

Abstract
Alzheimer’s disease (AD) is the most common form of dementia. During the recent decade, nanotechnology has been widely considered, as a promising tool, for theranosis (diagnosis and therapy) of AD. Here we first discuss pathophysiology and characteristics of AD with a focus on the amyloid cascade hypothesis. Then magnetic nanoparticles (MNPs) and recent works on their applications in AD, focusing on the superparamagnetic iron oxide nanoparticles (SPIONs), are reviewed. Furthermore, the amyloid–nanoparticle interaction is highlighted, with the scope to be highly considered by the scientists aiming for diagnostics and/or treatment of AD employing nanoparticles. Furthermore, recent findings on the “ignored” parameters (e.g., effect of protein “corona” at the surface of nanoparticles on amyloid-β (Aβ) fibrillation process) are discussed.
Keywords: Magnetic resonance imaging, SPION, amyloid-β, nanomedicine, nanotechnology
Alzheimer’s disease (AD) is named after Alois Alzheimer, who described the first case in a 55 year old female patient (i.e., Auguste Deter) in 1906. The description of Auguste’s pathology was featured by lifelong deteriorating memory, speaking, physical, and social abilities. After her death, the autopsy revealed uniform brain atrophy, atherosclerosis changes in larger cerebral vessels, neuronal loss, and numerous small foci perceivable even without staining distributed over the entire cortex. It took over 70 years to reveal that those foci consist of aggregates of extracellular loads of small peptides called amyloid-β (Aβ), that are considered today one of the hallmarks of the disease.1
AD is characterized by progressive deterioration of cognitive function, most commonly of memory, that increasingly interferes with patients’ daily activities leading to loss of independency (for details, see http://www.alz.org/downloads/facts_figures_2012.pdf). To date, no precise treatments have been clinically proven to avoid or prevent the progression of AD. Several different pharmacological agents can only ameliorate or provide temporary alleviation of the symptoms.2
In this review, we introduce clinical aspects and characteristics of AD with a focus on the amyloid cascade hypothesis. Magnetic nanoparticles (MNPs), as promising theranosis tools, are introduced, and recent reports on the potential applications of superparamagnetic iron oxide nanoparticles (SPIONs) in AD are summarized. It is worthwhile to note that the SPIONs are known as promising theranosis candidates for AD, due to their biocompatibility, unique magnetic properties and multifunctional application capability.3,4 The amyloid–nanoparticle interaction is highlighted, with the scope to be highly considered by the scientific community aiming for diagnostics and/or treatment of AD employing nanoparticles. Moreover, recent findings on the “ignored” parameters (e.g., the effect of protein “corona” at the surface of nanoparticles on Aβ fibrillation process) are discussed.
Clinical Aspects of AD
AD affects a person’s ability to carry out daily activities and is characterized by gradual memory loss, anomia (i.e., difficulties with word finding), apraxia (i.e., difficulties with complex movements), confusion, and general withdrawal. As the disease progresses, severe cognitive impairment, impaired executive functions and personality changes worsen the lifestyle condition. Patients in more advanced stages become very susceptible to infections, pneumonia, and decubitus ulcers. Life expectancy after a diagnosis of AD is usually between 7 and 10 years.5,6 Early diagnosis and early intervention have consistently emerged as key policy priorities in recently formulated national dementia strategies. Yearly updated guidelines define the criteria for a clinical diagnosis of AD. They include the use of cognitive test, such as the mini-mental state test,7 the clock-drawing test,8 and other sensory-related test (e.g., the odor identification test9).
In addition to these well recognized tests, very recently, Mahmoudi and Suslick showed that the molecular aspects of olfactory dysfunction can be also recognized as a hallmark of AD and there may even be a causal link through the disruption of multivalent metal ion transport and storage.10 Once cognitive deficits are demonstrated, cerebrospinal fluid biomarker evaluation and brain imaging are demanded for a more accurate diagnosis. Among various imaging methods in AD investigation, magnetic resonance imaging (MRI) and positron emission tomography (PET) are the most common techniques.
Macroscopic and Microscopic Changes in AD Brain
AD is neuropathologically characterized by extracellular accumulation of amyloid plaques, caused by Aβ protein aggregation, and the presence of intracellular neurofibrillary tangles (NFTs), formed by aggregation of hyperphosphorylated tau protein. The Aβ is generated by sequential cleavage of the transmembrane amyloid precursor protein (APP), via groups of enzymes named α-, β-, and γ-secretases.11,12 When the APP protein is cleaved by the α-secretase, a soluble amino (N)-terminal ectodomain (sAPPα) and a C-terminal fragments (CTF) are released. Thereby the formation of Aβ is prohibited, due to the cleaving that occurs inside the Aβ region.13 This is known as the “non-amyloidogenic” pathway. The “amyloidogenic” pathway generates Aβ when the precursor protein is cleaved by a β-secretase (BACE: β-site APP cleaving enzyme) at the N-terminal domain, releasing a soluble N-terminal fragment (sAPPb) and the remaining C-terminal part (b-CTF). The CTF and b-CTF are then cleaved in the transmembrane domain by the γ-secretase to release either extracellular p2 or Aβ, respectively. Cleavage by γ-secretase takes place at position 40 or 42 of the protein, which generates amyloid-β 40 (Aβ40) or amyloid-β 42 (Aβ42), respectively. Approximately 90% of the residues consist of Aβ40. However, the Aβ42 is considered the more toxic form and aggregates more readily. Therefore, Aβ42 is the most abundant isoform in amyloid plaques.
Besides the parenchymal deposition of Aβ in the brain, Aβ deposits are also found in the cerebral vasculature.14 Amyloid deposition first occurs in arterioles of the pia mater surrounding the cerebrum and subsequently spreads to the cortical arterioles, capillaries, and vessels in other brain areas.15 This progressive vascular Aβ accumulation leads to cerebral amyloid angiopathy (CAA), although its extent may vary among AD brains and its pathophysiological contribution to the disease progression remains unclear.
It has been suggested that CAA alters the cerebrovascular perfusion by damaging the vascular wall, causing endothelial dysfunction and impairing the vessel reactivity with an enhanced production of free radicals.16,17 This might ultimately lead to chronic hypoperfusion of specific brain regions resulting in associated cerebrovascular dysfunction and subsequent neuronal loss and degeneration.18−20 In addition, Aβ deposits have been found in the olfactory bulbs, followed by subsequent deposition in the olfactory cortex and hippocampus.21
It was commonly thought that amyloid plaque formations in various areas of the brain cause the symptoms of AD,1 but recently attention has shifted toward the oligomeric soluble Aβ that is considered as toxic form that causes neuronal loss.22−24
Synaptic Dysfunction in AD
The pathogenic process of AD probably starts decades before clinical onset of the disease. During this preclinical period, often designated as mild cognitive impairment (MCI), there is a gradual synaptic dysfunction and neuronal loss revealed, most often, by impaired episodic memory.
Alteration and loss of synapses in a specific circuitry of the medial temporal lobe system is a normal age-related process believed to initiate memory loss in the elderly.25 These processes occur first in the entorhinal cortex and rapidly affect the molecular layer of the dentate gyrus and the nucleus basalis of Meynert.26 However, in normal aging, this loss of connections is not likely to precede or provoke loss of neurons, as the neuronal number is consistently preserved.27 Instead, in the case of MCI and AD, a higher selective vulnerability of neurons in these areas seems to strongly contribute to the neurodegenerative cascade and regional atrophy.28 Soluble and fibrillar Aβ formation together with development of NFTs accompany these processes, although the reports on their mutual interaction are not consistent.
Some studies in transgenic mouse models for AD with mutation in genes encoding APP or presenilins 1 or 2, reported the occurrence of synaptic loss, decreased dendritic spine deficits and memory impairments much earlier than Aβ plaque deposition in the limbic system.29,30 Others, instead, described a spatial relationship between synapse density loss and Aβ plaques in the hippocampus and entorhinal cortex, suggesting a direct causal mechanism of Aβ in the process of neurodegeneration.31 Nevertheless, the severity of cognitive impairments appears to correlate better with loss of synapses, rather than with the presence of amyloid deposits and NFTs in the brain.
Etiology of AD
The AD consists of two major types termed early onset or presenile AD and late onset or sporadic AD. Patients are classified as early onset AD when the first symptoms of AD occur at an age earlier than 65 (see: http://www.alz.org/downloads/facts_figures_2012.pdf). Early onset AD accounts for 5% of all AD cases worldwide and is caused by genetic mutations, as the persons affected generally have a positive family history.2 Several mutations in APP on chromosome 21, in presenilin 1 (PS1) on chromosome 12 and presenilin 2 (PS2) on chromosome 1 have shown to be autosomal-dominant inheritable.32 Mutation in PS1 and PS2 lead to an increased production of the strong self-aggregating Aβ42 peptide by elevating the γ-secretase activity. This causes the most aggressive form of AD with an early onset that can occur even earlier than age of 40. However, mutations in APP, PS1, and PS2 have been found to cause less than 30% of the early onset AD. Other genetic disorders responsible for the remaining cases of early onset are poorly known.33
Patients with late onset AD account for the remaining 95% of AD incidence. The exact causes of late onset AD are still elusive. A large number of risk factors for the development of AD have been identified and can be attributed to environmental- and lifestyle- risk factors, and gene mutations affecting AD pathology. However, aging is known as the most important risk factor for AD.34 Other risk factors include smoking, dietary intake, stroke, hypertension, heart disease, hypercholesterolemia, and diabetes mellitus.35,36 The only gene that has currently been identified as a risk factor for the late onset AD is the ε4 allele of the cholesterol transport protein, apolipoprotein E (apoE).37,38
ApoE is a 34-kDa cholesterol transport glycoprotein that exists in three isoforms: apoE-ε2, -ε3, and -ε4. The highest expression of apoE is found in the liver, followed by the brain where it is mainly expressed by astrocytes and to a certain extent by microglia.39,40 Neurons are also capable of producing apoE under certain conditions, but in a much less extent than astrocytes.41,42 Extensive studies in genetic epidemiology demonstrated that the inheritance of one apoE-ε4 allele is associated with increased risk of developing AD by 2–5-fold and the inheritance of two ε4 alleles rise this risk by 4–10-fold compared to expressing the most common isoform ε3.43 The ε4 allele is also associated with decreased age of the AD onset and with an increased conversion from MCI to AD.44,45 On the contrary, inheritance of the ε2 allele appears to protect against AD pathogenesis.46 The mechanism by which apoE-ε4 promotes the disease is still an argument of research.
ApoE is produced after injury to promote neuron protection or to repair injured neurons by transporting cholesterol to the membrane; however, apoE4 appears to be less effective in this signaling process due to its susceptibility to intraneuronal proteolysis.47 Apart from its cholesterol transporting capacity, apoE can bind to Aβ and form stable complexes, which can be transported from the brain to periphery and vice versa. A study showed that this active brain to blood clearance of Aβ is less efficient in the apoE4 isoform compared to apoE2 and apoE3.48 The fast clearance of free Aβ is redirected, by binding of apoE4 from the lipoprotein-receptor-related protein 1 (LRP-1) to the very-low-density-lipoprotein receptor (VLDLR), which internalizes apoE4 and apoE4-Aβ complexes at the blood-brain barrier (BBB) at a lower rate than LRP1. ApoE2-Aβ and apoE3-Aβ complexes are removed by both LRP-1 and VLDLR at a significantly higher rate than apoE4-Aβ complexes.49−51 This might explain why mice expressing human apoE4 have higher levels of Aβ deposition, compared to mice expressing human apoE3.52 These experimental data well resemble human neuropathological analyses, where subjects carrying one or two apoE4 alleles show significantly higher level of Aβ, even in the absence of neurological AD symptoms.53 The ApoE4 isoform is also strongly associated with the presence of Aβ in the vascular walls and the occurrence of CAA.54
To date, many theories about the causes of AD have been postulated; among them, the “amyloid cascade hypothesis” is still the most widely accepted one which is discussed in the next section.
Amyloid Cascade Hypothesis
As described above, genetic risk factors for AD include mutations in genes expressing APP, PS1, PS2, and ApoE4. Most of these mutations result in an elevated Aβ peptide production or failure of Aβ clearance mechanisms, leading to an abnormal accumulation of Aβ in the brain. Furthermore, patients with Down syndrome develop AD early in life and show overproduction of Aβ and plaque deposition in the brain before tangles and other AD lesions.55
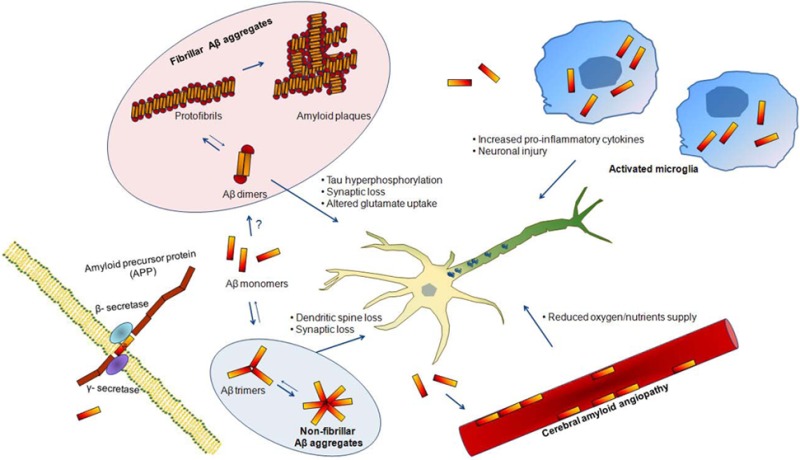
Based on these observations, the accumulation of Aβ in the brain was described by Hardy and Allsop as the key event in the pathogenesis of AD more than 20 years ago.56 The so-called “amyloid cascade hypothesis” suggested that the accumulation and aggregation of Aβ, specifically Aβ42, would trigger further pathological events such as formation of NFTs, disruption of synaptic connections with decreased neurotransmitters release, microglial and astrocytes activation in a chronic inflammation response, initiates neuronal loss and ultimately leads to dementia. However, later studies revealed that the amount of fibrillar Aβ deposits in AD brain poorly correlates with the severity of cognitive impairment.57,58 Instead, nonfibrillar oligomeric Aβ species were found to be highly neurotoxic and their levels much better correlated with severity of disease and synaptic loss.59,60 These findings were the basis for a revision of the amyloid hypothesis by Hardly and Selkoe, pointing toward the oligomeric Aβ species as the initiator culprits of AD rather than Aβ plaques61 (see Figure 1). It has been confirmed that soluble Aβ oligomers inhibit neuronal function,62 induce dendritic spine loss,63 and alter neuronal plasticity.64 A recent study from Selkoe and colleagues reported that Aβ dimers could trigger tau hyperphosphorylation and tau-dependent cytoscheletal microtubule abnormalities in primary neurons.22
Figure 1.

Basis for a revision of the amyloid hypothesis. Amyloid-β (Aβ) monomers are formed by β- and γ-secretase clevage of the amyloid precursor protein (APP). These monomers accumulate in the extracellular space and trigger several processes associated with AD pathophisiology. Aβ monomers can initiate the formation of fibrillar Aβ aggregates, possibly by an initial aggregation in the form of Aβ dimers, or nonfibrillar Aβ aggregates, by an initial aggregation in the form of Aβ trimers. Accumulation of Aβ also results in an activation of microglial cell which increase the production of cytokines that can damage neuronal health when expressed in high concentrations. Furthermore, Aβ can aggregate in the cerebral blood vessels causing cerebral amyloid angiopathy (CAA), impairing the neurovascular unit function and decreasing oxygen and nutrients supply to the brain cells.
Intracellular Trafficking of APP
In 1984, Glenner and Wong successfully purified and analyzed cerebral Aβ deposits.65 They probed the same concept in AD pathology by focusing on the amyloid in meningeal vessel walls; after careful considerations, they could chromatographically enrich the amyloid subunit and reported the partial amino acid sequence of the hydrophobic peptide, the “β-protein”. A year after, Masters and colleagues confirmed the existence of 4.2 kDa “β-protein” and extended the rest of amino acid sequences.1 Using the findings of Glenner’s66,67 in 1987, based on “reverse genetic” strategy, several laboratories cloned the human APP gene independently.68−71
Genetically, APP maps to chromosome 21(21q21.2–3) and expresses ubiquitously in all tissue types in a cell specific manner. As chromosome 21 is duplicated in individuals with trisomy 21 (Down syndrome), it is reasonable to think of an interesting immediate correspondence to the invariant development of AD pathology. Historically, the first mutations implicated in inherited forms of familial AD and a related inherited condition were identified in the APP gene.72 All genes encode members of a glycosylated type I single spanning transmembrane proteins, which carry a large ectodoplasmic N-terminal region and a tiny C-terminal tail.73 Importantly, only APP, and not any other APP related gene, contains sequence encoding the Aβ domain. Therefore, APLP1 and APLP2 are not the precursors to the peptide and their contribution in AD pathogenesis is not well-known yet.
Unlike many cell surface receptors, the time that full length APP settles at the cell surface is not considerably long. Therefore, the holo-APP molecules, which are not discharged from the cell surface, experience endocytosis phenomenon within minutes of arrival. This internalization seems to happen due to the presence of the “YENPTY” motif near the carboxyl terminus of APP. This domain regulates clathrin-coated pit74 internalization through a series of binding partners. Following internalization, depending on the type of the marker incorporated on the membrane of early endosome, APP is delivered to sort endosomes and some fraction traffics back to the cell surface by recycling endosomes. It has been shown that remarkable amounts of internalized APP might undergo degradation in lysosomes.75 The YENPTY domain is 100% reserved in the entire APP family gene and the mutation of this motif has been shown to selectively impair APP internalization and then decrease the Aβ generation.76 Many cytosolic adaptors with phosphotyrosine-binding domain, including Fe65, Mint1, Dab1, sorting nexin 17 and many other binding partners interact with this motif and the nearby region. In the case of nervous system, it has been shown that the overexpression of some of these binding partners may utterly influence the processing fate of APP. For example, in transgenic mice, overexpression of Fe65 and Mint1 reduces the generation of Aβ and its deposition, strongly implying a physiological role for these adaptors in regulating the amyloidogenic processing of APP.77,78
In case of neurons, scientists are willing to better understand how APP is destined and also how it is processed to its cleavage products during intracellular trafficking. Many studies have shown that neurons with their structural polarization into axons, dendrites and soma demand much more sophisticated sorting mechanisms with various proteins and lipids.77 Subdivision of neuronal structure to dendritic shafts, dendritic spines, axonal shafts, and neuronal endings can worse off the situation to rule all these sorting. Taking together, a complex system composed of microtubules, kinesine, and dynein motor proteins along with specific sorting signals facilitate proper delivery of the protein. Importantly, minor malfunction in this complex system may interfere with the pathogenesis of AD.79
Another pathway is soma sorting pathway, which is the same as the non-neuronal cells (i.e., the transportation from ER to Golgi and TGN). Although APP, as a membrane receptor along with kinesin-1 microtubular motor protein, reaches an unprecedented fast and unidirectional axonal transportation, however, the exact nature of APP carriers into axons and dendrites is not fully recognized. Recently, it has been reported that the sorting signal of APP toward dendritic and axonal compartments seems to follow no specific pathways,80 which implies the distinct behavior of neurons, compared to the other peripheral cells, for handling polarized trafficking of APP.
Amyloid-β Aggregates and Their Role in AD
Misfolding (toxic folding)81 of the Aβ protein is recognized as a major cause of AD. Misfolded Aβ molecules form β-sheet containing structures that assemble into a variety of polymorphic oligomers, i.e., annulus, dodecamers or amyloshperoides,82,83 protofibers, and fibers ranging from dimers to 24-mers, or even higher molecular weights (in the range of 10–100 kDa).84
Detection of the Aβ deposits in the healthy subjects, not suffering from dementia,85 allows to conclude that soluble Aβ aggregates might be responsible for creation of severe neuronal toxicity in brain cells rather than fibrillar forms.24,86 It seems that the size of the oligomers has a crucial role in their correspondence toxicities; for instance, it has been shown that the tetramers have 13-fold and dimers have 3-fold toxic effects higher than the monomeric form of Aβ.87
Although extensive research efforts are performed in characterizing assembly states, conformations, and formation mechanisms, but their correspondence toxicological role on the neurodegenerative diseases is not well recognized. However, the most dominant biological effects of the Aβ species, as upstream of the pathogenesis cascade in AD, are supposed to be synaptic failure (known as an earliest event), activation of apoptotic signals, receptor-mediated toxicity, pore formation, and sequestration of crucial proteins and autophagy dysfunction.2,88−90 The proposed toxicity mechanisms of these “invisible toxins” have been discussed in detail elsewhere.24,87,91−93
In parallel to conformational changes of the Aβ monomers, distribution of free multivalent cations (e.g., Cu2+, Fe2+, Zn2+, and Al3+) can potentially induce the fibrillation process of the negatively charged monomers.94,95 In order to better understand the interaction mechanism of the Aβ monomers with cations, Laurent et al.95 empolyed the molecular dynamic (MD) simulation method (see Figure 2 for details). They found that the multivalent cations (and not monovalent cation such as Na+) can play a bridging role between the amyloid monomers and induce the first seeding of the oligomerization process. From this observation, one can conclude that the existence of free multivalent cationic ions in the brain could be considered as main risk factor for AD. Nowadays, it is clear that individual ions have sophisticated transporting system(s) (e.g., by proteins) and the trafficking network is dependent on the protein ligands chemistries, coordination geometries, and other physicochemical properties of ions and transporters. Interestingly, based on standard chemometric approach (hierarchical clustering analysis), Mahmoudi’s group found that the serum concentrations of an array of such multivalent cations can be a fingerprint for identification of AD patients.96 This may pave the way for a reliable, efficient, and cheap method for early detection and treatment of AD.
Figure 2.

(a) Display of two amyloids in the presence of Ca2+ where two cations are placed on one amyloid and another cation is placed on another amyloid. (b) Distance between the centers of mass of amyloids as a function of simulation length for different cations: Na+ (red line), Ca2+ (blue line); the figure was adapted from ref (95) with permission from The Royal Society of Chemistry.
There are strict controlling systems (e.g., cells recruit protein-based components as metal signal pathway soldiers); some serve as import/export channels (transporter and channel proteins) as exchangers, as escorting proteins (the Chaperones), metalloinsertase, endocytosis elements and in some cases as pumps) for ion homeostasis and handling the right free ion concentrations (perfect trafficking) in the human body. Among these various beneficial pathways in regulating the metal ions levels, there are some detrimental pathways perturbing the metal ion concentration balance inside the cell. For example, it has been shown that metal sites of individual proteins are not sufficiently selective to a specific metal ion in a way to exclude the others.97 Oxidative stress (OS; such as reactive oxygen species) has a significant role in unbalancing of the cellular ion transporting systems; for instance, combination of Fenton reactions (inevitable component of the OS) with Harber-Weiss reactions (a metal ions release circle forms) results in release of free radical-containing reactions and, consequently, uncontrolled metal ion liberations.
Importance of Nanotechnology in AD
Due to the lack of an early diagnostic and therapeutic approach, theranosis is highly challenging in AD. Numerous rodent studies have reported that Aβ plaques can be imaged by MRI, without using contrast agents and by just employing advanced imaging techniques.98,99 Such experiments mainly have been done at high-field with long scan times which would be problematic in the routine clinical human imaging. Therefore, having an exogenous biocompatible agent for AD theranosis is greatly appreciated. Nanomedicine is a cutting-edge technique representing effective impact on the early detection and treatment of neurodegenerative disorders, such as AD. Figure 3 schematically illustrates the ideal passage from nanotechnology to the neurodegenerative diseases.
Figure 3.

Scheme of the ideal passage from nanotechnology to the neurodegenerative diseases.
Recently, scientists have been developing novel nanomaterials as imaging contrast agents and drug delivery systems for early detection and treatment of the central nervous system (CNS) diseases, respectively. However, drug delivery into the brain is complicated by restrictive mechanisms imposed at the BBB which protects the brain against harmful substances, for example, viruses. Thus, in order to elucidate any neurological disorder such as AD, one needs to overcome the BBB.
Various strategies and approaches, to efficiently transport the drug delivery to the CNS, have been studied in a number of publications.100−102 Roney et al.103 discussed biodegradable polymeric nanoparticles with appropriate surface modifications and ability to deliver drugs beyond the BBB for theranosis of neurological disorders, such as AD. Tanifum and co-workers synthesized an Aβ-targeted lipid conjugate and incorporated it in stealth liposomal nanoparticles targeted to amyloid plaque deposits in a preclinical AD model. Their compound crossed the BBB and bind to Aβ plaque deposits, by labeling parenchymal amyloid deposits and vascular amyloid characteristic of cerebral amyloid angiopathy.104 Because MRI is a noninvasive technique, development of magnetic nanoparticles (MNPs) has been increasingly considered for AD theranosis.105−109
Magnetic Nanoparticles
The MNPs are promising candidates for a wide range of biomedical applications. They are consisting of a magnetic core, for example, maghemite, and a biocompatible coating, for example, polyethylene glycol (PEG). MNPs become more interesting and applicative when getting functionalized, that is, incorporating them with biological vectors, luminescent labels, antibodies, drugs, and so forth.110 Ideal MNPs are nontoxic to the cells/tissues and stable for a long storage time.
Magnetic Nanoparticles as MRI Contrast Agent
MRI is one of the most noninvasive diagnostic techniques that would benefit a lot from nanotechnology, that is, by employing MNPs. Despite the relatively high quality MR images of the soft tissues, in some cases it is not possible to have enough image contrast to diagnose the pathology of interest. In such cases, generally, a contrast agent (CA) is used. The CAs in MRI shortens the spin–lattice T1 and/or spin–spin T2 relaxation times of the water protons within the tissues/regions where they are delivered to, thus enhancing the image contrast. Therefore, what is imaged is not the CA compound but rather its effect on the relaxivity of the adjacent water protons, predominantly through the dipolar interaction.
Most commonly, the contrast is made by a gadolinium-based compound (paramagnetic CA) administration.111−114 They enhance the MR signal intensity, and for this reason they are called “positive” CAs. Superparamagnetic CAs, are iron oxide-based MNPs.115−118 Such agents commonly decrease the MR signal intensity of the regions where they are delivered to and thus those regions appear darker in the image, and therefore they are called “negative” CA. Both positive and negative CAs are being considered to be used as smart probes for biomedical applications such as molecular targeting of Alzheimer’s amyloid plaques.
Magnetic Nanoparticles for Molecular Imaging of Aβ
MNPs have great potential to noninvasively be used for molecular imaging and therapy. They can be used as target-specific agents, to selectively enhance the contrast in molecular level, if functionalized, for instance by incorporating them with antibodies. Targeted compounds improve the lesion detectability (as MRI CAs) of certain pathologies and more importantly provide the localized therapy (as drug delivery systems).
Targeted MNPs have been used to detect Aβ plaques of AD. For instance, Poduslo et al. targeted Aβ plaques of AD using a Gadolinium-loaded molecular probe.108 Their smart system was able to cross the BBB, following intravenous injection, and specifically bind to the plaques and was imaged in MRI. According to their report, they gained more than 9-fold enhancement in regions of cortex and hippocampus in AD transgenic mice at 7 T. Such systems not only have great potentials for early diagnosis but also can be used for direct measurement of the efficacy of antiamyloid therapies. In another study, several magnetic CAs were introduced to detect amyloid plaques in AD transgenic mice.105 The authors used ultrasmall superparamagnetic iron oxide nanoparticles (USPION) and were able to detect and well identify amyloid plaques by T2*-weighted MRI in transgenic mice compared to the wild-type. In another interesting study, Sillerud et al. synthesized antibody-conjugated SPIONs targeted to the Aβ plaques in AD transgenic mice and were able to detect the plaques in MR images;109 the detectibility was twice higher than control cases.
Due to some limitations such as uncertainty in signal quantification of the probes, nuclei other than 1H, e.g. fluorine-19 (19F), have been considered for molecular/cellular imaging. 19F nucleus has 100% natural abundance, spin 1/2 and sensitivity close to that of 1H. Furthermore, there is no in vivo background signal of 19F and this allows unambiguous signal localization and quantification. 19F MRI, after injection of fluorinated contrast media, is carried out by acquiring anatomical 1H MR image followed by a 19F image. In this case, both 1H and 19F images are acquired after injection of the fluorinated compound and precontrast images are not required. 19F MRI technique has been used to detect Aβ in AD.119,12019F MRI technique has been introduced elsewhere.121
Magnetic Nanoparticles Against Aβ Fibrillation
Functionalized MNPs are highly being considered to be used not only for Aβ detection and longitudinal studies of the therapeutic response noninvasively, but also as promising drug delivery systems. Targeted multifunctional nanocarriers for diagnostics, drug delivery and targeted treatment across BBB have been nicely argued in a review by Bhaskar and colleagues.122
To date, there is no effective prevention or treatment for AD. Recently Mahmoudi et al.123 probed the physicochemical effects of the SPIONs on the Aβ fibrillation process. They found that SPION size has significant effects on the Aβ fibrillation process; thus, the size distribution of MNPs is a key issue. Beside nanoparticles size, surface area and charge showed “dual” effects on the fibrillation kinetics; more specifically, lower concentrations of SPIONs inhibited Aβ fibrillation rate, while higher concentrations enhanced it,123 an effect also observed for nonmagnetic nanoparticles (i.e., polystyrene nanoparticles).124 Surface charge influenced the concentration at which the acceleratory effects were observed, with the positively charged SPIONs promoting fibrillation at significantly lower particle concentrations than both negatively charged and essentially uncharged (plain) SPIONs. This suggested that in addition to the presence of SPIONs affecting the concentration of monomeric protein in solution (and thereby the nucleation time), there were also effects of binding on the Aβ conformation, which was mainly detected with the positively charged SPIONs.123 Furthermore, one can conclude that SPIONs designed for medical imaging applications should consider using a negatively charged or uncharged surface coating preferentially, as these are less likely to induce undesired side effects such as protein fibrillation, while maintaining the desired magnetic function; interestingly, this matter has been also shown in the in vivo simulated situations (i.e., protein coated nanoparticles).125
Although there are extensive reports on effects of nanoparticles on the kinetics of amyloid fibrillation, there are several crucial “ignored matters” which are poorly understood and need to be extensively considered in future. These ignored matters, which mainly introduced by the group of Prof. Morteza Mahmoudi at Tehran University of Medical Sciences, are as follows: (i) the effect of “corona” coated nanoparticles, (ii) effect of slight temperature changes (i.e., in the physiological range) on the amyloid fibrillation process, and (iii) cell “vision” effect on cellular toxicity of the amyloid oligomers/fibrils.
It is now well recognized that the surface of nanoparticles is covered by biomolecules (e.g., proteins) upon their entrance to the biological medium.126−129 Thus, what the biological entities (e.g., Aβ) actually “see” in the biological medium, is the protein coated nanoparticles (so-called “protein corona”); in this case, Mahmoudi and co-workers130,131 showed that the presence of the nanomaterials protein corona inhibits the formation of Aβ fibrils for various types of nanomaterials (e.g., silica (100 and 200 nm), polystyrene with carboxyl surface modification (100 nm), multiwalled carbon nanotubes (CNT; diameter, 10–40 nm; length, 0.1–10 μm), and graphene) whereas the same nanomaterials could accelerate the rate of Aβ fibrillation when bare.
Besides the protein corona, it has been shown that the temperature has a crucial role on corona composition132 and the amyloid fibrillation process;133 more specifically, changes in the incubation temperature (i.e., at physiological range), can have “dual” effects on the amyloid fibrillation process, in the presence of hydrophilic (silica) and hydrophobic (polystyrene) nanoparticles; the reason is that the core hydrophobic backbone of Aβ monomers can be more available for interactions by increasing the temperature from 37 to 42 °C. In such a case, the hydrophobic nanoparticles (i.e., polystyrene) have the capability to show dual effects (i.e., acceleratory and inhibitory) on the fibrillation process by slight temperature enhancement; however, for hydrophilic nanoparticles (i.e., silica), the acceleratory effects on the fibrillation process can be significantly increased (see Figure 4).
Figure 4.

Transmission electron microscopy (TEM) images of (a) polystyrene and (b) silica nanoparticles with various magnifications. (c,d) Representative schemes showing the exposure of the amyloids’ hydrophilic and hydrophobic backbone to the free amyloid monomers after interaction with polystyrene and silica particles at 42 °C, respectively. (e,f) TEM images of the amyloid interacted proteins with polystyrene and silica particles at 42 °C, respectively. As seen, there is no trace of fibrillation in (e); however, severe fibrillation (red arrows as examples) was observed in (f). In (e) and (f), left and right images correspond to interaction of amyloid with nanoparticles at 20 and 400 min, respectively. Obviously, at 42 °C, fibrillation at the surface of silica nanoparticles is considerably enhanced by increasing the interaction time, compared to that of the polystyrene. The scale bar is 100 nm. Reprinted with permission from ref (133). Copyright 2013 American Chemical Society.
Cell “vision” is also considered as additional crucial ignored factor for the cellular toxicity evaluations of amyloid oligomers/fibrils. Cell vision is the numerous detoxification strategies that any particular cell can utilize in response to toxins (e.g., amyloid oligomers/fibrils).134−138 The uptake and defense mechanism could be considerably different according to the cell type. Thus, what the cell “sees”, when it is faced with amyloid oligomers/fibrils, is most likely dependent on the cell type, which should be considered for interpretation of the toxic effects of amyloid oligomers/fibrils.
Challenges and Limitations
Due to their high biocompatibility, superparamagnetic properties, and their high capacity for using them as multimodal contrast agents, functionalized SPIONs are recognized as the most promising MNPs in nanomedicine.139−142 However, although their application is still extremely limited,143−146 they have been proposed for neurodegenerative diseases.147−149 Moreover, their potential with high affinity for circulating Aβ forms to induce a “sink effect” and, thus, ideally ameliorate AD has been reported.150 Therefore, deep understanding of their interactions with Aβ, and other amyloidogenic proteins is of crucial importance to ensure that their medical use does not inadvertently contribute to amyloid related disease progression.
There are couple of reports on the interaction of SPIONs with amyloidogenic proteins,123,136,151−156 but there have been very few studies on the physicochemical properties of SPIONs on Aβ fibrillation process. Importantly, all of the performed experiments on the nanoparticle–Aβ interactions have been focused on the pristine coated SPIONs, which is not the real state of SPIONs in vivo; thus, the main direction of future research should be focused on the interaction of SPIONs with the corona coated nanoparticles at various temperatures (as the temperature has crucial role in amyloid fibrillation as well as cellular signaling pathways).133,157,158
Very recently, in a study, SPIONs were administered intravenously in APP transgenic mouse models of AD, to investigate cerebral amyloid angiopathy.106 In this case, iron labeled macrophages were found in the brain, despite the fact that the BBB was intact as verified using gadolinium-contrast enhanced MRI. Circulating monocytes migrate from the lumen into the vessel wall159 and are able to transmigrate the BBB when attracted by chemokines produced by Aβ-stimulated cells.160,161 A study by Beckmann et al.106 suggested that monocytes take up the SPIONs in the circulation and then penetrate the brain. Further in vivo experiments on the SPIONs with various surface charges are required to assess how the monocytes uptake can influence the effects of SPIONs on the fibrillation process.
Summary
Owing to their unique physical, chemical, and biological properties, compared to their bulk structures, nanoparticles have attracted rapidly growing interest in biomedical sciences. One of the promising subjects is the interactions of nanoparticles with amyloid proteins, which are consistent features in the pathogenesis of several neurodegenerative diseases (e.g., AD). The use of nanoparticles as theragnostic agents, to inhibit fibril formation, could be a potential treatment for such diseases. In this case, previous reports have showed that nanoparticles of various surface chemistries and sizes could both accelerate and inhibit Aβ fibrillation in solution. Among various types of nanoparticles, MNPs such as SPIONs, are recognized as promising nanoparticles due to their multi task capabilities (e.g., drug delivery, hyperthermia, and imaging within the same nanosystem).139,148
Very recent results have shown that the slight temperature changes (which can be induced by hyperthermia capability of magnetic nanoparticles) can have significant effects on the fibrillation process.133 While these are important observations, in order to understand the fate of Aβ exposed to a nanoparticle in a biological context, very recent reports confirmed that the real state of the nanoparticles in biological medium, so-called corona coated nanoparticles, has been ignored.130,131 Thus, an important step forward to better understand the Aβ–nanoparticle interactions in more biologically relevant conditions is the consideration of the fact that nanoparticle surface gets covered with proteins and thus potentially becomes less accessible to the Aβ. Therefore, the effect of protein corona coated nanoparticles on Aβ fibrillation is an important issue to be investigated in the future studies.
One may expect that various types of corona, including those that are themselves potentially pro-fibrillar (e.g., those containing unfolded proteins), lead to an NF-κB response,162 but this aspect should be investigated. Importantly, despite the reduction in the amount of protein fibrillation for corona nanoparticle complexes, there are still significant levels of oligomerization of Aβ on the nanoparticle–corona surface and in the bulk solution (albeit at higher Aβ concentrations than expected in vivo). For instance, recent results confirmed that the interaction between Aβ and fibrinogen modifies fibrinogen’s structure, which may then lead to abnormal fibrin clot formation.163 In this case, one can conclude that the presence of fibrinogen in the composition of hard corona might be harmful due to the formation of fibrin clot (more investigation is required to validate this issue).
Author Contributions
H.A. and V.Z. designed the review and directed its implementation. K.S., P.B., and V.Z. contributed in literature search and drafting the pathophysiological aspects of the disease. H.A., A.M., and M.G. participated in literature search, drafting the applications of magnetic nanoparticles as theranostic agents, and reviewed the article. All authors read and approved the final manuscript.
The authors declare no competing financial interest.
References
- Masters C. L.; Simms G.; Weinman N. A.; Multhaup G.; McDonald B. L.; Beyreuther K. (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J.; Schenk D. (2003) Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol. 43, 545–584. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Shokrgozar M. A. (2012) Multifunctional stable fluorescent magnetic nanoparticles. Chem. Commun. 48, 3957–3959. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Serpooshan V. (2012) Silver-coated engineered magnetic nanoparticles are promising for the success in the fight against antibacterial resistance threat. ACS Nano 6, 2656–2664. [DOI] [PubMed] [Google Scholar]
- Helzner E. P.; Scarmeas N.; Cosentino S.; Tang M. X.; Schupf N.; Stern Y. (2008) Survival in Alzheimer disease: a multiethnic, population-based study of incident cases. Neurology 71, 1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Brayne C.; Matthews F. E.; (2008) Survival times in people with dementia: analysis from population based cohort study with 14 year follow-up. BMJ 336, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein M. F.; Folstein S. E.; McHugh P. R. (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198. [DOI] [PubMed] [Google Scholar]
- Berger G.; Frolich L.; Weber B.; Pantel J. (2008) Diagnostic accuracy of the clock drawing test: the relevance of ″time setting’’ in screening for dementia. J. Geriatr. Psychiatry Neurol. 21, 250–260. [DOI] [PubMed] [Google Scholar]
- Wilson R. S.; Arnold S. E.; Tang Y.; Bennett D. A. (2006) Odor identification and decline in different cognitive domains in old age. Neuroepidemiology 26, 61–67. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Suslick K. S. (2012) Protein fibrillation and the olfactory system: speculations on their linkage. Trends Biotechnol. 30, 609–610. [DOI] [PubMed] [Google Scholar]
- Van Dam D.; De Deyn P. P. (2006) Drug discovery in dementia: the role of rodent models. Nat. Rev. Drug Discovery 5, 956–970. [DOI] [PubMed] [Google Scholar]
- Allinson T. M.; Parkin E. T.; Turner A. J.; Hooper N. M. (2003) ADAMs family members as amyloid precursor protein alpha-secretases. J. Neurosci. Res. 74, 342–352. [DOI] [PubMed] [Google Scholar]
- Wisniewski T.; Sigurdsson E. M. (2010) Murine models of Alzheimer’s disease and their use in developing immunotherapies. Biochim. Biophys. Acta, Mol. Basis Dis. 1802, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller R. O.; Nicoll J. A. (2003) Cerebral amyloid angiopathy: pathogenesis and effects on the ageing and Alzheimer brain. Neurol. Res. 25, 611–616. [DOI] [PubMed] [Google Scholar]
- Maat-Schieman M.; Roos R.; van Duinen S. (2005) Hereditary cerebral hemorrhage with amyloidosis-Dutch type. Neuropathology 25, 288–297. [DOI] [PubMed] [Google Scholar]
- Crawford F.; Suo Z.; Fang C.; Mullan M. (1998) Characteristics of the in vitro vasoactivity of beta-amyloid peptides. Exp. Neurol. 150, 159–168. [DOI] [PubMed] [Google Scholar]
- Niwa K.; Porter V. A.; Kazama K.; Cornfield D.; Carlson G. A.; Iadecola C. (2001) A beta-peptides enhance vasoconstriction in cerebral circulation. Am. J. Physiol.: Heart Circ. Physiol. 281, H2417–2424. [DOI] [PubMed] [Google Scholar]
- de la Torre J. C. (2000) Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer’s pathogenesis. Neurobiol. Aging 21, 331–342. [DOI] [PubMed] [Google Scholar]
- Crawford F.; Soto C.; Suo Z.; Fang C.; Parker T.; Sawar A.; Frangione B.; Mullan M. (1998) Alzheimer’s beta-amyloid vasoactivity: identification of a novel beta-amyloid conformational intermediate. FEBS Lett. 436, 445–448. [DOI] [PubMed] [Google Scholar]
- Thomas T.; Sutton E. T.; Hellermann A.; Price J. M. (1997) Beta-amyloid-induced coronary artery vasoactivity and endothelial damage. J. Cardiovasc. Pharmacol. 30, 517–522. [DOI] [PubMed] [Google Scholar]
- Wesson D. W.; Levy E.; Nixon R. A.; Wilson D. A. (2010) Olfactory dysfunction correlates with amyloid-beta burden in an Alzheimer’s disease mouse model. J. Neurosci. 30, 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M.; Shepardson N.; Yang T.; Chen G.; Walsh D.; Selkoe D. J. (2011) Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart J. C.; Bales K. R.; Gannon K. S.; Greene S. J.; DeMattos R. B.; Mathis C.; DeLong C. A.; Wu S.; Wu X.; Holtzman D. M.; Paul S. M. (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat. Neurosci. 5, 452–457. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Burke S. N.; Barnes C. A. (2010) Senescent synapses and hippocampal circuit dynamics. Trends Neurosci. 33, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel W.; Masliah E.; Hill L. R.; Butters N.; Terry R. (1994) Hippocampal connectivity and Alzheimer’s dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology 44, 2081–2088. [DOI] [PubMed] [Google Scholar]
- Rapp P. R.; Gallagher M. (1996) Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc. Natl. Acad. Sci. U.S.A. 93, 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranahan A. M.; Mattson M. P. (2010) Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neural Plast. 2010, 108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L.; Masliah E.; Yu G. Q.; Mallory M.; Rockenstein E. M.; Tatsuno G.; Hu K.; Kholodenko D.; Johnson-Wood K.; McConlogue L. (2000) High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen J. S.; Wu C. C.; Redwine J. M.; Comery T. A.; Arias R.; Bowlby M.; Martone R.; Morrison J. H.; Pangalos M. N.; Reinhart P. H.; Bloom F. E. (2006) Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5161–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H.; Martin M. V.; Chambers S.; Csernansky J. G. (2007) Spatial relationship between synapse loss and beta-amyloid deposition in Tg2576 mice. J. Comp. Neurol. 500, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R.; Cruts M.; Van Broeckhoven C. (2003) Genetics of early-onset Alzheimer dementia. Sci. World J. 3, 497–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzi R. E.; Bertram L. (2001) New frontiers in Alzheimer’s disease genetics. Neuron 32, 181–184. [DOI] [PubMed] [Google Scholar]
- Mayeux. (2003) Epidemiology of Neurodegeneration. Annu. Rev. Neurosci. 26, 81–104. [DOI] [PubMed] [Google Scholar]
- Van Duijn C. M.; Clayton D. G.; Chandra V.; Fratiglioni L.; Graves A. B.; Heyman A.; Jorm A. F.; Kokmen E.; Kondo K.; Mortimer J. A.; et al. (1994) Interaction between genetic and environmental risk factors for Alzheimer’s disease: a reanalysis of case-control studies. EURODEM Risk Factors Research Group. Genet. Epidemiol. 11, 539–551. [DOI] [PubMed] [Google Scholar]
- Kivipelto M.; Helkala E. L.; Laakso M. P.; Hanninen T.; Hallikainen M.; Alhainen K.; Soininen H.; Tuomilehto J.; Nissinen A. (2001) Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ 322, 1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder E. H.; Saunders A. M.; Strittmatter W. J.; Schmechel D. E.; Gaskell P. C.; Small G. W.; Roses A. D.; Haines J. L.; Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Strittmatter W. J.; Saunders A. M.; Schmechel D.; Pericak-Vance M.; Enghild J.; Salvesen G. S.; Roses A. D. (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grehan S.; Tse E.; Taylor J. M. (2001) Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J. Neurosci. 21, 812–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitas R. E.; B. J.; Lee S. H.; Hui D.; Weisgraber K. H. (1987) Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J. Biol. Chem. 6, 63–75. [PubMed] [Google Scholar]
- Xu P. T.; Schmechel D.; Qiu H. L.; Herbstreith M.; Rothrock-Christian T.; Eyster M.; Roses A. D.; Gilbert J. R. (1999) Sialylated human apolipoprotein E (apoEs) is preferentially associated with neuron-enriched cultures from APOE transgenic mice. Neurobiol. Dis. 6, 63–75. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Bernardo A.; Walker D.; Kanegawa T.; Mahley R. W.; Huang Y. (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley R. W.; Rall S. C. Jr. (2000) Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537. [DOI] [PubMed] [Google Scholar]
- Blacker D.; Haines J. L.; Rodes L.; Terwedow H.; Go R. C.; Harrell L. E.; Perry R. T.; Bassett S. S.; Chase G.; Meyers D.; Albert M. S.; Tanzi R. (1997) ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology 48, 139–147. [DOI] [PubMed] [Google Scholar]
- Tierney M. C.; Szalai J. P.; Snow W. G.; Fisher R. H.; Tsuda T.; Chi H.; McLachlan D. R.; St George-Hyslop P. H. (1996) A prospective study of the clinical utility of ApoE genotype in the prediction of outcome in patients with memory impairment. Neurology 46, 149–154. [DOI] [PubMed] [Google Scholar]
- Corder E. H.; Saunders A. M.; Risch N. J.; Strittmatter W. J.; Schmechel D. E.; Gaskell P. C. Jr.; Rimmler J. B.; Locke P. A.; Conneally P. M.; Schmader K. E.; et al. (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7, 180–184. [DOI] [PubMed] [Google Scholar]
- Mahley R. W.; Weisgraber K. H.; Huang Y. (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5644–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu M. J.; Falduto M. T.; Manelli A. M.; Reardon C. A.; Getz G. S.; Frail D. E. (1994) Isoform-specific binding of apolipoprotein E to beta-amyloid. J. Biol. Chem. 269, 23403–23406. [PubMed] [Google Scholar]
- Deane R.; Sagare A.; Hamm K.; Parisi M.; Lane S.; Finn M. B.; Holtzman D. M.; Zlokovic B. V. (2008) apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell R. D.; Sagare A. P.; Friedman A. E.; Bedi G. S.; Holtzman D. M.; Deane R.; Zlokovic B. V. (2007) Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. 27, 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; Ohtsuki S.; Kamiie J.; Nezu Y.; Terasaki T. (2007) Cerebral clearance of human amyloid-beta peptide (1–40) across the blood-brain barrier is reduced by self-aggregation and formation of low-density lipoprotein receptor-related protein-1 ligand complexes. J. Neurochem. 103, 2482–2490. [DOI] [PubMed] [Google Scholar]
- Holtzman D. M.; Bales K. R.; Tenkova T.; Fagan A. M.; Parsadanian M.; Sartorius L. J.; Mackey B.; Olney J.; McKeel D.; Wozniak D.; Paul S. M. (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polvikoski T.; Sulkava R.; Haltia M.; Kainulainen K.; Vuorio A.; Verkkoniemi A.; Niinisto L.; Halonen P.; Kontula K. (1995) Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. New Engl. J. Med. 333, 1242–1247. [DOI] [PubMed] [Google Scholar]
- Kinnecom C.; Lev M. H.; Wendell L.; Smith E. E.; Rosand J.; Frosch M. P.; Greenberg S. M. (2007) Course of cerebral amyloid angiopathy-related inflammation. Neurology 68, 1411–1416. [DOI] [PubMed] [Google Scholar]
- Lemere C. A.; Blusztajn J. K.; Yamaguchi H.; Wisniewski T.; Saido T. C.; Selkoe D. J. (1996) Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 3, 16–32. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Allsop D. (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388. [DOI] [PubMed] [Google Scholar]
- Terry R. D.; Masliah E.; Salmon D. P.; Butters N.; DeTeresa R.; Hill R.; Hansen L. A.; Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. [DOI] [PubMed] [Google Scholar]
- Dickson D. W.; Crystal H. A.; Bevona C.; Honer W.; Vincent I.; Davies P. (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 16, 285–298discussion 298–304.. [DOI] [PubMed] [Google Scholar]
- McLean C. A.; Cherny R. A.; Fraser F. W.; Fuller S. J.; Smith M. J.; Beyreuther K.; Bush A. I.; Masters C. L. (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866. [DOI] [PubMed] [Google Scholar]
- Lue L. F.; Kuo Y. M.; Roher A. E.; Brachova L.; Shen Y.; Sue L.; Beach T.; Kurth J. H.; Rydel R. E.; Rogers J. (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. (2007) A beta oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. [DOI] [PubMed] [Google Scholar]
- Shankar G. M.; Bloodgood B. L.; Townsend M.; Walsh D. M.; Selkoe D. J.; Sabatini B. L. (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Hong S.; Shepardson N. E.; Walsh D. M.; Shankar G. M.; Selkoe D. (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner G. G.; Wong C. W. (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890. [DOI] [PubMed] [Google Scholar]
- Glenner G. G. (1989) Amyloid beta protein and the basis for Alzheimer’s disease. Prog. Clin. Biol. Res. 317, 857–868. [PubMed] [Google Scholar]
- Ikeda S.; Allsop D.; Glenner G. G. (1989) Morphology and distribution of plaque and related deposits in the brains of Alzheimer’s disease and control cases. An immunohistochemical study using amyloid beta-protein antibody. Lab. Invest. 60, 113–122. [PubMed] [Google Scholar]
- Goldgaber D.; Lerman M. I.; McBride O. W.; Saffiotti U.; Gajdusek D. C. (1987) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 235, 877–880. [DOI] [PubMed] [Google Scholar]
- Tanzi R. E.; Gusella J. F.; Watkins P. C.; Bruns G. A.; St George-Hyslop P.; Van Keuren M. L.; Patterson D.; Pagan S.; Kurnit D. M.; Neve R. L. (1987) Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235, 880–884. [DOI] [PubMed] [Google Scholar]
- St George-Hyslop P. H.; Tanzi R. E.; Polinsky R. J.; Haines J. L.; Nee L.; Watkins P. C.; Myers R. H.; Feldman R. G.; Pollen D.; Drachman D.; et al. (1987) The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science 235, 885–890. [DOI] [PubMed] [Google Scholar]
- Kang J.; Lemaire H. G.; Unterbeck A.; Salbaum J. M.; Masters C. L.; Grzeschik K. H.; Multhaup G.; Beyreuther K.; Muller-Hill B. (1987) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. [DOI] [PubMed] [Google Scholar]
- Levy E.; Carman M. D.; Fernandez-Madrid I. J.; Power M. D.; Lieberburg I.; van Duinen S. G.; Bots G. T.; Luyendijk W.; Frangione B. (1990) Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (1998) The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 8, 447–453. [DOI] [PubMed] [Google Scholar]
- Bonifacino J. S.; Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447. [DOI] [PubMed] [Google Scholar]
- Yamazaki T.; Koo E. H.; Selkoe D. J. (1996) Trafficking of cell-surface amyloid beta-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J. Cell Sci. 109(Pt 5), 999–1008. [DOI] [PubMed] [Google Scholar]
- Koo E. H.; Squazzo S. L.; Selkoe D. J.; Koo C. H. (1996) Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J. Cell Sci. 109(Pt 5), 991–998. [DOI] [PubMed] [Google Scholar]
- Haass C.; Kaether C.; Thinakaran G.; Sisodia S. (2012) Trafficking and proteolytic processing of APP. Cold Spring Harbor Perspect. Med. 2, a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien R. J.; Wong P. C. (2011) Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin G. B.; Goldstein L. S. (2006) Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 75, 607–627. [DOI] [PubMed] [Google Scholar]
- Tienari P., D. S. B., Ikonen E., Simons M., Weidemann A., Czech C., et al. (1996) The beta-amyloid domain is essential for axonal sorting of amyloid precursor protein., EMBO J. 4. [PMC free article] [PubMed] [Google Scholar]
- Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. (2009) Amyloid beta-protein assembly and Alzheimer disease. J. Biol. Chem. 284, 4749–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J. E.; Ruotolo B. T.; Robinson C. V.; Bowers M. T. (2009) Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colletier J. P.; Laganowsky A.; Landau M.; Zhao M.; Soriaga A. B.; Goldschmidt L.; Flot D.; Cascio D.; Sawaya M. R.; Eisenberg D. (2011) Molecular basis for amyloid-beta polymorphism. Proc. Natl. Acad. Sci. U.S.A. 108, 16938–16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (2011) Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 17, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Rahimi F.; Shanmugam A.; Bitan G. (2008) Structure-function relationships of pre-fibrillar protein assemblies in Alzheimer’s disease and related disorders. Curr. Alzheimer Res. 5, 319–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakono M.; Zako T. (2010) Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 277, 1348–1358. [DOI] [PubMed] [Google Scholar]
- Palop J. J.; Mucke L. (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L.; Selkoe D. J. (2012) Neurotoxicity of Amyloid beta-Protein: Synaptic and Network Dysfunction. Cold Spring Harbor Perspect. Med. 2, a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves C. A.; Glabe C. G.; Kayed R. (2011) Amyloid-beta annular protofibrils evade fibrillar fate in Alzheimer disease brain. J. Biol. Chem. 286, 22122–22130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I.; Karran E.; De Strooper B. (2012) The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. [DOI] [PubMed] [Google Scholar]
- Caughey B.; Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. [DOI] [PubMed] [Google Scholar]
- Glabe C. G.; Kayed R. (2006) Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66, S74–78. [DOI] [PubMed] [Google Scholar]
- Williams R. (2001) Chemical selection of elements by cells. Coord. Chem. Rev. 216, 583–595. [Google Scholar]
- Laurent S.; Ejtehadi M. R.; Rezaei M.; Kehoe P. G.; Mahmoudi M. (2012) Interdisciplinary challenges and promising theranostic effects of nanoscience in Alzheimer’s disease. RSC Adv. 2, 5008–5033. [Google Scholar]
- Azhdarzadeh M.; et al. (2013) Serum Multivalent Cationic Pattern: Speculation on the reliable Approach for Detection of Alzheimer’s Disease. Sci. Rep. 3, 2783. 10.1038/srep02782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tottey S., Harvie D. R., and Robinson N. J. (2007) Understanding how cells allocate metals. In Molecular microbiology of heavy metals (Nies D. H., and Silver S., Eds.), Springer-Verlag, Berlin, Heidelberg. [Google Scholar]
- Chamberlain R.; Reyes D.; Curran G. L.; Marjanska M.; Wengenack T. M.; Poduslo J. F.; Garwood M.; Jack C. R. Jr. (2009) Comparison of amyloid plaque contrast generated by T2-weighted, T2*-weighted, and susceptibility-weighted imaging methods in transgenic mouse models of Alzheimer’s disease. Magn. Reson. Med. 61, 1158–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbi V.; Jansen D.; Dederen P. J.; Veltien A.; Hamans B.; Liu Y.; Heerschap A.; Kiliaan A. J. (2012) Microvascular cerebral blood volume changes in aging APP(swe)/PS1 (dE9) AD mouse model: a voxel-wise approach. Brain Struc. Funct. 218, 1805–1898. [DOI] [PubMed] [Google Scholar]
- Fazil M.; Shadab, Baboota S.; Sahni J. K.; Ali J. (2012) Nanotherapeutics for Alzheimer’s disease (AD): Past, present and future. J. Drug Targeting 20, 97–113. [DOI] [PubMed] [Google Scholar]
- Begley D. J. (2004) Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol. Ther. 104, 29–45. [DOI] [PubMed] [Google Scholar]
- Gabathuler R. (2010) Approaches to transport therapeutic drugs across the blood-brain barrier to treat brain diseases. Neurobiol. Dis. 37, 48–57. [DOI] [PubMed] [Google Scholar]
- Roney C.; Kulkarni P.; Arora V.; Antich P.; Bonte F.; Wu A.; et al. (2005) Targeted nanoparticles for drug delivery through the blood-brain barrier for Alzheimer’s disease. J. Controlled Release 108, 193–214. [DOI] [PubMed] [Google Scholar]
- Tanifum E. A.; Dasgupta I.; Srivastava M.; Bhavane R. C.; Sun L.; Berridge J.; Pourgarzham H.; Kamath R.; Espinosa G.; Cook S. C.; Eriksen J. L.; Annapragada A. (2012) Intravenous delivery of targeted liposomes to amyloid-beta pathology in APP/PSEN1 transgenic mice. PloS One 7, e48515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Wadghiri Y. Z.; Hoang D. M.; Tsui W.; Sun Y.; Chung E.; Li Y.; Wang A.; de Leon M.; Wisniewski T. (2011) Detection of amyloid plaques targeted by USPIO-Abeta1–42 in Alzheimer’s disease transgenic mice using magnetic resonance microimaging. NeuroImage 55, 1600–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann N.; Gerard C.; Abramowski D.; Cannet C.; Staufenbiel M. (2011) Noninvasive magnetic resonance imaging detection of cerebral amyloid angiopathy-related microvascular alterations using superparamagnetic iron oxide particles in APP transgenic mouse models of Alzheimer’s disease: application to passive Abeta immunotherapy. J. Neurosci. 31, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiet A.; Santin M.; Bertrand A.; Wiggins C. J.; Petit F.; Houitte D.; Hantraye P.; Benavides J.; Debeir T.; Rooney T.; Dhenain M. (2012) Gadolinium-staining reveals amyloid plaques in the brain of Alzheimer’s transgenic mice. Neurobiol. Aging 33, 1533–1544. [DOI] [PubMed] [Google Scholar]
- Poduslo J. F.; Wengenack T. M.; Curran G. L.; Wisniewski T.; Sigurdsson E. M.; Macura S. I.; Borowski B. J.; Jack C. R. Jr. (2002) Molecular targeting of Alzheimer’s amyloid plaques for contrast-enhanced magnetic resonance imaging. Neurobiol. Dis. 11, 315–329. [DOI] [PubMed] [Google Scholar]
- Sillerud L. O.; Solberg N. O.; Chamberlain R.; Orlando R. A.; Heidrich J. E.; Brown D. C.; Brady C. I.; Vander Jagt T. A.; Garwood M.; Vander Jagt D. L. (2013) SPION-Enhanced Magnetic Resonance Imaging of Alzheimer’s Disease Plaques in AbetaPP/PS-1 Transgenic Mouse Brain. J. Alzheimer’s Dis. 34, 349–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W.; Chen X. (2007) Nanoplatforms for targeted molecular imaging in living subjects. Small 3, 1840–1854. [DOI] [PubMed] [Google Scholar]
- Cao C. Y.; Shen Y. Y.; Wang J. D.; Li L.; Liang G. L. (2013) Controlled intracellular self-assembly of gadolinium nanoparticles as smart molecular MR contrast agents. Sci. Rep. 3, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanal E. (2012) Gadolinium-based magnetic resonance contrast agents for neuroradiology: an overview. Magn. Reson. Imaging Clin. North Am. 20, 625–631. [DOI] [PubMed] [Google Scholar]
- Othman M.; Desmaele D.; Couvreur P.; Vander Elst L.; Laurent S.; Muller R. N.; Bourgaux C.; Morvan E.; Pouget T.; Lepetre-Mouelhi S.; Durand P.; Gref R. (2011) Synthesis and physicochemical characterization of new squalenoyl amphiphilic gadolinium complexes as nanoparticle contrast agents. Org. Biomol. Chem. 9, 4367–4386. [DOI] [PubMed] [Google Scholar]
- Laurent S.; Vander Elst L.; Fu Y.; Muller R. N. (2004) Synthesis and physicochemical characterization of Gd-DTPA-B(sLex)A, a new MRI contrast agent targeted to inflammation. Bioconjugate Chem. 15, 99–103. [DOI] [PubMed] [Google Scholar]
- Amiri H.; Bustamante R.; Millan A.; Silva N. J.; Pinol R.; Gabilondo L.; Palacio F.; Arosio P.; Corti M.; Lascialfari A. (2011) Magnetic and relaxation properties of multifunctional polymer-based nanostructured bioferrofluids as MRI contrast agents. Magn. Reson. Med. 66, 1715–1721. [DOI] [PubMed] [Google Scholar]
- Amiri H.; Mahmoudi M.; Lascialfari A. (2011) Superparamagnetic colloidal nanocrystal clusters coated with polyethylene glycol fumarate: a possible novel theranostic agent. Nanoscale 3, 1022–1030. [DOI] [PubMed] [Google Scholar]
- Hosseini F.; P. A.; Adeli M.; Amiri H.; Lascialfari A.; Orsini F.; Doschak M. R.; Mahmoudi M. (2013) Synthesis of pseudopolyrotaxanes-coated Superparamagnetic Iron Oxide Nanoparticles as new MRI contrast agent. Colloids Surf., B 652–657. [DOI] [PubMed] [Google Scholar]
- Wang Y. X. (2011) Superparamagnetic iron oxide based MRI contrast agents: Current status of clinical application. Quant. Imaging Med. Surg. 1, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M.; Iwata N.; Matsuba Y.; Sato K.; Sasamoto K.; Saido T. C. (2005) 19F and 1H MRI detection of amyloid beta plaques in vivo. Nat. Neurosci. 8, 527–533. [DOI] [PubMed] [Google Scholar]
- Yanagisawa D.; Amatsubo T.; Morikawa S.; Taguchi H.; Urushitani M.; Shirai N.; Hirao K.; Shiino A.; Inubushi T.; Tooyama I. (2011) In vivo detection of amyloid beta deposition using (1)(9)F magnetic resonance imaging with a (1)(9)F-containing curcumin derivative in a mouse model of Alzheimer’s disease. Neuroscience 184, 120–127. [DOI] [PubMed] [Google Scholar]
- Houshang Amiri M. S., Andor Veltein Mark J., van Uden I., Jolanda M., and de Vries Arend Heerschap. (2013) Cell Tracking by 19F MRI:Technical Aspects and Challenges towards Clinical Applications. Submitted for publication. [DOI] [PubMed]
- Krol S.; Macrez R.; Docagne F.; Defer G.; Laurent S.; Rahman M.; Hajipour M. J.; Kehoe P. G.; Mahmoudi M. (2013) Therapeutic benefits from nanoparticles: the potential significance of nanoscience in diseases with compromise to the blood brain barrier. Chem. Rev. 113, 1877–1903. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Quinlan-Pluck F.; Monopoli M. P.; Sheibani S.; Vali H.; Dawson K. A.; Lynch I. (2013) Influence of the physiochemical properties of superparamagnetic iron oxide nanoparticles on amyloid beta protein fibrillation in solution. ACS Chem. Neurosci. 4, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabaleiro-Lago C.; Quinlan-Pluck F.; Lynch I.; Dawson K. A.; Linse S. (2010) Dual effect of amino modified polystyrene nanoparticles on amyloid beta protein fibrillation. ACS Chem. Neurosci. 1, 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri H.; Bordonali L.; Lascialfari A.; Wan S.; Monopoli M. P.; Lynch I.; Laurent S.; Mahmoudi M. (2013) Protein corona affects the relaxivity and MRI contrast efficiency of magnetic nanoparticles. Nanoscale 5, 8656–8665. [DOI] [PubMed] [Google Scholar]
- Dorota Walczyk B. F.; Monopoli M. P.; Lynch I.; Dawson K. A. (2010) What the cell ″sees″ in bionanoscience. J. Am. Chem. Soc. 5761–5768. [DOI] [PubMed] [Google Scholar]
- Monopoli M. P.; Walczyk D.; Campbell A.; Elia G.; Lynch I.; Bombelli F. B.; Dawson K. A. (2011) Physical-chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 133, 2525–2534. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Lynch I.; Ejtehadi M. R.; Monopoli M. P.; Bombelli F. B.; Laurent S. (2011) Protein-nanoparticle interactions: opportunities and challenges. Chem. Rev. 111, 5610–5637. [DOI] [PubMed] [Google Scholar]
- Lesniak A.; Fenaroli F.; Monopoli M. P.; Aberg C.; Dawson K. A.; Salvati A. (2012) Effects of the presence or absence of a protein corona on silica nanoparticle uptake and impact on cells. ACS Nano 6, 5845–5857. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Monopoli M. P.; Rezaei M.; Lynch I.; Bertoli F.; McManus J. J.; Dawson K. A. (2013) The protein corona mediates the impact of nanomaterials and slows amyloid beta fibrillation. ChemBioChem 14, 568–572. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Akhavan O.; Ghavami M.; Rezaee F.; Ghiasi S. M. (2012) Graphene oxide strongly inhibits amyloid beta fibrillation. Nanoscale 4, 7322–7325. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Abdelmonem A. M.; Behzadi S.; Clement J. H.; Dutz S.; Ejtehadi M. R.; Hartmann R.; Kantner K.; Linne U.; Maffre P.; Metzler S.; Moghadam M. K.; Pfeiffer C.; Rezaei M.; Ruiz-Lozano P.; Serpooshan V.; Shokrgozar M. A.; Nienhaus G. U.; Parak W. J. (2013) Temperature: The “Ignored” Factor at the NanoBio Interface. ACS Nano 7, 6555–6562. [DOI] [PubMed] [Google Scholar]
- Ghavami M.; Rezaei M.; Ejtehadi R.; Lotfi M.; Shokrgozar M. A.; Abd Emamy B.; Raush J.; Mahmoudi M. (2013) Physiological temperature has a crucial role in amyloid beta in the absence and presence of hydrophobic and hydrophilic nanoparticles. ACS Chem. Neurosci. 4, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi M.; Laurent S.; Shokrgozar M. A.; Hosseinkhani M. (2011) Toxicity evaluations of superparamagnetic iron oxide nanoparticles: cell “vision” versus physicochemical properties of nanoparticles. ACS Nano 5, 7263–7276. [DOI] [PubMed] [Google Scholar]
- Laurent S.; Burtea C.; Thirifays C.; Rezaee F.; Mahmoudi M. (2013) Significance of cell “observer” and protein source in nanobiosciences. J. Colloid Interface Sci. 392, 431–445. [DOI] [PubMed] [Google Scholar]
- Rauch J.; Kolch W.; Mahmoudi M. (2012) Cell type-specific activation of AKT and ERK signaling pathways by small negatively-charged magnetic nanoparticles. Sci. Rep. 2, 868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi M.; Saeedi-Eslami S. N.; Shokrgozar M. A.; Azadmanesh K.; Hassanlou M.; Kalhor H. R.; Burtea C.; Rothen-Rutishauser B.; Laurent S.; Sheibani S.; Vali H. (2012) Cell “vision”: complementary factor of protein corona in nanotoxicology. Nanoscale 4, 5461–5468. [DOI] [PubMed] [Google Scholar]
- Laurent S.; Burtea C.; Thirifays C.; Hafeli U. O.; Mahmoudi M. (2012) Crucial ignored parameters on nanotoxicology: the importance of toxicity assay modifications and “cell vision”. PloS one 7, e29997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi M.; Hosseinkhani H.; Hosseinkhani M.; Boutry S.; Simchi A.; Journeay W. S.; Subramani K.; Laurent S. (2011) Magnetic resonance imaging tracking of stem cells in vivo using iron oxide nanoparticles as a tool for the advancement of clinical regenerative medicine. Chem. Rev. 111, 253–280. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; M. A.; Stroeve P. (2010) Surface Architecture of Superparamagnetic Iron Oxide Nanoparticles for Application in Drug Delivery and Their Biological Response: A Review. Int. J. Biomed. Nanosci. Nanotechnol. 164–201. [Google Scholar]
- Mahmoudi M.; Shokrgozar M. A.; Sardari S.; Moghadam M. K.; Vali H.; Laurent S.; Stroeve P. (2011) Irreversible changes in protein conformation due to interaction with superparamagnetic iron oxide nanoparticles. Nanoscale 3, 1127–1138. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; S. A.; Imani M. (2010) Recent Advances in Surface Engineering of Superparamagnetic Iron Oxide Nanoparticles for Biomedical Applications. J. Iran. Chem. Soc. 7, S1–S27. [Google Scholar]
- Nighoghossian N.; Wiart M.; Cakmak S.; Berthezene Y.; Derex L.; Cho T. H.; Nemoz C.; Chapuis F.; Tisserand G. L.; Pialat J. B.; Trouillas P.; Froment J. C.; Hermier M. (2007) Inflammatory response after ischemic stroke: a USPIO-enhanced MRI study in patients. Stroke 38, 303–307. [DOI] [PubMed] [Google Scholar]
- Saleh A.; Schroeter M.; Ringelstein A.; Hartung H. P.; Siebler M.; Modder U.; Jander S. (2007) Iron oxide particle-enhanced MRI suggests variability of brain inflammation at early stages after ischemic stroke. Stroke 38, 2733–2737. [DOI] [PubMed] [Google Scholar]
- Dousset V.; Brochet B.; Deloire M. S.; Lagoarde L.; Barroso B.; Caille J. M.; Petry K. G. (2006) MR imaging of relapsing multiple sclerosis patients using ultra-small-particle iron oxide and compared with gadolinium. AJNR Am. J. Neuroradiol. 27, 1000–1005. [PMC free article] [PubMed] [Google Scholar]
- Tourdias T.; Roggerone S.; Filippi M.; Kanagaki M.; Rovaris M.; Miller D. H.; Petry K. G.; Brochet B.; Pruvo J. P.; Radue E. W.; Dousset V. (2012) Assessment of disease activity in multiple sclerosis phenotypes with combined gadolinium- and superparamagnetic iron oxide-enhanced MR imaging. Radiology 264, 225–233. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Sahraian M. A.; Shokrgozar M. A.; Laurent S. (2011) Superparamagnetic iron oxide nanoparticles: promises for diagnosis and treatment of multiple sclerosis. ACS Chem. Neurosci. 2, 118–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi M.; Sant S.; Wang B.; Laurent S.; Sen T. (2011) Superparamagnetic iron oxide nanoparticles (SPIONs): development, surface modification and applications in chemotherapy. Adv. Drug Delivery Rev. 63, 24–46. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M, Stroeve P., Milani A. S., and Arbab A. S. (2010) Superparamagnetic iron oxide nanoparticles for biomedical applications, Nova Science Publishers, Hauppauge, NY. [Google Scholar]
- Brambilla D.; Verpillot R.; Le Droumaguet B.; Nicolas J.; Taverna M.; Kona J.; Lettiero B.; Hashemi S. H.; De Kimpe L.; Canovi M.; Gobbi M.; Nicolas V.; Scheper W.; Moghimi S. M.; Tvaroska I.; Couvreur P.; Andrieux K. (2012) PEGylated nanoparticles bind to and alter amyloid-beta peptide conformation: toward engineering of functional nanomedicines for Alzheimer’s disease. ACS Nano 6, 5897–5908. [DOI] [PubMed] [Google Scholar]
- Skaat H.; Belfort G.; Margel S. (2009) Synthesis and characterization of fluorinated magnetic core-shell nanoparticles for inhibition of insulin amyloid fibril formation. Nanotechnology 20, 225106. [DOI] [PubMed] [Google Scholar]
- Skaat H.; Margel S. (2009) Synthesis of fluorescent-maghemite nanoparticles as multimodal imaging agents for amyloid-beta fibrils detection and removal by a magnetic field. Biochem. Biophys. Res. Commun. 386, 645–649. [DOI] [PubMed] [Google Scholar]
- Skaat H.; Sorci M.; Belfort G.; Margel S. (2009) Effect of maghemite nanoparticles on insulin amyloid fibril formation: selective labeling, kinetics, and fibril removal by a magnetic field. J. Biomed. Mater. Res., Part A 91, 342–351. [DOI] [PubMed] [Google Scholar]
- Andersson B. V.; Skoglund C.; Uvdal K.; Solin N. (2012) Preparation of amyloid-like fibrils containing magnetic iron oxide nanoparticles: effect of protein aggregation on proton relaxivity. Biochem. Biophys. Res. Commun. 419, 682–686. [DOI] [PubMed] [Google Scholar]
- Bellova A.; Bystrenova E.; Koneracka M.; Kopcansky P.; Valle F.; Tomasovicova N.; Timko M.; Bagelova J.; Biscarini F.; Gazova Z. (2010) Effect of Fe3O4 magnetic nanoparticles on lysozyme amyloid aggregation. Nanotechnology 21, 065103. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Hofmann H.; Rothen-Rutishauser B.; Petri-Fink A. (2012) Assessing the in vitro and in vivo toxicity of superparamagnetic iron oxide nanoparticles. Chem. Rev. 112, 2323–2338. [DOI] [PubMed] [Google Scholar]
- Conti B.; Hansen M. (2013) A cool way to live long. Cell 152, 671–672. [DOI] [PubMed] [Google Scholar]
- Xiao R.; Zhang B.; Dong Y.; Gong J.; Xu T.; Liu J.; Xu X. Z. (2013) A Genetic Program Promotes C. elegans Longevity at Cold Temperatures via a Thermosensitive TRP Channel. Cell 152, 806–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinters H. V.; Natte R.; Maat-Schieman M. L.; van Duinen S. G.; Hegeman-Kleinn I.; Welling-Graafland C.; Haan J.; Roos R. A. (1998) Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). Acta Neuropathol. 95, 235–244. [DOI] [PubMed] [Google Scholar]
- El Khoury J.; Toft M.; Hickman S. E.; Means T. K.; Terada K.; Geula C.; Luster A. D. (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438. [DOI] [PubMed] [Google Scholar]
- Hickman S. E.; El Khoury J. (2010) Mechanisms of mononuclear phagocyte recruitment in Alzheimer’s disease. CNS Neurol. Disord.: Drug Targets 9, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z. J.; Liang M.; Monteiro M.; Toth I.; Minchin R. F. (2011) Nanoparticle-induced unfolding of fibrinogen promotes Mac-1 receptor activation and inflammation. Nature Nanotechnol. 6, 39–44. [DOI] [PubMed] [Google Scholar]
- Ahn H. J.; Zamolodchikov D.; Cortes-Canteli M.; Norris E. H.; Glickman J. F.; Strickland S. (2010) Alzheimer’s disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. U.S.A. 107, 21812–21817. [DOI] [PMC free article] [PubMed] [Google Scholar]