

Abstract
We report a new methodology for direct visualization of superoxide production in the dopaminergic area of the brain in Parkinson’s disease, based on the redox cycle of mito-TEMPO, a blood-brain barrier-, cell-, and mitochondria-penetrating nitroxide derivative with superoxide scavenging properties and T1 magnetic resonance imaging (MRI) contrast. The experiments were conducted on healthy and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. In healthy mice, the nitroxide-enhanced MRI signal was weak and short-lived (half-life ∼ 40 s; duration ∼ 80 s). The profile of the histograms indicated a high reducing activity of normal brain tissues against mito-TEMPO. In MPTP-treated mice, the nitroxide-enhanced MRI signal was strong and long-lived (half-life > 20 min; duration > 20 min), especially in the dopaminergic area of the brain. The histograms indicated a high oxidative activity in dopaminergic tissues of MPTP-treated mice. The results show directly, on intact mammals, that superoxide is a major inducer and/or mediator of neurodegenerative damage in Parkinson’s disease. The high oxidative status of brain tissue in Parkinson’s disease was also confirmed on isolated tissue specimens, using total reducing capacity assay and ROS/RNS assay.
Keywords: Mito-TEMPO, methoxy-TEMPO, TEMPOL, superoxide, nitroxide-enhanced magnetic resonance imaging, Parkinson’s disease
Parkinson’s disease (PD) is a common neurodegenerative disorder of unknown cause. PD symptoms are attributed to the degeneration of dopamine-containing neurons in the substantia nigra pars compacta (SNpc) and a deficit in brain dopamine.1−3 In most instances of PD, however, neither the cause nor the mechanisms underlying the loss (death) of the nigrostriatal dopaminergic neurons are known. Thus, the outstanding questions are why and how these specific neurons die in PD. Although investigations of post-mortem PD tissues have their place in the quest to answer these questions, they typically provide a fixed image of end-stage PD.1 This approach is not suitable to define early neurodegenerative events and their temporal evolution.
In the experimental medicine, one of the widely used models of PD is the “MPTP mouse model”.1,2 MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) is a byproduct of the chemical synthesis of a meperidine analogue with heroin-like effects that can induce a parkinsonian syndrome in humans almost indistinguishable from PD.4 MPTP is the only known dopaminergic neurotoxin capable of causing a reliable and reproducible lesion of the nigrostriatal dopaminergic pathway and a clinical picture of parkinsonism in various mammalian species (e.g., human, nonhuman primates, mice).2 For the past 20 years, MPTP mouse model of PD has generated important insights into PD pathogenesis. Alterations in energy metabolism (e.g., mitochondrial dysfunction), generation of reactive oxygen species (ROS), and perturbation in calcium homeostasis occur within hours of MPTP administration, before any significant neuronal death has occurred.5−7 These initial events could be intracellular signals that can set into motion deleterious molecular cascades (e.g., apoptosis activation), which are responsible for the death of dopamine neurons.
It is worthwhile noting that post-mortem brain samples from patients with PD show a selective defect in the mitochondrial electron transport chain complex that is affected by MPTP.1,2,8,9 The molecular mechanism is shown in Figure 1. It is well-described by Vila and Przedborski.2 After its systemic administration, MPTP, which is a generally nontoxic pro-toxin, rapidly crosses the blood-brain barrier (BBB) and is metabolized to 1-methyl-4-phenyl-2,3-dihydropyrolidine (MPDP+) by the monoamine oxidase B (MAO-B) in nondopamine cells, and then, probably by spontaneous oxidation, to 1-methyl-4-phanylpyrolidine (MPP+). MPP+ is the active toxic compound with high affinity to dopamine transporters. Thus, MPP+ can be delivered in the dopamine neurons. MPP+ is concentrated within the mitochondria, where it inhibits complex-I of the electron transport chain and impairs mitochondrial respiration. The inhibition of complex-I impedes the flow of electrons along the mitochondrial electron transport chain, resulting in an increased production of ROS by mitochondria (probably, superoxide radicals and/or hydrogen peroxide), subsequent activation of NADPH-dependent oxidases (NOX), which causes oxidative stress and activation of apoptosis and necrosis of neuron.
Figure 1.
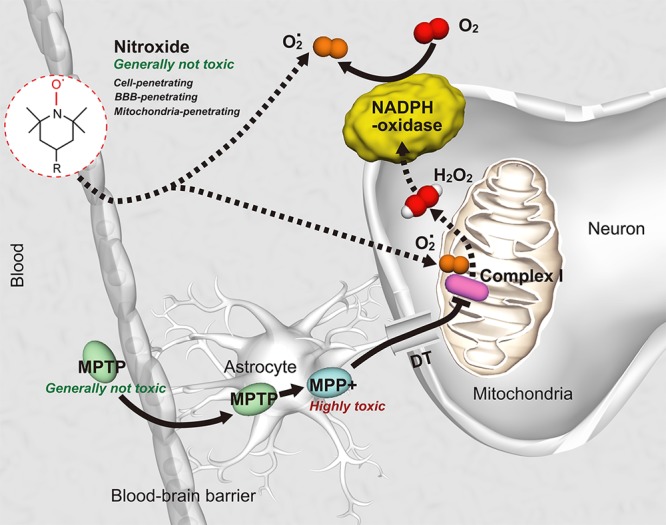
Molecular mechanism of MPTP-induced Parkinson’s disease [according to Vila and Przedborski2] and detection of superoxide generation using BBB-penetrating and mitochondria-penetrating nitroxide as a molecular sensor. DT, dopamine transporter; MPP+, 1-methyl-4-phenylpyridinium; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; O2·, superoxide radical; H2O2, hydrogen peroxide.
Currently, there is no universal methodology for direct visualization of the initial oxidative events in dopaminergic area of the brain in vivo (on intact mammals), caused by MPTP administration.
In earlier publications, based on cancer models, it was demonstrated that some cell-penetrating nitroxide derivatives (TEMPOL, TEMPO-labeled nitrosourea, methoxy-PROXYL) are appropriate molecular sensors for imaging of ROS (in particular, superoxide generation) in living tissues (in vivo), using magnetic resonance imaging (MRI) or electron-paramagnetic resonance imaging (EPRI).10−14 These nitroxide derivatives penetrate BBB and are also appropriate molecular sensors for imaging of superoxide generation in the brain.15−18 The method is based on nitroxide redox cycle shown in Figure 2.
Figure 2.

Nitroxide redox cycle as a sensing platform for imaging of superoxide generation in the brain: principle of the method. (A) In vitro studies demonstrate that nitroxide radical, which is characterized by MRI/EPR contrast enhancement, can be converted to the noncontrast-enhancing hydroxylamine and/or oxoammonium by different compounds in cells, body fluids, and tissues (e.g., free ions of transition metals, hydroperoxyl radicals, ubiquinols, NAD(P)H, ascorbate, glutathione, etc.). The contrast-enhancing radical form can be recovered by interaction of hydroxylamine with the superoxide/hydroperoxyl radical at physiological pH (7.4). It seems that in vivo nitroxide will exist primarily in two forms: as a radical and as a hydroxylamine. Various reducers and oxidizers are involved (directly or indirectly) in the formation of hydroxylamine, but only the interaction of hydroxylamine to dominate in vivo as the process that restores the nitroxide radical and its MRI/EPR contrast. Thus, nitroxide-enhanced MRI/EPR signal follows the reduction/oxidation of the nitroxide derivative and could serve as a marker of superoxide generation. (B) In vivo studies demonstrate that half-life (τ1/2) of nitroxide-enhanced MRI signal in healthy brain is less than 2 min, which indicates a high reducing activity of brain tissues. In cancer-bearing brain, and especially in cancer tissue, the half-life of nitroxide-enhanced MRI signal is over 15 min. The MRI images were obtained and processed after intravenous injection of TEMPO-labeled nitrosourea in healthy mouse or glioma-bearing mouse (details are described in refs (10) and (17)).
In vitro studies demonstrate that the nitroxide radical (which is characterized by T1 contrast) could be converted rapidly to noncontrast hydroxylamine and/or oxoammonium by different cellular compounds (free ions of transition metals, hydroperoxyl radicals, ubiquinols, NAD(P)H, glutathione, ascorbate, etc.).13,14,19−23 Hydroxylamine and oxoamonium are “superoxide dismutase-mimetics” and could recover the nitroxide radical.19,21,24,25 It was found that the interaction of oxoammonium with superoxide occurs very fast (rate constant ∼ 109 M–1·s–1).25 Under physiological conditions, the oxoamonnium could be reduced effectively to hydroxylamine by NAD(P)H.21 Hydroxylamine also interacts effectively with hydroperoxyl radicals at physiological pH ∼ 7.4 with recovery of the contrast form.25 Therefore, in vivo various reducers and oxidizers could be involved (directly or indirectly) in the formation of oxoammonium and/or hydroxylamine, but only the interaction of both compounds with superoxide/hydroperoxyl radical seems to be a dominant process in vivo that can restore the nitroxide radical and MRI/EPR contrast.
In vivo studies (on intact mammals) demonstrate that nitroxide-enhanced MRI/EPR signal follows the reduction/oxidation of the nitroxide derivative in tissues and body fluids.10−19 Therefore, this parameter could serve as a marker of superoxide generation and oxidative stress in vivo. A short-lived nitroxide-enhanced MRI/EPR signal indicates a high reducing capacity of the respective tissue, while a long-lived nitroxide-enhanced MRI/EPR signal indicates a high oxidative capacity. This can be seen very well in experimental cancer models (Figure 2B).10−12 Using BBB- and cell-penetrating TEMPOL and TEMPO-labeled nitrosourea, we found that nitroxide-enhanced MRI signal in the brain of healthy mammals had a very short half-life (τ1/2), less than 2 min, and there was a difference between the kinetic curves of MRI signal decay in different parts of the brain.17,18 In contrast, in cancer-bearing brain, which is characterized by elevated ROS levels, the nitroxide-enhanced MRI signal was long-lived (τ1/2 > 15 min) (Figure 2B).10−12
In the present study, we applied the described methodology for visualization of superoxide production in the dopaminergic area of the brain of MPTP-treated mice, using mito-TEMPO. Mito-TEMPO is BBB- and cell-penetrating nitroxide derivative with prevalent mitochondrial accumulation,26 superoxide scavenging properties26,27 and comparatively high T1 MRI contrast. This makes it a useful molecular sensor for detection of superoxide production in the dompaniergic area of the brain, using MRI (Figure 1).
Figure 3 shows typical images of the extracted nitroxide-enhanced MRI signal (normalized to the baseline) in healthy (B) and MPTP-treated mice (C). Two ROIs were selected: (i) the whole brain area (ROI1) and (ii) the dopaminergic area (ROI2) (Figure 3A).
Figure 3.

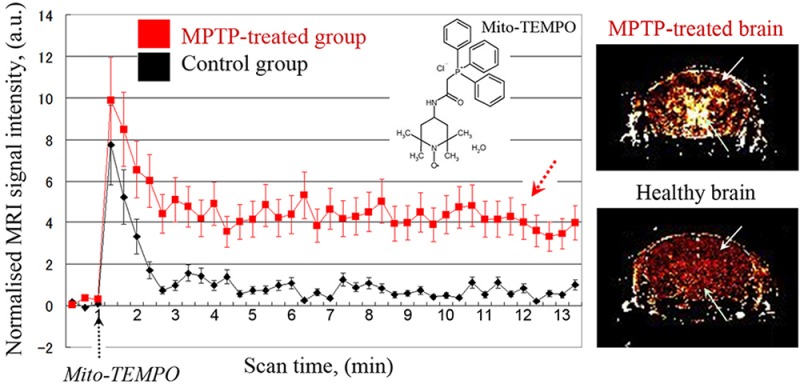
Nitroxide-enhanced MRI in healthy and MPTP-treated mice, using mito-TEMPO as a contrast agent. (A) MR images indicating region-of-interest (ROI; dotted lines). (B) MR images of brain of healthy mouse: (a) MR image of mouse brain; (b) extracted MRI signal enhancement, obtained before injection of mito-TEMPO; (c–e) extracted MRI signal enhancement, obtained 1, 5, and 15 min after injection of mito-TEMPO, respectively. (C) MR images of brain of MPTP-treated mouse: (a) MR image of mouse brain; (b) extracted MRI signal enhancement, obtained before injection of mito-TEMPO; (c–e) extracted MRI signal enhancement, obtained 1, 5, and 15 min after injection of mito-TEMPO, respectively. The MRI signal intensity in dopaminergic area is markedly higher in comparison with control. In (A), (B), and (C), the images were obtained by T1-weighted gradient-echo sequence. (D) Kinetic curves of nitroxide-enhanced MRI signal in the brain (ROI1) of healthy mice (black line) and MPTP-treated mice (red line). (E) Kinetic curves of nitroxide-enhanced MRI signal in the dopaminergic area of the brain (ROI2) of healthy mice (black line) and MPTP-treated mice (red line). In (D) and (E), the data are the mean ± SD from 6 or 10 animals for control group or MPTP-treated group, respectively.
In control mice, in both ROIs, the signal increased slightly after the injection of mito-TEMPO followed by a rapid decrease to the baseline (Figure 3B and D1, D2, black lines). The enhancement of the MRI signal in the beginning is due to the penetration and accumulation of mito-TEMPO in the brain tissues, whereas the decrease is due to its reduction to noncontrast hydroxylamine, which occurs predominantly inside the brain cells. The half-life of the nitroxide-enhanced MRI signal was approximately 45 s and the duration of the signal was ∼1 min 20 s. The profile of the histograms indicates a high reducing activity of normal brain tissues against nitroxide radical mito-TEMPO.
In the MPTP-treated mice, a strong and long-lived nitroxide-enhanced MRI signal was detected in the dopaminergic area of the brain after injection of mito-TEMPO (Figure 3C). The kinetic curves of the nitroxide-enhanced MRI signal in the brain (Figure 3D1, D2, red lines) were completely different from the reference profiles, recorded for the control group (Figure 3D1, D2, black lines). In both ROIs of MPTP-treated mice, the signal increased after injection, after which the intensity decreased by ∼50% and reached a plateau without further decrease to the baseline within 15 min. Using long-term scanning, it was established that the duration of the nitroxide-enhanced MRI signal was over 20 min (p < 0.001 vs control). The images in Figure 3C show that the enhancement of MRI signal in the cortex of MPTP-treated mice was negligible. Therefore, the observed differences between the kinetic curves of healthy mice and MPTP-treated mice are due to the different tissue redox activity against mito-TEMPO in the dopaminergic area of the brain. The histograms indicate a high oxidative activity in mitochondria-rich dopaminergic area of MPTP-treated mice due to abnormal production of mitochondrial superoxide.
The data suggest that mito-TEMPO, which is an well-known mitochondria-targeted superoxide scavenger,26−28 penetrates into the brain and participates in redox transformations. The redox cycle of mito-TEMPO consists of two antagonistic processes in respect of its MRI contrast properties. The reduction of mito-TEMPO to hydroxylamine form is accompanied with loss of nitroxide-enhanced MRI signal. In contrast, the interaction of the hydrohylamine form with superoxide recovers the radical form of mito-TEMPO and nitroxide-enhanced MRI contrast.
Similar differences in the kinetics of nitroxide-enhanced MRI between healthy and MPTP-treated mice were obtained using other nitroxide derivatives, methoxy-TEMPO and hydroxy-TEMPO (TEMPOL) (Figure 4). Both nitroxides are BBB-penetrating and cell-penetrating. The intensity of nitroxide-enhanced MRI signal is higher in MPTP-treated brain than in healthy brain (Figure 4A). However, 10 min after injection of nitroxide probe, the enhancement is evenly distributed in all brain tissues (including cortex) and does not strongly localized in dopaminergic area of the brain (Figure 4B).
Figure 4.
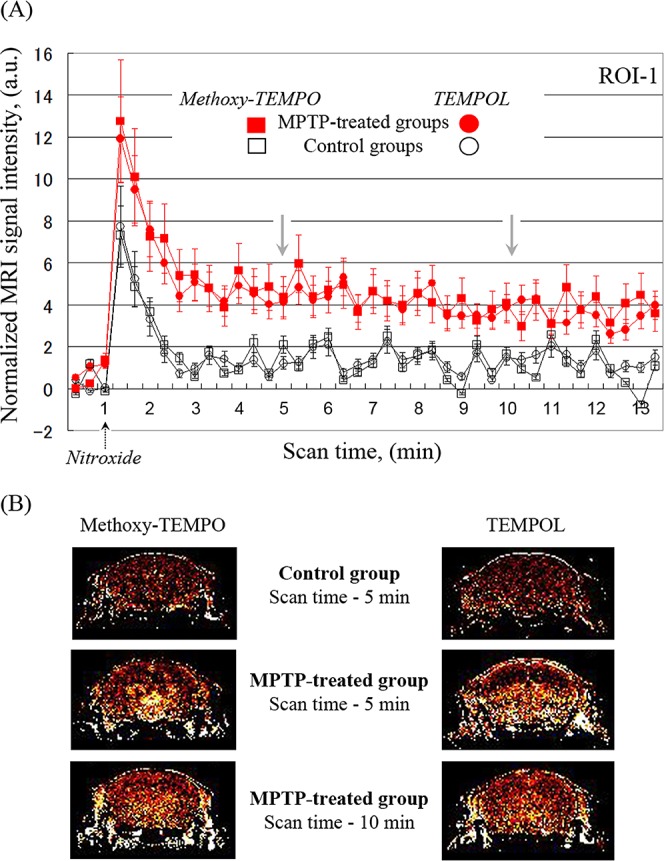
Nitroxide-enhanced MRI in healthy and MPTP-treated mice, using methoxy-TEMPO or hydroxy-TEMPO (TEMPOL) as a contrast agent. (A) Kinetic curves of nitroxide-enhanced MRI signal in the brain (ROI1) of healthy mice (black lines) and MPTP-treated mice (red lines). In the case of methoxy-TEMPO, the data are the mean ± SD from 4 and 7 animals for control group and MPTP-treated group, respectively. In the case of TEMPOL, the data are the mean ± SD from 4 and 5 animals for control group and MPTP-treated group, respectively. (B) Extracted MRI signal enhancement in the brain of healthy mice or MPTP-treated mice, obtained 5 and 10 min after injection of subsequent nitroxide probe.
The treatment with MPTP affected the general physiological conditions of mice: it led to hypotension and slight decrease of the body temperature (data are not shown). However, it did not affect the nitroxide distribution in the brain. The average amount of mito-TEMPO in healthy brain tissue (estimated by EPR spectroscopy on tissue specimens isolated 10 min after injection) was 8.4 ± 2.1 pmol/g tissue. In MPTP-treated brain, the amount was almost same 8.1 ± 1.7 pmol/g tissue.
The data suggest that mito-TEMPO is appropriate redox sensor for MR imaging of superoxide generation in dopaminergic area of the brain in Parkinson’s disease. Obviously, in MPTP-treated brain, the oxidation of mito-TEMPO dominates over reduction as a result of abnormal production of mitochondrial superoxide in dopaminergic tissues. The results show directly, on intact mammals, that mitochondrial superoxide is a major inducer and/or mediator of neurodegenerative damage in Parkinson’s disease. The high oxidative status of brain tissue in Parkinson’s disease was also confirmed on isolated tissue specimens, using total antioxidant (reducing) capacity assay and ROS/RNS assay (Figure 5).
Figure 5.

(A) Total antioxidant capacity (TAC) of the brain tissue of healthy and MPTP-treated mice. The data are the mean ± SD from 4 animals for each group. In the control group, TAC was considered 100%, which corresponded to 17 μM uric acid equivalents per milligram of protein (∼37 μM copper reducing equivalents per milligram of protein). (B) ROS/RNS level in the brain tissue of healthy and MPTP-treated mice, estimated by DFC fluorescence intensity at 530 nm. A higher DCF fluorescence intensity is a marker for a higher ROS/RNS level in tissue specimens. The data are the mean ± SD from 4 animals for each group. **p < 0.01, vs control group.
The described methodology could be also applied for noninvasive analysis of brain oxidative stress and redox imbalance in other neurodegenerative conditions, such as Alzheimer’s disease, schizophrenia, epilepsy, and so forth.
Methods
Chemicals
MPTP and urethane (99%) were purchased from Sigma-Aldrich. Mito-TEMPO was purchased from SantaCruz Biotechnology, Inc. 4-Methoxy-TEMPO and 4-hydroxy-TEMPO were purchased from Sigma-Aldrich.
MPTP Mouse Model of Parkinson’s Disease
All experiments were conducted in accordance with the guidelines of the Physiological Society of Japan and were approved by the Animal Care and Use Committee of the National Institute of Radiological Sciences, Chiba, Japan.
The acute MPTP mouse model of Parkinson’s disease was developed following strictly the protocol (A) (“MPTP injection”), described by Jackson-Lewis and Przedborski in Nature Protocols.1
The mice (C57Bl/6; male) were separated in two groups: Control group (healthy mice) and MPTP-treated group (mice with Parkinson’s disease). In both groups, the mice were the same age (10–12 weeks of age), almost the same weight (28 ± 2 g), and maintained under the same conditions.
MPTP was dissolved in sterile saline solution. It was injected intraperitoneally (i.p.). One common regimen involves one injection of MPTP every 2 h for a total of four doses over an 8 h period in 1 day. Each dose consisted of 20 mg/kg body weight (b.w.), and the injected volume was 150 μL per one dose. It has been reported that under this regimen striatal dopamine can depleted by at least 40–50% in young adult C57Bl/6 mice, dopaminergic neurodegeneration occurs morphologically by a nonapoptotic form of death, and the loss of dopaminergic cell bodies in the substancia nigra is stable by 7 days after MPTP administration.1,29 Control mice were injected with saline solution at the same regimen. The mice were subjected to MRI measurements on the eighth hour after the first injection.
In Vivo MRI Measurements
MRI measurements were performed on 7.0 T horizontal magnet (Kobelco and Jastec, Kobe, Japan) interfaced to a Bruker Avance-I console (Bruker BioSpin, Ettlingen, Germany) and controlled with ParaVision 4.0.1 (Bruker BioSpin).
The mice were anesthetized by urethane (1.5–2.0 g/kg b.w.; i.p. injection) and placed in a head holder (Rapid Biomedical, Limper, Germany). A respiration sensor (SA Instruments, Stony Brook, NY) was placed on the chest of the mice. A temperature probe (FOT-M and FTI-10, FISO Technologies, Quebec, Canada) was used to monitor the rectal temperature. The tail vein was cannulated via polyethylene tube (PE-10, Becton-Dickinson, Franklin Lakes, NJ) for injection of nitroxide. The mouse was placed in the 1H-volume radio frequency (RF) resonator (Bruker BioSpin) with a surface RF receiver (1.5 cm, quadrature mouse brain coil, Rapid Biomedical), which was prewarmed using a body temperature controller (Rapid Biomedical). The resonator units, including the mouse, were placed in the magnet bore. The body temperature was kept at 36.5 ± 0.5 °C during the MRI measurements. Initially, for the selection of region-of-interest (ROI; substancia nigra), a high resolution MRI spin–echo sequence was used. After proper positioning of the slices in the area of substancia nigra (about −3.30 mm from Bregma), three control images of the mouse brain were obtained before injection of nitroxide, with the following parameters: T1-weighted incoherent gradient-echo sequence (fast low-angle shot; FLASH), repetition time = 75 ms; echo time = 5.135 ms; flip angle = 45°; field of view = 1.92 × 1.92 cm2; number of averages = 4; scan time = 19.2 s; matrix = 64 × 64; slice thickness = 0.5 mm; number of slices = 2. A solution of mito-TEMPO (dissolved in 10 mM phosphate-buffered saline, pH 7.4) (100 mM stock-solution) was injected via the tail vein (100 μL per 25 g mouse; 0.4 μmol/g b.w.) after 1 min from the start of the scan. T1-weighted images were acquired continuously within ∼20 min, using the parameters mentioned above. Mice injected with PBS only served as negative controls. None of mice died as a result of the i.v. injection of mito-TEMPO.
The MRI data were analyzed using ImageJ software (National Institute of Health, Bethesda, MD). The averaged value of the first three control sequences (registered before injection) was calculated, and each sequence of the kinetic curve was normalized to this averaged value by division, using the same algorithm as in Zhelev et al.17
In Vitro Total Antioxidant Capacity Assay
The mice were euthanized, and the brain was isolated and kept at −80 °C (without fixation). Tissue lysates were obtained by homogenation in cold PBS (1:20, w:v; ∼50 mg/mL), subsequent sonication (2 min at 20 kHz, on ice), and centrifugation at 2000g/10 min (4 °C).
The total antioxidant capacity (TAC) of isolated tissue specimens (brain tissue lysates) was analyzed using the OxiSelect Total Antioxidant Capacity Assay Kit (Cell Biolabs, Inc.), according to the manufacturer’s instructions. The method is based on the reduction of Cu2+ to Cu+ by the endogenous antioxidants and by the other reducing equivalents in the sample. Cu+ interacts with chromophore, which is converted to color product with absorbance maximum at 490 nm. The absorbance value is proportional to the total antioxidant (respectively reducing) capacity of tissue specimen. The samples were analyzed spectrophotometrically at 490 nm using the microplate reader “Tecan Infinite F200 PRO” (Tecan Austria GmbH).
The TAC was determined by calibration curve based on uric acid standards. The data were calculated in μM uric acid equivalents (UAE) or μM copper reducing equivalents (CRE). A concentration of 1 mM uric acid corresponds to 2189 μM CRE.
In Vitro ROS/RNS Assay
Brain tissue homogenates/lysates were prepared as it was described in the previous paragraph. The amount of ROS/RNS was analyzed using OxiSelect In vitro ROS/RNS Assay Kit, Green Fluorescence (Cell Biolabs, Inc.), according to the manufacturer’s instructions. The method is based on the use of fluorogenic probe 2′,7′-dichlorodihydrofluorescin DiOxyQ (DCFH-DiOxyQ), which is specific for ROS/RNS. In the cytosol, the probe is deacetylated to the nonfluorescent 2′,7′-dichlorodihydrofluorescin (DCFH). DCFH reacts rapidly with ROS and RNS (H2O2, ROO, NO, ONOO–) with the formation of the fluorescent product 2′,7′-dichlorodihydrofluorescein (DCF). The intensity of DCF fluorescence (λex = 480 nm, λem = 530 nm) is proportional to the amount of ROS/RNS in the biological specimen.
The amount of ROS/RNS was calculated by calibration curve, based on DCF standard solutions in PBS. The measurements were performed on the microplate reader “Tecan Infinite F200 PRO” (Tecan Austria GmbH).
In Vitro EPR Measurements
The mice were euthanized 10 min after i.v. injection of nitroxide derivative. The brain was isolated and kept at −80 °C (without fixation). Brain tissue was homogenized in a 4-fold volume of PBS. Protein concentration was measured using the Bradford method, and each homogenate was diluted to 10 mg protein/mL. A volume of 100 μL of potassium ferricyanide (10 mM stock-solution) was added to 400 μL of tissue homogenate and incubated within 15 min as it was described by Hyodo et al.13 The ferricyanide quantitatively converts the hydroxylamine (produced as a result of in vivo reduction of nitroxide radical) back to the oxidized form. The sample (100 μL) was placed into a glass capillary, and X-band EPR spectra were recorded on an X-band EPR instrument (Bruker) with a TE-mode cavity. The measurements were made under the following conditions: microwave frequency = 9.4 GHz; magnetic field strength = 336 mT; microwave power = 2.0 mW; field modulation frequency = 100 kHz; field modulation amplitude = 0.063 mT; time constant = 0.01 s; sweep width = 10 mT; scan time (sweep time) = 1 min.17 The EPR spectra were recorded before and after addition of ferricyanide. The concentration of nitroxide derivative and its redox status in the tissue were determined by calibration curve.
Statistical Analysis
The data were statistically analyzed by ANOVA using Student’s t test.
Acknowledgments
The participation of Ms. Sayaka Shibata and Mr. Nobuhiro Nitta (Molecular Imaging Center, NIRS, Japan) in the MRI measurements is gratefully acknowledged.
Author Contributions
Z.Z. and R.B. formulated the experimental paradigm and designed all experiments. R.B. performed all experiments. D.L. performed a part of in vitro experiments. Z.Z. and I.A. performed analysis of the data. All authors had the opportunity to discuss the results, participated in writing, and made comments on the manuscript.
The study was partially supported by a Grant-in-Aid (Kakenhi) from the Ministry of Education, Science and Technology of Japan.
The authors declare no competing financial interest.
References
- Jacksin-Lewis V.; Przedborski S. (2007) Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2, 141–151. [DOI] [PubMed] [Google Scholar]
- Vila M.; Przedborski S. (2003) Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 1–11. [DOI] [PubMed] [Google Scholar]
- Dauer W.; Przedborski S. (2003) Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. [DOI] [PubMed] [Google Scholar]
- Langston J. W.; Ballard P.; Irwin I. (1983) Chronic parkinsonism in humans due to a product of meperidine-analogue synthesis. Science 219, 979–980. [DOI] [PubMed] [Google Scholar]
- Przedborski S., and Tieu K. (2005) Toxic animal models. In Neurodegenerative Diseases: Neurobiology, Pathogenesis and Therapeutics (Beal M. F., Lang A. E., and Ludolph A., Eds.), Chapter 16, p 196, Cambridge University Press, Cambridge, UK. [Google Scholar]
- Chinta S. J.; Andersen J. K. (2008) Redox imbalance in Parkinson’s disease. Biochim. Biophys. Acta 1780, 1362–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M. H.; Wang X.; Zhu X. (2013) Mitochondrial defects and oxidative stress in Alzheimer’s disease and Parkinson’s disease. Free Radical Biol. Med. 62, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. S.; Bennett J. P. Jr. (1997) Mitochondrial toxins in models of neurodegenerative diseases. I: In vivo brain hydroxyl radical production during systemic MPTP treatment or following microdialysis infusion of methylpyridinium or azide ions. Brain Res. 765, 183–188. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman P. M.; Muqit M. M. K.; Wood N. W. (2006) Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 7, 207–219. [DOI] [PubMed] [Google Scholar]
- Bakalova R.; Zhelev Z.; Aoki I.; Saga T. (2013) Tissue redox activity as a hallmark of carcinogenesis: from early to terminal stages of cancer. Clin. Cancer Res. 19, 2503–2517. [DOI] [PubMed] [Google Scholar]
- Zhelev Z.; Aoki I.; Gadjeva V.; Nikolova B.; Bakalova R.; Saga T. (2013) Tissue redox activity as a sensing platform for imaging of cancer based on nitroxide redox cycle. Eur. J. Cancer 49, 1467–1478. [DOI] [PubMed] [Google Scholar]
- Zhelev Z.; Gadjeva V.; Aoki I.; Bakalova R.; Saga T. (2012) Cell-penetrating nitroxides as molecular sensors for imaging of cancer in vivo, based on tissue redox activity. Mol. BioSyst. 8, 2733–2740. [DOI] [PubMed] [Google Scholar]
- Hyodo F.; Matsumoto K.; Matsumoto A.; Mitchell J. B.; Krishna M. C. (2006) Probing the intracellular redox status of tumors with magnetic resonance imaging and redox-sensitive contrast agents. Cancer Res. 66, 9921–9928. [DOI] [PubMed] [Google Scholar]
- Davis R. M.; Matsumoto S.; Bernardo M.; Sowers A.; Matsumoto K.; Krishna M. C.; Mitchell J. B. (2011) Magnetic resonance imaging of organic contrast agents in mice: capturing the whole-body redox landscape. Free Radical Biol. Med. 50, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K.; Hyodo F.; Anzai K.; Utsumi H.; Mitchell J. B.; Krishna M. C. (2011) Brain redox imaging. Methods Mol. Biol. 711, 397–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyodo F.; Chuang K. H.; Goloshevsky A. G.; Sulima A.; Griffiths G. L.; Mitchell J. B.; Krishna M. C. (2008) Brain redox imaging using blood-brain barrier-permeable nitroxide MRI contrast agent. J. Cereb. Blood Flow Metab. 28, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhelev Z.; Bakalova R.; Aoki I.; Matsumoto K.; Gadjeva V.; Anzai K.; Kanno I. (2009) Nitroxyl radicals for labeling of conventional therapeutics and noninvasive magnetic resonance imaging of their permeability for blood-brain barrier: relationship between structure, blood clearance, and MRI signal dynamic in the brain. Mol. Pharmaceutics 6, 504–512. [DOI] [PubMed] [Google Scholar]
- Zhelev Z.; Bakalova R.; Aoki I.; Matsumoto K.; Gadjeva V.; Anzai K.; Kanno I. (2009) Nitroxyl radicals as low toxic spin-labels for non-invasive magnetic resonance imaging of blood-brain barrier permeability for conventional therapeutics. Chem. Commun. (Cambridge, U. K.) 1, 53–55. [DOI] [PubMed] [Google Scholar]
- Matsumoto K.; Hyodo F.; Matsumoto A.; Koretsky A. P.; Sowers A. L.; Mitchell J. B.; Krishna M. C. (2006) High-resolution mapping of tumour redox status by MRI using nitroxides as redox-sensitive contrast agents. Clin. Cancer Res. 12, 2355–2362. [DOI] [PubMed] [Google Scholar]
- Fuchs J.; Groth N.; Herrling T.; Zimmer G. (1997) Electron paramagnetic resonance studies on nitroxide radical 2,2,5,5-tetramethyl-4-piperidin-1-oxyl (TEMPO) redox reactions in human skin. Free Radical Biol. Med. 22, 967–976. [DOI] [PubMed] [Google Scholar]
- Batinic-Haberle I.; Reboucas J. S.; Spasijevic I. (2010) Superoxide dismutase mimetics: chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signaling 13, 877–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlhorn R. J. (1991) Ascorbate- and dehydroascorbic acid-mediated reduction of free radicals in the human erythrocytes. J. Biol. Chem. 266, 2724–2731. [PubMed] [Google Scholar]
- Bobko A. A.; Kirilyuk I. A.; Grigor’ev I. A.; Zweier J. L.; Khramtsov V. V. (2007) Reversible reduction of nitroxides to hydroxylamines: the roles for ascorbate and glutathione. Free Radical Biol. Med. 42, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A.; Matsumoto K.; Matsumoto S.; Hyodo F.; Sowers A. L.; Koscielniak J. W.; Krishna M. C.; Mitchell J. B. (2011) Intracellular hypoxia of tumour tissue estimated by noninvasive EPR oximetry technique using paramagnetic probes. Biol. Pharm. Bull. 34, 142–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman S.; Merenyi G.; Russo A.; Samuni A. (2003) The role of oxoammonium cation in the SOD-mimic activity of cyclic nitroxides. J. Am. Chem. Soc. 125, 789–795. [DOI] [PubMed] [Google Scholar]
- Dikalova A. E.; Bikineyeva A. T.; Budzyn K.; Nazarewicz R. R.; McCann L.; Lewis W.; Harrison D. G.; Dikalov S. I. (2010) Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S. I.; Kirilyuk I. A.; Voinov M.; Grigor’ev I. A. (2011) EPR detection of cellular and mitochondrial superoxide using cyclic hydrohylamines. Free Radical Res. 45, 417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarewicz R. R.; Dikalova A.; Bikineyeva A.; Ivanov S.; Kirilyuk I. A.; Grigor’ev I. A.; Dikalov S. I. (2013) Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signalling pathways?. Antioxid. Redox Signaling 19, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Lewis V.; Jakowec M.; Burke R. E.; Przedborski S. (1995) Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration 4, 257–269. [DOI] [PubMed] [Google Scholar]