalase overexpressing SH-SY5Y neuronal cells. (A) Alignment of Aβ 25–35 (shown in red) to similar regions of IAPP 20–29, PrP 106–126, ABri 1–34, and ADan 1–34. Red amino acids are identical to those in Aβ 25–35, blue amino acids have similar properties. The purple box highlights the Gly-Ala-Ile-Ile region of Aβ 25–35 that binds catalase. (B) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat; hatched columns) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect; closed columns) were exposed to 25 μM Aβ 25–35 (clear columns), IAPP 20–29 (red columns), PrP 106–126 (blue columns), ABri 1–34 (green columns), or ADan 1–34 (yellow columns) and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs PVect; one-way ANOVA. (C) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat; closed red circles) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect; closed blue circles) were exposed to 0–50 μM Aβ 25–35 and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs PVect; one-way ANOVA. (D) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector plus the pcDNA6/TR vector (PCatIND) were exposed to 1 μg/mL tetracycline (Tet; open hatched column), 50 μM Aβ 31–35 (closed red column) or 1 μg/mL Tet plus 50 μM Aβ 31–35 (hatched red column) and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs control; †P < 0.05 vs Aβ alone; one-way ANOVA. (E) Naïve (untransfected) SH-SY5Y neuronal cells were exposed to conditioned medium from PVect SH-SY5Y neuronal cells (blue columns) or PCat SH-SY5Y neuronal cells (red columns) plus 25 μM Aβ 25–35 alone (hatched columns) or 25 μM Aβ 25–35 and 50 mM 3-AT (stippled columns). Results are mean ± SEM; *P < 0.05 vs PVect alone; one-way ANOVA.
The PCatIND SH-SY5Y neuronal cells were susceptible to H2O2 toxicity and induction of catalase expression by pretreatment of the cells with Tet was neuroprotective against H2O2. This system was used to determine whether similar effects could be observed with neuroprotection against Aβ. Results showed that 50 μM Aβ 31–35 was toxic to the PCatIND SH-SY5Y neuronal cells and that the toxicity was reversed by pretreatment with 1 μg/mL Tet (Figure 4D), confirming that the overexpression of catalase was responsible for the neuroprotection against Aβ.
The observation that conditioned medium from PCat SH-SY5Y neuronal cultures was protective against H2O2 toxicity (Figure 2D) suggests that the catalase overexpressing cells release catalase into the medium. Catalase is well-known to be neuroprotective against Aβ when added to culture medium.12,43−46 Conditioned medium from PVect and PCat SH-SY5Y neuronal cultures was harvested after 24 h incubation with fresh medium. The effect of 24 h treatment with 25 μM Aβ 25–35 on naïve (untransfected) SH-SY5Y neuronal cells in the presence of conditioned medium from PVect or PCat SH-SY5Y neuronal cells was determined. The naïve (untransfected) SH-SY5Y neuronal cells treated with PVect conditioned medium were susceptible to Aβ 25–35 toxicity, and this effect was not blocked by 50 mM of the catalase inhibitor 3-AT (Figure 4E). The conditioned medium from PCat SH-SY5Y neuronal cells was significantly protective against 25 μM Aβ 25–35 toxicity compared to conditioned medium from PVect SH-SY5Y neuronal cells (Figure 4E) and 50 mM of the catalase inhibitor 3-AT did not alter this effect.
These results confirm that the increased catalase expression in the PCat SH-SY5Y neuronal cells is neuroprotective against amyloid peptide toxicity in agreement with previous studies using catalase overexpression.29 This suggests that the PCat SH-SY5Y neuronal model may be suitable for investigating the mechanism of catalase in neuroprotection against Aβ.
Effects of the Catalase Inhibitor 3-AT on Amyloid Peptide Toxicity in Catalase Overexpressing SH-SY5Y Neuronal Cells
A possible mechanism for catalase neuroprotection against amyloid peptides is via catalase breakdown of H2O2 and blockade of the oxidative stress induced by the amyloid peptides.27,28 The observation that 3-AT reverses the catalase overexpression neuroprotection against H2O2 toxicity (Figure 2B) suggests that 3-AT could be used to determine if catalase activity is an essential component of the neuroprotection against amyloid peptides. The observations of catalase immunoreactivity in the cells when 3-AT enhanced Aβ toxicity, led to the suggestion that the enhancement of Aβ toxicity was due to inhibition of endogenous catalase.27 The PVect cells contain catalase (Figure 1B) that is active (Figure 1D). The previous reports that catalase inhibition by 3-AT enhance Aβ toxicity complicate the interpretation of results, however by comparing the effects of 3-AT on PVect and PCat SH-SY5Y neuronal cells this effect can be accounted for in viability determinations.27,28
PVect and PCat SH-SY5Y neuronal cells were pretreated with 3-AT at a range of concentrations up to 50 mM. The Aβ 1–42 peptide (10 μM) was then added to the cells and incubated for a further 24 h prior to determination of cell viability. Results showed that doses above 10 mM the 3-AT significantly increased the neurotoxicity of Aβ 1–42 in PVect SH-SY5Y neuronal cells (Figure 5A). In the PCat SH-SY5Y neuronal cells the 3-AT significantly increased the neurotoxicity of Aβ 1–42 at doses of 40 mM and 50 mM. The increased neurotoxicity of Aβ 1–42 in the presence of 3-AT suggests that the compound is allowing Aβ to exert its toxic actions by inhibiting H2O2 breakdown. These results are in agreement with the observations that 3-AT enhances Aβ toxicity.27,28
Figure 5.
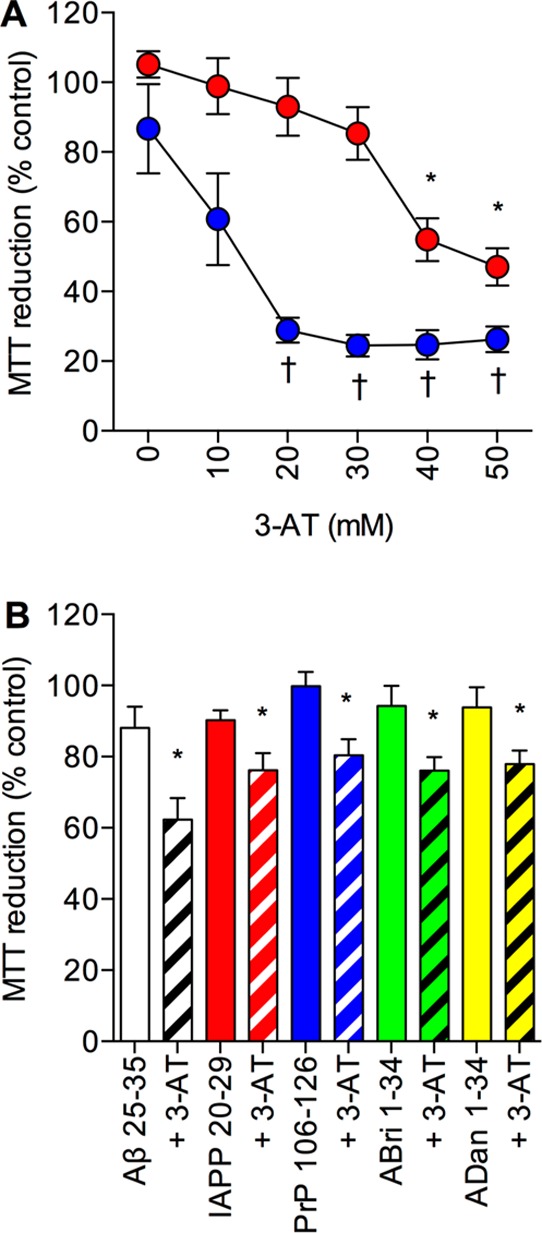
Effects of the catalase inhibitor 3-AT on amyloid peptide toxicity in catalase overexpressing SH-SY5Y neuronal cells. (A) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat; closed red circles) and stably expressing the pcDNA4/TO/myc–His expression vector (PVect; closed blue circles) were pretreated with 0–50 mM 3-AT prior to exposure to 10 μM Aβ and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs PCat plus Aβ alone; †P < 0.05 vs PVect plus Aβ alone; one-way ANOVA. (B) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) were pretreated with 50 mM 3-AT (hatched columns) or medium alone (closed columns) and exposed to 25 μM Aβ 25–35 (clear columns), IAPP 20–29 (red columns), PrP 106–126 (blue columns), ABri 1–34 (green columns), or ADan 1–34 (yellow columns) and cell viability determined by MTT reduction. Results are mean ± SEM; * = P < 0.05 vs amyloid peptide alone; one-way ANOVA.
The effects of the 50 mM 3-AT pretreatment prior to addition of 25 μM of the amyloid peptides, Aβ 25–35, ABri 1–34, ADan 1–34, IAPP 20–29, and PrP 106–126, was tested on PCat SH-SY5Y neuronal cells. Results showed that for all the amyloid peptides the 3-AT significantly increased the neurotoxicity (Figure 5B). This suggests that for all of the amyloid peptides the catalase overexpression is neuroprotective via an antioxidant mechanism and breakdown of H2O2. However, the 3-AT was not able to increase the toxicity of Aβ 1–42 in the PCat SH-SY5Y neuronal cells to the levels seen in PVect SH-SY5Y neuronal cells (Figure 5A), suggesting that there is some remaining catalase neuroprotection. The highest dose of 3-AT (50 mM) reduces catalase activity in extracts of PCat SH-SY5Y neuronal cells to undetectable (Figure 1D) raising the possibility that the catalase is also working via another mechanism. The failure of 50 mM 3-AT to abolish the neuroprotective effect of conditioned medium from PCat SH-SY5Y neuronal cells (Figure 4E) contrasts with its ability to reduce the neuroprotection in the PCat SH-SY5Y neuronal cells and suggests that the activity dependent component may involve intracellular catalase.
Effects of the BTA-EG4 Inhibitor of Catalase-Amyloid Interactions on Amyloid Peptide Toxicity in Catalase Overexpressing SH-SY5Y Neuronal Cells
Another possible mechanism for catalase neuroprotection against amyloid peptides is via a direct binding interaction that effectively blocks the amyloid actions leading to cell death.2,12 The BTA-EG4 compound is a well characterized amyloid binding compound that is known to disrupt catalase-amyloid interactions.18,20,22,23 The BTA-EG4 is a low molecular weight (416.18 Da) compound that has been demonstrated to be taken up by SH-SY5Y neuronal cells and to disrupt intracellular catalase-Aβ interactions.18 These results suggest that this compound may be effective for determining if catalase-amyloid interactions are involved in the neuronal catalase overexpression model. PCat SH-SY5Y neuronal cells were pretreated with BTA-EG4 at a range of concentrations up to 20 μM. The Aβ 25–35 peptide (50 μM) was then added to the cells and incubated for a further 24 h prior to determination of cell viability. Results showed that BTA-EG4 was not toxic over the 2.5–20 μM range (Figure 6A). At a dose of 20 μM the BTA-EG4 significantly increased the neurotoxicity of 50 μM Aβ 25–35 (Figure 6A). The increased neurotoxicity of Aβ 25–35 in the presence of BTA-EG4 suggests that the compound is disrupting catalase-Aβ interactions to allow Aβ to exert its toxic actions. This result contrasts with the observations that BTA-EG4 was significantly neuroprotective against the Aβ peptide at concentrations of 20 μM.18 The previous study preincubated the BTA-EG4 and Aβ for 12 h to allow the binding interaction to take place prior to addition to cells.18 In this study, cells were pretreated with BTA-EG4 to allow uptake into the cells prior to addition of Aβ. As such during the incubation with Aβ the cellular catalase and BTA-EG4 are competing for binding to Aβ, and this may explain the different effects observed on Aβ toxicity between the two studies.18 When PVect SH-SY5Y neuronal cells were pretreated with 20 μM BTA-EG4 prior to 50 μM Aβ 25–35 treatment, there was no significant difference in the toxicity compared to PVect SH-SY5Y neuronal cells treated with 50 μM Aβ 25–35 alone (Aβ alone 36.5 ± 5.7; BTA-EG4 plus Aβ 37.2 ± 11.7; MTT reduction (% control) mean ± SEM).
Figure 6.
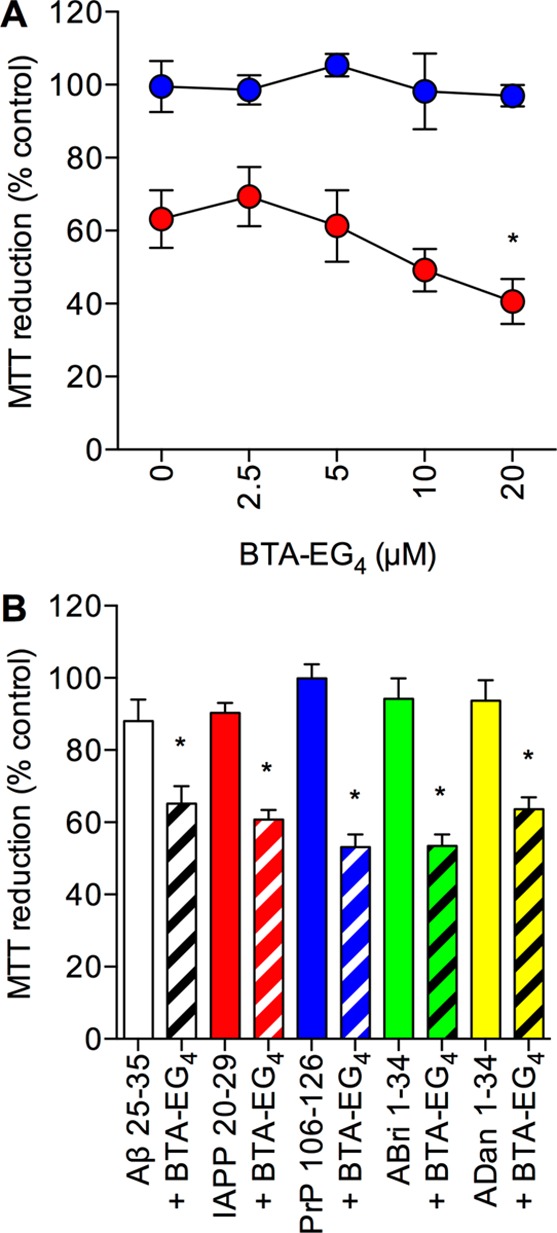
Effects of the BTA-EG4 inhibitor of catalase-amyloid interactions on amyloid peptide toxicity in catalase overexpressing SH-SY5Y neuronal cells. (A) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) were pretreated with 0–20 μM BTA-EG4 prior to exposure to medium alone (closed blue circles) or 50 μM Aβ (closed red circles) and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs Aβ alone; one-way ANOVA. (B) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) were pretreated with 20 μM BTA-EG4 (hatched columns) or medium alone (closed columns) and exposed to 25 μM Aβ 25–35 (clear columns), IAPP 20–29 (red columns), PrP 106–126 (blue columns), ABri 1–34 (green columns), or ADan 1–34 (yellow columns) and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs amyloid peptide alone; one-way ANOVA.
The effects of the 20 μM BTA-EG4 pretreatment prior to addition of 25 μM of the amyloid peptides; Aβ 25–35, ABri 1–34, ADan 1–34, IAPP 20–29, and PrP 106–126 was tested on PCat SH-SY5Y neuronal cells. Results showed that for all the amyloid peptides the BTA-EG4 significantly increased the neurotoxicity (Figure 6B). This suggests that for all of the amyloid peptides the catalase overexpression is neuroprotective via a direct binding interaction. However, the 20 μM BTA-EG4 was not able to increase the toxicity of 25 μM Aβ 25–35 in the PCat SH-SY5Y neuronal cells (BTA-EG4 plus Aβ 65.2 ± 4.9; MTT reduction (% control) mean ± SEM) to the levels seen in PVect SH-SY5Y neuronal cells (20 μM BTA-EG4 plus 25 μM Aβ 37.2 ± 11.7; MTT reduction (% control) mean ± SEM) suggesting that there is some remaining catalase neuroprotection. This could be due to the doses of BTA-EG4 used, higher doses were avoided in this study due to the increased neuroprotective actions of higher doses plus the reports that BTA-EG4 is toxic to SH-SY5Y neuronal cells at higher concentrations with an IC50 of 60 μM.18,47 The observation that 3-AT enhances the toxicity of the amyloid peptides is another mechanism for the catalase neuroprotection and the binding interaction is a component of the neuroprotection rather than the sole mechanism.
Effects of the Amyloid Binding Peptide KP 45–50 on Amyloid Peptide Toxicity in Catalase Overexpressing SH-SY5Y Neuronal Cells
The observation that Aβ binding compounds and inhibitors of Aβ generation can be neurotoxic in SH-SY5Y neuronal cells, an effect that can be reversed by addition of Aβ, suggests that the effects seen with BTA-EG4 in PCat SH-SY5Y neuronal cells could be due to effects on endogenous Aβ.48 The KP 45–50 peptide has recently been identified as an amyloid binding peptide that inhibits the toxicity of Aβ, IAPP, and PrP.24,25 The KP 45–50 peptide binds to the same region of Aβ, IAPP plus PrP as catalase, and this binding can be inhibited by catalase.24,25,49 The KP 45–50 plus BTA-EG4 compounds have similar molecular weights, KP 45–54 (828.88 Da) and BTA-EG4 (416.18 Da), and have similar actions in SH-SY5Y neuronal cells.18,24,25 The effects of pretreatment of naïve SH-SY5Y neuronal cells with 1 μM KP 45–50 or 10 μM KP 45–50 for 24 h prior to addition of 25 μM of the amyloid peptides were tested. Results showed that the 1 μM KP 45–50 had no significant effect on the toxicity of Aβ 25–35, IAPP 20–29, PrP 106–126, ABri 1–34, or ADan 1–34 (Figure 7A). However, the 10 μM KP 45–50 significantly reduced the toxicity of the Aβ 25–35, IAPP 20–29, and PrP 106–126, but had no effect on the toxicity of ABri 1–34 or ADan 1–34 (Figure 7B), in agreement with previous studies.24,25
Figure 7.
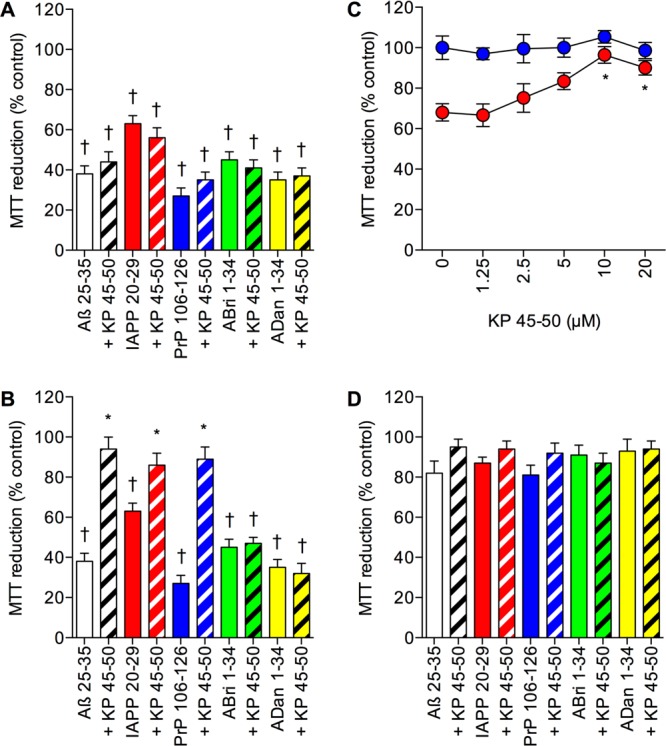
Effects of the amyloid binding peptide KP 45–50 on amyloid peptide toxicity in catalase overexpressing SH-SY5Y neuronal cells. (A) Naïve human SH-SY5Y neuronal cells were pretreated with medium alone (closed columns) or 1 μM KP 45–50 (hatched columns) prior to exposure to 25 μM Aβ 25–35 (clear columns), IAPP 20–29 (red columns), PrP 106–126 (blue columns), ABri 1–34 (green columns), or ADan 1–34 (yellow columns) and cell viability determined by MTT reduction. Results are mean ± SEM; †P < 0.05 vs control; one-way ANOVA. (B) Naïve human SH-SY5Y neuronal cells were pretreated with medium alone (closed columns) or 10 μM KP 45–50 (hatched columns) prior to exposure to 25 μM Aβ 25–35 (clear columns), IAPP 20–29 (red columns), PrP 106–126 (blue columns), ABri 1–34 (green columns), or ADan 1–34 (yellow columns) and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs amyloid peptide alone ; †P < 0.05 vs control; one-way ANOVA. (C) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) were pretreated with 0–20 μM KP 45–50 prior to exposure to medium alone (closed blue circles) or 50 μM Aβ (closed red circles) and cell viability determined by MTT reduction. Results are mean ± SEM; *P < 0.05 vs Aβ alone; one-way ANOVA. (D) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) were pretreated with 20 μM KP 45–50 (hatched columns) or medium alone (closed columns) and exposed to 25 μM Aβ 25–35 (clear columns), IAPP 20–29 (red columns), PrP 106–126 (blue columns), ABri 1–34 (green columns), or ADan 1–34 (yellow columns) and cell viability determined by MTT reduction. Results are mean ± SEM.
The effects of pretreatment of catalase overexpressing PCat SH-SY5Y neuronal cells with KP 45–50 (0–20 μM) for 24 h prior to addition of medium or 50 μM of Aβ 25–35 were tested. Results showed that KP 45–50 was not toxic over the 1.25–20 μM range (Figure 7C). When the PCat SH-SY5Y neuronal cells were pretreated with KP 45–50 (1.25–20 μM) for 24 h there was no significant enhancement of 50 μM Aβ 25–35 toxicity (Figure 7C). At doses of 10 and 20 μM the KP 45–50 significantly increased the neuroprotection against 50 μM Aβ 25–35 (Figure 7C). The pretreatment with 20 μM KP 45–50 for 24 h prior to addition of 25 μM of the amyloid peptides had no significant effect on the catalase neuroprotection against the amyloid peptides (Figure 7D). These results suggest that for all of the amyloid peptides the catalase overexpression is neuroprotective and that the KP 45–50 peptide is unable to block the neuroprotection. It is possible that the KP 45–50 inhibits catalase binding to amyloid peptides, however, this is not apparent in the neuroprotection studies, possibly due to the neuroprotective actions of KP 45–50 at higher doses. The results suggest that the enhancement of amyloid neurotoxicity in catalase overexpressing cells by BTA-EG4 is specific and not caused by the compound having a toxic effect as an endogenous Aβ binding compound.48
Effects of 3-AT, BTA-EG4, and KP 45–50 on Amyloid-β Binding to Catalase and Protection against H2O2 Toxicity
The results for 3-AT and BTA-EG4 pretreatment suggest that these two compounds enhance amyloid peptide toxicity in catalase overexpressing neuronal cells. In contrast, the KP 45–50 peptide had no significant effect on catalase overexpression neuroprotection against amyloid peptides. The proposed mechanisms of action are via 3-AT inhibition of catalase breakdown of H2O2 and BTA-EG4 inhibition of catalase-amyloid interactions. Both Aβ and 3-AT bind catalase and inhibit H2O2 breakdown, raising the possibility that the actions of 3-AT in the catalase overexpression system could be due to inhibition of the catalase-Aβ interactions.2,50 The effects of 3-AT, BTA-EG4 and KP 45–50 on the binding of biotinylated-Aβ to immobilized catalase were therefore determined. Results showed that both BTA-EG4 and KP 45–50 significantly inhibited Aβ binding to catalase while 3-AT had no effect (Figure 8A). The KP 45–50 peptide was more effective than the BTA-EG4 at inhibiting the binding with significantly greater inhibition of the interaction at concentrations between 1.25 and 10 μM compared to the mM concentrations of BTA-EG4 required to inhibit catalase-Aβ interactions (Figure 8A). The effect of BTA-EG4 at mM concentrations is in agreement with previous studies and the effects of 3-AT confirm that this compound is not acting to inhibit catalase-amyloid interactions.18,20,49 The results for KP 45–50 suggest that the failure to enhance amyloid peptide toxicity (Figure 7C and D) in catalase overexpressing cells is not due to an inability to displace catalase. It is therefore more likely that the neuroprotective effects of KP 45–50 are additive to those of catalase, as shown in Figure 7C.
Figure 8.
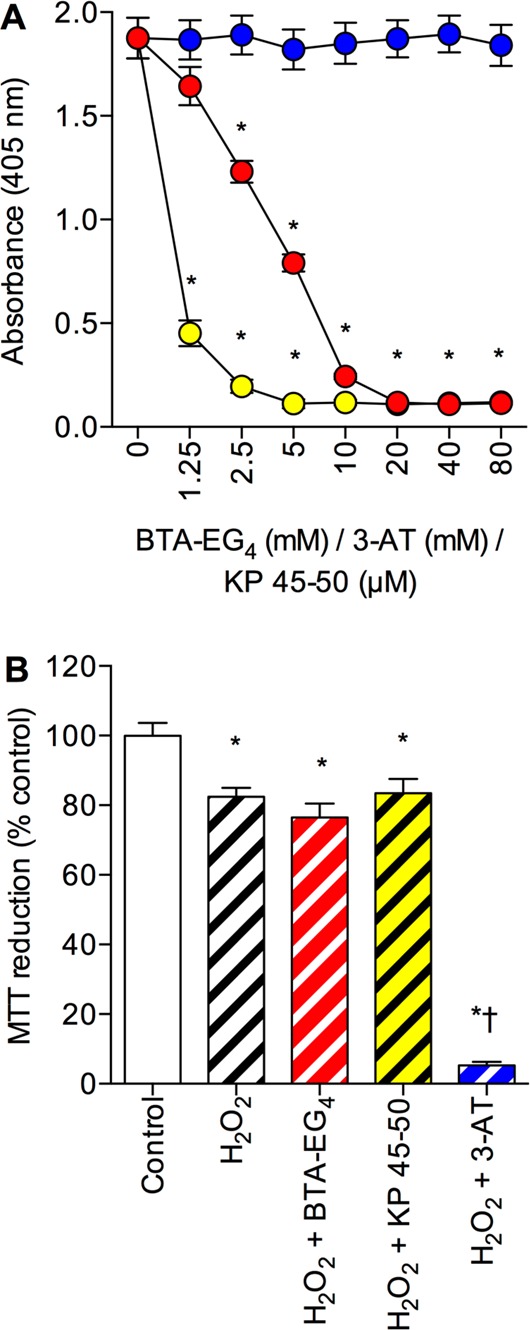
Effects of BTA-EG4, KP 45–50, and 3-AT on amyloid-β binding to catalase and protection against H2O2 toxicity. (A) Catalase coated plates were incubated with 1 μM biotinylated Aβ 1–42 in the presence of 0–80 mM 3-AT (closed blue circles), 0–80 mM BTA-EG4 (closed red circles), or 0–80 μM KP 45–50 (closed yellow circles) and bound material determined by EIA. *P < 0.05 vs biotinyl-Aβ 1–42 alone; one-way ANOVA. (B) Human SH-SY5Y neuronal cells stably expressing the catalase gene vector (PCat) were pretreated with medium alone (clear hatched column), 20 μM BTA-EG4 (red hatched column), 20 μM KP 45–50 (yellow hatched column), or 50 mM 3-AT (blue hatched column) prior to exposure to 500 μM H2O2 and cell viability determined by MTT reduction. Control was medium alone (clear column). Results are mean ± SEM; *P < 0.05 vs control; †P < 0.05 vs H2O2 alone; one-way ANOVA.
The observation that BTA-EG4 is toxic at higher concentrations than those used in the current study suggest that the compound could be acting generally to enhance toxicity rather than as an inhibitor of catalase-amyloid actions.47 The BTA-EG4 does not enhance the toxicity of Aβ in the PVect SH-SY5Y neuronal cells and a previous study has show that it does not enhance Aβ toxicity in a cell-line overexpressing the KiSS-1 metastasis-suppressor gene.25,51 To exclude BTA-EG4 acting as a general toxicity enhancer in the catalase overexpressing neuronal cells, and also to confirm that the mechanism of BTA-EG4 enhancement of amyloid peptide toxicity is not via inhibition of H2O2 breakdown, the effects of BTA-EG4 pretreatment on H2O2 toxicity were determined. The effects of KP 45–50 pretreatment were also determined. Results showed that 20 μM BTA-EG4 and 20 μM KP 45–50 had no significant effect on H2O2 toxicity (Figure 8B), pretreatment with 50 mM 3-AT significantly enhanced H2O2 toxicity. These results suggest that the enhancement of amyloid peptide toxicity in catalase overexpressing neuronal cells by BTA-EG4 is specific for amyloid peptides and likely to be due to inhibition of the catalase-amyloid binding interactions.
Conclusions
The results from this study demonstrate that catalase overexpression is neuroprotective against the Aβ, ABri, ADan, IAPP, and PrP amyloid peptides. The catalase neuroprotection against the Aβ, ABri, ADan, IAPP, and PrP amyloid peptides can be inhibited by either the catalase inhibitor 3-AT (Figure 5) or the catalase-amyloid interaction inhibitor BTA-EG4 (Figure 6). The mechanism of protection involves both catalase breakdown of H2O2 and also a direct binding catalase-amyloid interaction (Figure 9A). The catalase inhibitor 3-AT reduces the catalase overexpression neuroprotection by inhibiting H2O2 breakdown without altering the catalase-amyloid binding interaction (Figure 9B). The catalase-amyloid interaction inhibitor BTA-EG4 reduces the catalase overexpression neuroprotection by inhibiting catalase-amyloid binding interactions without altering the catalase breakdown of H2O2 (Figure 9C). The catalase activity dependent component may be intracellular since 3-AT failed to block neuroprotection of conditioned medium from catalase overexpressing cells (Figure 4E). Previous studies have shown that addition of inactive catalase to culture medium is neuroprotective, and it is unlikely that this catalase is taken up by cells.46 This suggests that the amyloid-binding dependent catalase neuroprotection may be mediated via an extra-cellular action to prevent amyloid peptide activating receptors or entering the cells.35,52−55 The ability of BTA-EG4 to enhance Aβ toxicity contrasts with previous reports that the BTA-EG4 compound is neuroprotective against Aβ and may be specific to the catalase overexpression model since the compound had no effects on Aβ toxicity in control neuronal cells or KiSS-1 overexpressing neuronal cells.18,20,22,23,25,51 The enhancement of Aβ toxicity is not seen with the KP 45–50 peptide, which has similar Aβ binding actions to the BTA-EG4 compound.18,20,22−25 The KP 45–50 has a similar affinity for amyloid peptides to catalase and has also been suggested to act via activation of a neuroprotective pathway involving activation of oxytocin/vasopressin and cyclo-oxygenase.24,25,51 The KP 45–50 neuroprotection in the naïve SH-SY5Y neuronal cells and enhancement of neuroprotection against Aβ in PCat SH-SY5Y neuronal cells confirms that additive effects were involved, that are not seen with the BTA-EG4 compound. The specificity of the BTA-EG4 enhancement of amyloid peptide toxicity may be due to targeting different sites on the amyloid peptides when compared to KP 45–50, which unlike the BTA-EG4 does not bind ABri or ADan.
Figure 9.

Diagram of proposed model for catalase neuroprotection against amyloid peptides. Panels represent (A) catalase neuroprotection, (B) reduced catalase in the presence of 3-AT, and (C) reduced catalase in the presence of BTA-EG4.
Neuronal cells that survive Aβ toxicity in cell culture models have been shown to have elevated catalase levels.27 In mouse models of AD, the levels of catalase change, with region specific increases at different time points within the disease progression.9,10 The study by Lovell et al. showed a 2–4 fold elevation of catalase activity in temporal regions of the AD brain including the hippocampus.8 As such, the catalase overexpression model with a 6-fold elevation of catalase activity may be closer to the situation observed in some regions of the AD brain than the control SH-SY5Y neuronal cells. This also suggests that the unwanted enhancement of Aβ toxicity by BTA-EG4 seen in the catalase overexpression model could be relevant to it is use in AD and other conditions where the levels of catalase are significantly elevated.22,23
Compounds that inhibit catalase–amyloid interactions have been suggested as having potential for development as therapeutics for AD, and possibly other amyloid associated diseases.2,11−14,18−26 The results from this study suggest that such compounds may have unwanted neurotoxic effects and that screening for catalase–amyloid interaction inhibitors may not therefore always yield beneficial compounds. Catalase interactions with amyloid peptides and its endogenous neuroprotection need to be considered in the search for effective therapeutic agents for the treatment of diseases such as AD, Familial British dementia, Familial Danish dementia, T2DM, and CJD which involve amyloid peptide induced neurodegeneration.35−41 The results from the present study also suggest that treatments which increase catalase expression, such as catalase gene therapy or the catalase gene activating peroxisome proliferator-activated receptor-γ agonists, may be beneficial in these neurodegenerative diseases.56−58
Methods
Test Compounds
Amyloid peptides (Aβ 1–42, Aβ 25–35, Aβ 31–35, ABri 1–34, ADan 1–34, IAPP 20–29, and PrP 106–126) were purchased from American Peptides, Bachem, and Sigma-Aldrich. N-Terminally biotinylated Aβ 1–42 was purchased from Bachem. Human erythrocyte catalase, H2O2, 3-AT, and BTA-EG4 were purchased from Sigma-Aldrich.
Cell Cultures
Human SH-SY5Y neuroblastoma cells were routinely grown in a 5% CO2 humidified incubator at 37 °C in a 1:1 mixture of Dulbecco’s modified Eagle’s medium and HAM’s F12 with Glutamax (Invitrogen) supplemented with 10% fetal calf serum (FCS), 1% nonessential amino acids, penicillin (100 units/mL), and streptomycin (100 mg/mL).28
Human Catalase Overexpression
The human catalase cDNA clone (NM_001752.3) was obtained from Origene and PCR cloned into the pcDNA4/TO/myc–His expression vector using forward (5′-AAGCTTATGGCTGACAGCCGGGAT-3′) and reverse (5′-GCGGCCGCCAGATTTGCCTTCTCCCTTGC-3′) oligonucleotides to create the PCat expression vector. SH-SY5Y cells were transfected with PCat or control pcDNA4/TO/myc–His expression vector (PVect) using lipofectamine (Invitrogen), and stably expressing clones were selected by culturing in 100 μg/mL Zeocin (Invitrogen). Immunocytochemistry, Western blot analysis, and RT-PCR were used to confirm catalase overexpression.
Immunocytochemistry of Catalase Expression
PVect and PCat SH-SY5Y neuronal cells were washed with PBS, fixed with 4% paraformaldehyde for 15 min, and permeabilized in ice cold methanol for 30 min. Cells were incubated in block solution (10% bovine serum albumin in PBS) for 15 min, followed by a 1 h incubation with primary CAT-505 mouse anti-catalase antibody (1:1000; 1 μg/mL final concentration) in block solution.59 Primary antibody was removed followed by 3 × 5 min washes in PBS, prior to incubation with goat anti-mouse IgG-Alexa-fluor 568 secondary (Abcam PLC, Cambridge; 1:500) in block solution for 45 min. Secondary antibody was removed, and cells were washed three times in PBS. Cells were incubated with 100 μg/mL RNase A for 20 min at 37 °C, followed by 3 × 5 min washes and incubation with 1 μM TO-PRO-3 iodide (642/661; Invitrogen) for 20 min. Cells were washed three times in PBS, and fluorescence was visualized by sequential scanning using a Leica TCS SP2 confocal system (Leica Microsystems, Milton Keynes, U.K.).60
Western Blot Analysis of Catalase
PVect and PCat SH-SY5Y neuronal cells were lysed on ice in 20 mM HEPES buffer supplemented with 1% Nonindet P-40, 1 mM EDTA, 150 mM sodium chloride (NaCl), 0.25% sodium deoxycholate, and protease inhibitors. Cell lysates were incubated for 1 h in lysis buffer and centrifuged at 12 000g for 10 min at 4 C. Total protein was measured by using the BCA assay. Supernatants were diluted to 1 mg/mL and resuspended in sample buffer before boiling for 5 min and separation of samples using a 15% SDS-PAGE gel. Proteins were then transferred to a nitrocellulose membrane and membranes blocked with 3% nonfat dried milk powder in PBS containing 0.1% Tween 20 (1 h at room temperature). Membranes were incubated overnight at 4 °C with CAT-505 mouse anti-catalase antibody.59 Unbound antibody was rinsed from the membranes before incubation with horseradish peroxidase-conjugated goat anti-mouse secondary antibody. Immunoreactivity was detected using an enhanced chemiluminescence substrate and UVP Bio Imaging system.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
To determine the steady-state levels of catalase mRNA, total RNA was isolated from PCat catalase overexpressing, and PVect control cells using a Qiagen RNeasy extraction kit (Cat No: 74104) according to the manufacturer’s instructions. RT-PCR was performed using the Qiagen one-step RT-PCR reagent kit (Cat No: 210210) with catalase forward (5′-AAGCTTATGGCTGACAGCCGGGAT-3′) and reverse (5′-GCGGCCGCCAGATTTGCCTTCTCCCTTGC-3′) primers. The level of β-actin was used to normalize loadings of total RNA.
Catalase Activity
PVect and PCat SH-SY5Y neuronal cells were lysed on ice in 20 mM HEPES buffer supplemented with 1% Nonindet P-40, 1 mM EDTA, 150 mM sodium chloride (NaCl), 0.25% sodium deoxycholate, and protease inhibitors. Cell lysates were incubated for 1 h in lysis buffer and centrifuged at 12 000g for 10 min at 4 °C. Total protein was measured by using the BCA assay. Lysates were diluted in phosphate buffer and catalase activity determined by mixing 50 μL sample with 50 μL of substrate (6.5 μmol H2O2 in phosphate buffer) for 60 s, adding 100 μL of 32.4 mM ammonium molybdate and measurement of absorbance change at 405 nm. Activity was calculated from a standard curve (0–100 kU/L) using purified human catalase (EC 1.11.1.6) from human erythrocytes of known activity. Activity was expressed as U/mg protein.27,61
Binding Studies
Immunoplates (96-well) were coated with catalase (1 μg/mL) in carbonate buffer, pH 9.6, and unoccupied sites blocked with 0.2% (w/v) marvel. Biotinylated Aβ peptides (200 pM) were added to plates alone or with test compounds (3-AT or BTA-EG4) in 50 mM TRIS (containing 0.1% BSA and 0.1% Triton X-100) and incubated at 4 °C for 16 h. After washing to remove unbound material an alkaline phosphatase polymer-streptavidin conjugate was added and incubated at 24 °C for 2 h. After washing to remove unbound material p-nitrophenylphosphate substrate was added and absorbance at 405 nm determined.58
Cell Viability
A 30% (w/v) H2O2 in H2O stock solution was diluted to the required concentration in culture medium immediately prior to addition to cells. Batches of synthetic Aβ 1–42, Aβ 25–35, Aβ 31–35, ABri 1–34, ADan 1–34, IAPP 20–29, and PrP 106–126 were dissolved in ddH2O at a concentration of 1.0 mg/mL and incubated at 37 °C for 24h, with constant oscillation. Following incubation, the formation of fibrils was confirmed by TEM or Congo red assay as previously described.14,21,25 The amyloid peptides were diluted to the required concentration in culture medium prior to addition to cells. Stock solutions of 3-AT, BTA-EG4, cobalt chloride, and KP 45–50 at least 100× the maximum required concentration for testing were prepared in ddH2O (3-AT and cobalt chloride) or DMSO (BTA-EG4 and KP 45–50) prior to dilution to the required concentration in culture medium. None of the vehicles used (ddH2O or DMSO) had a statistically significant effect on cell viability of PCat or PVect SH-SY5Y neuronal cells and vehicle controls were included in experiments.
For experiments involving testing the effects of 3-AT, BTA-EG4, or KP 45–50, the cells were pretreated with test compounds for 24 h prior to treatment with amyloid peptides or H2O2 as described. For experiments testing the effects of H2O2 toxicity, cells were incubated for 2 h prior to determination of cell viability. For experiments testing amyloid peptides or cobalt chloride, these were added directly to culture medium prior to incubation for 24 h. Cell viability was determined either by trypan blue dye exclusion with at least 100 cells counted per well or by MTT reduction.28 For MTT reduction determination, after incubation with test substances MTT (10 μL: 12 mM stock) was added and cells incubated for a further 4 h. Cell lysis buffer [100 μL/well; 20% (v/v) SDS, 50% (v/v) N,N-dimethylformamide, pH 4.7] was added and after repeated pipetting to lyse cells the MTT formazan product formation was determined by measurement of absorbance change at 570 nm. Control levels in the absence of test substances were taken as 100% and the absorbance in the presence of cells lysed with Triton X-100 at the start of the incubation period with test substances taken as 0%.
Data Analysis
All data are expressed as means ± SEM. For cytotoxicity experiments, data are expressed as % dead (trypan blue stained) cells or % control cells MTT reduction. Statistical analysis was performed by one-way analysis of variance (ANOVA) using GraphPad Prism software (version 6). Posthoc analysis was carried with Tukey or Dunnett multiple comparison based on the recommendations of GraphPad Prism software for the data sets concerned, with a P value of <0.05 considered statistically significant.
Acknowledgments
We would like to thank Marc Rhyan Puno for assistance with cloning and overexpression of catalase, Dr Anatoliy Markiv plus Dr Diluka Peiris for assistance with confocal image acquisition, and Amanda Nercessian for assistance with the kisspeptin experiments.
Author Contributions
N.G.N.M., A.C, and M.O. conceived and designed the experiments, performed the experiments, and analyzed the data. N.G.N.M. and A.C. wrote the paper. N.G.N.M., A.C., and M.O. critically reviewed the manuscript.
The work was funded by the University of Westminster plus grants from WestFocus (Park Board, PCF II 104 to N.G.N.M.) and the UK Department of Trade and Industry (LOT/0311684 to N.G.N.M.).
The authors declare the following competing financial interest(s): N.G.N.M. is named as the inventor on patent applications held by the University of Roehampton for the use of kissorphin peptides to treat Alzheimers disease, Creutzfeldt-Jakob disease or diabetes mellitus (Publication Numbers: GB2493313 A, WO 2011/144714 A1 and EP 2 388 012 A1); under the University of Roehampton rules he could benefit financially if the patent is commercially exploited. N.G.N.M. is also a shareholder and director of NeuroDelta Ltd (Company No: 06222473; http://www.neurodelta.co.uk).
References
- Kirkman H. N.; Gaetani G. F. (2007) Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem. Sci. 32, 44–50. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N. (1999) Amyloid-beta binds catalase with high affinity and inhibits hydrogen peroxide breakdown. Biochem. J. 344(Pt 2), 293–296. [PMC free article] [PubMed] [Google Scholar]
- Schriner S. E.; Linford N. J.; Martin G. M.; Treuting P.; Ogburn C. E.; Emond M.; Coskun P. E.; Ladiges W.; Wolf N.; van Remmen H.; Wallace D. C.; Rabinovitch P. S. (2005) Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909–1911. [DOI] [PubMed] [Google Scholar]
- Mao P.; Manczak M.; Calkins M. J.; Truong Q.; Reddy T. P.; Reddy A. P.; Shirendeb U.; Lo H.-H.; Rabinovitch P. S.; Reddy P. H. (2012) Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 21, 2973–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen R. H. J.; Johnson L. A.; Zuloaga D. G.; Limoli C.; Raber J. (2013) Enhanced hippocampus-dependent memory and reduced anxiety in mice over-expressing human catalase in mitochondria. J. Neurochem. 125, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharrazi H.; Vaisi-Raygani A.; Rahimi Z.; Tavilani H.; Aminian M.; Pourmotabbed T. (2008) Association between enzymatic and non-enzymatic antioxidant defense mechanism with apolipoprotein E genotypes in Alzheimer disease. Clin. Biochem. 41, 932–936. [DOI] [PubMed] [Google Scholar]
- Puertas M. C.; Martínez-Martos J. M.; Cobo M. P.; Carrera M. P.; Mayas M. D.; Ramírez-Expósito M. J. (2012) Plasma oxidative stress parameters in men and women with early stage Alzheimer type dementia. Exp. Gerontol. 47, 625–630. [DOI] [PubMed] [Google Scholar]
- Lovell M. A.; Ehmann W. D.; Butler S. M.; Markesbery W. R. (1995) Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 45, 1594–1601. [DOI] [PubMed] [Google Scholar]
- Belkacemi A.; Ramassamy C. (2012) Time sequence of oxidative stress in the brain from transgenic mouse models of Alzheimer’s disease related to the amyloid-β cascade. Free Radicals Biol. Med. 52, 593–600. [DOI] [PubMed] [Google Scholar]
- Cimini A.; Moreno S.; D’amelio M.; Cristiano L.; D’Angelo B.; Falone S.; Benedetti E.; Carrara P.; Fanelli F.; Cecconi F.; Amicarelli F.; Cerù M. P. (2009) Early biochemical and morphological modifications in the brain of a transgenic mouse model of Alzheimer’s disease: a role for peroxisomes. J. Alzheimer’s Dis. 18, 935–952. [DOI] [PubMed] [Google Scholar]
- Clementi M. E.; Martorana G. E.; Pezzotti M.; Giardina B.; Misiti F. (2004) Methionine 35 oxidation reduces toxic effects of the amyloid beta-protein fragment (31–35) on human red blood cell. Int. J. Biochem. Cell Biol. 36, 2066–2076. [DOI] [PubMed] [Google Scholar]
- Rensink A. A. M.; Verbeek M. M.; Otte-Höller I.; Donkelaar, ten H. T.; deWaal R. M. W.; Kremer B. (2002) Inhibition of amyloid-beta-induced cell death in human brain pericytes in vitro. Brain Res. 952, 111–121. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N.; Mayor N. P.; Rawlinson J. (2001) Identification of amyloid-β binding sites using an antisense peptide approach. NeuroReport 12, 2561–2566. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N.; Harris J. R. (2009) Polymorphism of amyloid-beta fibrils and its effects on human erythrocyte catalase binding. Micron 40, 800–810. [DOI] [PubMed] [Google Scholar]
- Chilumuri A.; Ashioti M.; Nercessian A. N.; Milton N. G. N. (2013) Immunolocalization of kisspeptin associated with amyloid-ß deposits in the pons of an Alzheimer’s disease patient. J. Neurodegener. Dis. 2013, 879710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappolla M. A.; Omar R. A.; Kim K. S.; Robakis N. K. (1992) Immunohistochemical evidence of oxidative [corrected] stress in Alzheimer’s disease. Am. J. Pathol. 140, 621–628. [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N. (2004) Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: implications for treatment. Drugs Aging 21, 81–100. [DOI] [PubMed] [Google Scholar]
- Habib L. K.; Lee M. T. C.; Yang J. (2010) Inhibitors of catalase-amyloid interactions protect cells from beta-amyloid-induced oxidative stress and toxicity. J. Biol. Chem. 285, 38933–38943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar P.; Yang J. (2006) Inhibiting protein-amyloid interactions with small molecules: a surface chemistry approach. Bioorg. Med. Chem. Lett. 16, 1076–1079. [DOI] [PubMed] [Google Scholar]
- Inbar P.; Li C. Q.; Takayama S. A.; Bautista M. R.; Yang J. (2006) Oligo(ethylene glycol) derivatives of thioflavin T as inhibitors of protein-amyloid interactions. ChemBioChem 7, 1563–1566. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N.; Harris J. R. (2010) Human islet amyloid polypeptide fibril binding to catalase: a transmission electron microscopy and microplate study. Sci. World J. 10, 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megill A.; Lee T.; DiBattista A. M.; Song J. M.; Spitzer M. H.; Rubinshtein M.; Habib L. K.; Capule C. C.; Mayer M.; Turner R. S.; Kirkwood A.; Yang J.; Pak D. T. S.; Lee H.-K.; Hoe H.-S. (2013) A Tetra(Ethylene Glycol) Derivative of Benzothiazole Aniline Enhances Ras-Mediated Spinogenesis. J. Neurosci. 33, 9306–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., and Inbar P. (2010) Compounds and methods for the diagnosis and treatment of amyloid associated diseases. US Patent 7,666,886, Feb. 23, 2010.
- Milton N. (2013) Kissorphin Peptides for Use in the Treatment of Alzheimer’s Disease, Creutzfeldt- Jakob Disease or Diabetes Mellitus. UK Pat. Appl. GB 2493313 A, Jan. 30, 2013.
- Milton N. G. N.; Chilumuri A.; Rocha-Ferreira E.; Nercessian A. N.; Ashioti M. (2012) Kisspeptin Prevention of Amyloid-β Peptide Neurotoxicity in Vitro. ACS Chem. Neurosci. 3, 706–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N.; Harris J. R. (2013) Fibril formation and toxicity of the non-amyloidogenic rat amylin peptide. Micron 44, 246–253. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N. (2001) Inhibition of catalase activity with 3-amino-triazole enhances the cytotoxicity of the Alzheimer’s amyloid-β peptide. Neurotoxicology 22, 767–774. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N. (2008) Homocysteine inhibits hydrogen peroxide breakdown by catalase. Open Enzyme Inhib. J. 1, 34–41. [Google Scholar]
- Sagara Y.; Dargusch R.; Klier F. G.; Schubert D.; Behl C. (1996) Increased antioxidant enzyme activity in amyloid beta protein-resistant cells. J. Neurosci. 16, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C.; Davis J. B.; Lesley R.; Schubert D. (1994) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77, 817–827. [DOI] [PubMed] [Google Scholar]
- Behl C. (2005) Oxidative stress in Alzheimer’s disease: implications for prevention and therapy. Subcell. Biochem. 38, 65–78. [DOI] [PubMed] [Google Scholar]
- Maczurek A. E.; Wild R.; Laurenti D.; Steele M. L.; Ooi L.; Münch G. (2013) Generation of hydrogen peroxide-resistant murine neuroblastoma cells: a target discovery platform for novel neuroprotective genes. J. Neural Transm. 120, 1171–1178. [DOI] [PubMed] [Google Scholar]
- Gáspár T.; Domoki F.; Lenti L.; Institoris Á.; Snipes J. A.; Bari F.; Busija D. W. (2009) Neuroprotective effect of adenoviral catalase gene transfer in cortical neuronal cultures. Brain Res. 1270, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake-Nara E.; Saida K. (2007) Characterization of CoCl2-induced reactive oxygen species (ROS): Inductions of neurite outgrowth and endothelin-2/vasoactive intestinal contractor in PC12 cells by CoCl2 are ROS dependent, but those by MnCl2 are not. Neurosci. Lett. 422, 223–227. [DOI] [PubMed] [Google Scholar]
- Buxbaum J. N.; Linke R. P. (2012) A molecular history of the amyloidoses. J. Mol. Biol. 421, 142–159. [DOI] [PubMed] [Google Scholar]
- Gibson G.; Gunasekera N.; Lee M.; Lelyveld V.; El-Agnaf O. M. A.; Wright A.; Austen B. (2004) Oligomerization and neurotoxicity of the amyloid ADan peptide implicated in familial Danish dementia. J. Neurochem. 88, 281–290. [DOI] [PubMed] [Google Scholar]
- Norrby E. (2011) Prions and protein-folding diseases. J. Intern. Med. 270, 1–14. [DOI] [PubMed] [Google Scholar]
- Revesz T.; Holton J. L.; Lashley T.; Plant G.; Frangione B.; Rostagno A.; Ghiso J. (2009) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 118, 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsachaki M.; Ghiso J.; Rostagno A.; Efthimiopoulos S. (2010) BRI2 homodimerizes with the involvement of intermolecular disulfide bonds. Neurobiol. Aging 31, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zraika S.; Hull R. L.; Verchere C. B.; Clark A.; Potter K. J.; Fraser P. E.; Raleigh D. P.; Kahn S. E. (2010) Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence?. Diabetologia 53, 1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson K., Barisone G. A., Diaz E., Jin L.-W., DeCarli C., and Despa F. (2013) Amylin deposition in the brain: A second amyloid in Alzheimer’s disease? Ann. Neurol., published online Jun. 22, 2013, DOI: 10.1002/ana.23956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M.; Kuroda Y.; Arispe N.; Rojas E. (2000) Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 275, 14077–14083. [DOI] [PubMed] [Google Scholar]
- Ciccotosto G. D.; Tew D.; Curtain C. C.; Smith D.; Carrington D.; Masters C. L.; Bush A. I.; Cherny R. A.; Cappai R.; Barnham K. J. (2004) Enhanced toxicity and cellular binding of a modified amyloid beta peptide with a methionine to valine substitution. J. Biol. Chem. 279, 42528–42534. [DOI] [PubMed] [Google Scholar]
- Jekabsone A.; Mander P. K.; Tickler A.; Sharpe M.; Brown G. C. (2006) Fibrillar beta-amyloid peptide Abeta1–40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J. Neuroinflammation 3, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.; Park E. J.; Jou I.; Kim J. H.; Joe E. H. (2001) Reactive oxygen species mediate A beta(25–35)-induced activation of BV-2 microglia. NeuroReport 12, 1449–1452. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Rydel R. E.; Drzewiecki G. J.; Fuson K.; Wright S.; Wogulis M.; Audia J. E.; May P. C.; Hyslop P. A. (1996) Amyloid beta-mediated oxidative and metabolic stress in rat cortical neurons: no direct evidence for a role for H2O2 generation. J. Neurochem. 67, 1595–1606. [DOI] [PubMed] [Google Scholar]
- Prangkio P.; Rao D. K.; Lance K. D.; Rubinshtein M.; Yang J.; Mayer M. (2011) Self-assembled, cation-selective ion channels from an oligo(ethylene glycol) derivative of benzothiazole aniline. Biochim. Biophys. Acta 1808, 2877–2885. [DOI] [PubMed] [Google Scholar]
- Plant L. D.; Boyle J. P.; Smith I. F.; Peers C.; Pearson H. A. (2003) The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J. Neurosci. 23, 5531–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N. and Harris J. R. (2013) Polymorphism of amyloid fibrils and their complexes with catalase. In Bio-nanoimaging: Protein Misfolding & Aggregation (Uversky V. N., and Lyubchenko Y. L., Eds.), Academic Press, Waltham, MA: (in press). [Google Scholar]
- Margoliash E.; Novogrodsky A.; Schejter A. (1960) Irreversible reaction of 3-amino-1:2:4-triazole and related inhibitors with the protein of catalase. Biochem. J. 74, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilumuri A.; Milton N. G. N. (2013) The role of neurotransmitters in protection against amyloid-β toxicity by KiSS-1 overexpression in SH-SY5Y neurons. ISRN Neurosci. 2013, 253210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Mucke L. (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida N.; Masters C. L.; Beyreuther K. (1996) Rapid cellular uptake of Alzheimer amyloid betaA4 peptide by cultured human neuroblastoma cells. FEBS Lett. 394, 174–178. [DOI] [PubMed] [Google Scholar]
- Jhamandas J. H.; Li Z.; Westaway D.; Yang J.; Jassar S.; MacTavish D. (2011) Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. Am. J. Pathol. 178, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. S.; Minogue A. M.; Connor T. J.; Lynch M. A. (2013) Amyloid-β-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J. Neuroimmun. Pharmacol. 8, 301–311. [DOI] [PubMed] [Google Scholar]
- Qi X.; Sun L.; Lewin A. S.; Hauswirth W. W.; Guy J. (2007) Long-term suppression of neurodegeneration in chronic experimental optic neuritis: antioxidant gene therapy. Invest. Ophthalmol. Visual Sci. 48, 5360–5370. [DOI] [PubMed] [Google Scholar]
- Okuno Y.; Matsuda M.; Miyata Y.; Fukuhara A.; Komuro R.; Shimabukuro M.; Shimomura I. (2010) Human catalase gene is regulated by peroxisome proliferator activated receptor-gamma through a response element distinct from that of mouse. Endocr. J. 57, 303–309. [DOI] [PubMed] [Google Scholar]
- Katsouri L.; Blondrath K.; Sastre M. (2012) Peroxisome proliferator-activated receptor-γ cofactors in neurodegeneration. IUBMB Life 64, 958–964. [DOI] [PubMed] [Google Scholar]
- Hwang T. S.; Choi H. K.; Han H. S. (2007) Differential expression of manganese superoxide dismutase, copper/zinc superoxide dismutase, and catalase in gastric adenocarcinoma and normal gastric mucosa. Eur. J. Surg. Oncol. 33, 474–479. [DOI] [PubMed] [Google Scholar]
- Markiv A.; Beatson R.; Burchell J.; Durvasula R. V.; Kang A. S. (2011) Expression of recombinant multi-coloured fluorescent antibodies in gor -/ trxB- E. coli cytoplasm. BMC Biotechnol. 11, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N. (2006) Interactions between amyloid-ß and enzymes. In Cell biology protocols (Harris J. R., Graham J. M., and Rickwood D., Eds.), pp 359–363, John Wiley & Sons Ltd, Chichester, England. [Google Scholar]