

Abstract
Objective
Acute respiratory distress syndrome/acute lung injury is a serious complication of burn patients with concomitant smoke inhalation injury. Nitric oxide has been shown to play a major role in pulmonary dysfunction from thermal damage. In this study, we have tested the hypothesis that inhibition of neuronal nitric oxide synthase could ameliorate the severity of acute lung injury using our well-established ovine model of cutaneous burn and smoke inhalation.
Design
Prospective, randomized, controlled, experimental animals study.
Setting
Investigational intensive care unit at university hospital.
Subjects
Adult female sheep
Interventions
Female sheep (n = 16) were surgically prepared for the study. Seven days after surgery, all sheep were randomly allocated into three study groups: sham (noninjured, nontreated, n = 6); control (injured, treated with saline, n = 6); and neuronal nitric oxide synthase (injured, treated with specific neuronal nitric oxide synthase inhibitor, ZK 234238 (n = 4). Control and neuronal nitric oxide synthase groups were given a cutaneous burn (40% of total body surface, third degree) and insufflated with cotton smoke (48 breaths, <40°C) under halothane anesthesia. Animals in sham group received fake injury also under halothane anesthesia. After injury or fake injury procedure, all sheep were placed on ventilators and resuscitated with lactated Ringer's solution. Neuronal nitric oxide synthase group was administered with continuous infusion of ZK 234238 started 1 hr postinjury with a dose of 100 μg/kg/hr. Sham and control groups received same amount of saline.
Measurements and Main Results
Cardiopulmonary hemodynamics monitored during the 24-hr experimental time period was stable in the sham group. Control sheep developed multiple signs of acute lung injury. This pathophysiology included decreased pulmonary gas exchange and lung compliance, increased pulmonary edema, and inflammatory indices, such as interleukin-8. Treatment of injured sheep with neuronal nitric oxide synthase inhibitor attenuated all the observed pulmonary pathophysiology.
Conclusions
The results provide definitive evidence that inhibition of neuronal nitric oxide synthase-derived excessive nitric oxide may be a novel and beneficial treatment strategy for pulmonary pathology in burn victims with smoke inhalation injury.
Keywords: acute lung injury, neuronal nitric oxide synthase, burn, smoke
Both the positive and negative aspects of nitric oxide have been extensively described in previous studies. Among the three isoforms of the nitric oxide synthase (NOS), constitutive NOS (endothelial and neuronal) is believed to have ben eficial effects, such as maintaining normal homeostasis (1, 2). In contrast, inducible NOS (iNOS) is responsible for the injurious effects nitric oxide (NO) under certain circumstances (3). However, recent studies (4–6), including our own work (7, 8), provide evidence that neuronal NOS (nNOS) may play a profound role in the pathophysiology of organ dysfunction in various pathologic conditions such as Gram-negative sepsis (8). The findings that all NOS isoforms are expressed in lung tissue, and nNOS-derived NO accounts for 40% of exhaled NO (9) strongly supports the hypothesis that nNOS may potentially contribute to pathologic alterations.
Pulmonary dysfunction and systemic hypoxemia with subsequent organ hypoxia is one of the crucial complications that determines the outcome for fire victims. A number of reports have previously described various pathologic factors, such as excessive NO, involved in the pathogenesis of multiple organ dysfunctions, including acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) in burn trauma (10). Using different iNOS inhibitors we have previously reported the important role of iNOS-associated NO in the pathophysiology of burn and smoke inhalation-induced multiple organ dysfunction (11–13). In this study, we tested the hypothesis that inhibition of nNOS-derived NO would ameliorate ALI in sheep exposed to combined burn and smoke inhalation injury, using a specific and potent nNOS inhibitor, ZK 234238 (3-methyl-2-amino-6,7,8,9-tetrahydro[2,1b]-1,4-oxazine).
MATERIALS AND METHODS
Animal Model
The model of this burn and smoke inhalation injury study has previously been described in detail (11, 12). Briefly, 16 adult female sheep weighing 30–40 kg were surgically prepared under halothane anesthesia. After a 7-day recovery period, sheep were randomly divided into three groups: sham (noninjured, nontreated; n = 6); control (injured, treated with saline; n = 6); and nNOS (injured, treated with nNOS inhibitor, n = 4). Then, control and nNOS groups were anesthetized with halothane and given flame burn (40% total body surface area [TBSA], third degree) and inhalation injury (48 breaths of cotton smoke, <40°C). Sham animals received a fake injury under halothane anesthesia. After burn and smoke inhalation or fake injury procedure, all sheep were awakened and placed on a ventilator with positive end expiratory pressure set to 5 cm H2O and tidal volume maintained at 15 mL/kg. The sheep were ventilated with 100% oxygen for the first 3 hrs after the injury for rapid clearance of carboxyhemoglobin. The fraction of inspired oxygen (Fio2) was adjusted according to blood gas analysis to maintain PaO2 above 80 mm Hg. Respiratory rate was initially set at 30 breaths/min and thereafter was adjusted to keep PaCO2 between 25 and 35 mm Hg. All sheep were fluid resuscitated with Ringer's solution using the formula 4 mL/kg/% burned body surface for 24 hrs. The experiment continued for 24 hrs. The treated group (nNOS) was given a continuous infusion of nNOS inhibitor, ZK 234238 (3-methyl-2-amino-6,7,8,9-tetrahydro[2,1b]-1,4-oxazine). The nNOS inhibitor was started 1 hr postinjury and infused for 24 hrs with a dose of 100 μg/kg/hr. Half maximum inhibitory concentration for nNOS, eNOS, and iNOS is 0.38 μM, 11.5 μM, and 200 μM, respectively. ZK 234238 was a kind gift from Schering AG, Berlin, Germany. The sham and control groups were treated with the same amount of normal saline as the treated group.
The Animal Care and Use Committee of the University Texas Medical Branch approved the experimental protocol, and all the animals were handled according to guidelines established by the American Physiology Society and the National Institutes of Health.
Measured Variables
Hemodynamics were monitored continuously. Arterial and mixed venous blood samples were taken at different time points for the measurement of blood gases (Blood gas analyzer 1302 interleukin [IL], Instrumental Laboratory, Lexington, MA). PaO2/FiO2 ratio was measured to assess pulmonary gas exchange. Pulmonary shunt fraction (Qs/Qt) was calculated using standard equations.
Lung Wet-to-Dry Weight Ratio
Sheep were killed under deep anesthesia 24 hrs after injury. The right lung was then removed and the lower half of the lower lobe was used for determination of bloodless wet-to-dry weight ratio (14).
Measurement of Plasma Nitrite/Nitrate
The plasma NO levels were evaluated by measuring the intermediate and end products, nitrate/nitrite as described previously (11). Briefly, for conversion of nitrate/nitrite to NO, the plasma samples were mixed with vanadium (III) at 95°C in the Nitrite/Nitrate (NOx) reduction assembly (ANTEK Model 745, Antek Instruments, Houston, TX). The resulting NO was then detected and quantitated using Nitric Oxide Analyzer (ANTEK Model 7020, Antek Instruments, Houston, TX). The NO content was expressed as μmol/L.
Lung Myeloperoxidase Activity
The myeloperoxidase (MPO) activity was evaluated in homogenized right lung with a commercially available assay (CytoStore, Calgary, AB, Canada) (15).
Measurement of Lung Tissue IL-8 messenger (mRNA) Levels
IL-8 mRNA (reverse transcriptase-polymerase chain reaction) was determined in lung tissue 24 hrs postinjury as described previously (16). All results were expressed as copy numbers per μg of total RNA.
Immunohistochemistry of Poly(ADP-Ribose) Polymerase (PARP) Activity
For the immunohistochemical detection of poly(ADP-ribose), mouse monoclonal anti-protease-activated receptor (PAR) antibody (Calbiochem, San Diego, CA) was used. The intensity of PAR staining of individual sections was determined by a blinded experimenter according to a semiquantitative PAR-positivity score from 1 to 10 (17, 18).
Western Blot Analysis
iNOS and 3-nitrotyrosine protein expression in lung tissue was determined using anti-iNOS/NOS2 N-20 (sc-651) and monoclonal biotinylated anti-3-nitrotyrosine antibodies, respectively, as described previously (19). Blots were quantified by National Institutes of Health IMAGE J (Image and Processing and Analysis in Java) scanning densitometry, and normalized to total actin expression.
Statistical Analysis
Data are presented as means ± sem. Results were compared between groups using repeated measures (analysis of variance) and the Newman-Keuls multiple comparison tests. A value of p < 0.05 was accepted as statistically significant.
RESULTS
All animals in the three study groups survived throughout the experimental time period. The average carboxyhemoglobin levels after the smoke inhalation was 65%±10% and 68%±7% (not statistically different) in inured treated and nontreated groups respectively, indicating a similar degree of injury in both injured groups.
Hemodynamics
Hemodynamic variables were stable in sham animals. Mean arterial and central venous pressures were similar in all three groups, and the values were not significantly altered compared to baseline. Heart rate and pulmonary artery pressure were significantly elevated in injured, nontreated control animals. Treatment with nNOS inhibitor tended to decrease these increased values. However, there were no statistical differences found between the groups. Cardiac index and left ventricle stroke work index was significantly decreased at 3 hrs postinjury in the control group compared to sham. nNOS inhibition did not significantly affect these changes. Treated animals showed a slightly higher left ventricle stroke work index than control animals (Table 1).
Table 1.
Cardiovascular variables
| Time (hr) |
||||||
|---|---|---|---|---|---|---|
| Variable/Group | Baseline | 3 | 6 | 12 | 18 | 24 |
| Mean arterial pressure (mm Hg) | ||||||
| Sham | 102 ± 4.0 | 111 ± 4.0 | 111 ± 4.4 | 107 ± 3.6 | 103 ± 2.2 | 103 ± 2.2 |
| Control | 97 ± 4.6 | 108 ± 8.1 | 112 ± 7.3 | 113 ± 9.8 | 113 ± 9.6 | 107 ± 7.3 |
| nNOS inhibitor | 96 ± 3.1 | 103 ± 2.5 | 110 ± 3.5 | 105 ± 5.4 | 101 ± 4.9 | 99 ± 4.3 |
| Mean pulmonary arterial pressure (mm Hg) | ||||||
| Sham | 20 ± 0.8 | 23 ± 0.4 | 24 ± 0.1 | 24 ± 0.5 | 24 ± 0.2 | 22 ± 0.7 |
| Control | 20 ± 0.4 | 23 ± 0.4 | 24 ± 0.7 | 25 ± 0.7 | 27 ± 1.6 | 28 ± 1.6a |
| nNOS inhibitor | 20 ± 0.3 | 25 ± 1.7 | 25 ± 0.9 | 27 ± 1.8 | 26 ± 1.9 | 25 ± 1.0 |
| Central venous pressure (mm Hg) | ||||||
| Sham | 8.1 ± 0.5 | 10.8 ± 0.4 | 10.1 ± 0.6 | 10.0 ± 0.9 | 10.1 ± 1.1 | 9.5 ± 0.5 |
| Control | 7.3 ± 0.8 | 9.0 ± 0.5 | 9.3 ± 0.8 | 8.3 ± 0.9 | 8.7 ± 1.0 | 8.5 ± 1.1 |
| nNOS inhibitor | 7.4 ± 1.1 | 9.5 ± 0.3 | 10.0 ± 0.9 | 10.2 ± 1.0 | 9.3 ± 1.1 | 10.5 ± 1.5 |
| Left ventricle stroke work index (g/m/m2) | ||||||
| Sham | 84 ± 4.9 | 97 ± 7.4 | 90 ± 4.8 | 84 ± 6.7 | 78 ± 4.3 | 79 ± 5.8 |
| Control | 77 ± 2.6 | 42 ± 4.8a | 58 ± 6.3a | 61 ± 7.0 | 59 ± 7.5 | 53 ± 7.3 |
| nNOS inhibitor | 85 ± 4.3 | 62 ± 6.6a | 77 ± 4.9 | 73 ± 5.4 | 67 ± 7.4 | 64 ± 3.9 |
| Cardiac index (L/min/m2) | ||||||
| Sham | 6.29 ± 0.07 | 7.11 ± 0.15 | 6.69 ± 0.24 | 6.10 ± 0.26 | 5.96 ± 0.15 | 6.00 ± 0.13 |
| Control | 6.32 ± 0.18 | 5.36 ± 0.30a | 6.06 ± 0.30 | 6.08 ± 0.50 | 6.24 ± 0.47 | 6.00 ± 0.42 |
| nNOS inhibitor | 6.41 ± 0.25 | 5.32 ± 0.26a | 5.79 ± 0.43 | 6.42 ± 0.15 | 6.01 ± 0.20 | 5.89 ± 0.13 |
| Heart rate (beats/min) | ||||||
| Sham | 94 ± 1.3 | 99 ± 4.2 | 98 ± 3.3 | 94 ± 5.2 | 102 ± 5.7 | 94 ± 4.8 |
| Control | 91 ± 2.4 | 151 ± 12a | 125 ± 12 | 114 ± 7.6 | 122 ± 6.7 | 131 ± 11a |
| nNOS inhibitor | 90 ± 2.0 | 111 ± 10 | 102 ± 8.1 | 115 ± 9.1 | 118 ± 12 | 121 ± 4.4 |
nNOS, neuronal nitric oxide synthase.
Values are means ± SE.
p < 0.05 vs. Sham group.
Effect of nNOS Inhibition on Vascular Leakage
Vascular leakage was indirectly evaluated by measuring plasma protein and oncotic pressure, hematocrit and fluid accumulation (Table 2). Despite similar fluid resuscitation, control animals displayed significantly increased hematocrit and fluid accumulation compared to sham animals. Although there was no statistical difference found vs. control group, the hematocrit was maintained at baseline in the nNOS inhibitor treated group. The animals treated with nNOS inhibitor showed a significantly lower fluid accumulation at 24 hrs postinjury vs. control group. The nNOS inhibitor significantly attenuated the decreased levels of plasma protein and oncotic pressure (p = 0.02, 0.04, and 0.03 at 12, 18, and 24 hrs postinjury, respectively) seen in control animals.
Table 2.
Vascular leakage
| Time (hr) |
||||||
|---|---|---|---|---|---|---|
| Variable/Group | Baseline | 3 | 6 | 12 | 18 | 24 |
| Plasma protein (g/dL) | ||||||
| Sham | 5.1 ± 0.2 | 4.5 ± 0.1 | 4.7 ± 0.2 | 4.7 ± 0.1 | 4.7 ± 0.2 | 4.9 ± 0.2 |
| Control | 5.2 ± 0.1 | 3.9 ± 0.2 | 3.4 ± 0.1a | 3.3 ± 0.1a | 3.1 ± 0.2a | 3.0 ± 0.2a |
| nNOS inhibitor | 5.4 ± 0.1 | 4.6 ± 0.2 | 4.3 ± 0.2 | 4.1 ± 0.3a,b | 4.4 ± 0.3† | 4.3 ± 0.2a,b |
| Plasma oncotic pressure (mmHg) | ||||||
| Sham | 22.5 ± 0.9 | 21.1 ± 0.4 | 21.5 ± 0.3 | 22.0 ± 0.5 | 22.1 ± 0.3 | 21.1 ± 0.3 |
| Control | 21.9 ± 0.8 | 15.6 ± 0.6a | 13.6 ± 0.6a | 13.4 ± 0.7a | 12.6 ± 1.0a | 11.8 ± 1.1a |
| nNOS inhibitor | 23.3 ± 1.2 | 16.7 ± 0.3a | 15.8 ± 0.2a | 16.0 ± 0.5a,b | 15.1 ± 0.7a,b | 15.0 ± 0.9a,b |
| Fluid net balance (mL/kg) | ||||||
| Sham | 0 ± 0 | 3.6 ± 2.9 | 27.6 ± 4.0 | 41.5 ± 2.8 | 42.4 ± 1.7 | 25.3 ± 2.4 |
| Control | 0 ± 0 | 17.2 ± 2.1 | 40.4 ± 4.6 | 60.8 ± 4.1a | 74.1 ± 7.2a | 86.6 ± 10.1a |
| nNOS inhibitor | 0 ± 0 | 20.4 ± 0.3 | 40.1 ± 2.1 | 50.3 ± 4.7 | 59.9 ± 5.8 | 61.2 ± 9.0a,b |
| Hematocrit (%) | ||||||
| Sham | 26.5 ± 0.6 | 25.3 ± 0.9 | 25.5 ± 0.6 | 26.0 ± 0.7 | 26.5 ± 0.7 | 26.5 ± 0.5 |
| Control | 26.6 ± 1.3 | 29.1 ± 1.2 | 28.5 ± 1.5 | 28.8 ± 1.0 | 30.5 ± 0.9a | 32.3 ± 0.8a |
| nNOS inhibitor | 27.7 ± 2.5 | 28.5 ± 2.9 | 26.0 ± 1.0 | 27.7 ± 1.3 | 27.5 ± 1.2 | 28.2 ± 0.7 |
nNOS, neuronal nitric oxide synthase.
Values are means ± SE.
p < 0.05 vs. Sham group
p < 0.05 vs. Control group.
Effect of nNOS Inhibition on Pulmonary Gas Exchange
Pulmonary gas exchange (PaO2/Fio2 and pulmonary shunt fraction) was stable in sham animals. Combined burn and smoke inhalation caused deterioration of pulmonary gas exchange, as evidenced by a reduced PaO2/Fio2 ratio (Fig. 1A) associated with a marked increase in pulmonary shunt fraction (Fig. 1B) in saline-treated control animals. PaO2/Fio2 ratio in these animals reached a sub300 level at 24 h postinjury, evidencing the presence of severe ALI. Treatment with nNOS inhibitor prevented the impaired gas exchange caused by burn and smoke inhalation as PaO2/Fio2 stayed above 400 in these animals throughout the experimental period. Furthermore, the increased pulmonary shunt fraction was almost reversed by nNOS inhibition (p < 0.001 at 18 and 24 hrs postinjury).
Figure 1.

Effects of neuronal nitric oxide synthase (nNOS) inhibitor on pulmonary gas exchange evaluated by measuring PaO2/FIO2 ratio (A) and pulmonary shunt fraction (Qs/Qt) (B). Data expressed as mean ± SEM *p < 0.05 vs. sham, and †p < 0.05 vs. saline.
Effect of nNOS Inhibition on Lung Water Content
Lung water content was evaluated by measuring bloodless lung wet-to-dry weight ratio. The lung wet-to-dry weight ratio was significantly increased in saline-treated control animals compared to sham animals. However, this increase in lung water content seen in control animals was significantly attenuated by treatment with nNOS inhibitor (Fig. 2).
Figure 2.

Effects of neuronal nitric oxide synthase (nNOS) inhibitor on lung wet-to-dry weight ratio, an index of lung water content. Data expressed as mean ± SEM *p < 0.05 vs. sham, and †p < 0.05 vs. saline.
Effect of nNOS Inhibition on Lung Tissue Expression of IL-8 mRNA and Leukocyte Accumulation
Lung tissue IL-8 mRNA was significantly increased in saline-treated control animals. The nNOS inhibitor reversed this increase to the normal level (Fig. 3A).
Figure 3.

Effects of neuronal nitric oxide synthase (nNOS) inhibitor on lung tissue interleukin (IL)-8 messenger myeloperoxidase RNA (A) and myeloperoxidase (MPO) activity. Data expressed as mean ± SEM *p < 0.05 vs. sham, and †p < 0.05 vs. saline.
Lung tissue leukocyte accumulation was estimated by measuring myeloperoxidase activity (Fig. 3B), which was significantly increased in control animals compared to those in sham group at 24 hrs after injury. However, the increase was significantly attenuated by post-treatment with nNOS inhibitor.
Effect of nNOS Inhibition on the Lung Tissue 3-Nitrotyrosine Level
Lung tissue 3-nitrotyrosine levels were markedly increased in control animals compared to sham. However, this increase was significantly inhibited by the nNOS inhibitor, ZK 234238 (Fig. 4).
Figure 4.
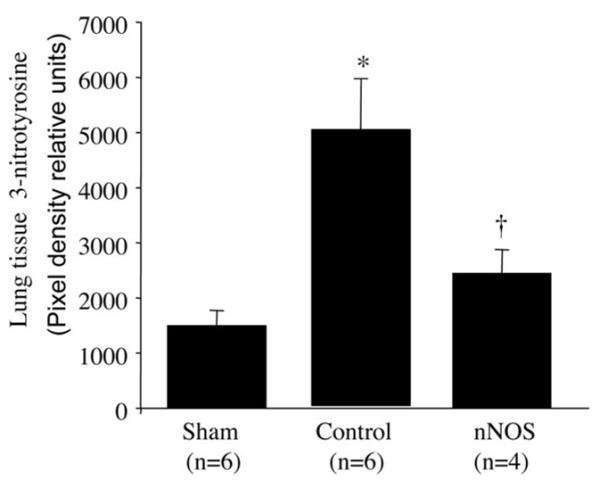
Effects of neuronal nitric oxide synthase (nNOS) inhibitor on lung tissue 3-nitrotyrosine level. Lung tissue 3-nitrotyrosine was measured by Western blotting assay and shown as densitometric values. Data expressed as mean ± SEM *p < 0.05 vs. sham and †p < 0.05 vs. saline.
Effect of nNOS Inhibition on Poly(ADP-Ribose) Polymerase Activity
Poly(ADP-ribose) polymerase (PARP) activity was determined by immunohistochemistry staining technique. Data are expressed as semiquantitative scores. As shown in Figure 5, PARP activity was significantly increased following combined burn and smoke inhalation injury in airway (Fig. 5A), airway glands (Fig. 5B), alveoli (Fig. 5C), and vessels (Fig. 5D). nNOS inhibitor significantly attenuated the above changes.
Figure 5.

Effect of neuronal nitric oxide synthase (nNOS) inhibitor on poly(ADP-ribose) polymerase activation in airway (A), airway gland (B), alveoli (C), and vessels (D). The poly(ADP-ribose) polymerase activation was evaluated by immunohstochemical technique, and shown as densitometric values. Data expressed as mean ± SEM *p < 0.05 vs. sham and †p < 0.05 vs. saline.
Effect of nNOS Inhibitor on Plasma Levels of NOx and Lung Tissue iNOS Protein Expression
Excessive production of NO was evaluated by measuring its stable metabolites, nitrite/nitrate. Plasma NOx started to increase in saline-treated control animals at 6 hrs after injury reaching a peak at 12 hrs and stayed elevated throughout the study period. Post-treatment with nNOS inhibitor reversed the increased levels of plasma NOx to normal values (Fig. 6A).
Figure 6.

Effect of neuronal nitric oxide synthase (nNOS) inhibitor on plasma levels of nitrite/nitrate, a stable metabolite of nitric oxide (A), and lung tissue inducible NOS (iNOS) protein expression (B). The iNOS protein level measured by Western blotting assay in lung tissue is shown as densitometric values. Data expressed as mean ± SEM *p < 0.05 vs. sham and †p < 0.05 vs. saline.
Figure 6B illustrates the effect of nNOS inhibitor, ZK 234238, on iNOS protein expression in lung tissue (Western blot) 24 hrs after injury. The increase in iNOS protein expression in control animals was blocked by nNOS inhibition.
DISCUSSION
ARDS is a significant cause of morbidity and mortality in critically ill patients including burn victims. In previous studies, we have demonstrated a potential role of iNOS-derived NO in the pathogenesis of burn and smoke-related cardiopulmonary morbidity (11–13). Despite the importance of iNOS in pathologic alterations, however, recent research has demonstrated that the constitutive NOS isoforms may also be important (4, 20). In our recent work, we have described a major role of nNOS-derived NO in sepsis-related cardiovascular collapse in sheep (8). In this study, we hypothesized that inhibition of nNOS-derived NO may also be beneficial in attenuating pulmonary dysfunction following cutaneous burn and smoke inhalation. For this purpose, we have tested the effects of potent and specific nNOS inhibitor, ZK 234238. Selectivity of the compound is ~30 and 500 times higher for eNOS and iNOS, respectively. At the concentration used in this study, we believe that the compound inhibited only nNOS-derived NO without significantly affecting the other isoforms. In support, ZK 234238 completely prevented the increase in plasma NOx seen in untreated animals. The nNOS inhibition also significantly attenuated pulmonary dysfunction, which was evidenced by decreased PaO2/Fio2 and increased pulmonary shunt fraction. It is worthwhile to note that nNOS inhibition resulted also in significantly less lung water content, associated with less accumulation of fluid and reduced hematocrit despite the similar fluid resuscitation. In addition, the treated group had a significant higher plasma protein and oncotic pressure, suggesting that nNOS inhibitor reduced the vascular hyperpermeability to protein. The exact mechanism by which nNOS inhibition reduced the vascular leakage is not completely understood. Excessive NO itself is recognized as a potent permeability factor (21, 22). It was also shown that NO causes the vascular hyperpermeability by promoting expression of potent permeability factor–vascular endothelial growth factor (23). We have recently reported that recombinant human antithrombin reduced pulmonary edema by inhibiting lung tissue vascular endothelial growth factor (24).
Previous studies have well documented the circumstantial evidence of the role of activated neutrophils in ALI and ARDS (25). We have also reported that depletion of the neutrophils with nitrogen mustard significantly decreased the smoke-induced increase in lung microvascular permeability to protein (26). Cytokines such as IL-8 play an important role in activation of neutrophils. IL-8, a major chemotactic factor for neutrophil, has been shown to mediate the lung tissue injury in various pathologic conditions. Laffon et al (27) reported that pre-treatment with a mAb to rabbit IL-8 prevented both lung endothelial and alveolar epithelial injury caused by smoke inhalation in rabbits. In this study we report that inhibition of excessive NO by specific nNOS inhibitor significantly inhibits both lung tissue neutrophil accumulation and IL-8 mRNA expression. Although the exact mechanism is not completely understood, nNOS inhibition may have attenuated the degree of lung tissue injury by inhibiting the accumulation of neutrophils through decreased IL-8 expression.
At high concentration, NO becomes a potential proinflammatory and cytotoxic factor by reacting with superoxide radicals to form a toxic product, peroxynitrite (28). Peroxynitrite, in turn, can oxidize/nitrate other molecules, or decay and produce other damaging species such as hydroxyl or carbonate radicals (29). Recently it was proposed that NO/ONOO−-mediated injury is related to DNA damage and consequent activation of the nuclear repair enzyme PARP (30). After activation by DNA strand breaks, PARP catalyzes ADP-ribose subunits to nuclear proteins. This process depletes the intracellular substrate NAD+ and slows the rate of glycolysis, electron transport, and ATP formation. Thus, excessive PARP activation in response to massive oxidant-induced DNA single-strand breakage leads to cell necrosis (30, 31). Previously, we have reported beneficial effects of a PARP inhibitor, INO-1001, which reduced the degree of ALI induced by cutaneous burn and smoke inhalation (32) and pneumonia/sepsis (33) in sheep. In addition, Endres et al (34) showed that nNOS is an important mediator of PARP activation in cerebral ischemia-reperfusion using nNOS knock-out mice. In this study, nNOS inhibition resulted in significant inhibition of PARP in lung tissue (airway, alveoli, mucus gland, and vessels). Thus, the results of previous and present studies strongly suggest that nNOS inhibitor, ZK 234238 ameliorates the degree of ALI by inhibiting excessive production of NO and neutrophil activation resulting in reduced formation of reactive nitrogen species, such as peroxynitrite, thereby preventing the excessive activation of PARP. Thus, excessive NO derived from nNOS, in part, could contribute to ALI in burn and smoke inhalation model through 1) pulmonary microcirculation disturbance; 2) activation of inflammatory cells; 3) formation of peroxynitrite or other toxic product(s); and 4) induction of PARP.
An important finding of this study is that the inhibition of nNOS resulted in suppression of iNOS protein expression in lung tissue. We have previously documented a major role for iNOS in cardio-pulmonary pathophysiology in the same model of cutaneous burn and smoke inhalation injury using a potent and selective iNOS dimerization inhibitor, BBS-2. BBS-2 significantly inhibited the increase in plasma NOx in that study (11). As mentioned in this study, the nNOS inhibitor almost completely prevented the production of excessive NO as well as iNOS protein expression. These results strongly suggest that nNOS may initiate the induction of iNOS expression. If this is the case, the nNOS inhibitor ZK 234238 may also ameliorate the degree of ALI in part, by preventing the subsequent iNOS induction. Our present results confirm the previous findings by Traber et al (7) that a reported specific nNOS inhibitor, 7-nitroindazole, blocked the lung tissue expression of iNOS mRNA in sheep exposed to combined burn and smoke inhalation injury. Recently, Iijima et al (4) found that nNOS is required for allergen-induced expression of iNOS in mice. The authors showed that ovalbumin significantly increased the iNOS activity in wild type mice, but not in nNOS knockout mice. They noted also that lipopolysaccharide challenge increased the iNOS activity in both wild type and nNOS knockout mice, confirming that nNOS-deficient mice can increase iNOS expression. These results suggest that interaction of nNOS and iNOS is stimulus specific.
Finally, the results of previous and our present studies provides a strong evidence that nNOS-derived NO may have a profound role in ALI in sheep exposed to cutaneous burn and smoke inhalation injury. Despite beneficial effects on ALI, nNOS inhibition did not significantly affect the burn and smoke inhalation induced hemodynamic changes. Although, the precise mechanism is not completely elucidated, we do not exclude the possible involvement of catecholamines (35) and various myocardial depressant factors.
Nevertheless, our data indicates that the use of specific nNOS inhibitors may be a novel alternative in the management ALI in burn victims and especially in those who have a concomitant smoke inhalation injury.
Acknowledgments
Supported, in part, by grants 84504, 8954, GM066312, GM060688 from Shrine's of North America.
Footnotes
See also p. 361.
The authors have not disclosed any potential conflicts of interest.
REFERENCES
- 1.Kerwin JF, Jr, Lancaster JR, Feldman PL. Nitric oxide: A new paradigm for second messengers. J Med Chem. 1995;38:4342–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- 3.Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 4.Iijima H, Tulic MK, Duguet A, et al. NOS 1 is required for allergen-induced expression of NOS 2 in mice. Int Arch Allergy Immunol. 2005;138:40–50. doi: 10.1159/000087356. [DOI] [PubMed] [Google Scholar]
- 5.Jang AS, Choi IS, Lee JU. Neuronal nitric oxide synthase is associated with airway obstruction in BALB/c mice exposed to ozone. Respiration. 2003;70:95–99. doi: 10.1159/000068406. [DOI] [PubMed] [Google Scholar]
- 6.Yonamine CM, Troncone LRP, Camillo MAP. Blocade of neuronal nitric oxide synthase abolishes the toxic effects of Tx2-5, a lethal phoneutria nigriventer spider toxin. Toxicon. 2004;44:169–172. doi: 10.1016/j.toxicon.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Traber DL, Hawkins HK, Enkhbaatar P, et al. The role of bronchial circulation in the acute lung injury resulting from burn and smoke inhalation. Pulm Pharmacol Ther. 2007;20:163–166. doi: 10.1016/j.pupt.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Enkhbaatar P, Murakami K, Shimoda K, et al. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole attenuates acute lung injury in an ovine model. Am J Physiol Regul Integr Comp Physiol. 2003;285:R366–R372. doi: 10.1152/ajpregu.00148.2003. [DOI] [PubMed] [Google Scholar]
- 9.De Sanctis GT, Mehta S, Kobzik L, et al. Contribution of type 1 NOS to expired gas NO and bronchial responsiveness in mice. Am J Physiol. 1997;273:L883–L888. doi: 10.1152/ajplung.1997.273.4.L883. [DOI] [PubMed] [Google Scholar]
- 10.Chen LW, Chang WJ, Wang JC, et al. Thermal injury-induced peroxynitrite production and pulmonary inducible nitric oxide synthase expression depend on JNK/AP-1 signaling. Crit Care Med. 2006;34:142–150. doi: 10.1097/01.ccm.0000190621.48720.8c. [DOI] [PubMed] [Google Scholar]
- 11.Enkhbaatar P, Murakami K, Shimoda K, et al. The inducible nitric oxide synthase inhibitor BBS-2 prevents acute lung injury in sheep after burn and smoke inhalation injury. Am J Respir Crit Care Med. 2003;167:1021–1026. doi: 10.1164/rccm.200209-1031PP. [DOI] [PubMed] [Google Scholar]
- 12.Enkhbaatar P, Murakami K, Shimoda K, et al. Inducible nitric oxide synthase dimerization inhibitor prevents cardiovascular and renal morbidity in sheep with combined burn and smoke inhalation injury. Am J Physiol Heart Circ Physiol. 2003;285:H2430–H2436. doi: 10.1152/ajpheart.00055.2003. [DOI] [PubMed] [Google Scholar]
- 13.Soejima K, Traber LD, Schmalstieg FC, et al. Role of nitric oxide in vascular permeability after combined burns and smoke inhalation injury. Am J Respir Crit Care Med. 2001;163:745–752. doi: 10.1164/ajrccm.163.3.9912052. [DOI] [PubMed] [Google Scholar]
- 14.Pearce ML, Yamashita J, Beazell J. Measurement of pulmonary edema. Circ Res. 1965;16:482–488. doi: 10.1161/01.res.16.5.482. [DOI] [PubMed] [Google Scholar]
- 15.Hollingsworth JW, Chen BJ, Brass DM, et al. The critical role of hematopoietic cells in lipopolysaccharide-induced airway inflammation. Am J Respir Crit Care Med. 2005;171:806–813. doi: 10.1164/rccm.200407-953OC. [DOI] [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 17.Toth-Zsamboki HE, Vargova K, Pankotai E, et al. Activation of poly(ADP-ribose) polymerase by myocardial ischemia and coronary reperfusion in human circulating leukocytes. Mol Med. 2006;12:221–228. doi: 10.2119/2006-00055.Toth-Zsamboki. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beller CJ, Radovits T, Kosse J, et al. Activation of the peroxynitrite-poly (adenosine diphosphate-ribose) polymerase pathway during neointima proliferation: A new target to prevent restenosis after endarterectomy. J Vasc Surg. 2006;43:824–830. doi: 10.1016/j.jvs.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 19.Gillett AM, Wallace MJ, Gillespie MT, et al. Increased expansion of the lung stimulates calmodulin 2 expression in fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2002;282:L440–L447. doi: 10.1152/ajplung.00202.2001. [DOI] [PubMed] [Google Scholar]
- 20.Vo PA, Lad B, Tomlinson JAP, et al. Autoregulatory role of endothelial-derived nitric oxide (NO) on lipopolysaccharide-induced vascular inducible NO synthase expression and function. J Biol Chem. 2005;280:7236–7243. doi: 10.1074/jbc.M411317200. [DOI] [PubMed] [Google Scholar]
- 21.Rumbaut RE, Huxley VH. Similar permeability responses to nitric oxide synthase inhibitors of venules from three animal species. Microvasc Res. 2002;64:21–31. doi: 10.1006/mvre.2002.2394. [DOI] [PubMed] [Google Scholar]
- 22.Huang Q, Yuan Y. Interaction of PKC and NOS in signal transduction of microvascular hyperpermeability. Am J Physiol Heart Circ Physiol. 1997;273:H2442–H2451. doi: 10.1152/ajpheart.1997.273.5.H2442. [DOI] [PubMed] [Google Scholar]
- 23.Brkovic A, Sirois MG. Vascular permeability induced by VEGF family members in vivo: Role of endogenous PAF and NO synthesis. J Cell Biochem. 2007;100:727–737. doi: 10.1002/jcb.21124. [DOI] [PubMed] [Google Scholar]
- 24.Enkhbaatar P, Cox RA, Traber LD, et al. Combined anticoagulants ameliorate acute lung injury in sheep after burn and smoke inhalation. Clin Sci (Lond) 2008;114:321–329. doi: 10.1042/CS20070254. [DOI] [PubMed] [Google Scholar]
- 25.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 26.Basadre JO, Sugi K, Traber DL, et al. The effect of leukocyte depletion on smoke inhalation injury in sheep. Surgery. 1988;104:208–215. [PubMed] [Google Scholar]
- 27.Laffon M, Pittet J-F, Modelska K, et al. Interleukin-8 mediates injury from smoke inhalation to both lung endothelial and the alveolar epithelial barriers in rabbits. Am J Respir Crit Care Med. 1999;160:1443–1449. doi: 10.1164/ajrccm.160.5.9901097. [DOI] [PubMed] [Google Scholar]
- 28.Hughes MN. Relationships between nitric oxide, nitroxyl ion, nitrosonium cation and peroxynitrite. Biochim Biophys Acta. 1999;1411:263–272. doi: 10.1016/s0005-2728(99)00019-5. [DOI] [PubMed] [Google Scholar]
- 29.Miranda K, Espey MG, Jourd'heuil D, et al. The chemical biology of nitric oxide. In: Ignarro LJ, editor. Nitric Oxide: Biology and Pathophysiology. Academic Press; San Diego: 2000. pp. 41–45. [Google Scholar]
- 30.Zhang J, Dawson VL, Dawson TM, et al. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 31.Szabo C, Cuzzocrea S, Zingarelli B, et al. Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J Clin Invest. 1997;100:723–735. doi: 10.1172/JCI119585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shimoda K, Murakami K, Enkhbaatar P, et al. Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep. Am J Physiol Lung Cell Mol Physiol. 2003;285:L240–L249. doi: 10.1152/ajplung.00319.2002. [DOI] [PubMed] [Google Scholar]
- 33.Murakami K, Enkhbaatar P, Shimoda K, et al. Inhibition of poly (ADP-ribose) polymerase attenuates acute lung injury in an ovine model of sepsis. Shock. 2004;21:126–133. doi: 10.1097/01.shk.0000108397.56565.4a. [DOI] [PubMed] [Google Scholar]
- 34.Endres M, Scott G, Namura S, et al. Role of peroxynitrite and neuronal nitric oxide synthase in the activation of poly(ADP-ribose) synthetase in a murine model of cerebral ischemia-reperfusion. Neurosci Lett. 1998;248:41–44. doi: 10.1016/s0304-3940(98)00224-9. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura T, Hayashida Y. Autonomic cardiovascular responses to smoke exposure in conscious rats. Am J Physiol. 1992;262:R738–R745. doi: 10.1152/ajpregu.1992.262.5.R738. [DOI] [PubMed] [Google Scholar]