

Abstract
Extensive experimental and computational studies have suggested that multiple Zn2+ binding modes in amyloid β (Aβ) peptides could exist simultaneously. However, consistent results have not been obtained for the effects of Zn2+ binding on Aβ structure, dynamics, and kinetics in particular. Some key questions such as why it is so difficult to distinguish the polymorphic states of metal ions binding to Aβ and what the underlying rationale is, necessitate elucidation. In this work, two 3N1O Zn2+ binding modes were constructed with three histidines (His6, His13, and His14), and Asp1/Glu11 of Aβ40 coordinated to Zn2+. Results from molecular dynamics simulations reveal that the conformational ensembles of different Zn2+-Aβ40 complexes are nonoverlapping. The formation of turn structure and, especially, the salt bridge between Glu22/Asp23 and Lys28 is dependent on specific Zn2+ binding mode. Agreement with available NMR observations of secondary and tertiary structures could be better achieved if the two simulation results are considered together. The free energy landscape constructed by combining both conformations of Aβ40 indicates that transitions between distinct Aβ40 conformations thar are ready for Zn2+ binding could be possible in aqueous solution. Markov state model analyses reveal the complex network of conformational space of Aβ40 modeulated by Zn2+ binding, suggesting various misfolding pathways. The binding free energies evaluated using a combination of quantum mechanics calculations and the MM/3D-RISM method suggest that Glu11 is the preferred oxygen ligand of Zn2+. However, such preference is dependent on the relative populations of different conformations with specific Zn2+ binding modes, and therefore could be shifted when experimental or simulation conditions are altered.
Keywords: Alzheimer’s disease, Markov state model, free energy calculation, metal ions, molecular dynamics simulations
Both amyloid β (Aβ) peptides and biological metal ions have been suggested to be associated with the pathogenesis of Alzheimer’s disease (AD).1−15 Aβ40 and Aβ42 peptides are the primary species in the structure of the senile plaques found in the brain tissues of AD patients.4−6 Metal ions like Zn2+ and Cu2+ have also been found in the vicinity of the plaques, and have been suggested to be able to modulate the aggregation of Aβ peptides in in vitro experiments.10,11,16−18 The N-terminal region of 1–16 amino acids (Aβ16) has been identified as the minimal metal binding site of Aβ.19−21 However, the exact coordination sphere seems to indicate a polymorphic property of metal binding in various experimental conditions.7−9,22 For example, solution NMR studies on the N-acetylated and C-amidated Aβ16 (PDB ID: 1ZE9) suggest that the Zn2+ binding sites include His6, Glu11, His13, and His14.19,20 Besides Glu11, recent NMR studies on Aβ28, Aβ40, and Aβ42 also imply that Asp1 is involved in the oxygen coordination of Zn2+.7,9,23−25 Other possible oxygen ligands include Glu3, Asp7, and H2O. Several experiments demonstrate that Arg5 and Tyr10 are unlikely to be oxygen ligands of Zn2+.7−9,23
Recent NMR studies on the effects of Zn2+ binding on the structural and dynamic properties of Aβ40/Aβ42 in monomeric form suggested that three histidines (His6, His13, and His14) are the nitrogen coordinate sites of Zn2+.23,24,26−28 A second but weaker binding site possibly involving residues Asp23 and Lys28 (Asp23, Val24, Asn26, and Lys28) has been proposed based on high-resolution NMR data.24 Another recent solid state NMR study with Aβ42 fibrils grown in physiological buffers also suggested that the binding of Zn2+ affects both N-terminus and the formation of the salt-bridge between Asp23 and Lys28.28 Complementary to experiments, computational studies provide valuable structural insights about the binding of Zn2+ on Aβ monomers and oligomers.29−33 Molecular dynamics (MD) simulations, especially replica-exchange MD (REMD) simulations, have been applied to explore the conformational distribution of Zn2+-bound Aβ40 monomer and oligomers.31−33 The polymorphic populations of Zn2+-bound monomers and oligomers have emerged from the above MD simulations. In addition, ab initio MD simulations have been performed to investigate the Aβ aggregation mechanism induced by Zn2+.29 Although these theoretical studies give deep insights into understanding of the metal–Aβ interactions, some key questions, such as the effects of different Zn2+ binding modes on the structural, dynamic, and kinetic properties of Aβ, the relative binding affinity of various Zn2+ binding modes, and the possible rationale for such affinity difference, still remain elusive.
In this work, two Zn2+-bound Aβ40 peptides were constructed. In addition to three histidines, either Asp1 or Glu11 was considered as the oxygen coordination ligand. Extensive REMD simulations were carried out to sample their conformational spaces,34,35 while quantum mechanical (QM) calculations were performed to compare the relative binding free energies of the two Zn2+ binding modes. Results from REMD simulations show that two Zn2+-Aβ40 systems display quite distinct structural properties. Dihedral principal component analyses, as well as Markov state models, provide molecular thermodynamic and kinetic pictures for two systems. Their preparation free energies that are required for the binding of Zn2+ overlap to a large extent. QM calculations suggest that Glu11 is the preferred oxygen ligand coordinated to Zn2+. However, such preference could be shifted when the preparation energy is added to the overall binding free energy.
Results and Discussion
Cluster Analysis of the Conformational Spaces of Zn2+-Aβ40 Peptides
First, it is interesting to investigate the effects of the hydrophobic region Aβ(17–40) on the Zn2+ binding conformations of Aβ16. The backbone root-mean-square deviations (RMSDs) of the N-terminal region (Aβ16) with respect to the NMR structure (PDB ID: 1ZE9)20,36 were calculated and shown in Figure 1. The RMSD distribution histogram shows that Aβ16 samples more conformations with two peaks located at different positions when Glu11 is coordinated to Zn2+ (Zn2+-Aβ40_m1), indicating different effects of the C-terminal region of Aβ(17–40) conformations on the dynamics of Aβ16.
Figure 1.

Distribution of the backbone RMSD of the N-terminal region of Aβ16.
The main structural features of each Zn2+-Aβ40 peptide could be captured by a few representative structures selected from its conformational space. Cluster analysis provides a convenient tool to separate the conformational ensemble into clusters with similar geometry. The method proposed by Daura et al.37 was applied here, using a cutoff of 3 Å with regard to backbone atoms of Aβ40. A total of 176 and 184 clusters were obtained for Zn2+-Aβ40_m1 and Zn2+-Aβ40_m2, respectively. However, the top five larger clusters contain about 80.2% and 82.6% of all conformations for the two systems, respectively. The central structures of these most populated clusters are shown in Figure 2. Apparently, the conformational ensemble of Zn2+-Aβ40_m1 was dominated by one cluster (57.8%). In contrast, the ensemble of Zn2+-Aβ40_m2 seems more heterogeneous, and three primary clusters cover 30.0%, 23.1%, and 20.7% of all conformations, respectively. The above results indicate that when the movement of Glu11 was constrained by coordinating to Zn2+, the overall plasticity of Aβ40 decreased significantly. However, in the case where Asp1 acted as the oxygen ligand, Aβ40 is still flexible enough to result in more small clusters (184 versus 176). Furthermore, if the two ensembles of Aβ40 conformations were mixed and the same clustering method was applied, a total of 360 clusters were obtained. Thus, there is no overlap between the two ensembles, excluding the possibility that Asp1 and Glu11 could be coordinated to Zn2+ simultaneously.
Figure 2.
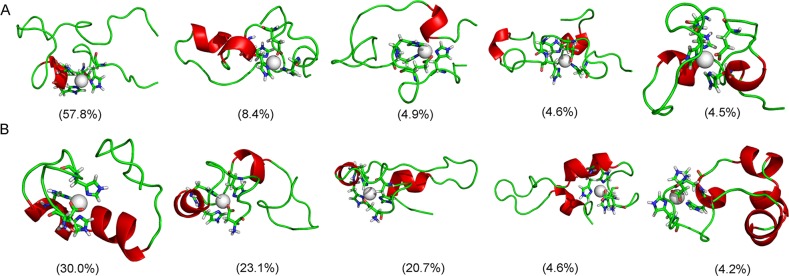
Representative conformations for the most populated cluster in Zn2+-Aβ40_m1 (A) and Zn2+-Aβ40_m2 (B).
Secondary Structure Analysis
The secondary structures of Zn2+-bound Aβ40 were then calculated and compared using DSSP program (Figure 3).38 For Zn2+-Aβ40_m1, residues at N- and C- termini are unstructured coil with more than 40% probability. Abundant helix (α-helix+310-helix) structures were observed in center hydrophobic core (CHC) 17–21 (∼42–71%), residues 26–29(∼44–53%), and residues 32–35 (∼65–73%). Such results were consistent with our previous REMD simulations where both termini of Aβ40 were capped.33 However, these observations may overestimate the helix contents in Zn2+-bound Aβ40 due to the force field used and DSSP program as discussed in the literatures.30,31,39−41 Because we are only interested in the relative values, these limitations would have little effect on our results and discussions. Notable turn structures form in regions 7–9 and 13–15, with probabilities varying from 43 to 58%. Residues 16 and 30 are more likely to display β strand with negligible probabilities (<15%) (Figure S1, Supporting Information).
Figure 3.

Content of secondary structures for two Zn2+-Aβ40 systems.
Compared to Zn2+-Aβ40_m1, the C-terminus of Zn2+-Aβ40_m2 (residues 34–40) adopts random coil structure more frequently, implying it is more dynamic. Residues 11–15 and 19–25 display pronounced helix structures, with probabilities varying from 50 to 83% and 38 to 50%, respectively. A very stable turn structure occurs in a region 3–5, with 82–91% probability.
Consistent results have not been obtained from various experimental studies of the effects of Zn2+ binding on the secondary structures of Aβ40. Based on high-resolution NMR studies of the Zn2+ binding to Aβ40 at 286 K and physiological pH, regions 2–5 and 7–12 could possibly display turn structures.24 Interestingly, our simulation results show that the region 2–5 exists as turn structure when Asp1 is the oxygen ligand of Zn2+ (Zn2+-Aβ40_m2), whereas the region 7–12 prefers turn structure when Glu11 is the oxygen ligand of Zn2+ (Zn2+-Aβ40_m1). Recent NMR studies performed at 278 K and pH 7.2 suggested that a turn like structure could be induced at a region 24–28 and stabilized by the salt bridge between Asp23 and Lys28.23 In contrast, according to the solid-state NMR studies of Zn2+ binding to Aβ42 fibrils carried out at room temperature and pH 7.4, major structural changes happened in the N-terminus and the loop region (residues 23–31).28 In particular, the salt bridge formed between Asp23 and Lys28 was found broken28 (see discussion below). With regard to this work, either helix or random coil structure was identified from our simulations, depending on whether Asp1 or Glu11 is the oxygen ligand of Zn2+.
Tertiary Structure Analysis
The tertiary structure analysis was performed by calculating the contact frequencies among all residues (Figure 4). We consider two amino acids are in contact if the center of mass between these two residues is within 6.5 Å. As expected, Zn2+ binding residues and residues flanking these amino acids are in contact. Especially, in Zn2+-Aβ40_m1, residues at the N-terminus (1–5) contact with C-terminal residues including Gly33, Leu34, Gly37, and Val38 with ∼20% probability. Two Zn2+ binding residue (His13 and His14) also contact with C-terminal residues like Ile32, Gly33, and Val40 with probabilities varying from 55% to 74%. The above two cooperative interactions may result in a higher turn content in the linking region 7–9. In the case of Zn2+-Aβ40_m2, the N-terminus contacts the C-terminus of Aβ40 more frequently than in the context of Zn2+-Aβ40_m1. In addition, several central residues (such as Phe19, Phe20, Asp23, and Val24) contact the C-terminal residues (Gly37, Gly38, and Val39) with ∼20% to 30% probabilities, which can be attributed to the flexible C-terminus of Aβ40. Moreover, a closer examination of the contacts between Glu22 or Asp23 and Lys28 in terms of the distance between the anionic and cationic residues suggests that a salt bridge formed between Asp23 and Lys28 occurs most frequently in the system Zn2+-Aβ40_m2 (Figure 5). On the other hand, the salt bridge formed between Glu22 and Lys28 is more frequent than it does between Asp23 and Lys28 in the system Zn2+-Aβ40_m1. Although Glu22 and Asp23 are neighboring residues, they seem to be competitors for the formation of salt bridge with Lys28. This phenomenon has also been observed for Zn2+-free Aβ40 previously.31 If we assume two Zn2+ binding modes coexist, it is not surprising to speculate that the formation of the salt bridge between Asp23 and Lys28 of Aβ40 could be either discernible or not, depending on which one is the most populated species under specific conditions. Because of the difficulties in the characterization of the polymorphic states of Zn2+ binding with Aβ peptides, fully consistent results obtained from experiments and simulations are not expected. Even so, our results demonstrate that using the results obtained from two independent simulations can provide better rationale for the observations from various experiments.
Figure 4.
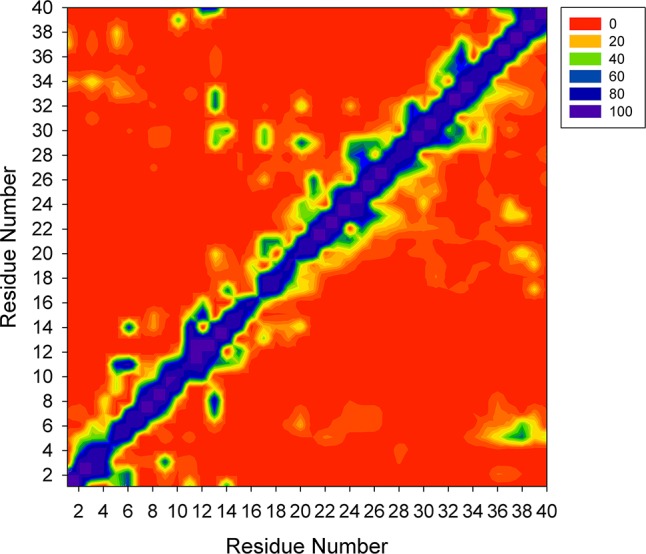
Contact maps for two Zn2+-Aβ40 systems. The contact map of Zn2+-Aβ40_m1 is shown in the upper half panel, and the contact map of Zn2+-Aβ40_m2 is shown in the lower half panel.
Figure 5.

Distance distributions between Cδ of Glu22 and Nξ of Lys28 and between Cγ of Asp23 and Nξ of Lys28.
Free Energy Surfaces
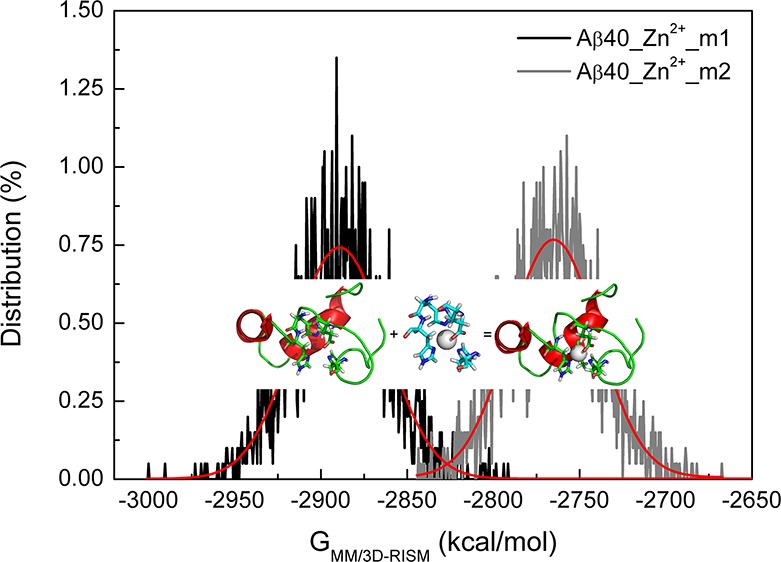
The two-dimensional free energy surfaces of Zn2+-bound Aβ40 peptides were constructed using dihedral principle component analysis (dPCA), which has been shown to be able to separate internal and overall motions of a protein and thus can characterize more complex features of the free energy landscape (FEL) of disordered proteins.42−44 Figure 6 shows that, for each system, the global energy minimum is separated from the other part by high energy barriers, which is more significant for system Zn2+-Aβ40_m2 (Figure 6B). Multiple local minima with comparable energies were identified to be separated by shallow energy barriers for each FEL. Conformational conversions within basins seem much easier for system Zn2+-Aβ40_m2 because each basin covers larger area in the surface (Figure 6B). These findings are consistent with the clustering results that the conformational space of Zn2+-Aβ40_m1 is dominated by one cluster whereas several comparable clusters exist in the ensemble of Zn2+-Aβ40_m2 conformations. Assuming two zinc binding modes are equally populated and combining the two trajectories of Aβ40, we constructed a third FEL as shown in Figure 6C. Similar to individual FEL, this surface is divided into two parts. According to their relative conformational energies (see discussion below), the global minimum corresponds to the lowest basin of Zn2+-Aβ40_m1. Of note, the other part of this surface is not so rugged as compared to individual FEL, suggesting that various Aβ40 conformations that are ready for Zn2+ binding can exist in aqueous solution simultaneously and conformational transitions may occur easily. However, such free energy scenario is not static but largely dependent on the relative populations of different Zn2+-bound Aβ40. From a statistical point of view (see discussion below), the average energy barrier needs to be crossed is ∼123 kcal/mol, indicating that conformational conversions between the two states might be difficult.
Figure 6.
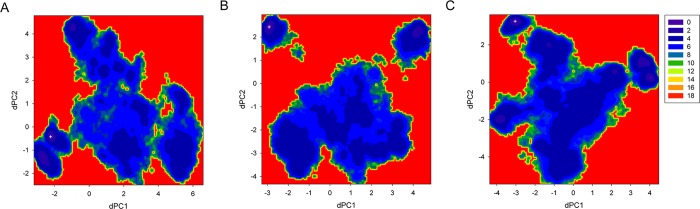
Conformational free energy surfaces for Zn2+-Aβ40 systems. Each was constructed by projecting its conformations onto the first two dihedral principle components (dPC1 and dPC2). The free energy values (in kcal/mol) were obtained by ΔG = −kBT[ln(Pi ) – ln(Pmax)], where Pi and Pmax are the probability distributions calculated from a histogram of individual REMD simulation trajectory. ln(Pi) – ln(Pmax) was used to ensure ΔG = 0 for the lowest free energy points (denoted as a cross symbol).
Markov State Model Analyses
The independent conformational states were further characterized by construction of Markov state models (MSM),45−47 which assume that the structural similarity implies a kinetic similarity, and conformations can interconvert rapidly within the same state. Network graphs containing 20 microstates are shown in Figures 7 and 8 for Zn2+-Aβ40_m1 and Zn2+-Aβ40_m2, respectively. Both networks display busy transitions from one states to another, with the most populated microstates serve as kinetic hubs whereas the less populated states are more likely to be metastable intermediates. In particular, conformations with β-strands are identified in the network of Zn2+-Aβ40_m2 (Figure 8), but their role in the whole transition network may not so significant because various alternative pathways that do not involve these states are available. The mean first passage times (MFPTs), which quantitatively estimate the average time needed to walk from all the other states to a give state, are calculated and compared for two Zn2+ binding modes (Figure S3, Supporting Information). In general, the MFPTs increase as the populations of microstates decrease, suggesting a simple population-dependent transition network for Zn2+-bound Aβ40. As intrinsically disordered proteins, there is no such native state that may act as kinetic hubs48 or traps;49 the role of each state in the kinetic network relies on its relative population and thus can be easily changed. In addition, both state populations (Figure S2, Supporting Information) and MFPTs (Figure S3, Supporting Information) of two systems are very close to each other, indicating that it is rather difficult to distinguish the kinetic behavior of different Zn2+-bound Aβ40.
Figure 7.

Markov state model constructed for Zn2+-Aβ40_m1. In the network graph, the node size is proportional to its corresponding equilibrium populations. Transition probabilities between microstates are colored according to their hot degrees; that is, darker color indicates larger transition probability. Representative conformations are shown for the top 10 microstates only. The Zn2+ binding sites including three His and one Glu residues are displayed as sticks, while Zn2+ is displayed as white van der Waals spheres.
Figure 8.

Markov state model constructed for Zn2+-Aβ40_m2. The representations are the same as shown in Figure 7.
Binding Free Energies
The calculated binding free energies of the two system are summarized in Tables 1 and 2. The Zn2+ binding free energies of the two model structures (Figure 9) are very close to each other in gas phase and in aqueous solution, suggesting there is no significant preference of Zn2+ for anionic Asp1 or Glu11. Previous studies of Cu2+ binding to Aβ40/Aβ42 peptides also found that Asp and Glu are comparably favorable ligands of Cu2+.50 To fully assess the capability of Zn2+ binding, the free energy change for the conformations of Aβ40 that are ready for Zn2+ binding is required. This preparation energy was first evaluated using the MM/GBSA method which has been shown to give satisfactory performance and efficiency.51 Because only the relative values are important, the conformational energy of free Aβ40 was taken as a reference and was not calculated. Adding the binding free energy from QM calculations and the preparation energy of Aβ40 gives the final Zn2+ binding free energy (Table 1). Compared to Glu11, Asp1 is a more favored Zn2+ ligand, with a relatively lower binding energy of 4.89 kcal/mol. Not surprisingly, the difference of enthalpy (8.73 kcal/mol) in the preparation energy is the predominant contribution, which can be further attributed to the solvation free energy (Table 2). As a result, different Zn2+ binding modes greatly influence the solvation property of Aβ40. Although entropy calculated using normal-mode analysis has been applied to Aβ and α-synuclein monomers,52−55 our results indicate that Aβ40 has almost the same entropy upon different Zn2+ binding modes may suggest that it is not an appropriate approach to characterize intrinsically disordered proteins (IDPs) like Aβ.56
Table 1. Binding Free Energies (in kcal/mol) for Zn2+-Bound Aβ40 Systems in Terms of the MM/3D-RISM Theorya.
| systems | ΔGbinding (QM,gas) | ΔGsolv (QM) | ΔGprep | ΔGbinding |
|---|---|---|---|---|
| Zn2+-Aβ40_m1 | –530.64 | 442.28 | –2888.69 – Gref | –2977.05 – Gref |
| Zn2+-Aβ40_m2 | –529.20 | 444.68 | –2765.23 – Gref | –2849.75 – Gref |
| ΔΔGbinding = ΔGbinding(m1) – ΔGbinding(m2) = −127.30 | ||||
ΔGbinding (QM,gas) is the binding energy calculated in gas phase based on QM model structures. ΔGsolv (QM) is the solvation free energy of QM model structures. ΔGbinding (QM) is the binding free energy of QM model structures. ΔGprep is the preparation energy calculated using the MM/3D-RISM method. Gref is the free energy of the reference system (Zn2+-free Aβ40). ΔGbinding is the total free energy of Zn2+ binding to Aβ40.
Table 2. Energy Components Used for the Calculation of Preparation Energy of Aβ40 Based on the MM/GBSA and MM/3D-RISM Methods.
| system | Zn2+-Aβ40_m1 | Zn2+-Aβ40_m2 |
|---|---|---|
| MM/GBSA method (energy unit: kcal/mol) | ||
| ⟨Sol.⟩ | –751.29 (4.48) | –703.67 (14.73) |
| ⟨H⟩ | –853.65 (1.81) | –863.25 (2.80) |
| ⟨TS⟩ | 443.72 (0.06) | 442.85 (0.07) |
| ⟨Gprep⟩ | –1297.37 (1.87) | –1306.10 (2.86) |
| MM/3D-RISM method | ||
| ⟨Sol.⟩ | 23.33 (1.69) | 85.27 (14.49) |
| ⟨H⟩ | –2151.46 (1.91) | –2142.43 (6.21) |
| ⟨TS⟩ | 737.23 (26.25) | 622.80 (10.34) |
| ⟨Gprep⟩ | –2888.69 (2.23) | –2765.23 (6.33) |
Figure 9.
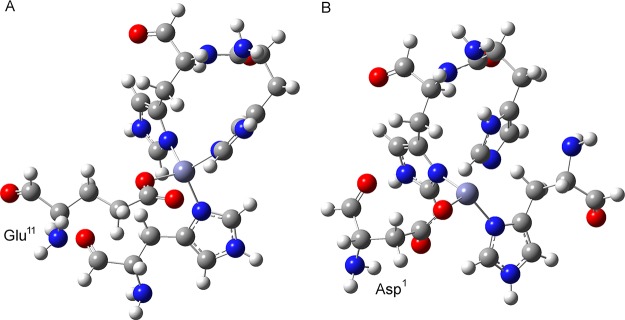
Small model structures used for quantum mechanical calculation of Zn2+ binding free energy when Glu11 (A) or Asp1 (B) is coordinated to Zn2+.
In contrast, results based on the MM/3D-RISM method indicate that Glu11 is more preferred oxygen ligand (Table 1), and the major contributions arise from the favorable enthalpy (including solvation free energy) and the entropy (Table 2). Moreover, the finding that the distribution of enthalpy can be well approximated by the Gaussian function (Figure S6, Supporting Information) is in agreement with previous studies of conformational entropy of Aβ42 and its mutants.57,58 The entropy of Aβ42 has been estimated to be 859.6 kcal/mol based on the 3D-RISM calculations,57 thus, the entropy loss of Aβ40 (assumed the same as Aβ42, and taken as a reference) is much significant in the case where Asp1 is coordinated to Zn2+ (Table 2).
Do the above results mean that we can clearly distinguish Zn2+ binding modes between Asp1 and Glu11 under the same conditions? Note that all the values obtained from the MM/3D-RISM calculations are block averaged. A Gaussian distribution is expected for the preparation energy of each system (Figure 10 and Supporting Information Figure S6). Of importance, in the whole energy range, the two sets of energy values overlap to some extents, suggesting the existence of Asp1 coordinated to Zn2+ cannot be excluded entirely. Overall, the different conformations of Aβ40 with different Zn2+ binding modes support the proposed concept of polymorphism in metal ions binding to Aβ peptides. Assuming the same population of conformations of Aβ40 (∼1%), the decrease of binding free energy (−127.30 kcal/mol) when Glu11 binds to Zn2+ relative to Asp1 implies an incredible increase of the binding affinity (∼1099). However, in the range of overlapped energy (−2850 to −2800 kcal/mol for example), the relative population of Zn2+-Aβ40 conformations with the same preparation energy determines which binding mode is preferred.
Figure 10.
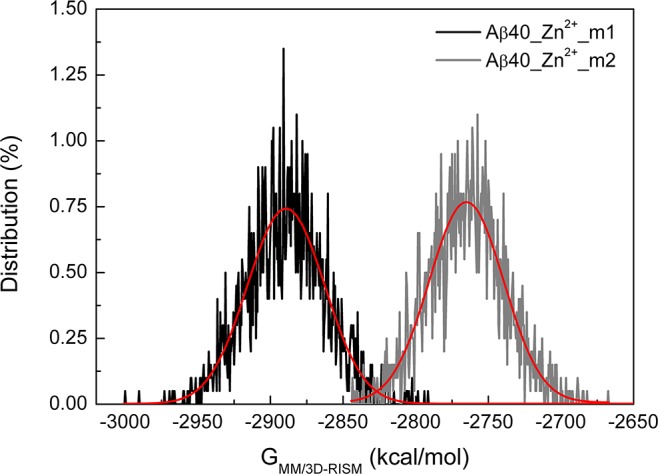
Distribution of the preparation free energies for Zn2+-Aβ40 systems. Red curves denote the fit by the Gaussian function.
Previous studies of interactions of small molecules with Aβ have suggested that multiple binding modes with different affinities are possible.59−61 Here, we show the same polymorphic property of binding of Zn2+ with disordered Aβ. The binding of metal ions to Aβ is very sensitive to environmental conditions such as temperature, ionic strength, peptide concentrations, and pH in particular,22,62,63 resulting in heterogeneous distribution of binding conformations.7,8,64 In addition, the binding mode is also dependent on the length of Aβ peptide.22 Note that the fragment of 1–16 of Aβ (Aβ16) has been considered as the metal binding domain.20,21,68 If Aβ16 was protected with N-terminus acetylated and C-terminus amidated, Glu11 has been identified as the only oxygen coordination of Zn2+, and the Zn2+-Aβ16 complex is soluble and stable. In contrast, the unprotected Aβ16 aggregated immediately upon addition of Zn2+ in physiological conditions.21 Our solvation free energy results based on the MM/3D-RISM also show that the Zn2+-Aβ40 complex is less soluble and consequently more prone to aggregate to form toxic oligomers when Asp1 is involved in the binding of Zn2+ (Table 2). Besides the polymorphic Zn2+-Aβ40 conformations, the interplay of different Zn2+ binding modes may further alter the conformations of Aβ40 and regulate their population shifts. As a result, the Zn2+ binding mode of Aβ40 is not just a problem with respect to Asp1 or Glu11, but greatly influenced by environmental variability.
Conclusions
In order to understand how the interactions of metal ions with Aβ influence the structural, thermodynamic, and kinetic properties of Aβ, a combination of REMD simulations, dihedral PCA, Markov state model, and QM calculations was applied to investigate the polymorphic states of Zn2+-bound Aβ40 peptides. In particular, the binding preference of two different Zn2+ binding modes, that is, Asp1 or Glu11, binds to Zn2+, was examined. Our results show that there is no overlap between conformational ensemble of Aβ40 with different residues (Asp1 or Glu11) coordinated to Zn2+. Using results obtained from two independent simulations, the formation of turn structure in the regions 3–5, 7–9, and 13–15 are consistent with available observations from various NMR studies. Notable, the formation of the salt bridge between Asp23 and Lys28 is more frequent when Asp1 is coordinated to Zn2+, whereas this salt bridge seems to be disrupted when Glu11 is coordinated to Zn2+ because the interactions of Glu22 with Lys28 are more competitive. These findings provide new insights into understanding the polymorphic properties of Aβ structures that coordinate with metal ions.
The binding preference of Zn2+ to Asp1 and Glu11 seems to be negligible according to our QM calculations of simplified model structures. However, Glu11 is found as a more favorable Zn2+ binding site in the framework of Aβ40 in terms of the more accurate MM/3D-RISM calculations, which is primarily attributed to the favorable enthalpy and entropy when Zn2+ binds to Glu11. Although the energy barrier is difficult to cross in the conformational transitions between different Zn2+-bound Aβ40, the overlap of the preparation free energies in a wide range indicates the co-occurrence of polymorphic Zn2+ binding modes under the same experimental conditions. The interplay between different binding modes further complicates their relative populations. The kinetic networks show the presence of numerous hub-like microstates that help to create diverse misfolding pathways. Strategies like chemical protection and mutation might not be useful to unambiguously probe the metal binding sites in Aβ peptides as have already been discussed.65
Methods
REMD Simulations
Because experimental structures of full-length Zn2+-bound Aβ40 peptides are not available, the Zn2+-Aβ40 complex was usually constructed by linking experimental structures of Zn2+-Aβ16 (PDB ID: 1ZE9)20,36 with C-terminal region of Aβ(17–40) extracted from Aβ fibril structure (PDB ID: 2BEG).31−33 Different from our previous studies,33,60 both N and C termini of Zn2+-Aβ16 were unprotected. In this work, two 3N1O Zn2+ binding modes were considered, that is, [His6, Glu11, His13, His14] (referred to as Zn2+-Aβ40_m1), and [Asp1, His6, His13, His14] (referred to as Zn2+-Aβ40_m2). The same force field parameters as used previously were applied in this work.33,60 Briefly, the peptide was represented by Amber ff99SB force fields,66 with the Zn2+ binding sites modeled by Amber force field parameters derived from QM calculations.33,60,67 The modified generalized Born model (Onufriev–Bashford–Case model) was used to implicitly account for the solvent effects.68 All REMD simulations were performed using Amber 12 software.69
MD simulations (including REMD simulations in particular) have become one of the most popular methods to explore the conformational spaces of intrinsically disordered proteins like Aβ peptides in various length and their interactoins with small molecules.31−33,36,41−44,70−80 In REMD simulations, N independent replicas are simulated at N different temperatures in parallel. At a specific time interval, the neighboring replicas attempt to exchange with an acceptance rate determined by the Metropolis criteria. Consistent with our previous studies,33,60 the temperature ranges from 280 to 400 K, and 16 replicas for each Zn2+-Aβ40 system were used. The average acceptance rates are about 53% and 52% for Zn2+-Aβ40_m1 and Zn2+-Aβ40_m2, respectively, in line with previous REMD simulations of Aβ monomers and oligomers with acceptance rates vary from 40% to 67%.45−48 Convergence of the REMD simulations was checked by the calculation of the cumulative helix content.33,60,81 It was found that each 250 ns REMD simulation reaches convergence after 100 ns (Figure S4, Supporting Information). Therefore, only the last 100 ns trajectories simulated at 280 K (experimental relevant temperature) were used for data analysis.
Direct comparisons with NMR observables such as chemical shifts, scalar coupling constants, and relaxation data are not feasible because rather limited NMR data of Zn2+-bound Aβ is available. In our recent studies of Zn2+-bound Aβ peptides,82 we attempted to reproduce the NMR 15N relaxation parameters including the longitudinal relaxation rates R1, transverse relaxation rates R2, and nuclear Overhauser effect rates (NOEs) using REMD simulation trajectories obtained at high temperature (408 K). In order to correct for the effect of elevated temperature, the time axis needs to be rescaled by optimizing a scaling factor that maximizes the agreement between simulated and experimentally determined relaxation data. Here, the same protocol was applied to compare the internal dynamics of different Zn2+ binding modes. It was found that using the same scaling factor (α = 260), Zn2+-Aβ40_m1 and Zn2+-Aβ40_m2 display similar trend for these relaxation data (Figure S5, Supporting Information). However, the significant discrepancy of NOE values between experiment and Zn2+-Aβ40_m2 may imply that different scaling factors are required to optimize the match between experimental and simulated relaxation data. As a result, the involvement of Asp1 or Glu11 in the binding of Zn2+ may affect the overall dynamics of Aβ in solution differently.
Markov State Model
The same protocol as applied in our recent work83 was used to build a coarse grained MSM for each Zn2+-Aβ40 system. Briefly, Ward’s algorithm was first applied to cluster conformations of each ensemble into small states named microstates based on the backbone dihedral angles. Then the transition probability matrix was calculated to determine the implied time scales that corresponds to the time scale required for transition between different microstates. The Perron cluster cluster analysis (PCCA) algorithm was used to group kinetically related microstates into different macrostates. MSMBuilder284 and MSMExplorer85 were used to construct and visualize MSMs.
Free Energy of Zn2+-Bound Aβ40
The preparation energy of Aβ40 is defined as the free energy difference between Zn2+-free conformations and conformations that are required for Zn2+ binding. Since the energy of Zn2+-free Aβ40 conformations was just taken as a reference, and we were concerned about the relative free energy, REMD simulations were not performed on Aβ40 peptide. The free energy of the conformations that are ready for Zn2+ binding is obtained using the conformational ensemble of Zn2+-bound Aβ40 after removing Zn2+. The MM/GBSA method was applied to estimated the preparation free energy as86
where EMM is the internal energy (a sum of electrostatic Eelec and van der Waals Evdw interaction energies). The solvation free energy Gsolv includes the electrostatic contribution and the nonpolar part that is proportional to the solvent accessible surface area of Zn2+-Aβ40. The entropy S was calculated using normal-mode analysis on trajectories obtained from REMD simulations. Each energy term was calculated using the block average method. That is, the 100 ns trajectory was divided into five 20 ns parts and the standard deviations were calculated.
Because of the drawback of the implicit model used to calculate the solvation free energy in the MM/GBSA method, the three-dimensional reference interaction site model (3D-RISM) was also applied to calculate the solvation free energy.87 This alternative approach is based on the integral-equation theory of liquids and is able to account for both polar and nonpolar features of the solvation structure.88,89 In this work, the KH closure approximation proposed by Kovalenko and Hirata was used to calculate the solvation free energy.90 In particular, a new method of computing the conformational entropy of IDPs like amyloid peptides has recently been derived from the 3D-RISM theory.57,58
where H is the enthalpy obtained from the 3D-RISM calculation and ⟨...⟩ denotes the average. In this work, the value of H was obtained by combining the protein internal energy (EMM) and the solvation free energy from the 3D-RISM calculations.
QM Calculations
Due to the size limitation for QM calculations, only a small model structure (Figure 9) containing the Zn2+ coordination sphere was used here. Note that His13 and His14 are linked together to accurately model adjacent hisditine residues in Aβ40. All QM calculations were carried out by DFT calculations implemented in Gaussian 09.91 The model structure was first optimized with B3LPY/6-31+G* basis set. Then frequency analysis was performed in order to verify that the optimized structure was at a stationary point of the potential energy surface. Finally, thermodynamic values such as zero-point energies, enthalpies, entropies, and Gibbs free energies were obtained. Solvation free energies were calculated by applying the self-consistent reaction field polarizable continuum model, as implemented in Gaussian 09 (with keywords scrf=cpcm).91 Previous studies on the binding of Cu2+ to Aβ40 suggested that the whole entropy of each Zn2+ ligand can be reasonably approximated using only the vibrational entropy of each ligand.50 Therefore, entropy contributions from translational and rotational components were neglected in our calculations. For metal ions, the semiempirical Sackur-Tetrode equation has been used to calculate the translational entropy of Cu2+, and the reported experimental solvation energy of Cu2+ was introduced as well.50 Since we are interested in the relative energy, both the translational entropy and solvation energy of Zn2+ were obtained from QM calculations.
The overall Zn2+ binding free energy was calculated as
and ΔGbinding (gas, QM) was calculated as
where Gmodel, GHis-His, GAsp/Glu, and GHis are the free energies of the small model structure, His-His, Asp/Glu, and His fragments, respectively. ΔGsolv (QM) is the QM solvation energy, and ΔGprep is the preparation energy relative to Zn2+-free Aβ40.
Acknowledgments
Computational time was provided by the High Performance Computing Department of Network and Information Center, Dalian University of Technology.
Supporting Information Available
β-Strand contents (Figure S1), populations of microstates (Figure S2), MFPTs (Figure S3), convergence test of REMD simulations (Figure S4), comparison of NMR observables (Figure S5), energy components for binding free energy calculations (Table S1), and distribution of preparation energy calculated by MM/3D-RISM theory (Figure S6). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
L.X. and X.W. performed research and analyzed data; L.X., X.W. and X.W. wrote the paper.
This work was supported by the Major State Basic Research Development Program (2009CB918501). L.X. is thankful for financial support from the Fundamental Research Funds for the Central Universities (Grant No. DUT12LK38).
The authors declare no competing financial interest.
Supplementary Material
References
- Bush A. I. (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci. 26, 207–214. [DOI] [PubMed] [Google Scholar]
- Goedert M.; Spillantini M. G. (2006) A century of Alzheimer’s disease. Science 314, 777–781. [DOI] [PubMed] [Google Scholar]
- Roberson E. D.; Mucke L. (2006) 100 years and counting: Prospects for defeating Alzheimer’s disease. Science 314, 781–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob-Roetne R.; Jacobsen H. (2009) Alzheimer’s disease: From pathology to therapeutic approaches. Angew. Chem., Int. Ed. 48, 3030–3059. [DOI] [PubMed] [Google Scholar]
- Buxbaum J. N.; Linke R. P. (2012) A molecular history of the amyloidoses. J. Mol. Biol. 421, 142–159. [DOI] [PubMed] [Google Scholar]
- Rauk A. (2009) The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 38, 2698–2715. [DOI] [PubMed] [Google Scholar]
- Faller P. (2009) Copper and zinc binding to amyloid-β: Coordination, dynamics, aggregation, reactivity and metal-ion transfer. ChemBioChem 10, 2837–2845. [DOI] [PubMed] [Google Scholar]
- Faller P.; Hureau C. (2009) Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-β peptide. Dalton Trans. 1080–1094. [DOI] [PubMed] [Google Scholar]
- Tõugu V.; Tiiman A.; Palumaa P. (2011) Interactions of Zn(ii) and Cu(ii) ions with Alzheimer’s amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics 3, 250–261. [DOI] [PubMed] [Google Scholar]
- Pithadia A. S.; Lim M. H. (2012) Metal-associated amyloid-β species in Alzheimer’s disease. Curr. Opin. Chem. Biol. 16, 67–73. [DOI] [PubMed] [Google Scholar]
- Kozlowski H.; Luczkowski M.; Remelli M.; Valensin D. (2012) Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord. Chem. Rev. 256, 2129–2141. [Google Scholar]
- Greenwald J.; Riek R. (2012) On the possible amyloid origin of protein folds. J. Mol. Biol. 421, 417–426. [DOI] [PubMed] [Google Scholar]
- Drago D.; Bolognin S.; Zatta P. (2008) Role of metal ions in the abeta oligomerization in Alzheimer’s disease and in other neurological disorders. Curr. Alzheimer Res. 5, 500–507. [DOI] [PubMed] [Google Scholar]
- Bush A. I. (2013) The metal theory of Alzheimer’s disease. J. Alzheimer’s Dis. 33(Suppl 1), S277–281. [DOI] [PubMed] [Google Scholar]
- Kepp K. P. (2012) Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 112, 5193–5239. [DOI] [PubMed] [Google Scholar]
- Duce J. A.; Bush A. I. (2010) Biological metals and Alzheimer’s disease: Implications for therapeutics and diagnostics. Prog. Neurobiol. 92, 1–18. [DOI] [PubMed] [Google Scholar]
- Alies B.; Pradines V.; Llorens-Alliot I.; Sayen S.; Guillon E.; Hureau C.; Faller P. (2010) Zinc(II) modulates specifically amyloid formation and structure in model peptides. J. Biol. Inorg. Chem. 16, 333–340. [DOI] [PubMed] [Google Scholar]
- Maret W. (2012) New perspectives of zinc coordination environments in proteins. J. Inorg. Biochem. 111, 110–116. [DOI] [PubMed] [Google Scholar]
- Zirah S.; Kozin S. A.; Mazur A. K.; Blond A.; Cheminant M.; Segalas-Milazzo I.; Debey P.; Rebuffat S. (2006) Structural changes of region 1–16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 281, 2151–2161. [DOI] [PubMed] [Google Scholar]
- Tsvetkov P. O.; Kulikova A. A.; Golovin A. V.; Tkachev Y. V.; Archakov A. I.; Kozin S. A.; Makarov A. A. (2010) Minimal Zn2+ binding site of amyloid-β. Biophys. J. 99, L84–L86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin S. A.; Zirah S.; Rebuffat S.; Hui Bon Hoa G.; Debey P. (2001) Zinc binding to Alzheimer’s Aβ(1–16) peptide results in stable soluble complex. Biochem. Biophys. Res. Commun. 285, 959–964. [DOI] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R. (2012) Metal binding sites in amyloid oligomers: Complexes and mechanisms. Coord. Chem. Rev. 256, 2245–2252. [Google Scholar]
- Rezaei-Ghaleh N.; Giller K.; Becker S.; Zweckstetter M. (2011) Effect of zinc binding on β-amyloid structure and dynamics: Implications for Aβ aggregation. Biophys. J. 101, 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson J.; Pierattelli R.; Banci L.; Gräslund A. (2007) High-resolution NMR studies of the zinc-binding site of the Alzheimer’s amyloid β-peptide. FEBS J. 274, 46–59. [DOI] [PubMed] [Google Scholar]
- Huang J.; Yao Y.; Lin J.; Ye Y.-H.; Sun W.-Y.; Tang W.-X. (2004) The solution structure of rat Aβ-(1–28) and its interaction with zinc ion: insights into the scarcity of amyloid deposition in aged rat brain. J. Biol. Inorg. Chem. 9, 627–635. [DOI] [PubMed] [Google Scholar]
- Kozin S. A.; Mezentsev Y. V.; Kulikova A. A.; Indeykina M. I.; Golovin A. V.; Ivanov A. S.; Tsvetkov P. O.; Makarov A. A. (2011) Zinc-induced dimerization of the amyloid-β metal-binding domain 1–16 is mediated by residues 11–14. Mol. BioSyst. 7, 1053–1055. [DOI] [PubMed] [Google Scholar]
- Valiente-Gabioud A. A.; Torres-Monserrat V.; Molina-Rubino L.; Binolfi A.; Griesinger C.; Fernández C. O. (2012) Structural basis behind the interaction of Zn2+ with the protein α-synuclein and the Aβ peptide: A comparative analysis. J. Biol. Inorg. Chem. 117, 334–341. [DOI] [PubMed] [Google Scholar]
- Mithu Venus S.; Sarkar B.; Bhowmik D.; Chandrakesan M.; Maiti S.; Madhu P. K. (2011) Zn2+ binding disrupts the Asp23-Lys28 salt bridge without altering the hairpin-shaped cross-β Structure of Aβ42 amyloid aggregates. Biophys. J. 101, 2825–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan S.; La Penna G. (2009) Modeling of the Zn2+ binding in the 1–16 region of the amyloid β peptide involved in Alzheimer’s disease. Phys. Chem. Chem. Phys. 11, 6468–6481. [DOI] [PubMed] [Google Scholar]
- Nguyen P. H.; Li M. S.; Derreumaux P. (2011) Effects of all-atom force fields on amyloid oligomerization: replica exchange molecular dynamics simulations of the Aβ16–22 dimer and trimer. Phys. Chem. Chem. Phys. 13, 9778–9788. [DOI] [PubMed] [Google Scholar]
- Li W.; Zhang J.; Su Y.; Wang J.; Qin M.; Wang W. (2007) Effects of zinc binding on the conformational distribution of the amyloid-β peptide based on molecular dynamics simulations. J. Phys. Chem. B 111, 13814–13821. [DOI] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R. (2010) Zinc ions promote Alzheimer A aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. U.S.A. 107, 9490–9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Perry G.; Coskuner O. (2012) Structures and free energy landscapes of aqueous zinc(II)-bound amyloid-β(1–40) and zinc(II)-bound amyloid-β(1–42) with dynamics. J. Biol. Inorg. Chem. 17, 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- Mitsutake A.; Sugita Y.; Okamoto Y. (2001) Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 60, 96–123. [DOI] [PubMed] [Google Scholar]
- Zirah S.; Kozin S. A.; Mazur A. K.; Blond A.; Cheminant M.; Segalas-Milazzo I.; Debey P.; Rebuffat S. (2006) Structural changes of region 1–16 of the Alzheimer disease amyloid beta-peptide upon zinc binding and in vitro aging. J. Biol. Chem. 281, 2151–2161. [DOI] [PubMed] [Google Scholar]
- Daura X.; Gademann K.; Jaun B.; Seebach D.; van Gunsteren W. F.; Mark A. E. (1999) Peptide folding: When simulation meets experiment. Angew. Chem., Int. Ed. 38, 236–240. [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Yang M.; Teplow D. B. (2008) Amyloid β-protein monomer folding: Free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 384, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cino E. A.; Choy W.-Y.; Karttunen M. (2012) Comparison of secondary structure formation using 10 different force fields in microsecond molecular dynamics simulations. J. Chem. Theory Comput. 8, 2725–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Buchete N.-V.; Hummer G. (2008) Are current molecular dynamics force fields too helical?. Biophys. J. 95, L07–L09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altis A.; Nguyen P. H.; Hegger R.; Stock G. (2007) Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 126, 244111–244120. [DOI] [PubMed] [Google Scholar]
- Mu Y.; Nguyen P. H.; Stock G. (2005) Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins 58, 45–52. [DOI] [PubMed] [Google Scholar]
- Maisuradze G. G.; Leitner D. M. (2007) Free energy landscape of a biomolecule in dihedral principal component space: sampling convergence and correspondence between structures and minima. Proteins 67, 569–578. [DOI] [PubMed] [Google Scholar]
- Bowman G. R.; Huang X.; Pande V. S. (2009) Using generalized ensemble simulations and Markov state models to identify conformational states. Methods 49, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande V. S.; Beauchamp K.; Bowman G. R. (2010) Everything you wanted to know about Markov State Models but were afraid to ask. Methods 52, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G. R.; Voelz V. A.; Pande V. S. (2011) Taming the complexity of protein folding. Curr. Opin. Struct. Biol. 21, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman G. R.; Pande V. S. (2010) Protein folded states are kinetic hubs. Proc. Natl. Acad. Sci. U.S.A. 107, 10890–10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson A.; Brooks C. L. (2013) Native states of fast-folding proteins are kinetic traps. J. Am. Chem. Soc. 135, 4729–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantri Y.; Fioroni M.; Baik M.-H. (2008) Computational study of the binding of CuII to Alzheimer’s amyloid-β peptide: Do Aβ42 and Aβ40 bind copper in identical fashion?. J. Biol. Inorg. Chem. 13, 1197–1204. [DOI] [PubMed] [Google Scholar]
- Hou T.; Wang J.; Li Y.; Wang W. (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskuner O.; Wise-Scira O.; Perry G.; Kitahara T. (2013) The Structures of the E22Δ Mutant-Type Amyloid-β Alloforms and the Impact of E22Δ Mutation on the Structures of the Wild-Type Amyloid-β Alloforms. ACS Chem. Neurosci. 4, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Aloglu A. K.; Dunn A.; Sakallioglu I. T.; Coskuner O. (2013) Structures and Free Energy Landscapes of the Wild-Type and A30P Mutant-Type α-Synuclein Proteins with Dynamics. ACS Chem. Neurosci. 4, 486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskuner O.; Wise-Scira O. (2013) Structures and Free Energy Landscapes of the A53T Mutant-Type α-Synuclein Protein and Impact of A53T Mutation on the Structures of the Wild-Type α-Synuclein Protein with Dynamics. ACS Chem. Neurosci 4, 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Dunn A.; Aloglu A. K.; Sakallioglu I. T.; Coskuner O. (2013) Structures of the E46K Mutant-Type α-Synuclein Protein and Impact of E46K Mutation on the Structures of the Wild-Type α-Synuclein Protein. ACS Chem. Neurosci 4, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirovitch H. (2007) Recent developments in methodologies for calculating the entropy and free energy of biological systems by computer simulation. Curr. Opin. Struct. Biol. 17, 181–186. [DOI] [PubMed] [Google Scholar]
- Chong S.-H.; Ham S. (2011) Configurational entropy of protein: A combined approach based on molecular simulation and integral-equation theory of liquids. Chem. Phys. Lett. 504, 225–229. [Google Scholar]
- Chong S.-H.; Ham S. (2013) Conformational Entropy of Intrinsically Disordered Protein. J. Phys. Chem. B 117, 5503–5509. [DOI] [PubMed] [Google Scholar]
- Chebaro Y.; Jiang P.; Zang T.; Mu Y.; Nguyen P. H.; Mousseau N.; Derreumaux P. (2012) Structures of Aβ17–42 trimers in isolation and with five small-molecule drugs using a hierarchical computational procedure. J. Phys. Chem. B 116, 8412–8422. [DOI] [PubMed] [Google Scholar]
- Xu L.; Gao K.; Bao C.; Wang X. (2012) Combining conformational sampling and selection to identify the binding mode of zinc-bound amyloid β peptides with bifunctional molecules. J.Comput.-Aided Mol. Des. 26, 963–976. [DOI] [PubMed] [Google Scholar]
- Convertino M.; Vitalis A.; Caflisch A. (2011) Disordered binding of small molecules to Aβ(12–28). J. Biol. Chem. 286, 41578–41588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawisza I.; Rózga M.; Bal W. (2012) Affinity of copper and zinc ions to proteins and peptides related to neurodegenerative conditions (Aβ, APP, α-synuclein, PrP). Coord. Chem. Rev. 256, 2297–2307. [Google Scholar]
- Faller P.; Hureau C.; Dorlet P.; Hellwig P.; Coppel Y.; Collin F.; Alies B. (2012) Methods and techniques to study the bioinorganic chemistry of metal–peptide complexes linked to neurodegenerative diseases. Coord. Chem. Rev. 256, 2381–2396. [Google Scholar]
- Pagel K.; Seri T.; von Berlepsch H.; Griebel J.; Kirmse R.; Bottcher C.; Koksch B. (2008) How metal ions affect amyloid formation: Cu2+- and Zn2+-sensitive peptides. ChemBioChem 9, 531–536. [DOI] [PubMed] [Google Scholar]
- Drew S. C.; Barnham K. J. (2011) The heterogeneous nature of Cu2+ interactions with Alzheimer’s amyloid-β Peptide. Acc. Chem. Res. 44, 1146–1155. [DOI] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F.; Wang R. (2010) Systematic derivation of AMBER force field parameters applicable to zinc-containing systems. J. Chem. Theory Comput. 6, 1852–1870. [DOI] [PubMed] [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55, 383–394. [DOI] [PubMed] [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Goetz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., and Kollman P. A. (2012) AMBER 12, University of California, San Francisco.
- Zhang T.; Zhang J.; Derreumaux P.; Mu Y. (2013) Molecular Mechanism of the Inhibition of EGCG on the Alzheimer Aβ1–42Dimer. J. Phys. Chem. B 117, 3993–4002. [DOI] [PubMed] [Google Scholar]
- Côté S.; Laghaei R.; Derreumaux P.; Mousseau N. (2012) Distinct Dimerization for Various Alloforms of the Amyloid-Beta Protein: Aβ1–40, Aβ1–42, and Aβ1–40(D23N). J. Phys. Chem. B 116, 4043–4055. [DOI] [PubMed] [Google Scholar]
- Fu Z.; Luo Y.; Derreumaux P.; Wei G. (2009) Induced β-Barrel Formation of the Alzheimer’s Aβ25–35 Oligomers on Carbon Nanotube Surfaces: Implication for Amyloid Fibril Inhibition. Biophys. J. 97, 1795–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebaro Y.; Derreumaux P. (2009) Targeting the early steps of Aβ16–22 protofibril disassembly by N-methylated inhibitors: A numerical study. Proteins 75, 442–452. [DOI] [PubMed] [Google Scholar]
- Côté S. b.; Derreumaux P.; Mousseau N. (2011) Distinct Morphologies for Amyloid Beta Protein Monomer: Aβ1–40, Aβ1–42, and Aβ1–40(D23N). J. Chem. Theory Comput. 7, 2584–2592. [DOI] [PubMed] [Google Scholar]
- Lockhart C.; Kim S.; Klimov D. K. (2012) Explicit Solvent Molecular Dynamics Simulations of Aβ Peptide Interacting with Ibuprofen Ligands. J. Phys. Chem. B 116, 12922–12932. [DOI] [PubMed] [Google Scholar]
- Wu C.; Scott J.; Shea J.-E. (2012) Binding of Congo Red to Amyloid Protofibrils of the Alzheimer Aβ9–40 Peptide Probed by Molecular Dynamics Simulations. Biophys. J. 103, 550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barz B.; Urbanc B. (2012) Dimer formation enhances structural differences between amyloid beta-protein (1–40) and (1–42): an explicit-solvent molecular dynamics study. PLoS One 7, e34345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. H.; Okamoto Y.; Derreumaux P. (2013) Communication: Simulated tempering with fast on-the-fly weight determination. J. Chem. Phys. 138, 61102–61105. [DOI] [PubMed] [Google Scholar]
- Attanasio F.; Convertino M.; Magno A.; Caflisch A.; Corazza A.; Haridas H.; Esposito G.; Cataldo S.; Pignataro B.; Milardi D.; Rizzarelli E. (2013) Carnosine Inhibits Aβ42 Aggregation by Perturbing the H-Bond Network in and around the Central Hydrophobic Cluster. ChemBioChem 14, 583–592. [DOI] [PubMed] [Google Scholar]
- Melquiond A.; Dong X.; Mousseau N.; Derreumaux P. (2008) Role of the region 23–28 in Abeta fibril formation: insights from simulations of the monomers and dimers of Alzheimer’s peptides Aβ40 and Aβ42. Curr. Alzheimer Res. 5, 244–250. [DOI] [PubMed] [Google Scholar]
- (62)Khandogin J.; Brooks C. L. 3rd. (2007) Linking folding with aggregation in Alzheimer’s beta-amyloid peptides. Proc. Natl. Acad. Sci. U.S.A. 104, 16880–16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.; Wang X.; Wang X. (2013) Characterization of the internal dynamics and conformational spaces of zinc-bound amyloid β peptides by replica-exchange molecular dynamics simulations. Eur. Biophys. J. 42, 575–586. [DOI] [PubMed] [Google Scholar]
- Xu L.; Shan S.; Wang X. (2013) Single point mutation alters the microstate dynamics of amyloid β-protein Aβ42 as revealed by dihedral dynamics analyses. J. Phys. Chem. B 117, 6206–6216. [DOI] [PubMed] [Google Scholar]
- Beauchamp K. A.; Bowman G. R.; Lane T. J.; Maibaum L.; Haque I. S.; Pande V. S. (2011) MSMBuilder2: Modeling conformational dynamics on the picosecond to millisecond scale. J. Chem. Theory Comput. 7, 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronkite-Ratcliff B.; Pande V. (2013) MSMExplorer: visualizing Markov state models for biomolecule folding simulations. Bioinformatics 29, 950–952. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. (2000) Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Luchko T.; Gusarov S.; Kovalenko A.; Ryde U. (2010) An MM/3D-RISM Approach for Ligand Binding Affinities. J. Phys. Chem. B 114, 8505–8516. [DOI] [PubMed] [Google Scholar]
- Kovalenko A.; Hirata F. (1999) Self-consistent description of a metal–water interface by the Kohn–Sham density functional theory and the three-dimensional reference interaction site model. J. Chem. Phys. 110, 10095–10102. [Google Scholar]
- Kovalenko A.; Hirata F. (2000) Potentials of mean force of simple ions in ambient aqueous solution. I. Three-dimensional reference interaction site model approach. J. Chem. Phys. 112, 10391–10405. [Google Scholar]
- Chong S. H.; Ham S. (2012) Impact of chemical heterogeneity on protein self-assembly in water. Proc. Natl. Acad. Sci. U.S.A. 109, 7636–7641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., and Fox D. J. (2009) Gaussian 09, revision A.1, Gaussian, Inc., Wallingford CT.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.