

Abstract
Recent experiments have shown that the mutation Tottori (D7N) alters the toxicity, assembly and rate of fibril formation of the wild type (WT) amyloid beta (Aβ) Aβ40 and Aβ42 peptides. We used all-atom molecular dynamics simulations in explicit solvent of the monomer and dimer of both alloforms with their WT and D7N sequences. The monomer simulations starting from a random coil and totaling 3 μs show that the D7N mutation changes the fold and the network of salt bridges in both alloforms. The dimer simulations starting from the amyloid fibrillar states and totaling 4.4 μs also reveal noticeable changes in terms of secondary structure, salt bridge, and topology. Overall, this study provides physical insights into the enhanced rate of fibril formation upon D7N mutation and an atomic picture of the D7N-mediated conformational change on Aβ40 and Aβ42 peptides.
Keywords: Amyloid simulation, Alzheimer’s disease, amyloid-β proteins, molecular dynamics, D7N mutation, monomer, dimer
Alzheimer’s disease (AD) is the most common form of dementia among the senior population.1 The patient with AD will lose memory,2 decay language,3 and experience problems with visual-spatial search,4 among other side-effects. AD is pathologically characterized by brain lesions that are senile plaques made of the beta amyloid (Aβ) peptides5 and neurofibrillary tangles inside neurons made of the tau protein.6 However, there is strong evidence that the Aβ peptides play a central role in AD.7,8 The Aβ peptides are proteolytic byproducts of the amyloid precursor protein and are most commonly composed of 40 (Aβ40) and 42 (Aβ42) amino acids. The Aβ monomers are mostly random coil in physiological buffers, but aggregate to form amyloid fibrils with a cross-β-sheet pattern.9−11
Presently, there are no efficient drugs against AD. One current strategy, based on the fact that the aesthetic of the cerebral defects in AD correlates with high levels of oligomers in the brain,12,13 is to design molecules preventing amyloid fibril formation and targeting the early formed toxic aggregates.5,7,14−18 The another strategy is to design molecules enhancing fibril formation and reducing therefore the lifetimes of Aβ oligomers.19 Unfortunately, because these assemblies are in dynamic equilibrium, experiments have failed thus far to provide atomic structures that would help design more efficient molecules in both scenarios.
Numerous pathogenic familial mutations are known to modulate fibril formation rates and enhance toxicity. There has been many experimental studies on the Flemish (A21G), Dutch (E22Q), Italian (E22K), Arctic (E22G), Iowa (D23N), and Osaka (ΔE22, deletion) variants.20−25 The English (H6R),26 Taiwanese (D7H),27 and Tottori (D7N)28 mutations that cause early onset familial AD have also been reported. The English and Tottori mutations do not affect Aβ production, but they accelerate fibril assembly in the absence of increased protofibril formation.29 A more recent experimental study showed that the Tottori mutation alters Aβ assembly at its earliest stages, monomer folding and oligomerization.30
While the effects of mutations at positions 21–23 on the Aβ monomer and oligomers have been extensively studied by computational means,31−36 the impact of mutations at the N-terminus has not been investigated theoretically. In this work, we study the effect of the Tottori mutation on the monomers and dimers of Aβ40 and Aβ42 using all-atom molecular dynamics (MD) simulations at 300 K in explicit solvent within the microsecond time scale. Our goal is not to determine the equilibrium structures of the WT and D7N monomers and dimers for both alloforms.37 Much longer time scales are required using all-atom MD33 or we must resort to enhanced conformational techniques such as replica exchange molecular dynamics (REMD),35,38−41 simulated tempering,42 or discontinuous molecular dynamics (DMD).43 In addition, repeating these extensive simulations on a total of eight systems is out of reach using standard computer resources.
Rather, here our aim is to understand at an atomic level of detail the effects of the mutation D7N on random coil monomeric structures and fibrillar-like dimeric structures of Aβ40 and Aβ42 in explicit solvent. By using simple reaction coordinates such as the β-strand content, global reaction coordinates and principal component analysis to construct free energy landscapes and studying the dynamics of Asp23-Lys28 salt bridge, we can provide structural and physical insights into the experimental observation that the D7N mutation modifies the early steps of aggregation (monomer and dimer) and enhances the rate of fibril formation.30 We also determine how our results fit with available low resolution experimental data from CD analysis and ion-mobility-based mass spectrometry and previous simulations of Aβ-WT monomers and dimers. Recently, the monomeric conformations of Aβ40-WT and Aβ42-WT were reevaluated by all-atom REMD41 and MD,44 but the resulting equilibrium ensembles are rather divergent from each other and from previous simulations.32,45 The Aβ40/42-WT monomer and dimer were also studied by all-atom Monte Carlo simulations with an implicit solvent model36 and the coarse-grained OPEP model coupled to REMD.46 The Aβ40/42-WT monomer and dimer conformations were also determined by using a multiscale approach, that is, by performing first DMD simulations with a coarse grained model and then all-atom MD simulations of 50 ns in explicit solvent starting from an ensemble of 40 DMD-generated structure for each system.43 However, coarse-grained models, which reduce all-atom side chains by one bead and thus accelerate convergence, are not designed yet to determine with high accuracy the impact of subtle mutations, such as D7N, that is, a change of a NH2 group by an OH group.47−50
Results and Discussion
For simplicity, the WT and D7N monomer and dimer will also be referred to as Aβ40-WT, Aβ40-D7N, 2Aβ40-WT, and 2Aβ42-D7N, and the Aβ peptide is segmented into four regions: the N-terminus (residues 1–16), the central hydrophobic core (CHC, residues 17–21), the fibril-loop region (residues 22–29), and the C-terminus (residues 30–42). Figure S1 in the Supporting Information shows the time evolution of the potential energies of the WT and D7N Aβ42 monomers and dimers. We see that the time for the energy to equilibrate varies from 100 (monomer) to 200 (dimer) ns. As a result, we used the last 650 ns for the monomers. For the dimers, we used the last 650 ns of Aβ40 and the last 600 ns of Aβ42 starting from the fibrillar state and the 300 ns generated six 50 ns MD simulations starting from the most populated states, unless specified.
Impact of D7N on the Secondary Structures, Salt Bridge Populations and Collision Cross Sections of Aβ40 and Aβ42 Monomers
The mean and standard deviation of secondary structures of the four monomers are reported in Table 1. The mutation does not change much the secondary structure of Aβ40. The coil and turn dominate and fluctuate around 40 and 53% in WT versus 35 and 60% in D7N, albeit higher standard deviations are observed in D7N. In both species, the β-strand and α-helix contents remain marginal, 5 and 1%, respectively. In contrast, the secondary structure of Aβ42 changes upon D7N mutation. While in WT, the coil dominates with 42%, followed by the turn (36%) and β-strand (21%), in D7N, the turn dominates with 52%, followed by the coil (28%) and the β-strand (18%). There is therefore an increase of 16% of turn and a decrease of 14% of coil upon D7N mutation. In both Aβ42 species, the α-helix content is marginal.
Table 1. Secondary Structure Compositions of Aβ Peptides in Monomer and Dimer Systemsa.
| Aβ40 |
Aβ42 |
2Aβ40 |
2Aβ42 |
|||||
|---|---|---|---|---|---|---|---|---|
| content (%) | WT | D7N | WT | D7N | WT | D7N | WT | D7N |
| β | 6 ± 2 | 4 ± 2 | 21 ± 1 | 18 ± 2 | 32 ± 3 | 36 ± 5 | 24 ± 2 | 11 ± 5 |
| α | 1 ± 2 | 1 ± 1 | 1 ± 1 | 2 ± 3 | 1 ± 1 | 0 ± 0 | 0 ± 1 | 0 ± 0 |
| turn | 53 ± 7 | 60 ± 16 | 36 ± 3 | 52 ± 3 | 29 ± 4 | 27 ± 4 | 48 ± 3 | 48 ± 6 |
| coil | 40 ± 7 | 35 ± 15 | 42 ± 3 | 28 ± 4 | 38 ± 6 | 37 ± 6 | 28 ± 3 | 41 ± 5 |
The data (mean and standard deviation) were obtained by using the last 650 ns for the monomers, 950 ns for the Aβ40 dimers, and 900 ns for Aβ42 dimers. Standard deviations are calculated using block analysis.
To further quantify the effect of D7N on Aβ40 and Aβ42 monomers, Figure 2 shows the β-strand, α-helix, and turn contents along the sequence. While the averaged propensities over all amino acids are rather similar in both Aβ40 species, the per residue propensities differ. Upon D7N mutation, residues Arg5 and Met35 have higher β contents, while residues Phe4 and Lys16 have lower β contents. Looking at Aβ42, Aβ42-WT is β-rich at residues Arg5, Lys6, Val18, Phe19, Asn27, Lys28, Val40, and Ile41, while Aβ42-D7N is β-rich at residues Val12, His13, Ile31, Ile32, Met35, and Gly38. The D7N mutation allows the formation of one turn of α-helix at the N-terminus (residues 2–4) with a population of 20%, and does not impact the helix at positions 14–16. Interestingly, in Aβ40, the turn increases in the regions 7–12, 23–35 and decreases in the CHC. In contrast, in Aβ42, D7N enhances the population of turns in the C-terminal (residues 33–40), the N-terminal (residues 1–8 (26%)) and for residues 28–30 (100%). Therefore, for both alloforms, one of the possible reasons for enhanced fibril formation is that D7N increases the turn character of the loop region (residues 23–29 to up 100% in Aβ40 and residues 22–30 up to 90% in Aβ42), rendering the conformations more favorable for aggregation.
Figure 2.

Probabilities of secondary structure in WT and D7N Aβ40, Aβ42 monomers. Results were obtained from the last 650 ns of MD simulations.
Both Aβ42-WT and Aβ40-WT species have six negatively charged residues (Asp1, Glu3, Asp7, Glu11, Glu22, and Asp23) and three positively charged residues residues (Arg5, Lys16 and Lys28) at neutral pH. We have studied the lifetimes and the population distributions of all possible 3 × 6 = 18 salt bridges (SB) in the WT sequences and all 3 × 5 = 15 in the D7N mutants. Figure 3 shows the population distributions of the distances between the Lys28-Glu22 and Lys28-Asp23 residues.
Figure 3.

Population of intra-28–22 (upper panels) and 28–23 (lower panels) salt bridge distances in Aβ40, Aβ42 WT and D7N monomers. Results were obtained from the last 650 ns of each MD simulation.
Table 2 reports the five SBs that have high lifetimes, namely Asp1-Arg5, Asp1-Lys16, Asp(Asn)7-Lys16, Lys28-Glu22 and Lys28-Asp23. It is found that the five SB have 96, 0, 0, 2 and 12% to be formed in Aβ40-WT vs 5, 75, 0, 74 and 1% in Aβ40-D7N. Thus upon D7N mutation, there is an interplay between the lifetimes of 1–5 and 1–16, and between the lifetimes of 23–28 and 28–22. Interestingly, in Aβ42-WT, only the salt bridge Asp1-Lys16 has a significant lifetime (42%), while in Aβ42-D7N only Asp1-Arg5 has a significant lifetime (48%), Lys16-Asn7 and Asp1-Lys16 being formed with lifetimes of 4% and 7%. As for Aβ40, there is an interplay between the salt bridges upon D7N mutation.
Table 2. Population (%) of Intramolecular Salt Bridges Formed in Aβ40 and Aβ42 Monomersa.
| Aβ40 |
Aβ42 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5–1 | 16–1 | 16–7 | 28–22 | 28–23 | 5–1 | 16–1 | 16–7 | 28–22 | 28–23 | |
| WT | 96 ± 2 | 0 ± 0 | 0 ± 0 | 2 ± 1 | 12 ± 3 | 1 ± 1 | 42 ± 9 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| D7N | 5 ± 3 | 75 ± 6 | 0 ± 0 | 74 ± 7 | 1 ± 1 | 54 ± 8 | 7 ± 2 | 4 ± 1 | 0 ± 0 | 0 ± 0 |
The salt bridge distances are calculated between the atom CG (in ASP1, ASP7, ASN7, or ASP23) or CD (in GLU3, GLU11, or GLU22) and the atom NH2 (in ARG5) or NZ (in LYS16 or LYS28). The cutoff distance used for considering one SB formed is 0.46 nm. Data were obtained using the last 650 ns of each MD and standard deviations using block analysis.
Finally, using dihdedral principal component analysis (dPCA), the free energy surface (FES) of each system was determined. The FES is characterized by four free energy minima for both Aβ40-WT and Aβ40-D7N. Their collision cross sections (CCS) are 755, 764, 753, and 729 Å2 for Aβ40-WT and 739, 796, 770, and 723 for Aβ40-D7N. In contrast, the FES of Aβ42-WT and Aβ42-D7N are characterized by three and four free energy minima, with CCS values of 777, 728, and 844, and 749, 753, 764, and 765 Å2, respectively. Note that the error bars for the calculated CCS is on the order of 1.5%.
Comparison of Secondary Structures and CCS with Experiments and Previous Simulations on Aβ40 and Aβ42 Monomers
By simply dehydrating and energy-minimizing the dominant structures, Aβ42-WT reduces its averaged CCS value to 765 Å2, Aβ42-D7N to 757 Å2, Aβ40-WT to 760 Å2 and Aβ40-D7N to 740 Å2, bringing the calculated results into the range of experimentally observed cross sections38 and indicating in agreement with ion-mobility-based mass spectrometry,51 that the mutant versions have cross sections similar to Aβ-WT. Note that Baumketner et al. also determined a CCS of 765 2 for Aβ42-WT using replica exchange MD with a generalized Born approximation.38
Using filtration through 10 000 molecular weight cutoff, circular dichroism (CD) of all low molecular weight Aβ40-WT aggregates gives 88% of random coil and turn, 12% of β-strand, and 0% of α-helix at 295 K, pH 7.5, and day 0.52 For all molecular weight Aβ42-WT aggregates, the same experiment leads to 11% of β-strand, 3% of α-helix, and 86% of coil+turn. In contrast, another preparation of Aβ40-WT aggregates gives a β-strand of 25% for the monomer.53 Our 93% of coil+turn for Aβ40-WT is in agreement with the first preparation (88%) and the DMD-MD multiscale approach (94%).43 Our 18% of β-strand for Aβ42-WT is consistent with the first experimental condition (11%), and the recent MD simulation carried out by Head-Gordon et al.44 (15%). The fact that D7N does not change the secondary structure composition of Aβ40 or the total content of α-helix and β-strand of Aβ42 is also consistent with CD analysis.30
Our higher β-strand content at the C-terminus and CHC in Aβ42-WT relative to Aβ40-WT is consistent with previous DMD,43 OPEP-based,32 and all-atom simulations.38,41,44,54 Our lifetime of 12% for the salt bridge Asp23-Lys28 salt in Aβ40-WT is also supported by previous all-atom computational studies of Aβ10–35-WT55,56 and Aβ40-WT43 peptides which revealed that this SB is not stable as its forming atoms are solvated. Our lifetime of 96% for the salt bridge Asp1-Arg5 in Aβ40-WT has also been discussed by two studies, albeit with lower values: REMD simulation using a GB implicit solvent (43%)35 and the DMD-MD multiscale approach (11%).43 In sharp contrast with the results reported by Coskuner et al. (31%)35 and Barz and Urbanc (27%),43 the salt bridge Asp1-Arg5 is formed 1% of the time in our simulation of Aβ42-WT, rather Asp1 prefers to interact with Lys16, while Asp1-Arg5 is formed 54% of the time in Aβ42-D7N. Note that Coskuner et al. use a generalized Born model; the termini are treated as NH3+ and CO2– by Urbanc vs NH2 and COOH in this study, and an extensive REMD simulation in explicit solvent with OPLS leads to a value of 15% only.40 Clearly additional simulations are required to fully understand the dynamics of the salt bridge Asp1-Arg5.
Impact of D7N on the Secondary Structure, Salt Bridge Population, and CCS in Aβ40 and Aβ42 Dimers
The β-strand decreases from 24% in 2Aβ42-WT to 11% 2Aβ42-D7N (Table 2). The N-terminus and CHC have higher β-strand content in 2Aβ42-WT than in 2Aβ42-D7N (Figure 4), reaching 33% and 12% using residues 1–21 in WT and D7N, respectively. The C-terminus is also more β-rich in WT than in the Tottori mutant, with the β-strand spanning residues 31–35 in D7N and residues 34, 36, and 38–41 in WT (Figure 4). The percentage of turn is similar in both Aβ42 species (46%), but there is less turn at positions 33–34 and more turn at positions 2–5 upon D7N mutation. The percentage of coil varies from 26% to 42% upon D7N mutation and impacts essentially the regions 6–9 and 33–41. In both species, the α-helix is almost zero.
Figure 4.

Distributions of secondary structure in WT and D7N Aβ40 and Aβ42 dimers. Results were obtained from 950 ns of the dimer Aβ40 systems and 900 ns of the dimer Aβ42 systems.
For the 2Aβ40-WT and 2Aβ40-D7N secondary structures, all means and standard deviations remain constant (Table 2). The mean percentage of β-strand, α-helix, turn, and coil is 36, 0, 27, and 37%, respectively, for D7N. We see, however, variations along the amino acid sequence for the β-strand and turn characters (Figure 4). The β-strand spans residues 13–20 in WT versus 11–19 in D7N. Upon D7N mutation, there is a decrease of turn at positions 6–9 (71% in WT vs 30% in D7N) and an increase of turn in the loop region (80% in D7N vs 65% in WT).
Looking at the four intramolecular salt bridges 5–1, 16–7, 28–22, and 28–23 in the dimers in Table 3 (Figure 5), we see significant changes upon D7N mutation. Upon D7N mutation, the lifetimes of the salt bridges 16–7, 28–22, and 28–23 decrease from 18 to 0%, 32 to 6%, and 18 to 8%, respectively, and that of the salt bridge 5–1 salt increases from 22 to 36% in 2Aβ40 (Table 3). Compared to the Aβ40-D7N monomer, the lifetimes of the salt bridges 28–22 and 5–1 are significantly reduced (73% in monomer) and increased upon dimerization (0.6% in monomer). We observe that, in 2Aβ42-D7N, the lifetime for the salt bridge 5–1 is higher than in 2Aβ40-D7N (52%), the lifetime for the salt bridge 28–22 increases to 18% (vs 3% in 2Aβ42-WT and 1.4% in 2Aβ40-WT), and the lifetime for the salt bridge 28–23 decreases to 14% vs 45% in 2Aβ42-WT and 0% in both Aβ42-WT and -D7N monomers.
Table 3. Population (%) of Intramolecular Salt Bridges Formed in Aβ40 and Aβ42 Dimersa.
| 2Aβ40 |
2Aβ42 |
|||||||
|---|---|---|---|---|---|---|---|---|
| 5–1 | 16–7 | 28–22 | 28–23 | 5–1 | 16–7 | 28–22 | 28–23 | |
| WT | 21 ± 4 | 22 ± 5 | 30 ± 7 | 17 ± 4 | 24 ± 3 | 2 ± 1 | 3 ± 2 | 53 ± 9 |
| D7N | 32 ± 7 | 0 ± 0 | 6 ± 1 | 8 ± 1 | 52 ± 4 | 0 ± 0 | 17 ± 3 | 14 ± 1 |
The data use 950 ns for the Aβ40 systems and 900 ns for the Aβ42 systems. Standard deviations are calculated using block analysis.
Figure 5.

Population of intra 28–22 (upper panels) and 28–23 (lower panels) salt bridge distances in WT and D7N Aβ40, Aβ42 dimers. Results were obtained from 950 ns for dimer Aβ40 systems and 900 ns for dimer Aβ42 systems.
Figures S2 and S3 in the Supporting Information show the populations of the intermolecular 28–23 and 28–22 salt bridge distances in the Aβ42 dimers. The SB 28–23, formed in the NMR Aβ42 fibril, has small lifetime in both WT (0.78%) and D7N (0.22%), while the SB 28–22 is formed 37% of the time in WT and 14% in D7N. Looking at the Aβ40 dimer results, the intermolecular salt bridges 28–22 and 28–23 are formed 32.1 and 18.1% of the time, respectively, in WT and 6.4 and 7.8% in D7N (Table 3).
Overall, we see that the enhanced fibril formation of the Aβ40 peptide can be explained in the dimer by the enhanced population of turns in the region 20–30, but it does not hold for the Aβ42 peptide, where the most significant change is the decrease of turn in the region 4–10.
Comparison of Secondary Structures with Experiments and Previous Simulations on Aβ40-WT and Aβ42-WT Dimers
CD and NMR studies of low molecular weight Aβ-WT, consisting of monomers and dimers in equilibrium with multimers as large as heptamers, give 15–25% of β-strand, and <10% of α helix.53,57,58 CD analysis using another preparation also reports a β-strand content of 38% for the Aβ40 dimer.53 Here we find that Aβ40-WT and Aβ42-WT dimers have 32% and 24% of β-strand, respectively, and upon dimerization, the β-strand content in Aβ42 does not change, while that in Aβ40 increases from 6% to 32%. Our amount of β-strand for Aβ42 is within the experimental values, while that for Aβ40 is either 1.3 times higher than that obtained using one experimental preparation or is consistent with another preparation. The higher β-strand in Aβ40 dimers relative to Aβ42 dimers, which has already been discussed by high temperature all-atom simulations,34 does not contradict the fact that Aβ42 forms fibrils faster than Aβ40. Indeed while simulations using the OPEP coarse grained force field led to 12% and 30% of β-strand for Aβ40 and Aβ42 dimers,32 several simulations do no show much difference in β-strand content of the dimers from one alloform to another. These include a DMD simulation which gives an averaged β-strand content of 20% for Aβ42-WT dimer and 18% for the Aβ40-WT dimer,59 a DMD-MD multiscale approach which gives a percentage of 6% of β-strand for both alloforms,43 and an extensive all-atom REMD simulations in explicit solvent with OPLS force field and 64 replicas, each of 200 ns, which gives only 8% of β-strand for the Aβ42-WT dimer.40 In addition, many other factors can contribute to enhanced aggregations rates, as discussed below.
Examining the β-strand propensity per residue, Aβ40-WT and Aβ42-WT dimers with DMD or multiscale simulations43,59 differ at the N-terminus where Ala2-Phe4 shows a β-strand propensity of up to 0.3 in Aβ40-WT dimer while it is Glu3-Arg5 in Aβ42-WT dimers. The simulations also show a higher β-strand signal at Val39-Val40 that is absent in Aβ40 dimers. Our simulations on Aβ42-WT show high β-strand signal at positions 1–6, up to 0.4, and higher β-strand signal at positions 38–42, up to 0.4, that are both absent in Aβ40.
Impact of D7N on the Free Energy Surfaces of Aβ40 and Aβ42 Dimer
The FES and the dominant structures of 2Aβ40-WT and 2Aβ40-D7N are shown in Figure 6 using, for each species, the full trajectory of 850 ns and six trajectories of 50 ns because we wish to understand the impact of the mutation on the fibrillar-like states. The FES of the Aβ40 D7N mutant is broader and more complex than that of the WT sequence, with seven free energy minima for D7N versus six for WT. Table 4 gives for each free energy minimum its population, the mean number of fibril contacts, the interpeptide distance between the centers of mass of residues 7, the total number of H-bonds, the secondary structure composition, the n-stranded β-sheet topology, and the CCS. The positions of the β-strands are given in Figure S7 in the Supporting Information.
Figure 6.
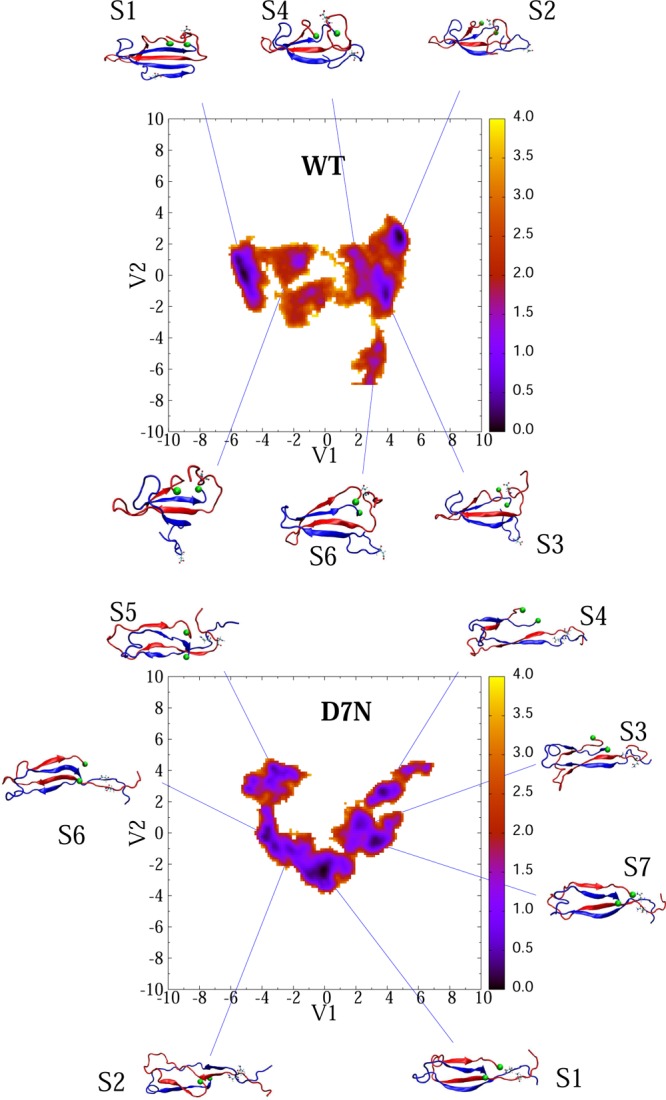
Free energy landscapes of 2Aβ40-D7N (lower) and 2Aβ40-WT (upper) as a function of the first two principal components V1 and V2 obtained from the PCA analysis on the inverse of inter-side-chain distances. The conformations corresponding to each free energy minimum are shown. Seventh residues are shown explicitly. The C-terminus amino acid of all structures is shown by a green ball. Units are in kcal/mol. The scalar product of the eigenvectors e1(WT) × e1(D7N) is 0.67, and the scalar product of the eigenvectors e2(WT) × e2(D7N) is −0.37.
Table 4. Characterization of the Conformational states (S) of the Aβ40 WT and D7N Dimersa.
| system | S | P | Nfb | dR7 | Hb | β | α | turn | coil | iPbs2s | oPbs2s | Pbs3s | Pbs4s | CCS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2Aβ40-WT | 1 | 36 | 16 | 2.85 | 19 | 33 | 1 | 30 | 36 | 29 | 0 | 4 | 67 | 1279 |
| 2 | 19 | 14 | 2.69 | 18 | 32 | 0 | 33 | 35 | 2 | 19 | 4 | 89 | 1347 | |
| 3 | 18 | 16 | 2.92 | 18 | 34 | 0 | 27 | 38 | 0 | 26 | 0 | 96 | 1211 | |
| 4 | 9 | 15 | 2.60 | 18 | 30 | 0 | 26 | 44 | 0 | 0 | 0 | 100 | 1297 | |
| 5 | 7 | 16 | 3.50 | 18 | 29 | 0 | 22 | 48 | 0 | 0 | 0 | 100 | 1387 | |
| 6 | 6 | 19 | 2.64 | 17 | 34 | 0 | 25 | 41 | 0 | 0 | 0 | 100 | 1275 | |
| 2Aβ40-D7N | 1 | 29 | 20 | 0.63 | 19 | 36 | 0 | 28 | 36 | 0 | 67 | 6 | 64 | 1450 |
| 2 | 16 | 19 | 0.63 | 16 | 30 | 0 | 21 | 49 | 0 | 100 | 14 | 22 | 1451 | |
| 3 | 14 | 22 | 0.73 | 18 | 37 | 0 | 29 | 34 | 0 | 126 | 15 | 34 | 1330 | |
| 4 | 12 | 23 | 0.79 | 18 | 37 | 0 | 29 | 34 | 0 | 121 | 25 | 21 | 1462 | |
| 5 | 10 | 23 | 0.71 | 12 | 30 | 0 | 20 | 49 | 0 | 41 | 11 | 60 | 1372 | |
| 6 | 10 | 15 | 0.64 | 12 | 25 | 0 | 24 | 51 | 0 | 195 | 0 | 0 | 1428 | |
| 7 | 8 | 22 | 0.66 | 20 | 42 | 0 | 27 | 31 | 0 | 116 | 0 | 0 | 1436 |
Shown are the population P (in %), the mean values of the total fibril contacts (Nfb), the center of mass distance (in nm) between the 7th residues of two chains (dR7), the secondary structure contents (in %) using residues 1–40, the populations (in %) of intramolecular two-stranded β-sheet (iPbs2s), intermolecular two-stranded β-sheet (oPbs), three-stranded β-sheet (Pbs3s), and four-stranded β-sheet (Pbs). The populations of higher-stranded β-sheets are almost zero. Lastly, the CCS values (in Å2) are given. Here, we use the full 1150 ns for analysis.
Looking at the first four minima, representing 90% and 78% of all conformations for the WT and D7N sequences, the mean number of fibril contacts averages 15 and 21 in WT and D7N sequences vs 51 in the fibril state, and the two N-termini are far apart in the WT dimer (averaged distance between the two residues 7 on the order of 2.8 nm), while they are close in proximity in D7N (four states have interpeptide distance between residues 7 below 0.65 nm). The seven D7N minima are more extended and less compact than the six WT minima, with the N-terminal (residues 1–10) more independent from the rest of the protein and upon D7N mutation, the β-strands are shifted from residues 12–21 to 10–20, and from 30–37 to 30–39. Using ion-mobility mass spectrometry, Bernstein et al.51 reported a CCS of 1200 Å2 for the dimer of Aβ40-D7N and 1142 Å2 for the dimer of Aβ40-WT with an accuracy of ±1%. The seven minima for Aβ40-D7N have CCS values that are 12–20% larger than the experimentally determined cross-section. In contrast, the minima S1, S3, and S6 of Aβ40-WT with CCS’s of 1279, 1211, and 1275 Å2 match the experiment within 1–6% (Table 4). These three minima are characterized by four-stranded β-sheets, with S1 displaying a β-hairpin at residues 9–11 and 14–21.
The free energy surfaces (FES) and the dominant structures of the 2Aβ42-WT and 2Aβ42-D7N dimers are shown in Figure 7 using, for each species, the full trajectory of 800 ns and six trajectories of 50 ns. As for 2Aβ40, the FES of D7N is more complex than that of WT, with 10 free energy minima for D7N versus 6 for WT. Table 5 describes the structural characteristics of each free minimum.
Figure 7.

Free energy landscapes of 2Aβ42-D7N (upper) and 2Aβ42-WT (lower) as a function of the first two principal components V1 and V2 obtained from the PCA analysis on the inverse of inter-side-chain distances. The conformations corresponding to the free energy minima are shown. Seventh residues are shown explicitly. The C-terminus amino acid of all structures is marked by green balls. Units are in kcal/mol. The scalar products of the eigenvectors e1(WT) × e2(D7N) or e2(WT) × Ve(D7N) are close to zero, indicating the absence of similarity in the two FES.
Table 5. Characterization of the Conformational States (S) of the Aβ42 WT and D7N Dimersa.
| system | S | P | Nfb | dR7 | β | α | turn | coil | iPbs2s | oPbs2s | Pbs3s | Pbs4s | CCS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2Aβ42-WT | 1 | 35 | 8 | 2.7 | 23 | 0 | 51 | 26 | 22 | 0 | 14 | 85 | 1342 |
| 2 | 24 | 9 | 2.3 | 24 | 1 | 47 | 28 | 27 | 0 | 12 | 87 | 1292 | |
| 3 | 9 | 12 | 2.3 | 26 | 0 | 43 | 31 | 23 | 0 | 9 | 90 | 1339 | |
| 4 | 8 | 8 | 3.1 | 25 | 0 | 51 | 24 | 20 | 2 | 10 | 88 | 1351 | |
| 5 | 7 | 24 | 2.9 | 27 | 0 | 23 | 50 | 0 | 99 | 0 | 0 | 1566 | |
| 6 | 6 | 18 | 2.9 | 25 | 0 | 37 | 38 | 24 | 2 | 76 | 20 | 1441 | |
| 2Aβ42-D7N | 1 | 24 | 12 | 2.1 | 17 | 0 | 41 | 41 | 2 | 92 | 12 | 0 | 1262 |
| 2 | 21 | 3 | 2.0 | 8 | 0 | 53 | 39 | 93 | 0 | 5 | 0 | 1261 | |
| 3 | 11 | 6 | 1.8 | 12 | 0 | 44 | 44 | 1 | 90 | 18 | 0 | 1314 | |
| 4 | 7 | 4 | 2.0 | 11 | 0 | 52 | 36 | 84 | 8 | 43 | 2 | 1297 | |
| 5 | 6 | 12 | 2.2 | 19 | 0 | 35 | 46 | 0 | 99 | 0 | 0 | 1297 | |
| 6 | 6 | 3 | 1.9 | 7 | 0 | 53 | 40 | 93 | 0 | 0 | 0 | 1303 | |
| 7 | 6 | 4 | 2.0 | 7 | 0 | 48 | 45 | 6 | 50 | 4 | 0 | 1320 | |
| 8 | 4 | 17 | 2.5 | 24 | 0 | 33 | 43 | 0 | 97 | 0 | 1 | 1517 | |
| 9 | 3 | 7 | 2.3 | 11 | 0 | 45 | 44 | 27 | 57 | 1 | 5 | 1311 | |
| 10 | 2 | 9 | 2.2 | 14 | 0 | 37 | 49 | 1 | 98 | 0 | 0 | 1311 |
Shown are the population P (in %), the mean values of the total fibril contacts (Nfb), the center of mass distance (in nm) between the 7th residues of two chains (dR7), the secondary structure contents (in %) using residues 1–42, the populations (in %) of intramolecular two-stranded β-sheet (iPbs2s), intermolecular two-stranded β-sheet (oPbs), three-stranded β-sheet (Pbs3s), and four-stranded β-sheet (Pbs). The populations of higher-stranded β-sheets are almost zero. Lastly, the CCS (in Å2) are given. Here we use the full 1150 ns for the analysis.
Looking at the first four minima, representing 85% and 59% of all conformations for the WT and D7N sequences, the number of fibril contacts averages 9 and 6 in WT and D7N sequences, respectively, vs 40 in the NMR fibril state, and the two N-termini are far apart in both sequences, in contrast to the Aβ40-D7N dimer. The β-strand positions for the WT and D7N dimers of Aβ42 are shown in Figure S6. In sharp contrast to the WT and D7N Aβ40 dimers and the Aβ42-D7N dimer, the percentage of β-strand is not negligible in the N-terminus region (residues 1–6 and 9–12) of the Aβ42-WT dimer. We also see that, upon D7N mutation, the β-strand becomes less populated in the CHC region and the C-terminal β-strand is shifted from residues 36–41 to 30–37.
Using ion-mobility mass spectrometry, Bernstein et al. reported a CCS of 1256 Å2 for Aβ42-WT dimer with an accuracy of ±1%. A value for the dimer of Aβ42-D7N was not obtained. The first 4 WT with CCSs varying between 1292 and 1351 Å2 are in reasonable agreement with experiment, and 8 D7N minima among 10 have similar CCS values. Looking at the S1–S4 WT minima, which feature turn-coil in the region 25–35 and β-strands in the CHC and C-terminus, they all display a β-hairpin in the N-terminus of one chain (residues 1–10). In contrast, the D7N minima, which overall show a 50% reduction of the β-strand content compared to the WT minima, have a propensity of 34% to display a β-hairpin involving the CHC region (residues 19–20) and residues 30–32 (states S2, S4, and S6). This finding is particularly interesting since OPEP simulations of the Aβ16–35 peptide39 and two recent MD simulations validated on a large ensemble of NMR data suggest that the Aβ42-WT monomer adopts transiently antiparallel β-hairpins, spanning residues 16–21 and 29–36 and compatible with known amyloid fibril forming regions,41,44 that can act be used as seeds for nucleation.
Impact of D7N on the Bending Free Energy and Fibril Formation
If we suppose that the mechanism of acceleration of fibril formation by mutation is governed by the constraint of the salt bridge 23–28 as suggested by other computational studies,56 and by the role of a lactam bridge connecting residues 23–28 that suppresses the lag phase prior to Aβ40 nucleation,60 we can estimate the bending free energy as follows:
| 1 |
where R23–28fib is the Cα distance between residues 23 and 28 in the fibril state, P(R23–28 < R23–28) and P(R23–28 > R23–28fib) are the probabilities of having the SB distance smaller and greater than R23–28, respectively. Note that Reddy and Thirumalai used the average distance ⟨R23–28⟩ instead of R23–28fib. In our opinion, the use of the latter is more suitable. The mutation shifts the bending free energy by ΔΔGbend = ΔGbend(D7N) – ΔGbend(WT). Using data obtained for the monomers, we obtain ΔΔGbend = 2.53kBT and 0.79kBT for Aβ40 and Aβ42, respectively. Since ΔΔGbend is related to the fibril formation rates κWT and κD7N as ΔΔGbend = kBT ln(KD7N/KWT), the mutation speeds up fibril formation by 12.5 and 2.2 times for Aβ40 and Aβ42, respectively. Our result for Aβ40 is in reasonable agreement with experiments, which point to a 12-fold rate increase for Aβ40 fibril by D7N mutation using ThT fluorescence.30 In contrast, our estimation of a twofold enhanced rate for Aβ42 fibril is lower than that observed experimentally which finds similar results between Aβ40 and Aβ42. However, we can also estimate the change in bending free energies for the dimers using the salt bridges 23–28 and 22–28. One can show that the mutation does not change the rate for 2Aβ40, but it speeds up the fibril formation for 2Aβ42 process by ≈9.3 and 11.3 times (see Table S1 in the Supporting Information). This result is in good agreement with experiments.
Conclusion
We have studied the influence of the D7N mutation on the Aβ40/42 monomers and dimers using long all-atom MD simulations in explicit solvent. Clearly repeating all-atom REMD simulations in explicit solvent for eight systems is out of reach using current computer resources.40 Though MD sampling in the microsecond time scale might not sufficient to converge to equilibrium and the present results must be confirmed by using other all-atom force fields, we are in a position to explain the contribution of the D7N mutation on the monomers and dimers of Aβ40 and Aβ42 and our main findings can be summarized as follows.
First, our results on the WT and D7N monomers and dimers are consistent with CD analysis at day 0 and in fair agreement with the experimental collision cross sections. While an increased β-strand from Aβ42-WT monomer (21%) to Aβ40-WT monomer (6%) correlates with the observation that Aβ42 forms fibrils faster than Aβ40, a reduction of β-strand content from Aβ42-WT dimer (24%) to Aβ40-WT dimer (32%), a constant β-strand content for Aβ40 dimer upon D7N mutation and a 50% reduction of β-strand content in Aβ42 dimer upon D7N mutation do not contradict the basic understanding of the differences between the two alloforms. Indeed, it is well established that other physicochemical factors such as charge and hydrophobicity contribute to enhanced aggregation rates,61 and what counts in explaining aggregation rate variation is not only the total β-strand content but the nucleation energy barrier that the system has to overcome for breaking any competiting β-strand alignments, side-chain contacts or loop conformations and adopting the fibril-prone state.44,56,62
Second, Ono et al. by following the secondary structures of Aβ as a function of time showed that D7N accelerates the conversion from random coil to β-sheet by 10-fold in Aβ40 and 5-fold in Aβ42 system.30 Our monomer simulations reveal a 5% and 14% reduction of coil in Aβ40 and Aβ42 upon mutation. In contrast, our dimer simulations show that either the coil content remains constant in Aβ40 or increases by 16% in Aβ42 upon D7N mutation. CD being averaging structures, and the solution consisting of monomers and dimers in equilibrium with larger aggregates, it is possible that dimeric structures exist with a richer coil-composition.
Third, Ono et al. proposed that the mutation D7N may affect the Ser8-Gly9 turn and from the effect on this turn, D7N impacts considerably the conformation of the N-terminus and the overall peptide assembly. This is supported by our simulations on all D7N species with an increased turn propensity at residues 5–12 (Aβ40 monomer) and 1–9 (Aβ42 monomer), as well as a decreased turn propensity at residues 6–9 in both dimers. As expected from removing a charged residue, the two N-termini and the two residues 7 which are far apart in the WT dimeric conformational ensemble of both alloforms, are close in proximity in 60% of the Aβ40–D7N dimer ensemble, but not in the Aβ42–D7N dimer ensemble. Removing the charge is also accompanied by a change in the lifetimes of the intramolecular salt bridges Asp1-Arg5, Asp1-Lys16, and Glu22-Lys28 in the monomers and dimers as well as intermolecular salt bridges in dimers. The interplay between the salt bridges Asp1-Arg5 and Asp1-Lys16 is particularly interesting since Lys16 has been shown to play a key role in Aβ assembly,63 it is a target for Alzheimer’s therapy,64 and a novel Aβ-K16N peptide has been discovered.65 Charge removal is also accompanied by an increased number of free energy minima for the dimers, with topologies differing from the WT ensemble, and a decrease in the β-strand propensity of the N-terminus (residues 7–12) of the Aβ42 dimer. Since this N-terminal strand was observed by solid-state NMR spectroscopy for a highly synaptoxic β-amyloid Aβ40 oligomer structure, (66) this suggests that the pathogenic conformation varies from WT to D7N.
Finally, our data support the experimental finding that the Tottori mutation accelerates fibril formation of Aβ40 and Aβ42 peptides. For both systems, we did not find an increase of β-strand content upon mutation. Rather we found that the enhanced formation rate of Aβ40 fibrils comes essentially from the formation of the loop Asp23-Lys28 in the monomer, although the propensity of the region 21–30 to adopt turn is enhanced in both the monomer and dimer upon mutation, and the population of fibrillar-like states, albeit marginal, is certainly higher in D7N dimer than in WT dimer.
In contrast, the enhanced rate of Aβ42 fibrils does not result from the formation of the loop Asp23-Lys28 in monomer, but may result from the formation of the same loop in dimer, the increased turn propensity of the residues 28–30 in monomer, and the higher population of β-hairpin conformations involving the residues 19–20 with residues 31–32 in the dimer. Note it is not surprising to provide different explanations for the enhanced rate of fibril formation of Aβ40-D7N and Aβ42-D7N, substitutions at position 22 and 23 producing different effects on Aβ assembly depending on whether they occur in Aβ40 or Aβ42.52
Overall, our all-atom simulations provide for the first time insights on the Aβ-D7N monomeric and dimeric structures in explicit solvent and an atomic picture of the D7N-mediated conformational change on Aβ40 and Aβ42 peptides.
Materials and Methods
Structures of the WT and D7N Aβ40 and Aβ42 Monomers and Dimers
The NMR structures of full-length WT Aβ40 (PDB code: 1BA4(67)) and Aβ42 (PDB code: 1Z0Q(68)) are rich in α-helix as they have been solved in a water–micelle solution. To obtain WT monomeric structures in aqueous solution, these conformations were subject to 5 ns MD simulations at 500 K, leading to random coil structures with the OPLS force field69 and TIP3P water model.70 The initial structure of the D7N mutation was obtained from the WT final structure by using the mutation tools in PyMOL.71
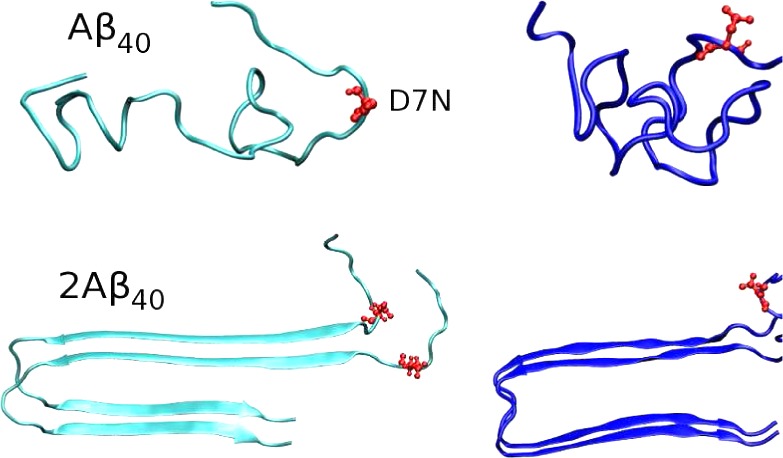
The starting structures of the WT Aβ40 and Aβ42 dimers were taken from the solid-state NMR structures of Aβ9–40 and Aβ17–4211,72 with addition of the residues 1–8 from the heated Aβ40 monomer, and addition of the residues 1–16 from the heated Aβ42 monomer. The structures for the D7N mutation were generated by using pymol.71 The starting configurations for all MD simulations are shown in Figure 1. In this study, each peptide was blocked by the NH2 and COOH groups, rather than the NH3+ and CO2– groups that are expected at the pH 7.4 of the extracellular milieu. These capping ends were selected because the γ-secretase generates a number of isoforms of 39–43 amino acids, and we believe that an uncharged C-terminus (and thus in turn an uncharged C-terminus to preserve neutrality) is preferable to transpose our Aβ42 results to the Aβ43 system. In addition, given the size of the system, we do expect little effects on the overall 2D and 3D structures of each system, albeit our capping ends may impact partially the populations of the salt bridges, and notably involving the negatively charged Asp1 residue.
Figure 1.
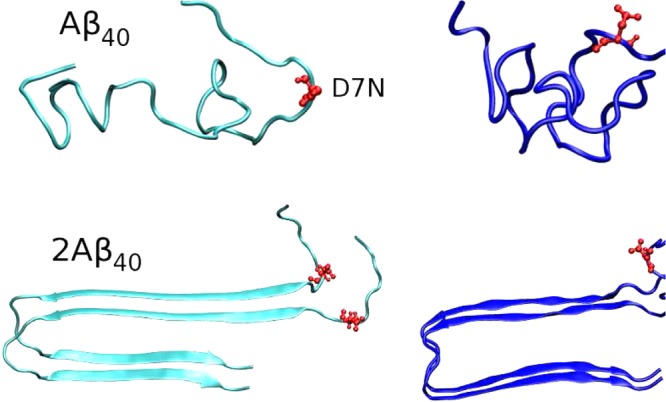
Initial structures for MD simulations of monomers and dimers. Residue 7, where the mutation is made, is shown in red. We use the same conformation for the WT and D7N sequences.
MD Simulations
The GROMACS 4.5.5 package73 was used with the TIP3P water model70 and the OPLS force field.69 The OPLS-AA force field was used because the OPLS-generated conformations for the Aβ40 and Aβ42 monomer match reasonably the NMR data.54 In addition, many studies have shown that OPLS is suitable for exploring the aggregation of several Aβ fragments in explicit water74,75 and gives results qualitatively similar to that found using CHARMM force field for the Aβ10–35 dimer.56 The equations of motion were integrated using a leapfrog algorithm76 with a time step of 2 fs. The LINCS algorithm77 was used to constrain the lengths of all covalent bonds with a relative geometrical tolerance of 10–4. Temperature was controlled by the Bussi–Donadio–Parrinello velocity rescaling thermostat with a relaxation time of 0.1 ps found to sample the canonical ensemble.78 The Berendsen pressure coupling method79 was applied at a pressure of 1 atm. The van der Waals (vdW) forces were calculated with a cutoff of 1.4 nm, and the particle mesh Ewald method80 was employed to treat the long-range Coulombic interactions with a cutoff of 1.0 nm. The nonbonded interaction pair list was updated every 10 fs.
The monomers were centered in octahedron boxes of 66 Å edges containing 4400 water molecules. The dimers were centered in cubic boxes of 90 Å sides containing 29 000 and 26 500 water molecules for the (WT and D7N) Aβ40 dimers and the (WT and D7N) Aβ42 dimers, respectively. Counter ions Na+ were added to neutralize each system. Each monomer and dimer were studied by running 750 and 800 ns MD simulations, respectively, at 300 K with periodic boundary conditions. To verify that proper sampling was achieved for the dimers, a total of six MD simulations, each of 50 ns, was also performed for each species starting the centers of the six most populated clusters obtained from the long MD simulation.
Analysis
The time dependence on the number of hydrogen bonds (HB), side chain–side chain (Sc–Sc) contacts, and salt bridges (SB) was monitored.81 One HB was formed if the distance between donor D and acceptor A is ≤3.5 Å and the D–H–A angle is ≥135°. A Sc–Sc contact was considered as formed if the distance between their centers of mass is ≤6.5 Å. This condition was used to define the number of native contacts in Aβ1–40 and Aβ1–42 fibrils using the residues 9–40 in Aβ40 and 17–42 in Aβ42, leading to 51 and 40 fibrillar-like contacts, respectively. A native contact between residue i of peptide 1 and residue j of peptide 2 was formed in MD snapshots if its distance dij < 0.65 nm. A salt bridge between two charged residues was considered formed if the distance between two specific atoms remains within 4.6 Å (see Tables 2 and 3 for further details).
To estimate the secondary structure of all peptides, we used the STRIDE algorithm.82 The percentage of β-strand in the solid-state NMR Aβ40 and Aβ42 WT fibrils is 57% (spanning residues 10–23 and 30–38) and 45% (spanning residues 18–26 and 31–40) using the full amino acid sequences, respectively.
The free-energy surface (FES) along the N-dimensional reaction coordinated V = (V1,...,VN) is given by ΔG(V) = −kBT[ln P(V) – ln Pmax], where P(V) is the probability distribution obtained from a histogram of MD data. Pmax is the maximum of the distribution, which is subtracted to ensure that ΔG = 0 for the lowest-free-energy minimum. For the monomers, we used dihedral principal component analysis (dPCA).83 For the dimers, we used the first two principal components obtained from principal component analysis using the inverse distances between the interpeptide side chain contacts. For detailed characterization of the free energy minima, we calculated several properties using all snapshots belonging to a state, and not just a single representative structure of a state. For the dimers, we also defined a topological descriptor ranging from a two-stranded β-sheet to a four-stranded β-sheet. Here, at least two interpeptide backbone H-bonds must be formed between consecutive β-strands to pass from a n-stranded β-sheet to a n+1-stranded β-sheet.
The collision cross section (CCS) of all dominant monomeric and dimeric structures was calculated using the MOBCAL software and the trajectory method84 which treats the molecule as a collection of atoms represented by a 12-6-4 potential and is often used for proteins.85
Acknowledgments
We are very thankful to R. Tycko for providing the structure of 6Aβ9–40. Allocation of CPU time at the supercomputer center TASK in Gdansk (Poland) is highly appreciated.
Supporting Information Available
Table S1 gives the relative rates of fibril formation upon D7N mutation. Figure S1 shows the potential energies of the dimeric and monomeric Aβ42 systems. Figures S2–S5 are populations of the intermolecular 22–28 and 23–28 salt-bridge distances in Aβ40 and Aβ42 systems. Figures S6 and S7 show the β-strand content along residues for each free energy minimum shown on the free energy landscapes of the dimer systems. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors analyzed the data. M.L. and P.D. wrote the manuscript.
The work was supported by Narodowe Centrum Nauki in Poland (Grant No. 2011/01/B/NZ1/01622) and Department of Science and Technology at Ho Chi Minh City, Vietnam. P.D. also acknowledges support from the ANR 12-BS07-0017-01. P.D. and M.L. also acknowledge funding from a Cooperation between CNRS/Polish Science Academy/Poland 2013–2014.
The authors declare no competing financial interest.
Supplementary Material
References
- Henderson A. S., and Jorm A. F. (2002) Dementia, Chapter 1, John Wiley & Sons Ltd, New York. [Google Scholar]
- Greene J. D. W.; Baddeley A. D.; Hodges J. R. (1996) Analysis of the episodic memory deficit in early Alzheimers Disease: Evidence from the doors and people test. Neuropsychologia 34, 537–551. [DOI] [PubMed] [Google Scholar]
- Price B. H.; Gurvit H.; Weintraub S.; Geula C.; Leimkuhler E.; Mesulam M. (1993) Neuropsychological patterns and language deficits in 20 consecutive cases of autopsy-confirmed Alzheimer’s disease. Arch. Neurol. 50, 931–937. [DOI] [PubMed] [Google Scholar]
- Esteban-Santillan C.; Praditsuwan R.; Ueda H.; Geldmacher D. S. (1998) Clock drawing test in very mild Alzheimer’s disease. J. Am. Geriatr. Soc. 46, 1266–1269. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. (2002) Medicine - The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Alonso A.; Zaidi T.; Novak M.; Grundke-Iqbal I.; Iqbal K. (2001) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M. (2004) Strategies for disease modification in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 677–685. [DOI] [PubMed] [Google Scholar]
- Aguzzi A.; O’Connor T. (2010) Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat. Rev. Drug. Discovery 9, 237–248. [DOI] [PubMed] [Google Scholar]
- Kirschner D. A.; Abraham C.; Selkoe D. J. (1986) X-ray-diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimers-disease indicates cross-beta conformation. Proc. Natl. Acad. Sci. U.S.A. 83, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J.; Antzutkin O.; Leapman R.; Delaglio F.; Tycko R. (2002) A structural model for Alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Doeli H.; Schubert D.; Riek R. (2005) 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L. F.; Kou Y. M.; Roher A. E. (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth H. W.; Laferla F. M. (2010) Mechanisms of disease Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. [DOI] [PubMed] [Google Scholar]
- Cummings J. L. (2004) Alzheimer’s Disease. N. Engl. J. Med. 351, 56–67. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. [DOI] [PubMed] [Google Scholar]
- Caughey B.; Lansbury P. T. (2003) Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. (2007) Aβ oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. [DOI] [PubMed] [Google Scholar]
- Chebaro Y.; Derreumaux P. (2009) Targeting the early steps of Aβ16–22 protofibril disassembly by N-methylated inhibitors: A numerical study. Proteins 75, 442–452. [DOI] [PubMed] [Google Scholar]
- Bieschke J.; et al. (2011) Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 8, 93–101. [DOI] [PubMed] [Google Scholar]
- Hendriks L.; van Duijin C. M.; Cras P.; Cruts M.; Hul W. V.; van Harskamp F.; Warren A.; McInnis M. G.; Antoarakis S. E.; Martin J.-J.; Hofman A.; Broekhoven C. V. (1992) Presenil dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β-amyloid precursor protein gene. Nat. Genet. 1, 218–221. [DOI] [PubMed] [Google Scholar]
- Levy E.; Carman M. D.; Fernandez-Madrid I. J.; Power M. D.; Lieberburg I.; van Duinen S. G.; Bots G. T. A. M.; Luyendijk W.; Frangione B. (1990) Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1, 1124–1126. [DOI] [PubMed] [Google Scholar]
- Bugiani O.; Padovani A.; Magoni M.; Andora G.; Sgarzi M.; Savoiardo M.; Bizzi A.; Giaccone G.; Rossi G.; Tagliavini F. (1998) An Italian type of HCHWA. Neurobiol. Aging 19, S238. [Google Scholar]
- Kamino K.; Orr H. T.; Payami H.; Wijman E. M.; Pulst S. M.; Anderson L.; O’dahl S.; Nemens E.; White J. A.; et al. (1992) Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am. J. Hum. Genet. 51, 998–1014. [PMC free article] [PubMed] [Google Scholar]
- Grabowski T. J.; Cho H. S.; Vonsattel J. P.; Rebeck G. W.; Greenberg S. M. (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705. [DOI] [PubMed] [Google Scholar]
- Tomiyama T.; Nagata T.; Shimada H.; Teraoka R.; Fukushima A.; Kanemitsu H.; Takuma H.; Kuwano R.; Imagawa M.; Ataka S.; Wada Y.; Yoshioka E.; Nishizaki T.; Watanabe Y.; Mori H. (2008) A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 63, 377–387. [DOI] [PubMed] [Google Scholar]
- Janssen J. C.; Beck J. A.; Campbell T. A.; Dickinson A.; Fox N. C.; Harvey R. J.; Houlden H.; Rossor M. N.; Collinge J. (2003) Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology 60, 235–239. [DOI] [PubMed] [Google Scholar]
- Chen W. T.; Hong C. J.; Lin Y. T.; Chang W. H.; Huang H. T.; Liao J. Y.; Chang Y. J.; Hsieh Y. F.; Cheng C. Y.; Liu H. C.; Chen Y. R.; Cheng I. H. (2012) Amyloid-beta (Aβ) D7H mutation increases oligomeric Aβ42 and alters properties of Aβ-Zinc/Copper assemblies. PLoS One 7, e35807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakutani Y.; Watanabe K.; Adachi Y.; Wada-Isoe K.; Urakami K.; Ninomiya H.; Saido T. C.; Hashimoto T.; Iwatsubo I.; Nakashima K. (2004) Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J. Neurol., Neurosurg. Psychiatry 75, 1039–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y.; Hashimoto T.; Wakutani Y.; Urakami K.; Nakashima K.; Condron M. M.; Tsubuki S.; Saido T. C.; Teplow D. B.; Iwatsubo T. (2007) The Tottori (D7N) and English (H6R) familial Alzheimer Disease mutations accelerate Aβ fibril formation without inscreasing protofibril formation. J. Biol. Chem. 282, 4916–4923. [DOI] [PubMed] [Google Scholar]
- Ono K.; Condron M. M.; Teplow D. B. (2010) Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid beta-protein assembly and toxicity. J. Biol. Chem. 285, 23186–23197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi F.; Straub J. E. (2001) Probing the origins of increased activity of the E22Q “Dutch” mutant Alzheimer’s beta-amyloid peptide. Biophys. J. 81, 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote S.; Derreumaux P.; Mousseau N. (2011) Distinct morphologies for amyloid beta protein monomer: Aβ1–40, Aβ1–42, and Aβ1–40(D23N). J. Chem. Theor. Comput. 7, 2584–2592. [DOI] [PubMed] [Google Scholar]
- Lin Y.-S.; Pande V. S. (2012) Effects of familial mutations on the monomer structure of Aβ42. Biophys. J. 103, L47–L49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet A.; Derreumaux P. (2006) Impact of the mutation A21G (Flemish variant) on Alzheimer’s β-amyloid dimers by molecular dynamics simulations. Biophys. J. 91, 3829–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskuner O.; Wise-Scira O.; Perry G.; Kitahara T. (2013) The structures of the E22Δ mutant-type amyloid-β alloforms and the impact of E22Δ mutation on the structures of the wild-type amyloid-β alloforms. ACS Chemical Neuroscience 4, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitternacht S.; Staneva I.; Hard T.; Irback A. (2010) Comparing the folding free-energy landscapes of Aβ42 variants with different aggregation properties. Proteins 78, 2600–2608. [DOI] [PubMed] [Google Scholar]
- Shea J.-E.; Urbanc B. (2012) Insights into Aβ aggregation: A molecular dynamics perspective. Curr. Top. Med. Chem. 12, 2596–61. [DOI] [PubMed] [Google Scholar]
- Baumketner A.; Bernstein S. L.; Wyttenbach T.; Bitan G.; Teplow D. B.; Bowers M. T.; Shea J. E. (2006) Amyloid β-protein monomer structure: A computational and experimental study. Protein Sci. 15, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebaro Y.; Mousseau N.; Derreumaux P. (2009) Structures and thermodynamics of Alzheimer’s amyloid- Aβ(16–35) monomer and dimer by replica exchange molecular dynamics simulations: Implication for full-length Aβ fibrillation. J. Phys. Chem. B 113, 7668–7675. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Zhang J.; Derreumaux P.; Mu Y. (2013) Molecular mechanism of the inhibition of EGCG on the Alzheimer Aβ1–42 dimer. J. Phys. Chem. B 117, 3993–4002. [DOI] [PubMed] [Google Scholar]
- Rosenman D. J.; Connors C.; Chen W.; Wang C.; Garcia A. E. (2013) Aβ monomers transiently sample oligomer and fibril-like configurations: ensemble characterization using a combined MD/NMR approach. J. Mol. Biol. 425, 3338–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. H.; Okamoto Y.; Derreumaux P. (2013) Communication: Simulated tempering with fast on-the-fly weight determination. J. Chem. Phys. 138, 061102. [DOI] [PubMed] [Google Scholar]
- Barz B.; Urbanc B. (2012) Dimer formation enhances structural differences between amyloid β-protein (1–40) and (1–42): An explicit-solvent molecular dynamics study. PLoS One 7, e34345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball K. A.; Phillips A. H.; Wemmer D. E.; Head-Gordon T. (2013) Differences in β-strand populations of monomeric Aβ40 and Aβ42. Biophys. J. 104, 2714–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball K. A.; Phillips A. H.; Nerenberg P. S.; Fawzi N. L.; Wemmer D. E.; Head-Gordon T. (2011) Homogeneous and heterogeneous tertiary structure ensembles of amyloid-β peptides. Biochemistry 50, 7612–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melquiond A.; Dong X.; Mousseau N.; Derreumaux P. (2008) Role of the region 23–28 in Aβ fibril formation: insights from simulations of the monomers and dimers of Alzheimer’s peptides Aβ40 and Aβ42. Curr. Alzheimer Res. 5, 244–50. [DOI] [PubMed] [Google Scholar]
- Melquiond A.; Boucher G.; Mousseau N.; Derreumaux P. (2005) Following the aggregation of amyloid-forming peptides by computer simulations. J. Chem. Phys. 122, 174904. [DOI] [PubMed] [Google Scholar]
- Derreumaux P. (1997) Folding a 20 amino acid alpha beta peptide with the diffusion process-controlled Monte Carlo method. J. Chem. Phys. 107, 1941–1947. [Google Scholar]
- Maupetit J.; Derreumaux P.; Tuffery P. (2010) A fast method for large-scale de novo peptide and miniprotein structure prediction. J. Comput. Chem. 34, 726–738. [DOI] [PubMed] [Google Scholar]
- Meral D.; Urbanc B. (2013) Discrete molecular dynamics study of oligomer formation by N-terminally truncated amyloid β-protein. J. Mol. Biol. 425, 2260–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessel M. M.; Bernstein S.; Kemper M.; Teplow D. B.; Bowers M. T. (2012) Familial Alzheimer’s Disease mutations differentially alter amyloid β-protein oligomerization. ACS Chem. Neurosci. 3, 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze M. D.; Condron M. M.; Teplow D. B. (2001) Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 312, 1103–1119. [DOI] [PubMed] [Google Scholar]
- Ono K.; Condron M. M.; Teplow D. B. (2009) Structure neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgourakis N. G.; Yan Y. L.; McCallum S. A.; Wang C. Y.; Garcia A. E. (2007) The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 368, 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarus B.; Straub J. E.; Thirumalai D. (2006) Dynamics of Asp23-Lys28 salt-bridge formation in Aβ10–35 monomers. J. Am. Chem. Soc. 128, 16159–16168. [DOI] [PubMed] [Google Scholar]
- Reddy G.; Straub J. E.; Thirumalai D. (2009) Influence of preformed Asp23-Lys28 salt bridge on the conformational fluctuations of monomers and dimers of Aβ peptides with implications for rates of fibril formation. J. Phys. Chem. B 113, 1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T.; Yang D.-S.; Plaskos N. P.; Go S.; Yip C. M.; Fraser P. E.; Chakrabartty A. (2000) Structural studies of soluble oligomers of the Alzheimer beta-amyloid peptide. J. Mol. Biol. 297, 73–87. [DOI] [PubMed] [Google Scholar]
- Bitan G.; Vollers S. S.; Teplow D. B. (2003) Elucidation of primary structure elements controlling early amyloid beta-protein oligomerization. J. Biol. Chem. 278, 34882–9. [DOI] [PubMed] [Google Scholar]
- Urbanc B.; Betnel M.; Cruz L.; Bitan G.; Teplow D. B. (2010) Elucidation of amyloid beta-protein oligomerization mechanisms: discrete molecular dynamics study. J. Am. Chem. Soc. 132, 4266–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarretta K. L.; Gordon D. J.; Petkova A. T.; Tycko R.; Meredith S. C. (2005) Aβ40-Lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry 44, 6003–6014. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Derreumaux P.; Guo Z.; Mousseau N.; Wei G. (2009) Thermodynamics and dynamics of amyloid peptide oligomerization are sequence dependent. Proteins 75, 954–63. [DOI] [PubMed] [Google Scholar]
- Co N. T.; Li M. S. (2012) New method for determining size of critical nucleus of fibril formation of polypeptide chains. J. Chem. Phys. 137, 095101. [DOI] [PubMed] [Google Scholar]
- Sinha S.; Lopes D. H. J.; Bitan G. (2012) A key role for lysine residues in amyloid β-protein folding, assembly, and toxicity. ACS Chem. Neurosci. 3, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M.; Murakami K.; Uno M.; Nakagawa Y.; Katayama S.; ichi Akagi K.; Masuda Y.; Takegoshi K.; Irie K. (2013) Site-Specific inhibitory mechanism for amyloid-β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 288, 23212–23224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaden D.; Harmeier A.; Weise C.; Munter L. M.; Althoff V.; Rost B. R.; Hildebrand P. W.; Schmitz D.; Schaefer M.; Lurz R.; Skodda S.; Yamamoto R.; Arlt S.; Finckh U.; Multhaup G. (2012) Novel APP/Aβ mutation K16N produces highly toxic heteromeric Aβ oligomers. EMBO Mol. Med. 4, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Amo J. M. L.; Schmidt M.; Fink U.; Dasari M.; Fandrich M.; Reif B. (2012) An asymmetric dimer as the basic subunit in Alzheimer’s disease amyloid β fibrils. Angew. Chem., Int. Ed. 51, 6136–6139. [DOI] [PubMed] [Google Scholar]
- Coles M.; Bicknell W.; Watson A. A.; Fairlie D. P.; Craik D. J. (1998) Solution structure of amyloid beta-peptide(1–40) in a water-micelle environment. Is the membrane-spanning domain where we think it is?. Biochemistry 37, 11064–11077. [DOI] [PubMed] [Google Scholar]
- Tomaselli S.; Esposito V.; Vangone P.; van Nuland N. A.; Bonvin A. M.; Guerrini R.; Tancredi T.; Temussi P. A.; Picone D. (2006) The α-to-β conformational transition of Alzheimer’s Aβ-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of β conformation seeding. ChemBioChem 7, 257–267. [DOI] [PubMed] [Google Scholar]
- Kaminski G. A.; Friesner R. A. (2001) Evaluation and reparametrization of the OPSL-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 105, 6474–6487. [Google Scholar]
- Jorgensen J. W.; Chandrasekhar J.; Madura J. D.; Imprey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.3, Schrodinger, LLC.
- Petkova A. T.; Yau W. M.; Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer’s β-amyloid Fibrils. Biochemistry 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Nguyen P. H.; Derreumaux P.; Li M. S. (2011) Effects of all-atom force fields on amyloid oligomerization: Replica exchange molecular dynamics simulations of the Aβ16–22 dimer and trimer. Phys. Chem. Chem. Phys. 13, 9778. [DOI] [PubMed] [Google Scholar]
- Nam H. B.; Kouza M.; Zung H.; Li M. S. (2010) Relationship between population of the fibril-prone conformation in the monomeric state and oligomer formation times of peptides: Insights from all-atom simulations. J. Chem. Phys. 132, 165104. [DOI] [PubMed] [Google Scholar]
- Hockney R. W.; Goel S. P.; Eastwood J. (1974) Quiet high resolution computer models of plasma. J. Comput. Phys. 14, 148–158. [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Vangunsteren W. F.; Dinola A.; Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Viet M. H.; Li M. S. (2012) Amyloid peptide Aβ40 inhibits aggregation of Aβ42: Evidence from molecular dynamics simulations. J. Chem. Phys. 136, 245105. [DOI] [PubMed] [Google Scholar]
- Heinig M.; Frishman D. (2004) STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 32, W500–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y.; Nguyen P. H.; Stock G. (2005) Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins 58, 45–52. [DOI] [PubMed] [Google Scholar]
- Mesleh M. F.; Hunter J. M.; Shvartsburg A. A.; Schatz G. C.; Jarrold M. F. (1996) Structural information from ion mobility measurements: Effects of the long range potential. J. Phys. Chem. 100, 16082. [Google Scholar]
- Chong S.-H.; Ham S. (2012) Atomic-level investigations on the amyloid-β dimerization process and its driving forces in water. Phys. Chem. Chem. Phys. 14, 1573–1575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.