

Abstract
Pentameric glycine receptors (GlyRs) couple agonist binding to activation of an intrinsic ion channel. Substitution of the R271 residue impairs agonist-induced activation and is associated with the human disease hyperekplexia. On the basis of a homology model of the α1 GlyR, we substituted residues in the vicinity of R271 with cysteines, generating R271C, Q226C, and D284C single-mutant GlyRs and R271C/Q226C and R271C/D284C double-mutant GlyRs. We then examined the impact of interactions between these positions on receptor activation by glycine and modulation by the anesthetic propofol, as measured by electrophysiological experiments. Upon expression in Xenopus laevis oocytes, D284C-containing receptors were nonfunctional, despite biochemical evidence of successful cell surface expression. At R271C/Q226C GlyRs, glycine-activated whole-cell currents were increased 3-fold in the presence of the thiol reductant dithiothreitol, whereas the ability of propofol to enhance glycine-activated currents was not affected by dithiothreitol. Biochemical experiments showed that mutant R271C/Q226C subunits form covalently linked pentamers, showing that intersubunit disulfide cross-links are formed. These data indicate that intersubunit disulfide links in the transmembrane domain prevent a structural transition that is crucial to agonist-induced activation of GlyRs but not to modulation by the anesthetic propofol and implicate D284 in the functional integrity of GlyRs.
Keywords: Ligand-gated ion channel, transmembrane domain, activation mechanism, propofol, cross-linking, Cys-loop receptor
Ligand-gated ion channels (LGICs) are membrane-bound proteins that couple neurotransmitter binding in a large extracellular domain (ECD) with the opening of an intrinsic ion channel in a transmembrane domain (TMD). Thus, they rapidly convert extracellular chemical signals into electrical signals at the cell membrane, determining excitation or inhibition of further signal transmission.1 On the basis of distinct structural features, LGICs can be divided into families of trimeric ATP-activated channels, tetrameric glutamate-activated channels, and the large family of pentameric LGICs (“pLGICs” or “Cys-loop receptors”). The latter includes inhibitory glycine receptors (GlyRs), glycine-activated chloride channels that are widely distributed in the spinal cord and some higher centers, where they contribute to motor control and various other functions.2 Human GlyRs consist of five homologous subunits, either five α1, α2, and α3 subunits or a mix of α and β subunits, arranged in 5-fold symmetry around a central pore.2 Single GlyR subunits consist of a large N-terminal ECD and a C-terminal TMD comprised of four membrane-spanning helices (M1–M4). The ligand binding domain (LBD) is formed at the interface of adjacent ECDs, and the channel is formed by the central apposition of five M2 helices. This arrangement is well-conserved throughout the family, which also includes the excitatory nicotinic acetylcholine receptors (nAChRs) and 5-HT type 3 receptors (5-HT3Rs) and the inhibitory γ-aminobutyric acid type A receptors (GABAARs).
Numerous mutagenesis studies have identified amino acid residues that determine ligand recognition in the LBD and ion conductance in the TMD of GlyRs; however, the coordinated movement of individual residues required by ligand activation of, for example, nAChRs3 is relatively unresolved in GlyRs. This rearrangement of individual residues is of great physiological importance, as exemplified by the arginine residue at position 271 (R271) at the extracellular end of M2 in the α1 GlyR. In recombinantly expressed receptors, substitution of this residue weakens both the preference of the channel for states of high conductance4,5 and the transition from closed to open states subsequent to glycine binding.5,6 Also in recombinant receptors, the fluorescent signal of a thiol-reactive fluorophore bound to an introduced cysteine at this position is shifted to lower wavelengths, suggesting that during activation by glycine, the residue at position 271 enters a more hydrophobic environment.7In vivo, the decreased level of activation of R271-mutated α1 GlyRs results in neuronal hyperexcitability and the condition known as hyperekplexia or startle disease.8 The arrangement of this residue within the receptor is also of pharmacological significance, as side chain properties of R271 and other vicinal residues may contribute to GlyR sensitivity to widely used drugs such as ethanol and the anesthetic propofol, which both enhance responses to glycine.9,10 Notably, enhancement by propofol remains intact in R271 mutant α1 GlyRs11,12 and is capable of restoring normal function on R271 mutant receptors, both in recombinant settings and in hyperekplexic mice.11
We sought to establish the structural arrangement of R271 and its rearrangement in both activation by agonists and enhancement of activation by propofol, as a basis for understanding the importance of this position in receptor function. We employed a site-directed cross-linking technique,13 whereby open-channel or closed-channel conformations can be mimicked by introducing a pair of vicinal cysteine residues and promoting disulfide links.14 We present electrophysiological and biochemical evidence of a disulfide link between introduced cysteines at the M2-R271 and M1-Q226 positions of adjacent subunits, implying that these positions are in the proximity of each other in functional α1 GlyRs. We find that the linking of two subunits via these positions decreases the efficiency of channel activation by full and partial agonists but has no effect on the enhancement by propofol.
Results and Discussion
Positions R271 in M2 and Q226 in M1 of the α1 GlyR Are in the Proximity of Each Other
The introduction of a cysteine pair into α1 GlyR subunits introduces 10 cysteine residues into the mature pentameric protein. Therefore, reaching a conclusion about which two cysteines are linked requires exhaustive experiments and/or high-resolution structural data. Although the atomic structure of GlyRs is unknown, ∼3 Å resolution data of prokaryotic and invertebrate pLGICs have recently been published,15−17 providing a structural template for assessing GlyR cross-linking results. In estimating the arrangement of R271 in the α1 GlyR, we used a previously published model of the α1 GlyR18 based on the crystal structure of the glutamate- and ivermectin-bound Caenorhabditis elegans α glutamate-gated chloride channel (GluCl).16 In the model, the R271 side chain is directed away from the central pore of the pentamer, toward the intersubunit cavity, such that its guanidino carbon is 4.1 Å from the carboxyl carbon of D284 in M3 of the same subunit and 3.6 Å from the amide carbon of Q226 in M1 of the adjacent subunit (Figure 1a). To test the possibility that in the α1 GlyR, R271 is in the proximity of D284 or Q226, we substituted these residues for cysteine, alone and in pairs, generating wild-type (WT), R271C, Q226C, D284C, R271C/Q226C, and R271C/D284C α1 GlyR constructs. Each construct also incorporated the C290S mutation, which was functionally silent (Figure 1 of the Supporting Information) and eliminated possible interactions between introduced cysteines and this endogenous M3 cysteine. We expressed each construct in Xenopus laevis oocytes and measured current responses to saturating concentrations of glycine alone or in the presence of either dithiothreitol (DTT) or HgCl2, which keep cysteine residues reduced or bridge unlinked cysteine residues within sufficient proximity, respectively.19,20
Figure 1.
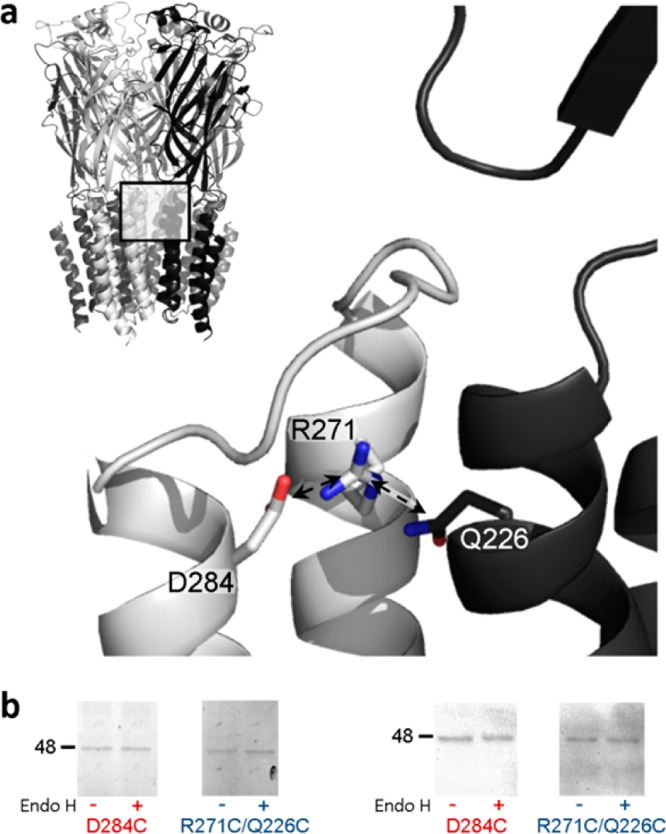
(a) Position of R271 and proximal residues in the α1 GlyR homology model. The pentameric structure is shown (top left), and the interface of two adjacent subunits (boxed area) is shown in greater detail, with one subunit colored gray and one black. Arrows indicate the 4.1 and 3.6 Å separation of M2-R271 ζC from M3-D284 γC and M1-Q226 δC, respectively. The distance from the M2-R271 ζC atom to that from adjacent subunits is 20.5 Å (not shown). The model, which was described previously,18 used as a template the glutamate- and ivermectin-activated C. elegans α GluCl crystal structure16 (Protein Data Bank entry 3RIF). (b) Cell surface expression of mutant α1 GlyR subunits. Oocytes were treated with mutant D284C or R271C/Q226C α1 GlyR cRNAs and either rinsed with the membrane-impermeable fluorophore Cy5, purified, separated by SDS–PAGE, and imaged (left) or separated by SDS–PAGE, subjected to α1 GlyR-specific Western blotting, and imaged. Both experiments identified single α1 GlyR subunits (∼48 kDa protein bands) that were insensitive to Endo H cleavage, indicating cell surface expression. Electrophysiological experiments showed that D284C α1 GlyRs were not responsive to glycine, whereas R271C/Q226C α1 GlyRs were.
Oocytes treated with D284C or R271C/D284C α1 GlyR cRNA showed no response to glycine, alone or in the presence of DTT, HgCl2, or propofol (n = 7–10 over three batches of oocytes). (This was also the case when the C290S mutation was absent; see the legend of Figure 1 of the Supporting Information.) This indicates that the D284C mutation either prevents responses to glycine in expressed receptors or prevents the expression or assembly of receptors. To establish which possibility is correct, we incubated D284C α1 GlyR-expressing oocytes with the membrane-impermeable fluorophore Cy5 NHS ester and subsequently purified and imaged GlyRs under denaturing conditions. This revealed ∼48 kDa bands of protein that were insensitive to Endo H cleavage (Figure 1b, left panel), indicative of cell surface-expressed α1 GlyR subunits.21 As Cy5 labels yielded a weak fluorescent signal, we performed a similar experiment but probed for α1 GlyR expression with an anti-α1 GlyR primary antibody and a horseradish peroxidase-conjugated secondary antibody. This too showed Endo H-insensitive expression of D284C α1 GlyRs (Figure 1b, right panel). The extent of expression seemed similar to the extent for R271C/Q226C α1 GlyRs (Figure 1b), which were used as a functional (as shown in electrophysiological experiments, below) control. Thus, the lack of agonist responses at mutant D284C α1 GlyRs is due to the inability to function after successful cell surface expression. We note that substitutions of the equivalent aspartate residue in α1 or β2 subunits of GABAARs also result in little or no response to the agonist.22
The peak magnitude of responses of R271C/Q226C receptors to glycine was significantly increased in the presence of 2 mM DTT and unaffected by 10 μM HgCl2, whereas WT and single-mutant receptors were not significantly affected by either treatment (Figure 2a,b). This indicates that in R271C/Q226C receptors, the introduced cysteines form a disulfide bond; the presence of DTT breaks this bond, resulting in a greater whole-cell current response to glycine. Thus, Cα atoms of positions 271 and 226 are likely separated by approximately 6 Å in at least one functional state of the receptor.23,24 Although not significant, there was a trend toward stronger responses at R271C receptors in the presence of HgCl2 (Figure 2b), raising the possibility that nonsymmetrical movement of adjacent M2 helices brings two position 271 Cα atoms within 8 Å of each other.19,25 We also treated oocytes with a 1:1 mix of single-mutant R271C and Q226C cRNAs. Assuming that R271C subunits and Q226C subunits are expressed with similar efficiency, this produces a mix of R271C homomers, R271C–Q226C heteromers of varying stoichiometry, and Q226C homomers. None of these receptors contain two introduced cysteines in a single subunit, and they thus provide a means of testing whether the observed disulfide link occurs within single subunits or across adjacent subunits.26 At these oocytes, responses to glycine were significantly enhanced in the presence of DTT, to an extent similar to that of oocytes expressing homomeric R271C/Q226C receptors (Figure 2b). Because DTT had little, if any, effect on R271C and Q226C homomers (Figure 2b), it is likely that this increase in current size is mediated by heteromeric receptors, which can contain only one or two R271C–Q226C interfaces. Thus, the linking of only one or two intersubunit interfaces is sufficient to inhibit maximal current responses to glycine. This interpretation makes the assumption that the detected interaction is caused by an intersubunit 271–226 interaction and not an intersubunit 271–271 or 226–226 interaction in R271C–Q226C heteromers. We see two arguments against the latter possibility. First, in the α1 GlyR model, the distances between two adjacent R271 side chains and between two adjacent Q226 side chains are 20.5 and 18.8 Å, respectively (not shown), which are at the upper limit of distances that receptor positions traverse to form disulfide bonds.27,28 Second, given that the promotion of 271–271 or 226–226 interactions (by HgCl2 at R271C and Q226C homomers) tends to increase current size (Figure 2b), it is unlikely that a DTT-induced increase in current is due to breaking of 271–271 or 226–226 interactions.
Figure 2.
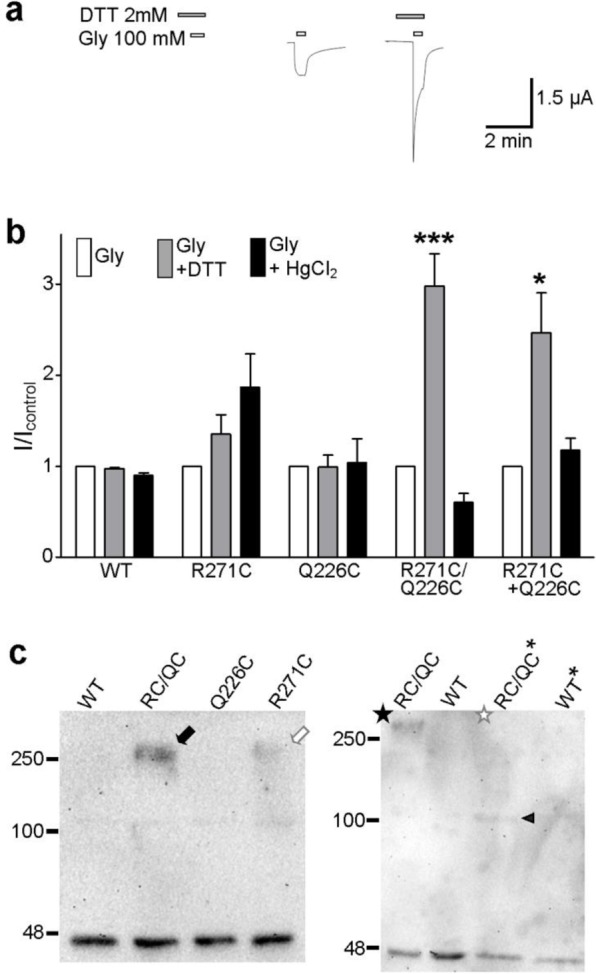
R271C/Q226C α1 GlyRs show altered function indicative of an intersubunit disulfide link. (a) Exemplary responses of R271C/Q226C cRNA-treated oocytes to 100 mM glycine alone (white bars) or in the presence of 2 mM DTT (gray bars). (b) Averaged responses (±SEM; n = 3–9) to glycine (100 mM for mutants and 3 mM for WT) alone or in the presence of 2 mM DTT or 10 μM HgCl2, normalized to the response to glycine alone (I/Icontrol) after experiments illustrated in panel a. Compared to the response to glycine alone with ANOVA and Dunnett post hoc analysis, *P < 0.05 and ***P < 0.001. (c) Western blotting of purified α1 GlyRs. In the left panel, purified WT, R271C/Q226C (RC/QC), Q226C, and R271C α1 GlyRs separate into ∼48 kDa bands indicative of single subunits but only R271C/Q226C (black arrow) and to a lesser extent R271C (white arrow) proteins retain an ∼250 kDa oligomer, indicative of a pentameric form. In the right panel, the pentameric form of R271C/Q226C subunits (black star, RC/QC) is abolished by 10% β-mercaptoethanol (white star, RC/QC*); WT subunits run only as monomers in the absence (WT) and presence (WT*) of 10% β-mercaptoethanol. The black triangle denotes an ∼100 kDa band for β-mercaptoethanol-treated R271C/Q226C subunits.
If our interpretations described above were correct, adjacent R271C/Q226C subunits would associate with greater stability than WT subunits, because of the introduced intersubunit disulfide link. To verify this, WT and mutant GlyRs were purified without DTT or β-mercaptoethanol and subjected to SDS–PAGE and Western blotting. This identified large amounts of ∼45 kDa protein for each construct (Figure 2c), indicating the denaturation of pentameric receptors into single α1 GlyR subunits, which have a molecular mass of 48 kDa.29 For R271C/Q226C subunits, however, a clear agglomeration of ∼250 kDa was also identified (Figure 2c, black arrow; Figure 2 of the Supporting Information), indicating that an intersubunit link between the introduced cysteines stabilizes the pentameric form of R271C/Q226C receptors. A faint 250 kDa agglomeration was also observed for R271C subunits (Figure 2c, white arrow), indicating a pentameric form of R271C receptors. As R271C receptors contain only five TMD cysteines, the stabilization of the pentameric form cannot be a result of five intersubunit links in the TMD (which would require a total of 10 TMD cysteines). Rather, we interpret this as the combined result of one to two intersubunit disulfide 271–271 links and various noncovalent intersubunit associations that can occur under mildly denaturing conditions. This interpretation is supported by the weak protein band at ∼100 kDa for WT subunits (black arrow in Figure 2 of the Supporting Information), indicative of α1 GlyR subunit dimers.
To confirm that the association of R271C/Q226C subunits was due to a disulfide interaction, we compared association in the presence or absence of the reducing agent β-mercaptoethanol; 10% β-mercaptoethanol abolished the pentameric form of R271C/Q226C subunits (in Figure 2c, compare the black and white stars), leaving only monomeric (∼45 kDa) and dimeric (∼100 kDa) bands (Figure 2c, black triangle). This confirms that intersubunit noncovalent interactions stabilize α1 GlyR dimers and intersubunit disulfide links between introduced cysteines stabilize α1 GlyR pentamers. We acknowledge here an alternate interpretation of the pentameric R271C/Q226C protein band, that it is simply a more robust manifestation of intersubunit 271–271 interactions induced by the Q226C substitution. Because the electrophysiological data suggest that 271–271 interactions enhance currents whereas 271–226 interactions inhibit currents (as discussed above), we reason that the Q226C substitution would actually counteract 271–271 interactions, and we exclude this alternate interpretation. Thus, the biochemical experiments confirm that intersubunit disulfide links occur between subunits of R271C/Q226C α1 GlyRs and that the increased current size at R271C/Q226C α1 GlyRs in the presence of DTT is due to the release of these intersubunit M2–M1 links.
The Separation of M2 and M1 Positions in R271C/Q226C α1 GlyRs Increases during Activation
If two α1 GlyR positions are in the proximity of each other in a resting state, introduced cysteines may link spontaneously, such that reduction by DTT induces currents directly or enhances current responses to agonists.14,30 Conversely, introduced cysteines at some α1 GlyR positions link only upon repeated channel activation, suggesting that the relevant positions are brought together during the activation process.26,31 To establish if the intersubunit link in R271C/Q226C receptors is formed in a resting state or at a later stage of the activation process, we compared responses to repeated applications of glycine, before and after the application of DTT (Figure 3a), and we tested for enhancement of glycine responses by DTT when applied in the complete absence of glycine (Figure 3b). We found that the initial response to saturating glycine was not significantly different from the second or third responses, and a subsequent application of glycine in the presence of 2 mM DTT caused a significant increase in current (Figure 3a). Subsequent applications of glycine alone saw the current size decrease to its original level. Also, when DTT was applied to oocytes in the absence of glycine, subsequent responses to glycine were significantly enhanced (Figure 3b). This suggests that the intersubunit M2–M1 link is formed in a resting state. This is in contrast, for example, to an intrasubunit M2–M3 link in the α1 GlyR, which requires repeated applications of glycine to form.31 This also contrasts the intersubunit M2–M1 link shown in the α1/β2/γ2 GABAAR, which forms after repeated agonist application.22 The GABAAR M1 residue in that study equates to α1 GlyR I225, next to Q226 at the intersubunit interface but further from R271 and facing the surrounding membrane.22
Figure 3.

Formation of the intersubunit link in R271C/Q226C receptors does not depend on repeated activation by glycine or on the concentration of glycine. (a) The left panel shows exemplary responses of R271C/Q226C α1 GlyR-expressing oocytes to repeated application of 100 mM glycine (white bars) before and after application in the presence of 2 mM DTT (gray bars). Oocytes were rinsed for ∼10 min between subsequent glycine or DTT applications. The right panel shows responses were normalized to that in the presence of DTT and averaged (±SEM; n = 6). Compared to the initial response with ANOVA and Tukey post hoc analyis, **P < 0.01 and ***P < 0.001. (b) The left panel shows responses to 100 mM glycine (white bars) at R271C/Q226C α1 GlyR-expressing oocytes are enhanced by 2 mM DTT (gray bars) in the absence of glycine and when co-applied with glycine. The right panel shows responses were normalized to the initial glycine response and averaged (±SEM; n = 5). Compared to the initial response with ANOVA and Tukey post hoc analyis, *P < 0.05 and **P < 0.01. (c) Glycine concentration–response data for WT and mutant α1 GlyR-expressing oocytes. Responses to each concentration of glycine were normalized to the maximal glycine-activated response (I/Imax) and fit with nonlinear regression giving the parameters listed in Table 1. (d) Effects of DTT and HgCl2 on glycine concentration–response data for R271C/Q226C α1 GlyR-expressing oocytes. Responses were normalized to the maximal response to glycine alone (I/Icontrol); for responses to glycine alone, data are from panel c and therefore are indicated only as a dotted line. Glycine EC50 values were 3.4 ± 0.67 mM (n = 7), 3.8 ± 0.66 mM in the presence of DTT (n = 6), and 4.1 ± 0.56 mM in the presence of HgCl2 (n = 3), which were not significantly different when compared via ANOVA.
As α1 GlyRs are activated with increasing efficiency by one to five molecules of glycine (i.e., dependent on glycine concentration),32 we next questioned if breaking the M2–M1 link enhanced currents similarly at various glycine concentrations. If the enhancement were different at low and high concentrations, this could imply that the M2–M1 separation is differentially required by partially and fully liganded GlyR activation. We first established the glycine sensitivity of WT and mutant receptors in the absence of DTT and HgCl2 (Figure 3c). Glycine EC50 values for R271C, Q226C, and R271C/Q226C receptors were all significantly higher than for WT receptors (Table 1), indicating significantly decreased apparent glycine affinity. R271C/Q226C receptors yielded a (nonsignificantly) lower EC50 value than the single-mutant receptors, showing that the deleterious effects of the two single mutations on glycine sensitivity were not additive. We applied to these results double-mutant cycle analysis, which holds that mutations at two sites of no energetic or structural interaction will be multiplicative in their reduction of function, as indicated by an Ω value [(EC50,WT × EC50,double mutant)/(EC50,mutant A × EC50,mutant B)] close to unity.33 The Ω value for R271C/Q226C α1 GlyRs was 4.3 × 10–3 (Table 1), indicating that an energetic coupling of C271 and C226 affects the apparent glycine affinity. [In comparison, the combinations of C290S and R271C mutations or C290S and Q226C mutations yielded Ω values of 0.89 and 1.8, respectively (see Figure 1 of the Supporting Information).] If this energetic coupling was a direct effect of the disulfide link, the glycine EC50 value of R271C/Q226C receptors should be increased (Ω brought closer to unity) by the presence of DTT. However, DTT enhanced responses to all concentrations of glycine similarly and thus had no significant effect on the glycine EC50 value for R271C/Q226C receptors (Figure 3d). This would suggest that the energetic coupling of these positions, with regard to the apparent glycine affinity, does not lie in the disulfide interaction; however, a possible DTT-induced increase in EC50 might be masked by desensitization during responses to high concentrations at unlinked receptors (compare traces 1 and 4 in Figure 3a). That DTT similarly enhanced responses to all concentrations of glycine suggests that the linking of M2 and M1 (at one or two intersubunit interfaces) prevents activation of both partially and fully liganded α1 GlyRs. Given that the EC50 is determined by agonist affinity and gating efficacy, this also reveals the likely mechanism by which M2–M1 links decrease whole-cell currents: one to two M2–M1 links in individual receptors prevent ligand-induced activation, decreasing the number of receptors that contribute to the whole-cell current without affecting the agonist affinity or gating efficacy; reduction by DTT allows the separation of upper M2 from M1 and thus channel activation in individual receptors, increasing the number of receptors that contribute to the whole-cell current. The fact that effects of R271C and Q226C mutations on the agonist EC50 are coupled, as revealed by double-mutant cycle analysis, and this coupling is insensitive to DTT reiterates that upper M2 and upper M1 are both part of the conventional agonist-induced channel activation pathway,6,34 and that their linking in this study simply precludes any channel activation.
Table 1. Nonlinear Regression Parameters for Glycine Activation of α1 GlyRsa and Double-Mutant Cycle Analysis Resultsb.
| α1 GlyR | EC50 (mM) | nH | Imax (μA) | Ωb |
|---|---|---|---|---|
| WT | 0.04 ± 0.01 | 2.0 ± 0.1 | 6.5 ± 0.7 | |
| R271C | 6.8 ± 0.47c | 1.5 ± 0.1 | 1.8 ± 0.3c | |
| Q226C | 4.7 ± 0.59c | 1.6 ± 0.5 | 1.6 ± 0.7c | |
| R271C/Q226C | 3.4 ± 0.67c | 1.3 ± 0.1 | 2.2 ± 0.4c | 4.3 × 10–3 |
All values are means ± SEM from three to seven experiments.
The coupling coefficient, Ω, was calculated with the equation Ω = (EC50,WT × EC50,R271C/Q226C)/(EC50,R271C × EC50,Q226C).
Significantly different from the WT value for that parameter (P < 0.001, ANOVA with Tukey post hoc analysis).
Activation of R271C/Q226C α1 GlyRs by Partial Agonists
β-Alanine and taurine are partial agonists at GlyRs, in that even at saturating concentrations, the maximal open probability they elicit at GlyR channels is less than that elicited by glycine.35 β-Alanine and taurine bind to R271 mutant GlyRs but barely activate any detectable current,5,11 although the presence of propofol permits the activation of large currents by β-alanine.11 We tested if DTT enhanced β-alanine- and taurine-activated currents at R271C/Q226C α1 GlyRs, to establish if the M2–M1 separation is similarly required by activation by full and partial agonists. Average current responses to 100 mM β-alanine and 100 mM taurine were 13 ± 3 (n = 4) and 6 ± 1 nA (n = 5), respectively, which were both substantially smaller than those activated by 100 mM glycine in the same series of experiments [0.4 ± 0.1 μA (n = 5); P < 0.001, ANOVA with Dunnett post hoc analysis]. In the presence of 2 mM DTT, average current responses to β-alanine and taurine were increased to 32 ± 9 and 19 ± 7 nA (n = 7) (Figure 4a), which are significantly greater than the initial responses (Figure 4b). Thus, the M2–M1 link has a similar inhibitory effect on both partial and full agonist activation, providing further evidence that transitions between different agonist-bound states are not affected by the link.
Figure 4.

Currents activated by partial agonists are significantly enhanced by DTT. (a) Exemplary responses of R271C/Q226C α1 GlyR-expressing oocytes to 100 mM β-alanine alone (white bars) and 2 mM DTT (gray bars) or 500 μM propofol (hashed bars). Propofol was used only to confirm robust activation by β-alanine.11 (b) Currents in response to 100 mM β-alanine (n = 4) and 100 mM taurine (n = 5) alone (white columns) and in the presence of DTT (gray columns) were normalized to the response to partial agonists alone (I/Icontrol); error bars indicate the SEM. Compared to the control response with a paired t test, *P < 0.05 and **P < 0.01.
Enhancement by Propofol Is Not Affected by the M2–M1 Link
The intravenous general anesthetic propofol depresses neuronal function by enhancing the activation of GlyRs and GABAARs.36−38 On the basis of homology with anesthetic-bound eukaryotic pLGICs and dynamic simulations of model GlyRs, it is predicted that propofol and functionally related drugs bind at the interface of adjacent subunits in the TMD, stabilizing the open channel.9,10 However, the binding site and the particular conformational changes influenced by propofol binding in the GlyR are unknown. In a final set of experiments, we therefore sought to establish if enhancement by propofol is affected by the linking of adjacent subunits in R271C/Q226C α1 GlyRs. At M2–M1-linked receptors, i.e., R271C/Q226C receptors in the absence of DTT, 500 μM propofol enhanced responses to a range of glycine concentrations, shifting the glycine EC50 value from to 3.4 ± 0.67 to 1.3 ± 0.50 mM (Figure 5a,b). At M2–M1-unlinked receptors, R271C/Q226C receptors in the presence of DTT, 500 μM propofol caused essentially identical enhancement of responses to glycine (compare filled and empty symbols in Figure 5b), shifting the glycine EC50 value from 3.8 ± 0.66 to 1.5 ± 0.48 mM. This suggests that the intersubunit link has no effect on the ability of propofol to enhance channel activation. We reach a similar conclusion when we compare the individual increases in current caused by DTT and propofol to the total increase in current caused by the combined presence of both DTT and propofol. Currents activated by 1 mM Gly (approximately an EC20 concentration) were enhanced 4.0 ± 1.3-fold by 2 mM DTT and 8.1 ± 1.4-fold by 500 μM propofol (Figure 5c). The combined presence of 2 mM DTT and 500 μM propofol caused a 20.8 ± 2.6-fold increase, significantly greater than the increase caused by either treatment alone (Figure 5c). The same combined effect was observed with saturating glycine concentrations (Figure 5c), implying that mechanisms of DTT- and propofol-mediated enhancement remain distinct at all glycine concentrations. Considering that (1) DTT unlinks individual receptors, thereby increasing the number of channels contributing to the whole-cell current, (2) propofol affects the apparent glycine affinity and agonist efficacy,11 and (3) the whole-cell current is proportional to both the number of functional receptors and the open probability of individual receptors, it is logical that the effects of DTT and propofol are additive (Figure 5c).
Figure 5.

Enhancement of α1 GlyR currents by propofol is not affected by the intersubunit link. (a) Exemplary responses of oocytes treated with R271C/Q226C α1 GlyR cRNA to 1 mM glycine alone (white bars) or in the presence of 500 μM propofol (hashed bars), 2 mM DTT (gray bars), or 500 μM propofol and 2 mM DTT. (b) Responses to glycine alone (□) or in the presence of propofol (○) were normalized to the maximal response to glycine alone, and responses to glycine in the presence DTT (■) and in the combined presence of DTT and propofol (●) were normalized to the maximal response to glycine in the presence of DTT. Average glycine EC50 values [mean ± SEM (n = 4–7)] under these conditions were 3.4 ± 0.67 (□), 1.3 ± 0.50 (○), 3.8 ± 0.66 (■), and 1.5 ± 0.48 mM (●). (Data for glycine alone or in the presence of DTT are essentially repeated from Figure 3, and fits are therefore shown as dashed lines.) (c) Responses to 1 and 100 mM glycine alone (white columns) and in the presence of 2 mM DTT, 500 μM propofol, or both, normalized to the response to glycine alone (I/IGly). For both glycine concentrations, the responses in the presence of DTT or propofol were compared to the response in the combined presence of DTT and propofol by ANOVA and Dunnett post hoc analysis (**P < 0.01; ***P > 0.001).
Binding sites for propofol in GlyRs have not been identified, although several molecular determinants of propofol sensitivity have been identified in the receptor TMD. A serine–isoleucine mutation at the M2-S267 position of α1 GlyRs does not affect enhancement by propofol but abolishes the direct activation by high concentrations of propofol.58 In α1β2γ2 GABAARs, an asparagine–methionine mutation at the equivalent position in the β2 subunit decreases the sensitivity to enhancement by propofol,39 and in α2β1γ2 GABAARs, a methionine–tryptophan substitution at the β1 position equivalent to α1 GlyR M3-288 decreases the sensitivity to enhancement by propofol.40 The side chains of both of these positions are likely oriented into the intersubunit cavity, according to GlyR and GABAAR models based on both prokaryotic pLGICs and the C. elegans α GluCl.18,22,41 Taken together, it seems that the enhancing actions of propofol at GlyRs and GABAARs depend on side chains in the intersubunit cavity but not on the ability of the R271 position to separate from the Q226 position during activation by glycine.
Functional Rearrangement of the M2-R271 Position during pLGIC Activation
As a mechanistic explanation for these results, we envisage that the separation of the M2-271 and M1-226 positions is minimal at rest and then increases either during an early stage of the activation process, such as ligand-induced “flipping” or “priming” of channel opening,42,43 or during channel opening. This is based on the following reasons. First, we observed no spontaneous activation of R271C/Q226C receptors in the absence of agonist (in the absence or presence of HgCl2), suggesting that the proximity of the two positions does not correspond to an open-channel state. Second, we showed that activation does not bring the positions closer together; they are already in the proximity of each other at rest, and enforced or prolonged proximity prevents channel activation. Finally, the M2 position in nAChRs equivalent to R271 in α1 GlyRs is suggested to rearrange shortly after ligand binding but before M2 residues in the channel pore rearrange to open the channel,44 suggesting by homology that rearrangement of this position in the α1 GlyR couples conformational changes in the LBD to conformational changes in the channel domain by rearranging before the channel gate opens.44 It is interesting that the linking of one or two M2–M1 interfaces (in R271C–Q226C heteromers) prevented activation to an extent similar to the extent of potential linking of five interfaces (in R271C/Q226C homomers). This implies that the contributions of five intersubunit interfaces to channel activation do not sum linearly. This reflects the nonsymmetrical contribution of agonist binding subunits to agonist-induced channel activation and contrasts with the symmetrical contribution of deeper, pore-lining M2 residues to channel conductance in nAChRs and nAChR–5-HT3R chimeras,45,46 further indicative of a role for position 271 of α1 GlyR in coupling agonist binding with channel activation.
If R271 is moved away from Q226 during activation, to where is it moved? Our site-directed cross-linking approach failed to establish if position 271 interacts with position 284 in M3, as mutant D284C subunits were expressed but did not respond to ligands. This nonetheless identifies M3-D284 as a crucial component of the ligand activation pathway. Notably, in a prokaryotic pLGIC activated by pH, the disulfide linking of positions equivalent to positions 271 and 284 of α1 GlyR activated currents in the absence of agonist.14 This suggests that in this homologue, activation involves an increased level of association of M2 and M3 helices, which could be the very mechanism by which protons activate this pLGIC.47 Although caution must be exercised in likening amino acid rearrangement in the GlyR to that in other pLGICs,48 a rigid-body, outward tilting of the upper half of M2 helices seems to be a common feature of GABAARs, nAChRs, 5-HT3Rs, and prokaryotic pLGICs.15,17,49−53 We acknowledge that across different receptors, this outward tilting could involve different relations to other helices, illustrated by the fact that the R271–Q226 Cα separation (α1 GlyR numbering) does not correlate with channel state in currently available pLGIC crystal structures (9.9 Å in closed-channel ELIC, 7.5 Å in open-channel GLIC, and 9.0 Å in open-channel GluCl).15−17 These results nonetheless confirm that the open-channel GluCl provides a suitable template for establishing the proximity of α1 GlyR amino acid residues and, in combination with site-directed cross-linking, if proximity corresponds to closed- or open-channel states.
In the α1 GlyR, outward movement of the upper portion of M2 during channel activation would mean that the set of M3 (in the same subunit) and M1 (in the adjacent subunit) side chains that neighbor M2-271 would differ in resting/closed versus activated/open (or intermediary) channel states. This is reflected in the findings that the signal of a fluorophore attached to position 271 shifts during glycine activation of α1 GlyRs to indicate a more hydrophobic environment,7 glycine activation of α1 GlyRs increases the strength of association of introduced cysteines at M2-267 (one helical turn internal to R271) and M3-288 (one helical turn internal to D284),31 and the arginine side chain at position 271 is no longer important to glycine potency if the surrounding microenvironment is significantly altered by numerous amino acid substitutions.12 The transition of the R271 side chain to a more hydrophobic environment is intriguing when compared to results at α1/β2/γ2 GABAARs, where activation by GABA induces linking of the M2-271 position to the M1-225 position (both α1 GlyR numbering). The M1-225 position is farther from the pore than Q226, is closer to M3 and the surrounding membrane, and is occupied by an isoleucine residue in the α1 GlyR.22 We speculate that during agonist-induced activation, the R271 side chain is moved from a hydrophilic environment close to the Q226 side chain to a more hydrophobic environment deeper in the intersubunit cavity and closer to M3. A rotation of the M2 helix along its long axis, as is suggested for nAChRs and GABAARs,25,54 would foreseeably facilitate this rearrangement. Conservation of this rearrangement across GlyRs and GABAARs could explain the similar decreases in agonist potency caused by the mutation of α1 R271 in heteromeric α1/β GlyRs and of β2 R269 in heteromeric α1/β2/γ2 GABAARs.4,22
Concluding Remarks
These results show that in cysteine-substituted α1 GlyRs, side chains at positions M2-271 and M1-226 of adjacent subunits are in the proximity of each other at rest and that their covalent linking at one to two intersubunit interfaces prevents channel activation by agonists. Mutant proteins differ from WT proteins in the range of conformations they are likely to adopt,55 but we liken the rearrangement of the mutated M2-271 position to that in WT α1 GlyRs and indeed to those in other pLGICs, based on evidence that at various pLGICs, the upper half of M2 moves away from the pore axis,17 M2 interactions with M3 (which is farther from the pore than M2) are enhanced,31,53 and the relation of M2 to M1 of the adjacent subunit is altered16,22 in the activation process. Our results show that, in contrast to activation by agonists, enhancement by propofol does not rely on the separation of adjacent M2 and M1 helices in the TMD. Therefore, the enhancing effect of propofol is likely via an allosteric pathway different from that activated by glycine,12 and despite possibly binding in this vicinity,10 propofol must target some other rearrangement related to channel activation and/or conductance.
Methods
Chemicals
Glycine, NaCl, KCl, CaCl2, MgCl2, HEPES, Tris, Na2HPO4, dodecyl maltoside, protease inhibitor, dimethyl sulfoxide, Rotiphorese gel mix, and skim milk powder were purchased from Carl Roth GmbH (Karlsruhe, Germany). NaOH was from AppliChem GmbH (Darmstadt, Germany). Nickel-NTA agarose beads were from Qiagen (Munich, Germany). Propofol (2,6-diisopropylphenol), dithiothreitol (DTT), HgCl2, tricaine, gentamycin, type IIA collagenase, imidazole, Triton X-100, and deoxycholate were from Sigma-Aldrich (Munich, Germany). Polyvinylidene fluoride (PDVF) membranes were from Bio-Rad (Hilden, Germany). Stocks of glycine (1 M), DTT (400 mM), and HgCl2 (10 mM) were prepared in a bath solution (components in Electrophysiological Experiments), and 1 M stocks of propofol were prepared in dimethyl sulfoxide. Stocks were stored at −20 °C, and experimental solutions were prepared from stocks on the day of the experiments.
Site-Directed Mutagenesis and cRNA Synthesis
The template cDNA for site-directed mutagenesis was a C-terminally His-tagged human GlyR α1 subunit in the pNKS2 vector.21 Mutant cDNAs were generated with the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Böblingen, Germany), and for all generated clones, the appropriate sequence of the entire α1 GlyR insert was confirmed (Eurofins MWG Operon, Ebersberg, Germany). cDNAs were linearized with NotI (New England Biolabs GmbH, Frankfurt, Germany), and cRNAs were synthesized with the mMESSAGE mMACHINE SP6 Kit (Life Technologies GmbH, Darmstadt, Germany).
As approved by the Technical University of Darmstadt (Agreement V54-19c20/15 DA8/Anz. 20), oocytes were removed from female Xenopus laevis frogs anesthetized with 0.3% Tricaine and transferred to frog Ringer’s solution [96 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES (pH 7.4 with NaOH)] supplemented with 50 mg/mL gentamycin. Oocytes were prepared as described by Laube et al.56 Briefly, stage V or VI oocytes were isolated manually, defolliculated by a 2 h incubation in type IIA collagenase, rinsed with Ca2+-free frog Ringer’s solution, and stored in frog Ringer’s solution at 18 °C. Oocytes were treated with 4 ng of cRNA (at 100 ng/μL); in experiments in which oocytes were treated with two different constructs, 2 ng of each was injected. Treated oocytes were then incubated in frog Ringer’s solution at 18 °C until the experiments were conducted.
Electrophysiological Experiments
Two to five days after injection, oocytes were transferred to a recording chamber where they were continuously perfused with a bath solution [115 mM NaCl, 1 mM KCl, 1.8 mM CaCl2, and 10 mM Hepes (pH 7.4 with NaOH)] or drugs dissolved in a bath solution. For two-electrode voltage-clamp recordings, microelectrodes were filled with 3 M KCl, oocytes were clamped at −70 mV, and currents were recorded at 200 Hz with a Geneclamp 500B amplifier, a Digidata 1322A interface, and Clampex version 9.2 (Molecular Devices, Sunnyvale, CA). In glycine dose–response experiments, peak current responses to glycine at increasing concentrations were measured, and these were plotted against concentration and fit with variable slope nonlinear regression (Prism 4, GraphPad Software Inc., San Diego, CA), generating EC50 and nH values. These were averaged for each construct and reported as means ± SEM. In a test of the effects of DTT and HgCl2, the oocyte was perfused in either 2 mM DTT or 10 μM HgCl2 for 100 s before application of glycine in the presence of 2 mM DTT or HgCl2. The effects of propofol were tested by perfusing the oocyte with 500 μM propofol for 30 s before applying glycine in the presence of 500 μM propofol. Increases or decreases in the peak current response to glycine caused by DTT, HgCl2, or propofol were generally expressed as the fraction of the response to glycine alone. This fraction was calculated for each oocyte, and the mean ± SEM was calculated for each construct. In all experiments, each construct was tested in at least two batches of oocytes, alongside at least two other constructs.
Statistical Analysis
For glycine dose–response experiments, EC50, nH, and Imax values (mean ± SEM) were compared by ANOVA and a Tukey post hoc analysis (Prism 4). In a test of the modulation of current responses by DTT and HgCl2, each construct was statistically analyzed separately: the averaged fractional responses (mean ± SEM) after the application of DTT or HgCl2 were compared by ANOVA and a Dunnett post hoc analysis to control responses for the respective construct (Prism 4). Statistical significance was interpreted as P values of <0.05 or as otherwise indicated in the text and tables.
Western Blotting and Surface Labeling
After expression for 2–3 days, 10 oocytes per construct were rinsed three times with frog Ringer’s solution and incubated for 10 min in frog Ringer’s solution containing glycine at an EC50 concentration. For surface labeling, oocytes were then incubated at 4 °C in frog Ringer’s solution containing Cy5 NHS ester (GE Healthcare, Freiburg, Germany) for 1 h. Oocytes were then lysed by repeated pipetting in 100 μL of a lysis solution [50 mM Tris, 150 mM NaCl, 0.5% Triton X-100, and 0.5% deoxycholate (pH 7.5)] and 20 brief steps of sonication on ice. The resulting homogenate was spun for 10 min at 15000g and 4 °C to isolate protein. His-tagged protein was bound to Ni2+ beads by rinsing protein for 30 min at 4 °C in wash buffer [100 mM Na2HPO4, 0.5% dodecyl maltoside, 0.1% protease inhibitor, and 30 mM imidazole (pH 8.0)] containing 10% Ni2+ beads. Protein was rinsed in wash buffer (containing 30 mM imidazole) and eluted in a solution containing 20 mM Tris, 0.5% dodecyl maltoside, 200 mM imidazole, and 10 mM EDTA (pH 8.0), as described by Haeger et al.21 Protein was then isolated by spinning for 2 min at 15000g and 4 °C and stored at −80 °C. For Western blotting, a third of the total protein from 10 oocytes was added to 40 mM Tris-HCl, 2% SDS, bromphenol blue, and, where appropriate, 10% β-mercaptoethanol, if indicated treated for 1 h at 37 °C in Endo H (New England Biolabs GmbH), heated for 5 min at 95 °C, and added to an 8% Rotipherese gel for separation. If prestained with Cy5, the gel was imaged via ChemiDoc MP (Bio-Rad). For Western blots, the gel was transferred to PDVF membranes for blotting. Membranes were first blocked with 5% skim milk, subsequently incubated in mAb2b, a primary antibody that is specific for α1 GlyRs,57 and finally incubated in horseradish peroxidase-conjugated goat anti-mouse IgG (Bio-Rad). Membranes were developed via ChemiDoc MP.
Acknowledgments
We thank Brett Giolma for help with electrophysiological experiments and Adriana Längle and Dr. Wibke Wagner for help with biochemical experiments.
Glossary
Abbreviations
- ANOVA
analysis of variance
- 5-HT3Rs
5-HT type 3 receptors
- DTT
dithiothreitol
- ECD
extracellular domain
- GABA
γ-aminobutyric acid
- GABAAR
γ-aminobutyric acid type A receptor
- Gly
glycine
- GlyR
inhibitory glycine receptor
- LBD
ligand-binding domain
- M2
membrane-spanning helix 2
- M3
membrane-spanning helix 3
- nAChR
nicotinic acetylcholine receptor
- pLGIC
pentameric ligand-gated ion channel
- Prop
propofol (2,6-diisopropylphenol)
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SEM
standard error of the mean
- TMD
transmembrane domain
Supporting Information Available
Two figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
T.L. and A.K. designed and performed the experiments. T.L., A.K., and B.L. analyzed the data. T.L. and B.L. prepared the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Changeux J. P. (2010) Allosteric receptors: From electric organ to cognition. Annu. Rev. Pharmacol. Toxicol. 50, 1–38. [DOI] [PubMed] [Google Scholar]
- Betz H.; Laube B. (2006) Glycine receptors: Recent insights into their structural organization and functional diversity. J. Neurochem. 97, 1600–1610. [DOI] [PubMed] [Google Scholar]
- Purohit P.; Mitra A.; Auerbach A. (2007) A stepwise mechanism for acetylcholine receptor channel gating. Nature 446, 930–933. [DOI] [PubMed] [Google Scholar]
- Langosch D.; Laube B.; Rundstrom N.; Schmieden V.; Bormann J.; Betz H. (1994) Decreased agonist affinity and chloride conductance of mutant glycine receptors associated with human hereditary hyperekplexia. EMBO J. 13, 4223–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra S.; Lynch J. W.; Pierce K. D.; French C. R.; Barry P. H.; Schofield P. R. (1995) Mutation of an arginine residue in the human glycine receptor transforms β-alanine and taurine from agonists into competitive antagonists. Neuron 14, 169–175. [DOI] [PubMed] [Google Scholar]
- Lynch J. W.; Rajendra S.; Pierce K. D.; Handford C. A.; Barry P. H.; Schofield P. R. (1997) Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J. 16, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless S. A.; Dibas M. I.; Lester H. A.; Lynch J. W. (2007) Conformational variability of the glycine receptor M2 domain in response to activation by different agonists. J. Biol. Chem. 282, 36057–36067. [DOI] [PubMed] [Google Scholar]
- Shiang R.; Ryan S. G.; Zhu Y. Z.; Hahn A. F.; O’Connell P.; Wasmuth J. J. (1993) Mutations in the α1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat. Genet. 5, 351–358. [DOI] [PubMed] [Google Scholar]
- Murail S.; Wallner B.; Trudell J. R.; Bertaccini E.; Lindahl E. (2011) Microsecond simulations indicate that ethanol binds between subunits and could stabilize an open-state model of a glycine receptor. Biophys. J. 100, 1642–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L.; Howard R. J.; Malherbe L.; Lee U. S.; Corringer P. J.; Adron Harris R.; Delarue M. (2013) Structural basis for potentiation by alcohols and anaesthetics in a ligand-gated ion channel. Nat. Commun. 4, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea S. M.; Becker L.; Weiher H.; Betz H.; Laube B. (2004) Propofol restores the function of “hyperekplexic” mutant glycine receptors in Xenopus oocytes and mice. J. Neurosci. 24, 2322–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q.; Han L.; Lynch J. W. (2012) Function of hyperekplexia-causing α1R271Q/L glycine receptors is restored by shifting the affected residue out of the allosteric signalling pathway. Br. J. Pharmacol. 165, 2113–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke J. J.; Koshland D. E. Jr. (1987) Global flexibility in a sensory receptor: A site-directed cross-linking approach. Science 237, 1596–1600. [DOI] [PubMed] [Google Scholar]
- Prevost M. S.; Sauguet L.; Nury H.; Van Renterghem C.; Huon C.; Poitevin F.; Baaden M.; Delarue M.; Corringer P. J. (2012) A locally closed conformation of a bacterial pentameric proton-gated ion channel. Nat. Struct. Mol. Biol. 19, 642–649. [DOI] [PubMed] [Google Scholar]
- Bocquet N.; Nury H.; Baaden M.; Le Poupon C.; Changeux J. P.; Delarue M.; Corringer P. J. (2009) X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114. [DOI] [PubMed] [Google Scholar]
- Hibbs R. E.; Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf R. J.; Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379. [DOI] [PubMed] [Google Scholar]
- Lynagh T.; Webb T. I.; Dixon C. L.; Cromer B. A.; Lynch J. W. (2011) Molecular determinants of ivermectin sensitivity at the glycine receptor chloride channel. J. Biol. Chem. 286, 43913–43924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon R.; Shapira E. (1969) Crystalline papain derivative containing in intramolecular mercury bridge. J. Biol. Chem. 244, 1033–1038. [PubMed] [Google Scholar]
- Cleland W. W. (1964) Dithiothreitol, a New Protective Reagent for SH Groups. Biochemistry 3, 480–482. [DOI] [PubMed] [Google Scholar]
- Haeger S.; Kuzmin D.; Detro-Dassen S.; Lang N.; Kilb M.; Tsetlin V.; Betz H.; Laube B.; Schmalzing G. (2010) An intramembrane aromatic network determines pentameric assembly of Cys-loop receptors. Nat. Struct. Mol. Biol. 17, 90–98. [DOI] [PubMed] [Google Scholar]
- Bali M.; Akabas M. H. (2012) Gating-induced conformational rearrangement of the γ-aminobutyric acid type A receptor β-α subunit interface in the membrane-spanning domain. J. Biol. Chem. 287, 27762–27770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M.; Akabas M. H. (2006) State-dependent cross-linking of the M2 and M3 segments: Functional basis for the alignment of GABAA and acetylcholine receptor M3 segments. J. Neurosci. 26, 4492–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowdhamini R.; Srinivasan N.; Shoichet B.; Santi D. V.; Ramakrishnan C.; Balaram P. (1989) Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis. Protein Eng., Des. Sel. 3, 95–103. [DOI] [PubMed] [Google Scholar]
- Horenstein J.; Wagner D. A.; Czajkowski C.; Akabas M. H. (2001) Protein mobility and GABA-induced conformational changes in GABA(A) receptor pore-lining M2 segment. Nat. Neurosci. 4, 477–485. [DOI] [PubMed] [Google Scholar]
- McCracken L. M.; McCracken M. L.; Gong D. H.; Trudell J. R.; Harris R. A. (2010) Linking of Glycine Receptor Transmembrane Segments Three and Four Allows Assignment of Intrasubunit-Facing Residues. ACS Chem. Neurosci. 1, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careaga C. L.; Falke J. J. (1992) Thermal motions of surface α-helices in the d-galactose chemosensory receptor. Detection by disulfide trapping. J. Mol. Biol. 226, 1219–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horenstein J.; Riegelhaupt P.; Akabas M. H. (2005) Differential protein mobility of the γ-aminobutyric acid, type A, receptor α and β subunit channel-lining segments. J. Biol. Chem. 280, 1573–1581. [DOI] [PubMed] [Google Scholar]
- Grenningloh G.; Rienitz A.; Schmitt B.; Methfessel C.; Zensen M.; Beyreuther K.; Gundelfinger E. D.; Betz H. (1987) The strychnine-binding subunit of the glycine receptor shows homology with nicotinic acetylcholine receptors. Nature 328, 215–220. [DOI] [PubMed] [Google Scholar]
- Todorovic J.; Welsh B. T.; Bertaccini E. J.; Trudell J. R.; Mihic S. J. (2010) Disruption of an intersubunit electrostatic bond is a critical step in glycine receptor activation. Proc. Natl. Acad. Sci. U.S.A. 107, 7987–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo I. A.; Trudell J. R.; Harris R. A. (2004) Cross-linking of glycine receptor transmembrane segments two and three alters coupling of ligand binding with channel opening. J. Neurochem. 90, 962–969. [DOI] [PubMed] [Google Scholar]
- Beato M.; Groot-Kormelink P. J.; Colquhoun D.; Sivilotti L. G. (2002) Openings of the rat recombinant α1 homomeric glycine receptor as a function of the number of agonist molecules bound. J. Gen. Physiol. 119, 443–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleitsman K. R.; Shanata J. A.; Frazier S. J.; Lester H. A.; Dougherty D. A. (2009) Long-range coupling in an allosteric receptor revealed by mutant cycle analysis. Biophys. J. 96, 3168–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pless S. A.; Leung A. W.; Galpin J. D.; Ahern C. A. (2011) Contributions of conserved residues at the gating interface of glycine receptors. J. Biol. Chem. 286, 35129–35136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lape R.; Colquhoun D.; Sivilotti L. G. (2008) On the nature of partial agonism in the nicotinic receptor superfamily. Nature 454, 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks N. P.; Lieb W. R. (1994) Molecular and cellular mechanisms of general anaesthesia. Nature 367, 607–614. [DOI] [PubMed] [Google Scholar]
- Hales T. G.; Lambert J. J. (1991) The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br. J. Pharmacol. 104, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. T.; Li K. Y.; daGraca R. L.; Delphin E.; Xiong M.; Ye J. H. (2009) Behavior and cellular evidence for propofol-induced hypnosis involving brain glycine receptors. Anesthesiology 110, 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegwart R.; Krahenbuhl K.; Lambert S.; Rudolph U. (2003) Mutational analysis of molecular requirements for the actions of general anaesthetics at the γ-aminobutyric acidA receptor subtype, α1β2γ2. BMC Pharmacol. 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski M. D.; Koltchine V. V.; Rick C. E.; Ye Q.; Finn S. E.; Harrison N. L. (1998) Propofol and other intravenous anesthetics have sites of action on the γ-aminobutyric acid type A receptor distinct from that for isoflurane. Mol. Pharmacol. 53, 530–538. [DOI] [PubMed] [Google Scholar]
- Bertaccini E. J.; Wallner B.; Trudell J. R.; Lindahl E. (2010) Modeling anesthetic binding sites within the glycine α1 receptor based on prokaryotic ion channel templates: The problem with TM4. J. Chem. Inf. Model. 50, 2248–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzomato V.; Beato M.; Groot-Kormelink P. J.; Colquhoun D.; Sivilotti L. G. (2004) Single-channel behavior of heteromeric α1β glycine receptors: An attempt to detect a conformational change before the channel opens. J. Neurosci. 24, 10924–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtasimova N.; Lee W. Y.; Wang H. L.; Sine S. M. (2009) Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature 459, 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafna P. A.; Purohit P. G.; Auerbach A. (2008) Gating at the mouth of the acetylcholine receptor channel: Energetic consequences of mutations in the αM2-cap. PLoS One 3, e2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarca C.; Nowak M. W.; Zhang H.; Tang L.; Deshpande P.; Lester H. A. (1995) Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature 376, 514–516. [DOI] [PubMed] [Google Scholar]
- Rayes D.; De Rosa M. J.; Sine S. M.; Bouzat C. (2009) Number and locations of agonist binding sites required to activate homomeric Cys-loop receptors. J. Neurosci. 29, 6022–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. L.; Cheng X.; Sine S. M. (2012) Intramembrane proton binding site linked to activation of bacterial pentameric ion channel. J. Biol. Chem. 287, 6482–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty D. A. (2008) Cys-loop neuroreceptors: Structure to the rescue?. Chem. Rev. 108, 1642–1653. [DOI] [PubMed] [Google Scholar]
- Akabas M. H.; Kaufmann C.; Archdeacon P.; Karlin A. (1994) Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the α subunit. Neuron 13, 919–927. [DOI] [PubMed] [Google Scholar]
- Cymes G. D.; Ni Y.; Grosman C. (2005) Probing ion-channel pores one proton at a time. Nature 438, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paas Y.; Gibor G.; Grailhe R.; Savatier-Duclert N.; Dufresne V.; Sunesen M.; de Carvalho L. P.; Changeux J. P.; Attali B. (2005) Pore conformations and gating mechanism of a Cys-loop receptor. Proc. Natl. Acad. Sci. U.S.A. 102, 15877–15882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velisetty P.; Chalamalasetti S. V.; Chakrapani S. (2012) Conformational transitions underlying pore opening and desensitization in membrane-embedded Gloeobacter violaceus ligand-gated ion channel (GLIC). J. Biol. Chem. 287, 36864–36872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M.; Akabas M. H. (1993) Amino acids lining the channel of the γ-aminobutyric acid type A receptor identified by cysteine substitution. J. Biol. Chem. 268, 21505–21508. [PubMed] [Google Scholar]
- Unwin N. (1995) Acetylcholine receptor channel imaged in the open state. Nature 373, 37–43. [DOI] [PubMed] [Google Scholar]
- Kar G.; Keskin O.; Gursoy A.; Nussinov R. (2010) Allostery and population shift in drug discovery. Curr. Opin. Pharmacol. 10, 715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laube B.; Kuhse J.; Betz H. (2000) Kinetic and mutational analysis of Zn2+ modulation of recombinant human inhibitory glycine receptors. J. Physiol. (Oxford, U.K.) 522(Part 2), 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirzel K.; Muller U.; Latal A. T.; Hulsmann S.; Grudzinska J.; Seeliger M. W.; Betz H.; Laube B. (2006) Hyperekplexia phenotype of glycine receptor α1 subunit mutant mice identifies Zn2+ as an essential endogenous modulator of glycinergic neurotransmission. Neuron 52, 679–690. [DOI] [PubMed] [Google Scholar]
- Ahrens J.; Leuwer M.; Stachura S.; Krampfl K.; Belelli D.; Lamber J. J.; Haeseler G. (2008) A transmembrane residue influences the interaction of propofol with the strychnine-sensitive glycine receptor α1 and α1β receptor. Anesth. Analg. 107, 1875–1883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.