

Abstract
Apoptosis-resistance and metabolic imbalances are prominent features of cancer cells. We have recently reported on populations of human fibroblasts that exhibit resistance to mitochondrial-mediated apoptosis, acquired as a result of a single genotoxic exposure. The objective of the present study was to investigate the intrinsic bioenergetic profile of the death-resistant cells, as compared to the clonogenic control cells. Therefore, we analyzed the basic bioenergetic parameters including oxygen consumption and extracellular acidification rates, coupling efficiency, and spare respiratory capacity. Our data demonstrate a strong correlation between enhanced spare respiratory capacity and death-resistance, which we postulate to be indicative of the earliest stages of carcinogenesis
Keywords: Mitochondria, bioenergetics, spare respiratory capacity, death-resistance
1. Introduction
Several lines of evidence have indicated that carcinogenesis evolves from consecutive genetic and/or epigenetic alterations that provide cellular survival advantages, ultimately leading to the conversion of normal human cells to malignant cancer cells [for review, see (Hanahan and Weinberg, 2000; Hanahan and Weinberg, 2011)]. As evasion of apoptosis and unlimited replicative potential are hallmarks of cancer, two questions become inevitable when studying death-resistance and early-stage carcinogenesis: 1) how does a cell gain enhanced proliferative capacity, and 2) what fuels this unlimited replicative potential? In most cells under aerobic conditions, glucose is converted to pyruvate through glycolysis, which then enters the TCA cycle where the flavin nucleotide (FADH2) and NADH are produced. The respective reduced equivalents are further oxidized by the mitochondrially-localized electron transport chain (ETC), which ultimately produces ~80% of total cellular ATP. Conversely, early studies in tumor cells have found an upregulation of glycolysis for energy production, even in the presence of sufficient oxygen levels. This phenotype is known as the Warburg effect, after Dr. Otto Warburg who uncovered the phenomenon (Warburg, 1956). While the high glycolytic phenotype of cancer cells has been exploited for clinical use (i.e. positron emission tomography), the molecular basis of the Warburg effect remains unclear.
The bioenergetic profile of several cell types such as neurons, endothelial cells, and human carcinoma cell lines, has been characterized in an effort to uncover alterations that may be targeted for therapeutic purposes (Rodriguez-Enriquez et al., 2008; van der Windt et al., 2012; Wu et al., 2007; Xun et al., 2012). Of particular interest to the present study, is the role that spare respiratory capacity (SRC) may play in death resistance. The term, SRC, describes the reserve capacity that enables the production of energy in response to cellular stress (Nicholls, 2009). It has been hypothesized that in the face of oxidative stress, cell survival can be potentiated when a maximal reserve of ATP is maintained (Choi et al., 2009; Fern, 2003; Zhu et al., 2012).
We have previously generated sub-populations of BJ-hTERT human diploid foreskin fibroblasts, which have acquired resistance to cell death induced by hexavalent chromium [Cr(VI)], a broad-spectrum DNA-damaging agent (Pritchard et al., 2005). Fibroblasts are integral to the cellular microenvironment and have been associated with pathological conditions such as fibrosis and carcinogenesis (McAnulty, 2007; Vaheri et al., 2009). This system is unique in that it models initial molecular events that occur in a normal cell that survived a single, acute, initiating genotoxic challenge. While the selection model in this study was generated by Cr(VI) treatment, it also exhibits a cross-resistance to the well-known chemotherapeutic agent, cisplatin, as well as to H2O2 (Nickens et al., 2012). Long-term exposure to certain forms of Cr(VI) is associated with respiratory carcinogenesis (IARC, 1990). Our recent report investigated the death-sensitivity of subclonal populations derived from clonogenic survivors of BJ-hTERT cells treated with Cr(VI) (DR), or selected by dilution-based cloning without treatment (CC) (Nickens et al., 2012). Notably, our data suggested the presence of more resilient mitochondria in DR cells, and that death resistance can be acquired in normal human cells early after genotoxin exposure. Taken together, these data led us to postulate that resistance to mitochondrially-mediated cell death and mitochondrial dysregulation may be initial phenotypic alterations associated with early-stage carcinogenesis.
Here we report on the bioenergetic profile of the DR and CC subclonal cell lines. By employing the Seahorse Bioscience XF Extracellular Flux Analyzer, we simultaneously measured glycolysis by assessing the extracellular acidification rate (ECAR), as well as the rate of oxidative phosphorylation by measuring the cellular oxygen consumption rate (OCR) (Eklund et al., 2004). We tested the hypothesis that survival after genotoxic stress may involve the selection of cells with intrinsically altered bioenergetic regulation. Our data show that while there is no difference in basal ATP content, ECAR, or OCR, there is an increase in the SRC of the DR cells, as compared to the CC cells. Taken together, the present data show that a greater intrinsic SRC is coincident with death-resistance in our model system. Moreover, this enhanced capacity may be a mechanistic step in the acquisition of death resistance, which in turn may potentially foster neoplastic progression. Importantly, we show that the intrinsically enhanced SRC was observed in diploid human cells that have acquired a death resistant phenotype following only a single exposure to a carcinogen.
2. Materials and Methods
2.1. Subcloning, cell lines, and culture parameters
Subclonal populations were derived as previously described (Nickens et al., 2012). The cell lines used in the present study include untreated clonogenic control cell lines, CC1 and CC2, as well as clones derived from clonogenic survivors of Cr(VI) exposure, DR1, DR2, DR3, and DR4, which display an apoptosis resistant phenotype. As previously reported, all CC and DR cell lines were derived from human foreskin fibroblasts transfected with the hTERT gene (BJ-hTERT; Geron Corp.), and further subcloned (at passage 138) into both CC and DR cell lines (Nickens et al., 2012; Pritchard et al., 2005). The original hTERT-immortalized foreskin fibroblast cell line, BJ-5ta, was derived by transfecting the BJ foreskin fibroblast cell line with the pGRN145 hTERT-expressing plasmid (ATCC MBA-141) (Bodnar et al., 1998). The population doubling time of the respective death resistant and death sensitive sub-populations is similar, at around 22–26 h. For the present study, cells were passaged at 80% confluence, and were only used for 10 passages, before a fresh vial was selected.
All cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen Corporation, Carlsbad, CA), containing Medium 199 (Invitrogen Corporation) (4:1), 10% fetal bovine serum (Hyclone Laboratories, Inc., Logan, UT), 5 μg/ml gentamicin (Life Technologies, Gaithersburg, MD), and 0.75 μg/ml fungizone antimycotic (Invitrogen Corporation; DMEM complete). The hTERT transgene was selected for by the addition of 10 μg/ml hygromycin B (Life Technologies) to the medium. All cell lines were incubated in a 95% air and 5% CO2 humidified atmosphere at 37°C, and the medium was replaced every 48 h.
2.2. ATP content
Cells were seeded at a density of 4 × 105/60 mm dish and incubated at 37°C for 24 h prior to analysis. Following incubation, the cells were rinsed with 5 ml PBS and removed from the dish by gentle scraping in 60 μl of CHAPS cell lysis buffer (Cell Signaling Technology, Danvers, MA) containing 50 mM PIPES/HCl (pH 6.5), 2mM EDTA, 0.1% CHAPS, 20 μg/mL leupeptin, and 10 μg/mL aprotinin; supplemented with 50 mM NaF, 1 mM Na3V04, and 1 mM PMSF. The cells were lysed on ice, and the lysates were centrifuged at 14,000 rpm for 12 min at 2–8°C. A luciferin-luciferase based bioluminescent ATP determination kit (Molecular Probes) was used to analyze ATP content (Ahn et al., 2008), according to the manufacturer’s instructions. Briefly, ATP standards ranging from 0.5 to 25 μM were prepared from a 5 mM ATP stock solution in dH2O. Duplicate aliquots of standards and samples were pipetted into a flat-bottom black polystyrene 96-well assay plate (Corning Incorporated, Corning, NY). The reaction was started immediately before analysis by the addition of the standard reaction solution containing 20 X Reaction Buffer, 0.1 M DTT, 10 mM D-luciferin, and 5 mg/mL firefly luciferase. Luminescence was measured after a 200 ms integration time using 485 nm excitation and 538 nm emission wavelengths on the Fluoroskan Ascent FL (Thermo Fisher Scientific, Waltham, MA), followed by analysis using the Ascent Software. Background luminescence was subtracted from the generated values using wells containing only the standard reaction solution. Luminescence was normalized to protein content which was assessed using the BCA protein assay kit (Thermo Fisher Scientific).
2.3. Metabolic flux analysis
Cells were seeded in triplicate in 100 μl DMEM complete medium at a density of 1 × 104/well in XF24 24-well V7 cell culture plate (Seahorse Bioscience, North Billerica, MA), leaving appropriate temperature control wells empty, and incubated at 37°C for 1 h. Following cell attachment, an additional 150 μl of medium was added to each well, and incubated at 37°C overnight. The XF24 sensor cartridge (Seahorse Bioscience) was prepared by incubation of each sensor pair in 1 ml of Seahorse Bioscience XF24 Calibrant pH 7.4 (Seahorse Bioscience) at 37°C without CO2 for 24 h. Prior to analysis, the medium was gently removed from the adherent cells and the wells were washed with 1 ml of pre-warmed specially formulated Seahorse DMEM (Seahorse Bioscience) supplemented with 25 mM glucose (assay medium). A final volume of 450 μl assay medium was added to each well and the plate was incubated at 37°C without CO2 for 1 h. Either 1.5 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) or 5 μM oligomycin (Sigma-Aldrich, St. Louis, MO) was added to injection port A of the XF24 sensor cartridge in assay medium and equilibrated at least 15 min prior to analysis at 37°C without CO2. The XF24 sensor cartridge was calibrated in the Seahorse XF24 analyzer (Seahorse Bioscience), which was pre-warmed to 37°C. Following calibration, the cell culture plate was placed in the analyzer and the extracellular acidification rate (ECAR) and the oxygen consumption rates (OCR) were simultaneously measured via the following protocol: three cycles of mix (2 min), delay (2 min); seven cycles of measure (4 min), mix (2 min), delay (2 min); port A injection; six cycles of mix (2 min), delay (2 min), measure (4 min). After the final measurement the assay medium was removed from each well and the cells were washed twice with warm PBS then incubated at 37°C without CO2 in 4 μM calcein AM (BD Biosciences, San Jose, California) in PBS for 40 min. Fluorescence was measured by using 485 excitation and 530 nm emission wavelengths on the Microplate Reader-Infinite® M1000 (Tecan Group Ltd., Männedorf) followed by data collection on the Tecan i-control software (Tecan Group Ltd.). ECAR and OCR rates were normalized to cell number as assessed by calcein AM fluorescence. Basal values were taken from the last measurement prior to drug injection and the post-drug injection values were from the first measurement after injection. Post-drug injection values were normalized to respective basal value and expressed as either mpH/min/104 cells (ECAR) or pmoles/min/104 cells (OCR). Cell number was determined by calcein AM incorporation (Zhang et al., 2012).
2.4. Statistical analysis
To determine differences among experimental groups, statistical analyses were performed using GraphPad Prism version 4.00 (San Diego, CA). A one-way analysis of variance and either a Tukey or Bonferroni post test was used for multiple sample comparisons. For all experiments, results are presented as the mean ± standard error of the mean and for all statistical tests, p < 0.05 was considered to be statistically significant.
3. Results and Discussion
Two death-resistant cell lines (DR1, DR2), as well as a death-susceptible clonogenic control line (CC1) were the focus of a recently published report from our laboratory (Nickens et al., 2012). Our data showed that, as compared to the death-susceptible cells, the DR cells were resistant to apoptosis as indicated by lack of cleaved caspase 3 expression following exposure to both Cr(VI) and cisplatin. The mitochondrial-mediated pathway of apoptosis was inhibited in the DR cell, as indicated by the lack of both mitochondrial-membrane depolarization and cytochrome c release, as well as attenuated/abrogated release of SMAC/DIABLO following Cr(VI) treatment. Moreover, we observed a marked increase in Bcl-2 protein expression upon Cr(VI) exposure in the DR cells, which was significant in the DR1 cells. Furthermore, our data uncovered an intrinsic alteration in the incidence of mtDNA damage, as the DR1 cells displayed significantly less basal mtDNA damage than that of the CC1 cells (Nickens et al., 2012). To ensure that any alterations in bioenergetic parameters observed in our model system were consistent with the generation of death-resistant survivors following a single genotoxin exposure, we included the additional subclonal cell lines, DR3 and DR4 derived from the B-5Cr cells, as well as an additional clonogenic control cell line, CC2, in the present study. While the DR3 and DR4 cell lines are not as extensively characterized as the DR1 and DR2 cells, they do exhibit resistance to Cr(VI)-induced cleaved caspase 3-mediated apoptosis, similar to the DR1 and DR2 cell lines (Supplemental Figure 1).
Escape from or resistance to apoptosis or terminal growth arrest has been shown to be a requirement for cells with limitless replicative potential (Hahn and Weinberg, 2002). Moreover, the selection of cells with the ability to survive after exposure to apoptogenic levels of a DNA damaging agent may yield a precursor pool of cells from which neoplastic variants may emerge. Furthermore, the dysregulation of mitochondrially-mediated cellular mechanisms, such as apoptosis and energy metabolism are observed in malignant tumors, highlighting a connection between mitochondria, death-resistance and carcinogenesis (Hasenjager et al., 2004; Warburg, 1956). Mitochondria control cellular energy production and bioenergetics. These functions can be altered by mtDNA damage, and our previous report suggests their involvement in our death-resistant model system (Nickens et al., 2012). Therefore, we proposed that intrinsically dysregulated cellular bioenergetics may also correlate with the observed death-resistant phenotype.
Mitochondria produce approximately 80% of cellular ATP, and play a critical role in cellular energy metabolism. Therefore, we measured total basal ATP content using a luciferin-luciferase based bioluminescent assay in each cell line. We found that there was no difference in total basal ATP content among the cell lines (Figure 1).
Figure 1. Intrinsic alterations in ATP content are not coincident with the observed death resistant phenotype.
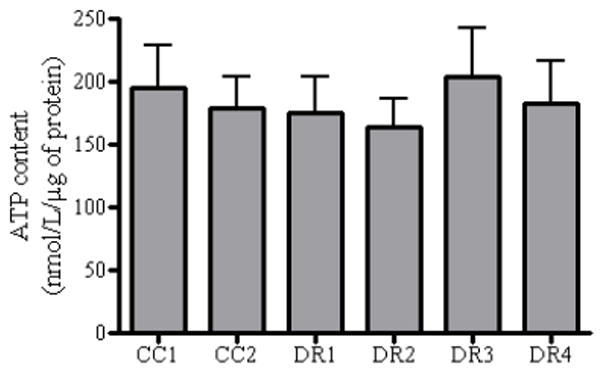
CC1, CC2, DR1, DR2, DR3, and DR4 cells were seeded and allowed to grow for 24 h prior to analysis, followed by total protein extraction. A luciferin-luciferase based bioluminescent ATP determination kit was used to analyze ATP content, according to the manufacturer’s instructions. Luminescence was measured after a 200 ms integration time using 485 excitation and 538 emission wavelengths on the Fluoroskan Ascent FL, followed by analysis using the Ascent Software. Background luminescence was subtracted from the generated values using wells containing only the standard reaction solution. Luminescence was normalized to protein content.
It has been suggested that an alteration in cellular bioenergetics is an important factor in carcinogenesis [for review, see (Brand and Nicholls, 2011; Weinberg and Chandel, 2009)]. Moreover, genotoxic stress has been associated with cellular energy crisis [for review, see (Surjana et al., 2010)]. The two primary ATP generating processes of the cell are glycolysis and oxidative phosphorylation. Therefore, to assess the potential contribution of altered cellular bioenergetics to death-resistance, we investigated both processes. The extracellular acidification rate (ECAR) was used as an indicator of glycolytic rate, and the oxygen consumption rate (OCR) as an indicator of oxidative phosphorylation/mitochondrial respiration (Choi et al., 2009; Fern, 2003; Gohil et al., 2010; Nicholls, 2009; Qian and Van Houten, 2010; Yadava and Nicholls, 2007). It is worth noting that ECAR is an indicator of lactate production, which is produced from excess pyruvate generated from glycolysis, but can also be generated from cellular processes such as fatty acid oxidation and the Krebs cycle. ECAR and OCR were measured simultaneously using the Seahorse XF24 metabolic flux analyzer. Our data show that basally, the ECAR (Figure 2A) and OCR (Figure 2B) were similar among all cell lines studied, with the DR1 cells showing a slight, but not significant, increase in OCR as compared to the other cell lines. These results correlated with the lack of alteration in basal ATP content among all of the cell lines studied. It has been well accepted that many tumor cells exhibit enhanced glycolysis, due to an increase in glucose uptake, and this indicator has been successfully correlated with malignancy, tumor stage, and progression (Bellance et al., 2009; Pedersen, 2007; Pelicano et al., 2006; Warburg, 1956; Zu and Guppy, 2004). The data from the present study suggest that basal differences in glycolysis and oxidative phosphorylation are not markers of death-resistance in our cellular model system.
Figure 2. Intrinsic alterations in the extracellular acidification rate and oxygen consumption rate are not coincident with the observed death resistant phenotype.
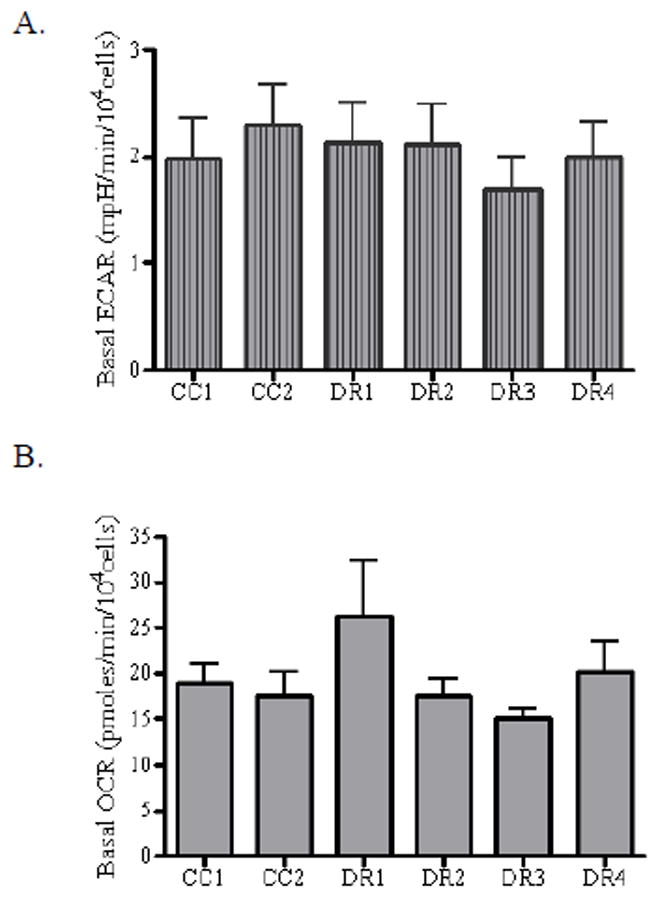
CC1, CC2, DR1, DR2, DR3, and DR4 cells were seeded in triplicate (104 cells/well) in XF24 24-well cell culture plates, and allowed to grow for 24 h at 37°C. Prior to analysis, the growth medium was removed from the plate and replaced with Seahorse assay DMEM supplemented with 25 mM glucose, and the cells were incubated at 37°C without CO2 for 1 h. The cell culture plate was placed in the Seahorse XF24 Analyzer and the ECAR was measured. (A) ECAR and (B) OCR values were normalized to cell number as assessed by calcein AM fluorescence. Fluorescence was measured using 485 excitation and 530 emission wavelengths on the Microplate Reader-Infinite® M1000. Data were expressed as ECAR (mpH/min/104 cells) or OCR (pmoles/min/104 cells), cell number was determined by calcein AM incorporation. Data are mean +/− SE of seven experiments (p < 0.05).
We analyzed ECAR and OCR after injection of 5 μM oligomycin, an ATP synthase inhibitor. Oligomycin blocks the Fo proton channel of ATP synthase, and prevents state 3 (phosphorylation) respiration. In our intact cellular model system, oligomycin treatment reveals the quantity of oxygen consumption needed for ATP synthesis and the quantity of oxygen consumption needed to surmount the naturally occurring proton leak across the inner mitochondrial membrane (Nicholls et al., 2010). . Following oligomycin treatment, and consequent inhibition of ATP synthesis via oxidative phosphorylation, the cells met their energy demand by tapping into glycolysis. This was demonstrated by an increase over baseline control in ECAR among all cell lines. Each cell line exhibited different percentages of ECAR post-oligomycin injection; however, while these data did not correlate with death sensitivity, they did reveal an intact response of the glycolytic pathway to the cellular energy demand. Specifically, the CC1 cells displayed a significant 1.8-fold increase, while the CC2 cells displayed a 1.2-fold increase in ECAR as compared to respective control, which was not significant. The DR1, DR2 and DR4 cell lines displayed statistically significant 1.7 to 1.8-fold increases in ECAR as compared to respective controls, while the DR3 cells exhibited a 1.4-fold increase, which was not significant (Figure 3A). Moreover, there were no differences in OCR when all cell lines were compared to each other. A significant 70% decrease in OCR as compared to the respective control was observed in each cell line as expected in response to an inhibition of ATP synthesis via oxidative phosphorylation, indicating that approximately 70% of oxygen consumption in this fibroblast model is coupled to ATP production (Figure 3B).
Figure 3. Intrinsic alterations in coupling efficiency are not coincident with the observed death resistant phenotype.
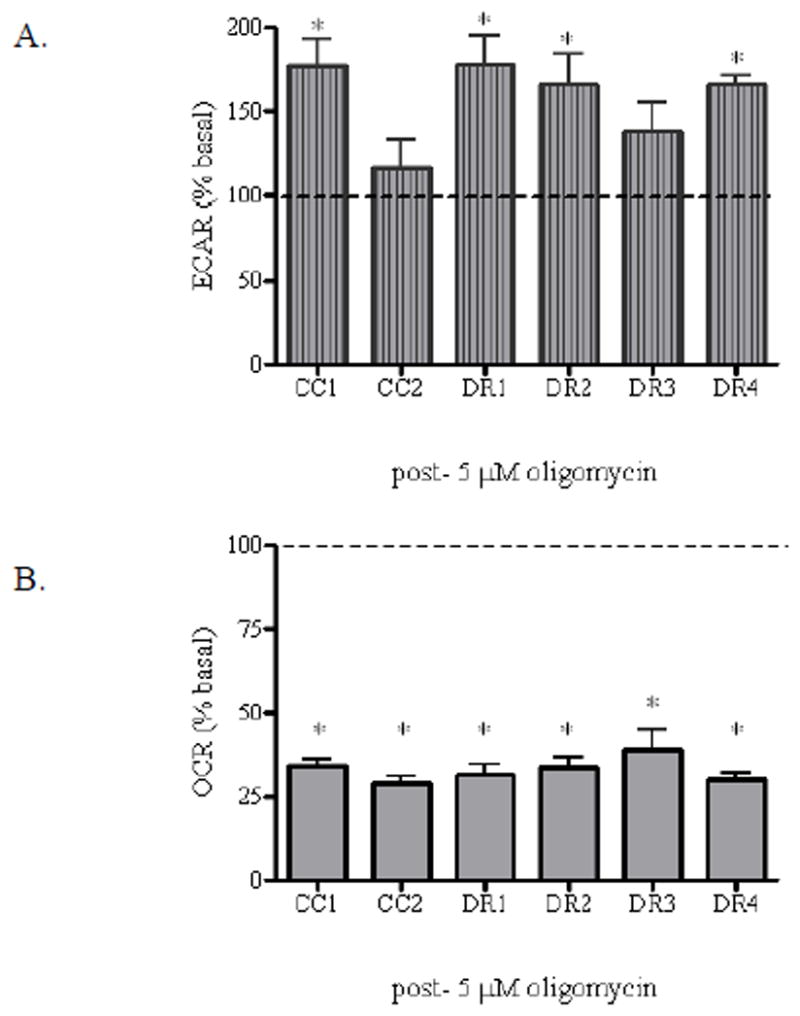
CC1, CC2, DR1, DR2, DR3, and DR4 cells were seeded in triplicate (104 cells/well) in XF24 24-well cell culture plates, and allowed to grow for 24 h at 37°C. Prior to analysis, the growth medium was removed from the plate and replaced with Seahorse assay DMEM supplemented with 25 mM glucose, and the cells were incubated at 37°C without CO2 for 1 h. 5 μM oligomycin was added to injection port A of the XF24 sensor cartridge in assay medium and equilibrated at least 15 min prior to analysis at 37°C without CO2. The cell culture plate was placed in the Seahorse XF24 Analyzer and the (A) ECAR and (B) OCR values were simultaneously measured. Data post-oligomycin injection were generated from rate levels directly after injection. Data were normalized to cell number as assessed by calcein AM fluorescence. Fluorescence was measured using 485 excitation and 530 emission wavelengths on the Microplate Reader-Infinite® M1000. Post-drug injection values were normalized to respective basal value and expressed as ECAR (% of basal) or OCR (% of basal). * indicates a statistically significant difference from control. Data are mean +/− SE of three experiments (p < 0.05).
Studies in neurons have implicated the involvement of mitochondrial metabolic dysfunction in the pathogenesis of age-related neurodegenerative disease, particularly oxidative stress and bioenergetic deficit (Yadava and Nicholls, 2007). It has been hypothesized that in the face of oxidative stress, neuronal cell survival can be potentiated when a maximal reserve of ATP is maintained. This concept has been coined as the “spare respiratory capacity” (SRC) or the “spare electron transport chain complex capacity” (Choi et al., 2009; Fern, 2003). Therefore, we measured the intrinsic SRC of the CC1, CC2, DR1, DR2, DR3, and DR4 cells. The SRC of cells can be quantified by measuring the percentage change between basal OCR and the maximal uncontrolled OCR following treatment by a respiratory chain uncoupling agent such as carbonylcyanide p trifluoromethoxyphenylhydrazone (FCCP). FCCP disrupts ATP synthesis by diverting hydrogen ions from the proton channel of ATP synthase and shuttling them across the mitochondrial membrane. The expected response to FCCP treatment is rapid energy consumption, resulting in an increase in both OCR and ECAR in order to maintain the cellular energy balance (Nicholls et al., 2010). Our data show that the ECAR post-1.5 μM FCCP injection was significantly increased in all cell lines as compared to respective pre-FCCP injection controls, but did not correlate with death sensitivity. The CC1 and CC2 cells exhibited 2.1 and 1.5-fold increases, respectively; while the DR1, DR2, DR3 and DR4 cells exhibited respective 1.7, 2.1, 1.8 and 2.0-fold increases as compared to control (Figure 4A). These results are in keeping with an anticipated increase in glycolytic function in order to compensate for the malfunction in energy production via oxidative phosphorylation (Stockl et al., 2007). Furthermore, our data show that immediately following FCCP injection, the CC1 and CC2 cells displayed a slight increase in OCR as compared to pre-FCCP injection basal controls, which was not significant. However, all DR cells exhibited a significant 1.3 to 1.5-fold increase as compared to their respective pre-FCCP injection controls (Figure 4B). Notably, the DR1 cells demonstrated the greatest SRC, which may also be reflected in the incremental trend in the basal OCR of these cells (Figure 2B). The difference in SRC observed in each cell line following FCCP treatment may be due to various bioenergetic factors, such as the capability of the cell to provide substrates to the mitochondria for energy production, and the functionality of enzymes involved in the electron transport chain (Nicholls et al., 2010).
Figure 4. Enhanced spare respiratory capacity is coincident with the observed death resistant phenotype.
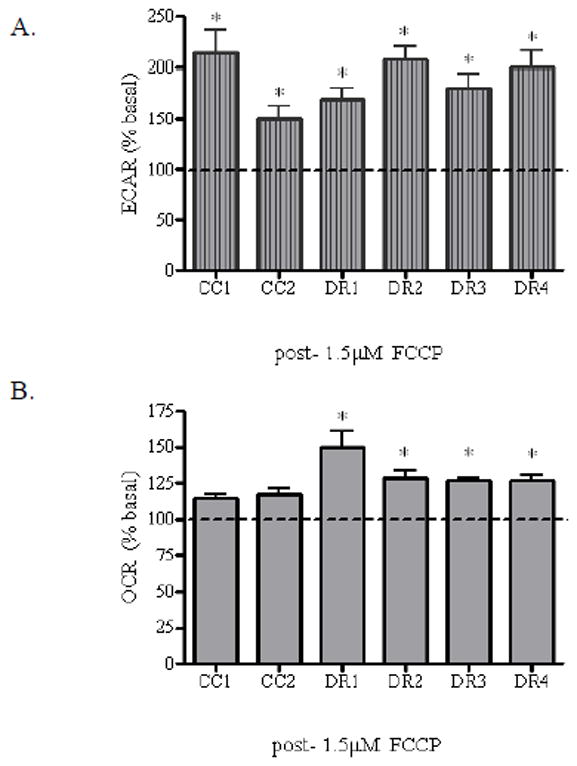
CC1, CC2, DR1, DR2, DR3, and DR4 cells were seeded in triplicate (104 cells/well) in XF24 24-well cell culture plates, and allowed to grow for 24 h at 37°C. Prior to analysis, the growth medium was removed from the plate and replaced with Seahorse assay DMEM supplemented with 25 mM glucose, and the cells were incubated at 37°C without CO2 for 1 h. 1.5 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) was added to injection port A of the XF24 sensor cartridge in assay medium and equilibrated at least 15 min prior to analysis at 37°C without CO2. The cell culture plate was placed in the Seahorse XF24 Analyzer and the (A) ECAR and (B) OCR values were simultaneously measured. Data post-FCCP injection were generated from rate levels directly after injection. Data were normalized to cell number as assessed by calcein AM fluorescence. Fluorescence was measured using 485 excitation and 530 emission wavelengths on the Microplate Reader-Infinite® M1000. Post-drug injection values were normalized to respective basal value and expressed as ECAR (% of basal) or OCR (% of basal). * indicates a statistically significant difference from control. Data are mean +/− SE of three experiments (p < 0.05).
The results of this study provide an understanding of the basic bioenergetic profiles of the cells in this unique death resistant model system. The data demonstrate the first direct indication that the maintenance of SRC may be related to apoptotic death-resistance and suggest that SRC may play a role in the enhanced survival of cells after a single, acute genotoxic challenge. Moreover, our data suggest a strong reliance on energy production via oxidative phosphorylation in death resistant cells, which has also been shown in metabolic profile analysis of melanoma tumor cells (Ho et al., 2012). The apparent importance of oxidative phosphorylation in the cell model system, while incongruent with the glyocolytic reliance of cancer cells described as the Warburg effect, may be suggestive of metabolic flexibility in response to cellular stress. Future studies are necessary to further investigate the mechanism(s) responsible for the intrinsically enhanced spare respiratory capacity observed in all DR cell lines.
It is important to note that several recent studies have demonstrated evidence that dysregulated bioenergetics play an important role in altered cellular phenotypes. Van der Windt et al. (2012) showed that SRC was enhanced in CD8+ memory T cells through the IL-15 regulation of mitochondrial biogenesis and carnitine palmitoyl transferase 1a (CPT1a) expression (a mitochondrial fatty acid oxidase enzyme). CPT1 activity may contribute to the high energy demands of cancer cells (Linher-Melville et al., 2011). Additionally, a recent study demonstrated the fundamental role that poly(ADP-ribose)1 (PARP1) plays in cellular bioenergetic homeostasis (Modis et al., 2012). The PARP family of proteins is involved in DNA damage and can trigger apoptosis. These authors show that PARP1 deficiency leads to an increase in mitochondrial respiratory reserve capacity in both endothelial and epithelial cell models, suggesting that cells with dysregulated cell death mechanisms may exhibit enhanced bioenergetics. Finally, a study by Caneba and colleagues (2012) investigating the metabolic variations among increasingly invasive ovarian cancer cell lines during detachment showed that high SRC (via increased OCR) was directly correlated with highly invasive ovarian cancer. Collectively, these and the present studies, underline the potential relationship between energy metabolism, cellular phenotypic alterations, and the associated physiological disease profiles.
In conclusion, our data suggest a strong physiologic correlation between intrinsic cellular death-resistance and enhanced mitochondrial spare respiratory capacity, which we postulate may represent the earliest phenotypic alterations of carcinogenesis. Taken together, these data show that resistance to apoptosis can be acquired in diploid human cells, following only a single exposure to a carcinogen; and that resistance to mitochondrial-mediated cell death and intrinsic mitochondrial dysregulation may be indicative of the earliest stages of carcinogenesis.
Supplementary Material
Highlights.
We have recently reported on populations of human fibroblasts that exhibit resistance to mitochondrial-mediated apoptosis, acquired as a result of a single genotoxic exposure.
We investigated the intrinsic bioenergetic profile of the death-resistant cells, as compared to the clonogenic control cells.
We found intrinsically enhanced spare respiratory human fibroblasts that have acquired a death resistant phenotype following only a single exposure to a carcinogen.
We postulate that the correlation between enhanced spare respiratory capacity and death-resistance may represent an early phenotypic alteration in the path to neoplastic transformation.
Acknowledgments
The authors would like to acknowledge Dr. Eric Kaldjian for assisting in this collaborative effort, as well as Dr. Travis O’Brien, Dr. Madhu Lal-Nag, and Dr. Tura Camilli for their helpful discussions and insight. This work was supported by the National Institutes of Health [R21ES017334 and R01CA107972 to S.C., R01CA107972 supplement to K.N., R01ES05304 and R01ES09961 to S.P.].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellance N, Lestienne P, Rossignol R. Mitochondria: from bioenergetics to the metabolic regulation of carcinogenesis. Front Biosci. 2009;14:4015–4034. doi: 10.2741/3509. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am J Physiol Endocrinol Metab. 2012;303:E1036–E1052. doi: 10.1152/ajpendo.00151.2012. [DOI] [PubMed] [Google Scholar]
- Choi SW, Gerencser AA, Nicholls DG. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: spare respiratory capacity and stochastic mitochondrial failure. J Neurochem. 2009;109:1179–1191. doi: 10.1111/j.1471-4159.2009.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund SE, Taylor D, Kozlov E, Prokop A, Cliffel DE. A microphysiometer for simultaneous measurement of changes in extracellular glucose, lactate, oxygen, and acidification rate. Anal Chem. 2004;76:519–527. doi: 10.1021/ac034641z. [DOI] [PubMed] [Google Scholar]
- Fern R. Variations in spare electron transport chain capacity: The answer to an old riddle? J Neurosci Res. 2003;71:759–762. doi: 10.1002/jnr.10553. [DOI] [PubMed] [Google Scholar]
- Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347:1593–1603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hasenjager A, Gillissen B, Muller A, Normand G, Hemmati PG, Schuler M, Dorken B, Daniel PT. Smac induces cytochrome c release and apoptosis independently from Bax/Bcl-x(L) in a strictly caspase-3-dependent manner in human carcinoma cells. Oncogene. 2004;23:4523–4535. doi: 10.1038/sj.onc.1207594. [DOI] [PubMed] [Google Scholar]
- Ho J, de Moura MB, Lin Y, Vincent G, Thorne S, Duncan LM, Hui-Min L, Kirkwood JM, Becker D, Van Houten B, Moschos SJ. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol Cancer. 2012;1:76. doi: 10.1186/1476-4598-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. Chromium, nickel and welding [erratum appears in IARC Monogr Eval Carcinog Risks Hum 1991;51:483] IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 1990;49:1–648. [PMC free article] [PubMed] [Google Scholar]
- Linher-Melville K, Zantinge S, Sanli T, Gerstein H, Tsakiridis T, Singh G. Establishing a relationship between prolactin and altered fatty acid beta-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer. 2011;11:56. doi: 10.1186/1471-2407-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39:666–671. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Modis K, Gero D, Erdelyi K, Szoleczky P, DeWitt D, Szabo C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem Pharmacol. 2012;83:633–643. doi: 10.1016/j.bcp.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Spare respiratory capacity, oxidative stress and excitotoxicity. Biochem Soc Trans. 2009;37:1385–1388. doi: 10.1042/BST0371385. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Darley-Usmar VM, Wu M, Jensen PB, Rogers GW, Ferrick DA. Bioenergetic profile experiment using C2C12 myoblast cells. J Vis Exp. 2010;46:e2511. doi: 10.3791/2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickens KP, Han Y, Shandilya H, Larrimore A, Gerard GF, Kaldjian E, Patierno SR, Ceryak S. Acquisition of mitochondrial dysregulation and resistance to mitochondrial-mediated apoptosis after genotoxic insult in normal human fibroblasts: a possible model for early stage carcinogenesis. Biochim Biophys Acta. 2012;1823:264–272. doi: 10.1016/j.bbamcr.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen PL. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i. e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- Pritchard DE, Ceryak S, Ramsey KE, O’Brien TJ, Ha L, Fornsaglio JL, Stephan DA, Patierno SR. Resistance to apoptosis, increased growth potential, and altered gene expression in cells that survived genotoxic hexavalent chromium [Cr(VI)] exposure. Mol Cell Biochem. 2005;279:169–181. doi: 10.1007/s11010-005-8292-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian W, Van Houten B. Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods. 2010;51:452–457. doi: 10.1016/j.ymeth.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Gallardo-Perez JC, Aviles-Salas A, Marin-Hernandez A, Carreno-Fuentes L, Maldonado-Lagunas V, Moreno-Sanchez R. Energy metabolism transition in multi-cellular human tumor spheroids. J Cell Physiol. 2008;216:189–197. doi: 10.1002/jcp.21392. [DOI] [PubMed] [Google Scholar]
- Stockl P, Zankl C, Hutter E, Unterluggauer H, Laun P, Heeren G, Bogengruber E, Herndler-Brandstetter D, Breitenbach M, Jansen-Durr P. Partial uncoupling of oxidative phosphorylation induces premature senescence in human fibroblasts and yeast mother cells. Free Radic Biol Med. 2007;43:947–958. doi: 10.1016/j.freeradbiomed.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Surjana D, Halliday GM, Damian DL. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J Nucleic Acids. 2010;2010:157591. doi: 10.4061/2010/157591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaheri A, Enzerink A, Rasanen K, Salmenpera P. Nemosis, a novel way of fibroblast activation, in inflammation and cancer. Exp Cell Res. 2009;315:1633–1638. doi: 10.1016/j.yexcr.2009.03.005. [DOI] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- Weinberg F, Chandel NS. Mitochondrial metabolism and cancer. Ann NY Acad Sci. 2009;1177:66–73. doi: 10.1111/j.1749-6632.2009.05039.x. [DOI] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- Xun Z, Lee DY, Lim J, Canaria CA, Barnebey A, Yanonne SM, McMurray CT. Retinoic acid-induced differentiation increases the rate of oxygen consumption and enhances the spare respiratory capacity of mitochondria in SH-SY5Y cells. Mech Ageing Dev. 2012;133:176–185. doi: 10.1016/j.mad.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava N, Nicholls DG. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J Neurosci. 2007;27:7310–7317. doi: 10.1523/JNEUROSCI.0212-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nuebel E, Wisidagama DR, Setoguchi K, Hong JS, Van Horn CM, Imam SS, Vergnes L, Malone CS, Koehler CM, Teitell MA. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat Protoc. 2012;7:1068–1085. doi: 10.1038/nprot.2012.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Gusdon AM, Cimen H, Van Houten B, Koc E, Chu CT. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 2012;3:e312. doi: 10.1038/cddis.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313:459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.