

Abstract
DNA methyltransferases (DNMTs) are promising epigenetic targets for the development of novel anticancer drugs and other diseases. Molecular modeling and experimental approaches are being used to identify and develop inhibitors of human DNMTs. Most of the computational efforts conducted so far with DNMT1 employ homology models of the enzyme. Recently, a crystallographic structure of the methyltransferase domain of human DNMT1 bound to unmethylated DNA was published. Following on our previous computational and experimental studies with DNMTs, we herein present molecular dynamics of the crystal structure of human DNMT1. Docking studies of established DNMT1 inhibitors with the crystal structure gave rise to a structure-based pharmacophore model that suggests key interactions of the inhibitors with the catalytic binding site. Results had a good agreement with the docking and pharmacophore models previously developed using a homology model of the catalytic domain of DNMT1. The docking protocol was able to distinguish active DNMT1 inhibitors from, for example, experimentally known inactive DNMT1 inhibitors. As part of our efforts to identify novel inhibitors of DNMT1, we conducted the experimental characterization of aurintricarboxylic acid (ATA) that in preliminary docking studies showed promising activity. ATA had a submicromolar inhibition (IC50 = 0.68 μM) against DNMT1. ATA was also evaluated for Dnmt3a inhibition showing an IC50 = 1.4 μM. This chapter illustrates the synergy from integrating molecular modeling and experimental methods to further advance the discovery of novel candidates for epigenetic therapies.
Keywords: Cancer, DNMT, Docking, Drug discovery, Epigenetics, Molecular dynamics, Pharmacophore modeling
I. Introduction
The genome contains genetic and epigenetic information. While the genome provides the blueprint for the manufacture of all the proteins required to create a living thing, the epigenetic information provides instruction on how, where, and when the genetic information should be used (Robertson, 2001). The major form of epigenetic information within the DNA molecule itself in mammalian cells is DNA methylation, that is, the covalent addition of a methyl group to the 5-position of cytosine, mostly within the CpG dinucleotides in somatic cells (Robertson, 2001). DNA methylation is involved in the control of gene expression, regulation of parental imprinting, and stabilization of X chromosome inactivation as well as maintenance of the genome integrity. It is also implicated in the development of the immune system, cellular reprogramming, and brain function and behavior (Jurkowska et al., 2011). DNA methylation is mediated by a family of DNA methyltransferase enzymes (DNMTs). In mammals, three DNMTs have been identified so far in the human genome, including the two de novo methyltransferases (DNMT3A and DNMT3B) and the maintenance methyltransferase (DNMT1), which is generally the most abundant and active of the three (Robertson, 2001, Yokochi and Robertson, 2002, Goll and Bestor, 2005). DNMT1 is responsible for duplicating patterns of DNA methylation during replication and is essential for mammalian development and cancer cell growth (Chen et al., 2007). These enzymes are key regulators of gene transcription, and their roles in carcinogenesis have been the subject of considerable interest over the past decade (Robertson, 2001, Jones and Baylin, 2007). Therefore, specific inhibition of DNA methylation is an attractive and novel approach for cancer therapy (Robertson, 2001, Lyko and Brown, 2005, Kelly et al., 2010, Portela and Esteller, 2010). DNA methylation inhibitors have also emerged as a promising strategy for the treatment of immunodeficiency and brain disorders (Zawia et al., 2009, Miller et al., 2010).
Human DNMT1 is a protein with 1616 amino acids whose structure can be divided into an N-terminal regulatory domain and a C-terminal catalytic domain (Cheng and Blumenthal, 2008, Lan et al., 2010, Jurkowska et al., 2011). The mechanism of DNA cytosine-C5 methylation is schematically depicted in Fig. 1 (Vilkaitis et al., 2001, Schermelleh et al., 2005, Sippl and Jung, 2009). DNMT forms a complex with DNA and the cytosine which will be methylated flips out from the DNA. The thiol of the catalytic cysteine acts as a nucleophile that attacks the 6-position of the target cytosine to generate a covalent intermediate. The 5-position of the cytosine is activated and conducts a nucleophilic attack on the cofactor S-adenosyl-l-methionine (AdoMet/SAM) to form the 5-methyl covalent adduct and S-adenosyl-l-homocysteine (AdoHcy/SAH). The attack on the 6-position is assisted by a transient protonation of the cytosine ring at the endocyclic nitrogen atom N3, which is stabilized by a glutamate residue. An arginine residue may participate in the stabilization of the intermediate making a hydrogen bonding interaction with the carbonyl oxygen of cytosine. The covalent complex between the methylated base and the DNA is resolved by deprotonation at the 5-position to generate the methylated cytosine and the free enzyme.
Fig. 1.
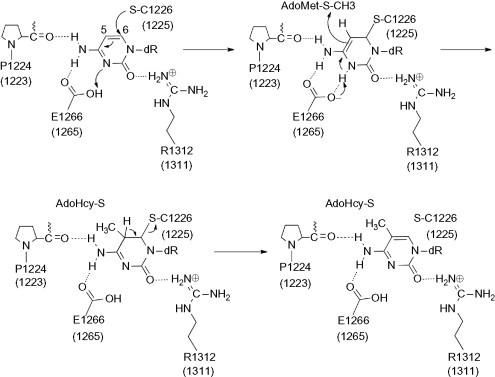
Mechanism of cytosine DNA methylation. Amino acid residue numbers are based on the crystal structure of hDNMT1. Equivalent residue numbers in parentheses correspond to the homology model.
The structure of mammalian DNMTs can be divided into two major parts: a large N-terminal regulatory domain of variable size which has regulatory functions, and a C-terminal catalytic domain which is conserved in eukaryotic and prokaryotic carbon-5 DNMTs (Fig. 2 ). The N-terminal domain guides the nuclear localization of the enzymes and mediates their interactions with other proteins, DNA, and chromatin. The smaller C-terminal domain harbors the active center of the enzyme and contains 10 amino acid motifs diagnostic for all carbon-5 DNMTs (Jurkowska et al., 2011). The catalytic domain has a core structure shared by all DNMTs. This core structure is termed the “AdoMet-dependent MTase fold” and consists of a six-stranded parallel β sheet with a seventh strand inserted in a parallel fashion between the fifth and sixth strands. Six helices are folded around the central β sheet. Motifs I and X of this domain are involved in the cofactor binding. Motifs IV, VI, and VIII are involved in catalysis. The target recognition domain (TRD) is a nonconserved region between motifs VIII and IX that is involved in DNA recognition and specificity (Fig. 2) (Jeltsch, 2002, Jurkowska et al., 2011).
Fig. 2.
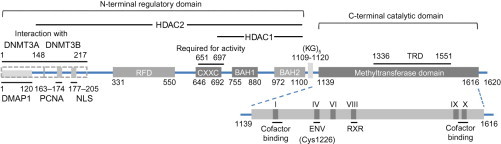
Schematic representation of human DNMT1. NLS, nuclear localization signal; RFD, replication foci-targeting sequence; BAH, bromo-adjacent homology domain; TRD, target recognition domain. Interaction domains of HDAC1, HDAC2, and the DNMT3s are indicated. The methyltransferase domain comprising six most conserved motifs is enlarged.
Three-dimensional structural information for human DNMT1 is limited. Just recently, a crystal structure of human DNMT1 bound to duplex DNA containing unmethylated cytosine–guanine (CG) sites was published (Song et al., 2011). However, the conformation of the crystal structure is in an “inactive state” (the target base from DNA is not flipped out) so that the geometry of the catalytic site does not reflect the catalytic mechanism. Further details of the structure of DNMTs and other available crystal structures of DNMTs are extensively reviewed elsewhere (Cheng and Blumenthal, 2008, Sippl and Jung, 2009, Lan et al., 2010).
DNA methylation inhibitors have been well characterized and tested in clinical trials (Issa and Kantarjian, 2009). To date, only 5-azacytidine and 5-aza-2′-deoxycytidine (decitabine) have been developed clinically. These two drugs are nucleoside analogues, which, after incorporation into DNA, cause covalent trapping and subsequent depletion of DNMTs (Schermelleh et al., 2005, Stresemann and Lyko, 2008). Aza nucleosides are approved by the Food and Drug Administration of the United States for the treatment of myelodysplastic syndrome, where they demonstrate significant, although usually transient, improvement in patient survival and are currently being tested in many solid cancers (Issa et al., 2005, Schrump et al., 2006). However, aza nucleosides have relatively low specificity and are characterized by substantial cellular and clinical toxicity (Stresemann and Lyko, 2008). Their exact mechanism of antitumor action also remains unclear (Issa, 2005, Palii et al., 2008, Fandy et al., 2009). Consequently, there is clear need to identify novel and more specific DNMT inhibitors that do not function via incorporation into DNA.
There are now an increasing number of substances that are reported to inhibit DNMTs (Lyko and Brown, 2005). Compounds such as RG108 and NSC14778 (see below) were identified using virtual screening (Siedlecki et al., 2006, Kuck et al., 2010b). Others are approved drugs for other indications such as hydralazine, procaine, and procainamide. These compounds and several natural products implicated in DNA methylation inhibition are extensively reviewed elsewhere (Hauser and Jung, 2008, Gilbert and Liu, 2010, Li and Tollefsbol, 2010, Medina-Franco and Caulfield, 2011).
Molecular docking, pharmacophore modeling, and molecular dynamics (MD) are some of the computational approaches that our and other research groups are using to explore the ligand-binding interactions of DNMT inhibitors (Evans and Bronowska, 2010, Caulfield and Medina-Franco, 2011, Medina-Franco and Caulfield, 2011, Yoo and Medina-Franco, 2011b, Yoo and Medina-Franco, 2011c). Due to the absence of a crystallographic structure of human DNMT1 that was published until recently (Song et al., 2011), molecular modeling studies into the catalytic binding site of DNMTs had been conducted using homology models (Medina-Franco and Caulfield, 2011)
Herein, we describe docking studies of known DNMT1 inhibitors in the catalytic site of the recently published crystal structure of DNMT1 (Song et al., 2011). Prior docking, the conformation of the catalytic site was modeled into an active conformation using MD. To our knowledge, this is the first molecular modeling study conducted with the catalytic binding site of the crystal structure of human DNMT1. Based on the docking results, we developed a structure-based pharmacophore model of DNMT1 inhibitors. In an effort to identify novel nonnucleoside DNMT1 inhibitors, we also report the experimental activity of aurintricarboxylic acid (ATA) that showed submicromolar inhibition against DNMT1. Molecular modeling studies of ATA with the catalytic site of the crystal structure of DNMT1 are also discussed.
II. Molecular Dynamics of the Crystallographic Structure
The recently published X-ray crystal structure of human DNMT1 corresponds to the enzyme bound to DNA-containing unmethylated CpG sites. In this structure, the catalytic loop has an open conformation such that the catalytic cysteine is far away from the binding site. Therefore, the geometry of the catalytic site of the crystal structure does not represent the active conformation of the enzyme that corresponds to the catalytic mechanism of DNA methylation. In contrast, in the X-ray structure of bacterial M.HhaI (Klimasauskas et al., 1994, Kumar et al., 1997) and in our previously developed homology model (Yoo and Medina-Franco, 2011b), the DNA is embedded in the catalytic core and the catalytic loops are also close to the inserted target cytosine in the active site (Fig. 3 ). We therefore used MD to model the crystal structure of the human DNMT1–DNA into an active state. To this end, the crystal structure of the DNMT1–DNA complex with bound AdoHcy was obtained from the Protein Data Bank (PDB code: 3PTA). The unmethylated DNA and other domains, except the methyltransferase domain, were removed. The DNA double helix reconstructed from the structure of M.HhaI (PDB: 1MHT) (Klimasauskas et al., 1994), in the catalytic core, and target cytosine inserted into the active site. The missing residues (1480–1483) were modeled using Prime (version 2.2, Schrödinger LLC, New York, NY, 2010). The refined model was treated according to the Protein Preparation Wizard implemented in Maestro, which optimizes H-bond networks and flip orientations/tautomeric states of Gln, Asn, and His residues, and performed a geometry optimization to a maximum root mean square deviation (RMSD) of 0.3 Å with OPLS2005 force field. We have recently employed a similar procedure to prepare the structure of other proteins (Hernández-Campos et al., 2010). The stability of the refined model was probed by an MD simulation using MacroModel (version 9.8, Schrödinger LLC, New York, NY, 2010). A stochastic dynamics (SD) method was used with the OPLS force field and implicit water solvation under the following conditions: (a) equilibration time 1.0 ps at 300 K, (b) 100 ps of simulation time at 300 K, (c) time steps = 1.5 fs, and (d) 100 conformations stored. To make a direct comparison of the energy of the conformers obtained by SD, the collection of SD conformations was submitted to a multiminimization step with the Polak-Ribiere Conjugate Gradient (PRCG). Convergence in the SD simulation was judged complete by the RMSD of the 100 stored structures and the interaction between key amino acid residues and target cytosine.
Fig. 3.
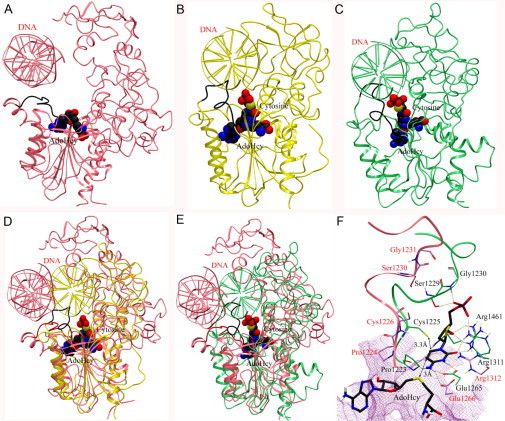
(A) Methyltransferase domain of human DNMT1 (PDB: 3PTA) (pink). (B) Published structure of the M.HhaI–DNA complex (PDB: 1MHT) (yellow). (C) Previously developed homology model of DNMT1 (green). In (A)–(C), the catalytic loops are in black. Superposition of the crystal structure of human DNMT1 (1135–1600)–DNA complex with (D) the M.HhaI–DNA complex and (E) the homology model. The catalytic loop of the crystal structure is in red. AdoHcy and the flipped cytosine are shown in space-filling view. (F) Binding model of deoxycytidine (carbon atoms in black) with key amino acid residues of the crystal structure (carbon atoms in pink) and the homology model (carbon atoms in green). Hydrogen bonding interactions are depicted with dashes.
Figure 4 shows the conformation of the modified X-ray structure with the catalytic loop in an active conformation. Although the RMSD of the Cα atoms of the initial and modified X-ray structure is 1.92 Å, the catalytic loop of the modified structure adopts a different conformation with respect to the X-ray structure. In the X-ray structure, the distance of superimposed target cytosine C6 to the sulfur atom of Cys1226 in the catalytic loop is 9.5 Å (Fig. 4D). In contrast, the catalytic loop of the modified structure is located above the cytosine as an “active site lid.” The target cytosine lies between the nucleophile Cys1226 and sulfur atom of AdoHcy. The distance of cytosine C6 to the sulfur atom of Cys1226 is 3.4 Å, and cytosine C5 atom is 3.5 Å away from the sulfur atom of AdoHcy. In addition, the N3 protonated form of cytosine can make hydrogen bond with the acidic side chain of Glu1266, Pro1224, Arg1310, and Arg1312 (Fig. 4E). The α-phosphate backbone of deoxycytidine makes a hydrogen bond network with Arg1312, Ser1230, Gly1231, and Lys1535. In addition, the 3′-OH of the sugar moiety forms hydrogen bonds with Thr1528 and Gly1577. Interestingly, the key interactions with Glu1266 in the ENV motif, Arg1312 in the RXR motif, Ser1230 and Gly1231 in the catalytic loop are observed in the homology model (Yoo and Medina-Franco, 2011b). The catalytic cores of the modified crystal structure and homology model of DNMT1 (Yoo and Medina-Franco, 2011b) have similar conformation of the catalytic loop. These insights suggest that the modified structure and homology model are reliable to explore the binding mode of inhibitors of human DNMT1.
Fig. 4.
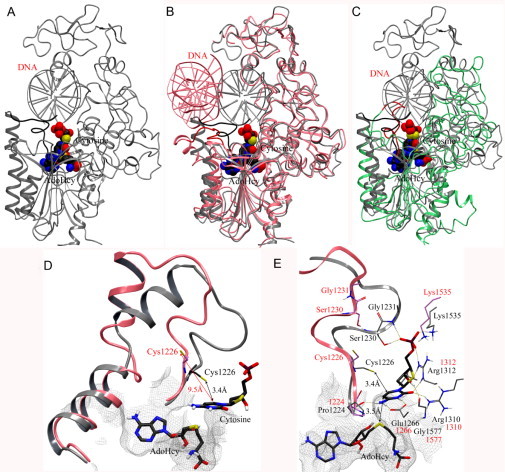
(A) Crystal structure of human DNMT1 (1135–1160)–DNA complex (gray) modified into an active state. The catalytic loop (residues 1224–1235) is highlighted with dark color. (B) Superposition of the modified crystal structure with the initial crystal structure of human DNMT1 (pink) and (C) homology model (green). The catalytic loops of the crystal structure and homology model are in red. AdoHcy and the flipped cytosine are shown in space-filling view. (D) Detail of the conformational change of the catalytic loop from “inactive” state (pink) into an active state (gray). (E) Binding interactions of modeled deoxycytidine (carbon atoms in black) with key amino acid residues in the modified crystal structure (carbon atoms in gray) and crystal structure (carbon atoms in pink). Hydrogen bonding interactions are depicted with dashes.
III. Docking
The docking of known active DNMT inhibitors into a homology model of DNMT1 has been published (Yoo and Medina-Franco, 2011b). In that study, the binding mode of 14 inhibitors (except ATA and NSC97317) was compared for the first time using the same molecular modeling protocol. Despite the fact that the inhibitors have different structural scaffolds, all compounds in that study showed common interactions with key amino acid residues such as Glu1265, Arg1311, Arg1461, Ser1229, and Gly1230. In addition, the insights of the molecular docking supported the proposed mechanism of inhibition of compounds such as curcumin and parthenolide that are putative covalent blockers of the catalytic site (Yoo and Medina-Franco, 2011b).
Building on our previous work, we performed the docking of 16 established inhibitors into the catalytic site of the X-ray structure of hDNMT1 modified to an active conformation (see previous section). For reference (as negative control), we also docked 19 compounds that previously have shown very weak or no enzymatic inhibitory activity or that were used as decoys (NSC4092, NSC21970, NSC57278, NSC19555, NSC27292, and NSC303530; Siedlecki et al., 2006, Kuck et al., 2010b). The chemical structures of both groups of compounds are shown in Fig. 5 . It is worth noting that all 35 compounds in Fig. 5 have not been screened under the same experimental conditions; therefore, a quantitative comparison between the docking scores and enzymatic inhibition is not feasible. However, it is possible to assess whether molecular docking is able to separate actives from inactives/decoys. Also, and more importantly in this case, we seek to explore the putative binding site of established inhibitors into the catalytic binding site using the crystallographic structure of DNMT1.
Fig. 5.
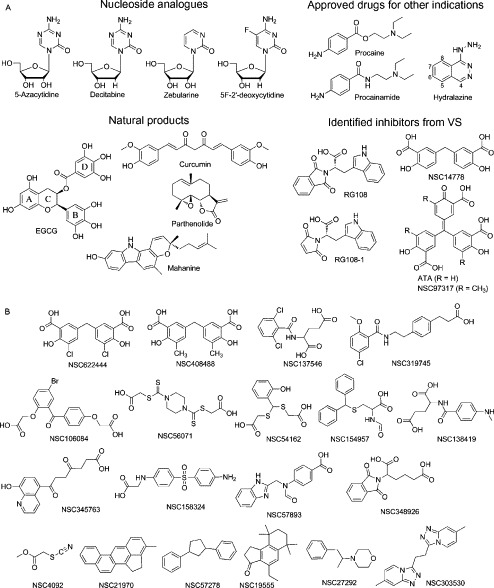
Compounds considered in this study that have been previously tested for inhibition of DNMT1: (A) active and (B) inactive/decoys.
To conduct the docking, the chemical structures of the 35 compounds were built in Maestro 9.1 (Schrödinger LLC, New York, NY, 2010) and minimized with the OPLS2005 force field and gradient termination at 0.001 kJ/mol-Å (MacroModel, 2010). Ligands were prepared with conformational search in MacroModel and LigPrep (version 2.4, Schrödinger LLC, New York, NY, 2010) to generate all the possible configurations (LigPrep, 2010). The conformational analysis was carried out with Monte Carlo Multiple Minimum and Low-Mode conformational search method, employing the OPLS force field using GB/SA water solvation model. The lowest energy conformation of each ligand was docked with the X-ray structure modified to an active conformation (see above) using the program Glide (version 5.6, Schrödinger LLC, New York, NY, 2010). Our group has successfully used Glide to develop binding models of inhibitors of DNMTs (Kuck et al., 2010a, Kuck et al., 2010b, Caulfield and Medina-Franco, 2011, Medina-Franco et al., 2011, Yoo and Medina-Franco, 2011b, Yoo and Medina-Franco, 2011c). Three flexible docking methods, including high-throughput virtual screening (HTVS), standard precision (SP), and extra precision (XP), implemented in Glide were used for comparison. For each method, a receptor grid box of 23 Å on each side, with a default inner box (10 × 10 × 10 Å3), was centered on the target cytosine. Input partial charges were used and no constrains were applied during docking. In the case of the HTVS and SP docking methods, one pose per ligand was intended for large database screening applications, and 10 poses per ligand for XP docking method was set to optimize more precise poses of the top-scoring ligands by HTVS and SP docking. The postdocking minimization was applied to investigate their effect on the accuracy in the three docking protocols: five docking poses per ligand were minimized for HTVS and SP, and 10 poses per ligand for XP. The best docking poses of XP docking were selected to generate the pharmacophore features (see below).
Table I summarizes the results of docking into the catalytic site of the crystal structure using Glide HTVS, SP, and XP. For comparison, the docking scores obtained using the previously reported homology model of DNMT1 (Yoo and Medina-Franco, 2011b) are shown. The docking results were able to predict energetically favorable binding modes with accurate scoring of docking conformations. Docking results with Glide HTVS and SP with the modified crystal structure and homology model showed significant impact on ranking score with remarkable consistency as shown in graph of Table I. In the docking results of HTVS and SP, the ranking of active and that of inactive have similar variations. Almost all active compounds have more stable energy values than inactives. Of note, all known inhibitors except procaine and procainamide ranked higher than decoys. Nucleoside inhibitors including 5-azacytidine and 5-aza-2′-deoxycytidine (decitabine) showed the best Glide XP docking scores using the modified crystallographic and homology models. It is clear from these studies that optimal docking and scoring combination will decrease the number of false positive and false negatives in virtual screening.
Table I.
Summary of Docking Results
| Compound | Homology model |
Crystal structure |
||||
|---|---|---|---|---|---|---|
| HTVS | SP | XP | HTVS | SP | XP | |
| Azacytidine | − 6.72 | − 6.53 | − 9.98 | − 7.58 | − 7.71 | − 11.53 |
| Decitabine | − 5.00 | − 6.03 | − 8.72 | − 7.56 | − 7.82 | − 11.17 |
| EGCG | − 4.67 | − 4.87 | − 9.37 | − 4.83 | − 5.92 | − 11.16 |
| Zebularine | − 5.77 | − 6.62 | − 10.12 | − 6.75 | − 7.17 | − 11.14 |
| 5F-2′-dC | − 6.61 | − 6.52 | − 8.84 | − 7.21 | − 7.65 | − 10.97 |
| Cytosine | − 5.13 | − 6.74 | − 10.03 | − 7.59 | − 7.82 | − 10.63 |
| ATA | − 3.85 | − 4.38 | − 9.52 | − 4.92 | − 6.52 | − 10.19 |
| NSC97317 | − 3.98 | − 4.21 | − 8.42 | − 5.21 | − 6.39 | − 8.89 |
| Curcumin | − 3.99 | − 3.86 | − 9.00 | − 4.68 | − 5.41 | − 8.56 |
| NSC14778 | − 4.25 | − 3.89 | − 7.45 | − 6.62 | − 7.31 | − 7.96 |
| RG108-1 | − 4.38 | − 5.47 | − 7.01 | − 5.04 | − 6.20 | − 6.90 |
| RG108 | − 4.08 | − 5.57 | − 6.67 | − 4.50 | − 5.30 | − 6.70 |
| Hydralazine | − 4.54 | − 4.65 | − 5.05 | − 4.63 | − 5.04 | − 5.51 |
| NSC408488 | − 3.16 | − 3.61 | − 6.22 | − 3.27 | − 3.57 | − 5.49 |
| NSC622444 | − 3.44 | − 3.10 | − 7.13 | − 2.99 | − 3.37 | − 5.25 |
| NSC54162 | − 3.02 | − 2.01 | − 5.90 | − 2.72 | − 1.60 | − 5.25 |
| NSC138419 | − 3.53 | − 4.09 | − 5.56 | − 1.80 | − 2.92 | − 5.19 |
| Mahanine | − 3.94 | − 4.58 | − 5.46 | − 3.93 | − 3.97 | − 5.08 |
| Procaine | − 3.25 | − 2.92 | − 4.83 | − 3.40 | − 3.26 | − 5.08 |
| NSC57893 | − 4.38 | − 3.91 | − 4.74 | − 3.83 | − 3.76 | − 5.05 |
| NSC137546 | − 1.98 | − 1.86 | − 4.34 | − 2.18 | − 3.25 | − 4.88 |
| Parthenolide | − 3.66 | − 3.67 | − 3.52 | − 3.99 | − 4.23 | − 4.77 |
| Procainamide | − 2.84 | − 3.05 | − 4.72 | − 3.04 | − 3.79 | − 4.70 |
| NSC56071 | − 3.95 | − 3.37 | − 4.98 | − 3.10 | − 3.20 | − 4.63 |
| NSC319745 | − 2.96 | − 1.62 | − 3.85 | − 1.52 | − 2.50 | − 4.57 |
| NSC106084 | − 3.37 | − 2.55 | − 1.90 | − 2.29 | − 2.92 | − 4.45 |
| NSC348926 | − 4.34 | − 3.40 | − 4.65 | − 2.57 | − 3.53 | − 4.42 |
| NSC19555 | − 2.66 | − 2.96 | − 3.46 | − 2.96 | − 3.60 | − 4.38 |
| NSC158324 | − 4.27 | − 3.46 | − 3.46 | − 2.46 | − 2.90 | − 4.32 |
| NSC303530 | − 2.41 | − 2.86 | − 3.26 | − 2.85 | − 2.90 | − 4.15 |
| NSC21970 | − 3.10 | − 3.10 | − 3.71 | − 3.31 | − 3.17 | − 3.78 |
| NSC154957 | − 2.80 | − 2.69 | − 1.81 | − 2.18 | − 2.72 | − 3.69 |
| NSC27292 | − 1.90 | − 1.89 | − 3.17 | − 1.91 | − 1.97 | − 3.59 |
| NSC345763 | − 1.93 | − 2.34 | − 3.72 | − 2.37 | − 2.64 | − 3.49 |
| NSC4092 | − 1.51 | − 1.46 | − 2.77 | − 1.49 | − 1.56 | − 2.91 |
 | ||||||
Figure 6 shows the corresponding binding mode of selected DNMT inhibitors generated with Glide XP into the catalytic binding site of the modified crystal structure. The binding modes of nucleoside inhibitors closely match with the target cytidine. The α-phosphate of 5-azacytidine and zebularine makes a hydrogen bond network with Ser1230 and Gly1231 in the catalytic loop, with Lys1535 in the TRD, and Arg1312. The ribose groups form additional hydrogen bonds with Thr1528, Gly1577, and Asn1578. The target base of most nucleoside inhibitors makes key interactions with the conserved glutamate residue in the ENV motif (motif IV: E/Glu1266, N/Asn1267, V/Val1268) and arginine residues in the RXR motif (motif VIII: R/Arg1310, X/1311, R/Arg1312), and Pro1224. Although zebularine does not form hydrogen bonds with Glu1266 and Pro1224 (due to the fact that zebularine lacks the amino group at the C4 position), the distance of 3.7 Å between Cys1226 and C6 is sufficient to start the catalytic mechanism. In addition, the N3 of zebularine and Oε1 of Glu1266 are in an available hydrogen bonding distance for N3 protonation (Fig. 6). These observations are in agreement with the docking poses in the catalytic site of homology model of hDNMT1 (Yoo and Medina-Franco, 2011b). In the homology model, nucleoside inhibitors make hydrogen bond networks with common key amino acid residues, Pro1223, Glu1265 Arg1311, Ser1229, and Gly1230. The RMSD values of the top-scored poses of nucleoside inhibitors between modified crystal structure and homology are less than 1.0 Å. These comparisons show that similar binding modes were obtained in the crystal structure modeled to an active state and the previously generated homology model of hDNMT1. Therefore, these results suggest that both models of DNMT1 are useful for scoring and pose selection for virtual screening. In the next few paragraphs, we describe in more detail the binding mode of representative inhibitors of DNMT1 into the catalytic site of the crystal structure of human DNMT1 modeled into an active conformation.
Fig. 6.
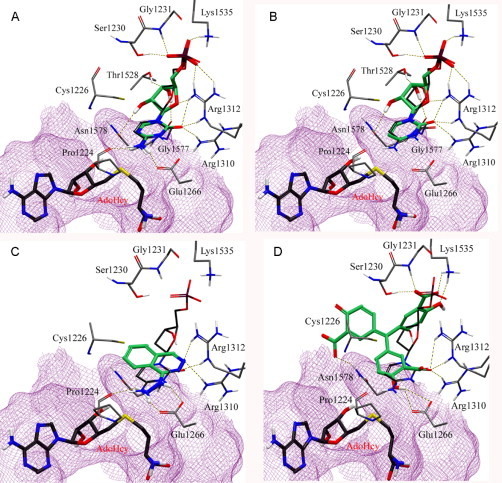
Binding mode of representative inhibitors of DNMT1 (carbon atoms in green) into the catalytic site of the modified X-ray structure of DNMT1. The binding mode of deoxycytidine (carbon atoms in dark gray) is shown for reference: (A) 5-azacytidine, (B) zebularine, (C) hydralazine, (D) ATA.
Hydralazine is an antihypertensive drug and has antitumor effect when used in combination with valproic acid (a histone deacetylase inhibitor) (Arce et al., 2006, Dueñas-González et al., 2008). Figure 6C shows the comparison of the binding modes of hydralazine with target cytidine. The amino group of hydralazine and cytidine matched well, forming a hydrogen bond with Glu1266 and Pro1224. The nitrogen of the phthalazine ring overlapped with the carbonyl oxygen and formed hydrogen bonds with Arg1310 and Arg1312 in the RXR motif. Similar binding mode of hydralazine with low RMSD (0.52 Å) was obtained using a homology model of DNMT1 with same docking protocol (Yoo and Medina-Franco, 2011b). According to the binding pose, the small structure of hydralazine could not occupy the site of the sugar ring and phosphate backbone of nucleoside analogues. We therefore can design analogues which are substituted at the C4 position of hydralazine with enhanced enzymatic affinity (Yoo and Medina-Franco, 2011b).
Natural products, such as epigallocatechin gallate (EGCG), curcumin, parthenolide, and mahanine; inhibitors identified from virtual screening, such as RG108 and its analogue RG108-1; and approved drugs for other indications, such as procaine and procainamide, were docked in the catalytic binding site of modified crystal structure and compared with the binding mode in the homology model. Table II summarizes the common amino acid residues participating in hydrogen bond interactions.
Table II.
Summary of the Interactions of the Docked Compounds in the Homology Model and the Modified Crystal Structure of Human DNMT1
| Compound | Homology model |
3PTA |
RMSD (Å) | Common amino acid residues participating in H-bonding |
||||
|---|---|---|---|---|---|---|---|---|
| XP | XP | Arg1312 (1311) | Glu1266 (1265) | Pro1224 (1223) | Ser1230 (1229) | Gly1231 (1230) | ||
| Azacytidine | − 9.98 | − 11.34 | 0.76–1.64 | HE, NH1, NH2 | OE2 | CO | OH | NH |
| 5F-dC | − 8.84 | − 11.22 | 0.52–0.88 | HE, NH1, NH2 | OE2 | CO | OH | NH |
| EGCG | − 9.37 | − 11.16 | 3.75–5.62 | HE, NH1 | OE1 | |||
| Decitabine | − 8.72 | − 11.00 | 0.40–0.63 | HE, NH1, NH2 | OE2 | CO | OH | NH |
| Zebularine | − 10.12 | − 10.97 | 0.81–1.47 | HE, NH1, NH2 | OE2 | OH | NH | |
| Cytosine | − 10.03 | − 10.73 | 0.35–0.72 | HE, NH1, NH2 | OE1, OE2 | CO | OH | NH |
| ATA | − 9.52 | − 10.19 | 0.57–4.13 | NH1 | OE1 | OH | ||
| NSC97317 | − 8.42 | − 8.89 | 1.48–5.02 | HE, NH1 | OE1 | OH | ||
| Curcumin | − 9.00 | − 8.56 | 2.90–3.57 | HE, NH1 | OE1 | |||
| NSC14778 | − 7.45 | − 7.96 | 1.47–2.44 | NH1 | OH | NH | ||
| RG108-1 | − 7.01 | − 6.90 | 2.82–4.37 | NH1, NH2 | OH | NH | ||
| RG108 | − 6.67 | − 6.70 | 2.96–4.55 | NH1, NH2 | OH | NH | ||
| Hydralazine | − 5.05 | − 5.51 | 0.52–0.92 | HE, NH1 | OE1, OE2 | CO | ||
| Mahanine | − 5.46 | − 5.08 | 1.24–1.79 | HE | OE1 | |||
| Procaine | − 4.83 | − 5.08 | 1.15–1.89 | OE2 | CO | |||
| Procainamide | − 4.72 | − 4.70 | 1.30–1.78 | OE2 | CO | |||
| Parthenolide | − 3.14 | − 3.75 | 0.17–0.22 | HE, NH1 | ||||
The sesquiterpene lactone parthenolide, which is identified in the anti-inflammatory medicinal herb feverfew, was suggested to be a covalent blocker. Previous docking studies with the homology model supported this hypothesis (Yoo and Medina-Franco, 2011b). The binding pose in the modified crystal structure overlaps with previous docking model at low RMSD (0.17 Å) and makes hydrogen bond with common arginine residue. The docking poses of procaine, procainamide, and mahanine in the two catalytic binding sites show similar interactions with key amino acid residues within 1.5 Å RMSD. Although the binding poses of EGCG, curcumin, RG108, and RG108-1 were different, they all form hydrogen bonds with common amino acid residues which are important to the catalytic mechanism.
IV. Pharmacophore Modeling
For 16 established DNMT inhibitors in Fig. 5A, we developed an energy-optimized pharmacophore (e-pharmacophore) hypothesis using the best-scoring pose for each compound and descriptors calculated with Glide XP. We employed the structure-based pharmacophore approach that we previously used to model DNMT inhibitors using a homology model of the catalytic site (Yoo and Medina-Franco, 2011b). This approach is based on mapping the energetic terms from the Glide XP scoring function onto atom centers and has the advantage of combining pharmacophore perception with protein–ligand energetic terms to rank the importance of the pharmacophore features (Salam et al., 2009). The docking models of the inhibitors were refined using Glide XP, the Glide XP scoring terms were computed, and the energies were mapped into atoms. Then, the pharmacophoric sites were automatically generated with Phase (version 3.2, Schrödinger, LLC, New York, NY, 2010) using the default set of six chemical features, namely, hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic (H), negative ionizable (N), positive ionizable (P), and aromatic ring (R) site. The Glide XP energies from the atoms that comprise each pharmacophoric site were summed. The pharmacophoric sites were then ranked based on the summed energies, and the most favorable sites were selected for the pharmacophore hypothesis (Salam et al., 2009). These features were used to develop a common pharmacophore model that was evaluated on its ability to reproduce known inhibitors using Phase. The distance matching tolerance was set to 2.0 Å. To account for protein flexibility and lessen the effects of minor steric clashes, excluded volume spheres were created for all receptor atoms within 5 Å around each ligand. Each sphere has a radius corresponding to 50% of the van der Waals radius of the receptor atom. Receptor atoms less than 1.5 Å from the ligand were ignored. The combination of best-scoring features that matched a minimum of two sites in each known inhibitor was chosen and regenerated (Salam et al., 2009).
Figure 7A shows the five-feature pharmacophore model for the 16 DNMT1 inhibitors. The energetic value and amino acid residues participating in the interaction are shown in the figure. The best-scoring feature is a negative charge (N) which is close to the side chains of Ser1230, Gly1231, Lys1535, and Arg1312. The second and third favorable features are a donor site (D) that is near the side chain of Gly1577 and an acceptor site (A) that is in close proximity to the side chain of Arg1310 and Arg1312 in the RXR motif. The fourth ranked feature is an aromatic ring (R) that stabilizes the binding conformations of ligands between AdoHcy and Cys1226. The fifth ranked feature is a donor site (D) that is near the side chain of Glu1266, which is a residue implicated in the mechanism of methylation (Fig. 7A). These pharmacophore features represent the most important interactions of the 16 DNMT1 inhibitors with the catalytic site. Notably, glutamate in the ENV motif and the arginines in the RXR motif are highly conserved residues in catalytic domains of DNMTs (Jurkowska et al., 2011). Interestingly, the five-feature pharmacophore model derived with the modified crystal structure of DNMT1 (Fig. 7A) is consistent with the pharmacophore hypothesis previously reported with the homology model of DNMT1, although the ranking of the features and nearby amino acids are slightly different. Most of the inhibitors matched several pharmacophore features considering a distance tolerance of 2 Å. Representative examples are shown in Fig. 7B–E. All nucleoside analogues except zebularine matched with all five pharmacophore features (Fig. 7B and C). Zebularine matches four features, but it does not satisfy the hydrogen bonding donor feature interacting with Glu1266 because zebularine does not have the amino group on the base ring. However, the donor feature (D) can match the N3 protonated form of zebularine.
Fig. 7.
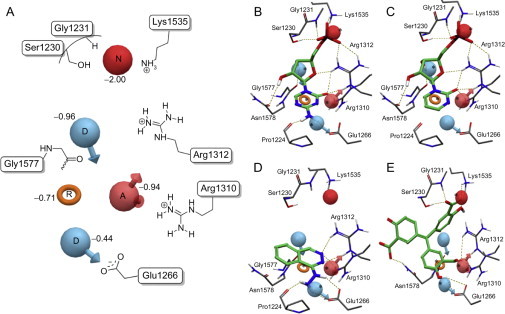
(A) Structure-based pharmacophore model using binding modes of known inhibitors in the catalytic binding site of the modified crystal structure. Red sphere negative ionizable (N), pink sphere hydrogen bond acceptor (A), blue sphere hydrogen bond donors (D), and orange ring aromatic ring (R). Selected amino acid residues in the catalytic site are schematically depicted for reference. Comparison between the binding mode and pharmacophore hypothesis for representative DNMT inhibitors: (B) 5-azacytidine, (C) zebularine, (D) hydralazine, (E) ATA.
Despite the fact that hydralazine has a small structure, it matches the aromatic ring (R), donor (D), and acceptor (A) features that are close to Glu1266, Arg1310, and Arg1312 (Fig. 7D). This is in agreement with our previous findings of the modeling of hydralazine with homology models of DNMT1 (Singh et al., 2009, Yoo and Medina-Franco, 2011b). Based on these results, it is expected that hydralazine analogues that match the top-ranked negative charge feature (N) will have improved potency, as we have suggested previously (Yoo and Medina-Franco, 2011a).
Interestingly, RG108 and RG108-1, both with a carboxylate functional group, matched the negative feature (N). The indole ring of both structures satisfied the ring feature (R). EGCG also matched with four pharmacophoric features; the hydroxyl group of ring B matched with the ring feature (R), acceptor (A) and donor (D) features close to glutamate and arginine residues; the ketone of the gallate moiety matched with negative feature (N). Curcumin and mahanine matched with the aromatic ring (R) and donor feature (D) nearby Arg1310 and Arg1312. Although parthenolide matched only the acceptor feature (A), it is possible that the γ-methylene lactone alkylates the catalytic Cys1226, as previously suggested (Liu et al., 2009). These results are similar to the insights previously reported with a homology model of the catalytic domain of DNMT1.
In contrast, the 19 compounds, inactive or previously used as decoys (Fig. 5B), did not match with any of the pharmacophore features or satisfied only one feature. Even if most of these molecules have a carboxylate group like known inhibitors, they did not match the negative feature (N) and the donor feature (D) interacting with Glu1266. Taken together, results of the structure-based pharmacophore modeling with the modified crystal structure were in good agreement with the docking studies described above and previously reported docking and pharmacophore modeling with a homology model of DNMT1.
V. Experimental and Molecular Modeling Studies of a Novel Inhibitor of Human DNMT1
It is well recognized that integration of computational and experimental approaches boosts the design of novel enzyme and other molecular target inhibitors and further advances drug candidates (López-Vallejo et al., 2011). This has been demonstrated in the area of inhibitors of DNMT1 (Medina-Franco and Caulfield, 2011). For example, virtual screening of large compound databases followed by experimental characterization leads to the identification of novel DNMT1 inhibitors such as RG108 and 5,5-methylenedisalicylic acid (NSC14478; Fig. 5A) (Siedlecki et al., 2006, Kuck et al., 2010b).
There is now an increased interest in the biological activity of 5,5-methylenedisalicylic acid and related compounds. For example, 5,5-methylenedisalicylic acid also emerged as an experimentally validated hit of a virtual screening with the viral NS5 RNA methyltransferase, a promising drug target against flaviviruses which are the causative agents of severe diseases such as dengue or yellow fever (Podvinec et al., 2010). In addition, in a separate virtual screening, a structural analogue of 5,5-methylenedisalicylic acid, ATA (Fig. 5A) was identified as a potent inhibitor of the methyltransferase activities on flaviviral methyltransferases (Milani et al., 2009). Also, a recent study shows that ATA inhibits two AdoMet-dependent RNA methyltransferases of the severe acute respiratory syndrome (SARS) coronavirus (Bouvet et al., 2010). A recent study showed that NSC97317, a structural analogue of ATA (Fig. 5A), is an inhibitor of DNMT1 (Yoo and Medina-Franco, 2011c). Preliminary docking studies of ATA with a homology model of DNMT1 strongly suggested that this compound would show enzymatic inhibitory activity. To experimentally test this hypothesis and, as part of an ongoing effort to identify novel inhibitors of DNMT1, we conducted the experimental characterization of ATA as an enzymatic inhibitor of DNMT1 using in vitro assays.
A. Enzymatic Inhibition of ATA
In order to experimentally test the hypothesis that ATA is an inhibitor of DNMT1, the enzymatic inhibitory activity of this compound was measured using a nonradioactive in vitro enzymatic assay. For this assay, full-length human DNMT1 was expressed by baculovirus infection of Sf9 insect cells for 48 h. DNMT1 in this system (FastBac-HT system, Invitrogen) is hexahistidine tagged and purified on nickel NTA resin as we have described previously (Yokochi and Robertson, 2002) and dialyzed to remove imidazole. DNMT1 was used with the colorimetric 96-well-plate-based DNMT activity kit from Epigentek (an ELISA-like assay using anti-5-methylcytosine antibody to detect methylation). After a 1-h incubation of DNMT1 with substrate plus or minus inhibitor, samples were read in a plate reader at 450 nm to determine activity. Figure 8A illustrates the dose–response plot over a range of DNMT1 concentrations for this assay. This figure (Fig. 8B–D) also shows inhibition plots for selected inhibitors that were used as positive controls, namely, SAH, the natural product EGCG, and the l-tryptophan derivative RG108. SAH and EGCG showed IC50 values of 8.6 and 0.7 μM, respectively. Under these assay conditions, RG108 showed only 11% inhibition at 100 μM. Figure 9A shows the inhibition plot for ATA, revealing an IC50 = 0.68 μM against DNMT1. Of note, the enzymatic inhibitory activity of ATA against DNMT1 was greater than the activity of SAH and RG108 and it was comparable with the activity of EGCG under the current assay conditions. We also measured the Dnmt3a inhibition by ATA that showed an IC50 = 1.4 μM (Fig. 9B), indicating that ATA is somewhat selective toward DNMT1 (approximately two-fold).
Fig. 8.
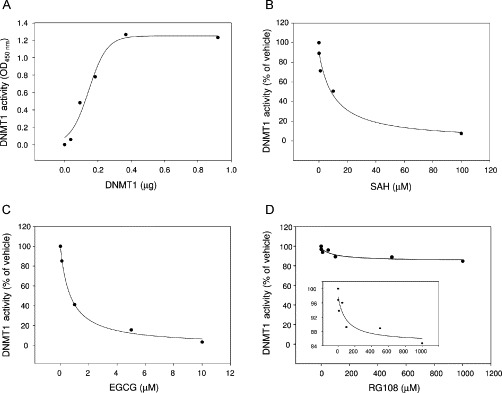
(A) Titration of recombinant DNMT1 enzymatic activity using a colorimetric assay. DNMT1 activity was measured using EpiQuik DNA methyltransferase activity/inhibition assay kit (Epigentek). (B–D) Dose–response plots for each compound against DNMT1. Data are presented in terms of percentage activity versus the results using vehicle only treated control, which was assigned a value of 100%. The IC50 concentrations of compounds were determined by enzyme assay under identical conditions (0.3 μg/1.6 pmol of DNMT1, incubated for 1 h).
Fig. 9.
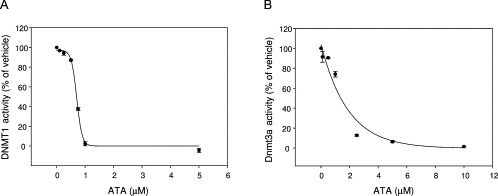
(A) Dose–response plots for ATA inhibition against human DNMT1. Data are presented in terms of percent of vehicle only and the IC50 concentration of ATA was determined by enzyme assay with 0.3 μg of DNMT1 (1.6 pmol). (B) Dose–response plots for ATA inhibition against murine Dnmt3a. Data are presented in terms of percent of vehicle only control and the IC50 concentration of ATA was determined by enzyme assay with 4.0 μg of DNMT3A (33.3 pmol).
B. Docking and Pharmacophore Modeling of ATA
In order to explore the binding mode of ATA with DNMT1 at the molecular level, we conducted molecular modeling of ATA. Docking of ATA with the crystal structure of DNTM1 modified into an active conformation suggested two binding poses. The first docking pose of ATA is shown in Fig. 6D as a deprotonated form (expected in aqueous solution). According to this binding model, ATA forms a strong hydrogen bond network with the same amino acid residues that interact with the nucleoside inhibitors. The hydroxybenzoic acid moieties of ATA form hydrogen bonds with Glu1266, Arg1310, Arg1312, Ser1230, Gly1231, and Lys1535. The other carboxylate group makes interactions with Asn1578. The structurally related 5,5-methylenedisalicylic acid (NSC14778) showed weak inhibitory activity against DNMT1 in a biochemical assay (Kuck et al., 2010b). Although the binding pose of NSC14778 partially overlaps ATA showing same interactions with Ser1230, Gly1231, and Lys1535, NSC14778 does not make key interactions with Glu1266 and Arg1312 (not shown). A similar binding model of NSC14778 was also determined in our previous study using the homology model (Yoo and Medina-Franco, 2011b). The second pose of ATA was consistent with the recently reported binding mode in the catalytic site of homology model (Yoo and Medina-Franco, 2011c). Of note, ATA shows a good docking score comparable to those of nucleoside inhibitors (Table I).
We also compared the binding model of ATA obtained with Glide XP with the pharmacophore model developed with the modified crystal structure (Fig. 7E). The binding position of ATA matches with the best-scoring feature, that is, negative charge (N), hydrogen bond acceptor (A) (interaction with Glu1266), donor (D) (interaction with Arg1310 and Arg1312), and aromatic ring (R) (Fig. 7E). In contrast, NSC14778 satisfies only one, the negative feature (N). Therefore, this model suggests that introduction of a substituent that could match the donor or acceptor feature associated with the interaction of glutamate or arginine residues would enhance the affinity of the analogue NSC14778. The binding position of NSC97317 matches with three pharmacophoric features, that is, negative charge (N), hydrogen bond acceptor (A), and aromatic ring (R). These findings are further supported by the experimental observation and analysis of docking energy.
VI. Conclusions and Perspectives
DNMTs are involved in epigenetic regulation of the genome and are promising targets for therapeutic intervention in cancer and other diseases. Computational approaches are increasingly being used to identify promising compounds as potential therapeutic agents or as research tools to study epigenetic mechanisms involving DNMTs. Also, computational methods are employed to better understand the mechanism of established inhibitors of DNA methylation. Herein, we describe molecular modeling studies with a recently published crystallographic structure of human DNMT1 that contains unmethylated DNA and the enzyme in an “inactive” conformation. The conformation of the crystallographic structure was modeled in an active state using molecular dynamics. Experimentally known DNMT1 inhibitors were docked in parallel into the modified crystal structure with Glide HTVS, SP, and XP. The binding modes were characterized by common interactions with amino acid residues that participate in the proposed mechanism of DNA C5-methylation and other key amino acid residues including Ser1230, Gly1231, Glu1266, Arg1310, Arg1312, and Lys1535. The docking scores were also able to distinguish active inhibitors from known inactive and previously used decoy compounds. Indeed, most of the established inhibitors of DNMT1 had high ranking scores. Based on the docking results, a structure-based pharmacophore model was developed that was characterized by five features, including a negative charge, an aromatic ring, a hydrogen bond acceptor, and two donor sites. The pharmacophore model represents the key protein–ligand interactions of the DNMT1 inhibitors obtained in docking. Results of the docking and pharmacophore modeling had a good agreement with the insights of the modeling studies published previously using a homology model of the catalytic domain of DNMT1. As part of an ongoing interest of identifying novel inhibitors, we experimentally tested ATA in an enzymatic inhibition assay. Preliminary docking studies of ATA with DNMT1 had strongly suggested that this structural analogue of 5,5-methylenedisalicylic acid would show enzymatic inhibitory activity. Indeed, ATA showed an IC50 = 0.68 μM against human DNMT1. We also measured the DNMT3A inhibition by ATA, which showed an IC50 = 1.4 μM, indicating that ATA is somewhat selective toward DNMT1 (approximately twofold).
A major follow-up study of this work is to perform virtual screening of large compound data sets using our validated docking protocol and pharmacophore model with the crystal structure of human DNMT1 modeled into an active conformation to identify potential novel DNMT1 inhibitors. In order to further explore and perhaps someday enhance the enzymatic selectivity toward DNMT1 over murine Dnmt3a, we are currently employing molecular docking and other modeling approaches of ATA with the published crystal structure of DNMT3A (Jia et al., 2007). Results of these insights from the molecular modeling will be published in a separate communication in due course.
Acknowledgments
This work was supported by the Menopause & Women's Health Research Center, the State of Florida, Executive Office of the Governor's Office of Tourism, Trade, and Economic Development, and R01 CA116028 (K. D. R.).
References
- Arce C., Segura-Pacheco B., Perez-Cardenas E., Taja-Chayeb L., Candelaria M., Dueñnas-Gonzalez A. Hydralazine target: from blood vessels to the epigenome. J. Transl. Med. 2006;4:10. doi: 10.1186/1479-5876-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M., Debarnot C., Imbert I., Selisko B., Snijder E.J., Canard B. In vitro reconstitution of SARS-Coronavirus mRNA cap methylation. PLoS Pathog. 2010;6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield T., Medina-Franco J.L. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J. Struct. Biol. 2011;176:185–191. doi: 10.1016/j.jsb.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Chen T.P., Hevi S., Gay F., Tsujimoto N., He T., Zhang B.L. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- Cheng X.D., Blumenthal R.M. Mammalian DNA methyltransferases: a structural perspective. Structure. 2008;16:341–350. doi: 10.1016/j.str.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueñas-González A., García-López P., Herrera L.A., Medina-Franco J.L., González-Fierro A., Candelaria M. The prince and the pauper. A tale of anticancer targeted agents. Mol. Cancer. 2008;7:82. doi: 10.1186/1476-4598-7-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans D.A., Bronowska A.K. Implications of fast-time scale dynamics of human DNA/RNA cytosine methyltransferases (DNMTs) for protein function. Theor. Chem. Acc. 2010;125:407–418. [Google Scholar]
- Fandy T.E., Herman J.G., Kerns P., Jiemjit A., Sugar E.A., Choi S.H. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114:2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert E.R., Liu D. Flavonoids influence epigenetic-modifying enzyme activity: structure-function relationships and the therapeutic potential for cancer. Curr. Med. Chem. 2010;17:1756–1768. doi: 10.2174/092986710791111161. [DOI] [PubMed] [Google Scholar]
- Goll M.G., Bestor T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Hauser A.T., Jung M. Targeting epigenetic mechanisms: potential of natural products in cancer chemoprevention. Planta Med. 2008;74:1593–1601. doi: 10.1055/s-2008-1081347. [DOI] [PubMed] [Google Scholar]
- Hernández-Campos A., Velázquez-Martínez I., Castillo R., López-Vallejo F., Jia P., Yu Y. Docking of protein kinase B inhibitors: implications in the structure-based optimization of a novel scaffold. Chem. Biol. Drug Des. 2010;76:269–276. doi: 10.1111/j.1747-0285.2010.01002.x. [DOI] [PubMed] [Google Scholar]
- Issa J.-P. Optimizing therapy with methylation inhibitors in myelodysplastic syndromes: dose, duration, and patient selection. Nat. Clin. Pract. Oncol. 2005;2(Suppl 1):S24–S29. doi: 10.1038/ncponc0355. [DOI] [PubMed] [Google Scholar]
- Issa J.-P.J., Kantarjian H.M. Targeting DNA methylation. Clin. Cancer Res. 2009;15:3938–3946. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa J.P.J., Kantarjian H.M., Kirkpatrick P. Azacitidine. Nat. Rev. Drug Discov. 2005;4:275–276. doi: 10.1038/nrd1698. [DOI] [PubMed] [Google Scholar]
- Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. ChemBioChem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Jia D., Jurkowska R.Z., Zhang X., Jeltsch A., Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–251. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.A., Baylin S.B. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowska R.Z., Jurkowski T.P., Jeltsch A. Structure and function of mammalian DNA methyltransferases. ChemBioChem. 2011;12:206–222. doi: 10.1002/cbic.201000195. [DOI] [PubMed] [Google Scholar]
- Kelly T.K., De Carvalho D.D., Jones P.A. Epigenetic modifications as therapeutic targets. Nat. Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimasauskas S., Kumar S., Roberts R.J., Cheng X.D. Hhal methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- Kuck D., Caulfield T., Lyko F., Medina-Franco J.L. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010;9:3015–3023. doi: 10.1158/1535-7163.MCT-10-0609. [DOI] [PubMed] [Google Scholar]
- Kuck D., Singh N., Lyko F., Medina-Franco J.L. Novel and selective DNA methyltransferase inhibitors: docking-based virtual screening and experimental evaluation. Bioorg. Med. Chem. 2010;18:822–829. doi: 10.1016/j.bmc.2009.11.050. [DOI] [PubMed] [Google Scholar]
- Kumar S., Horton J.R., Jones G.D., Walker R.T., Roberts R.J., Cheng X. DNA containing 4′-thio-2′-deoxycytidine inhibits methylation by HhaI methyltransferase. Nucleic Acids Res. 1997;25:2773–2783. doi: 10.1093/nar/25.14.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Hua S., He X.N., Zhang Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim. Biophys. Sin. 2010;42:243–252. doi: 10.1093/abbs/gmq015. [DOI] [PubMed] [Google Scholar]
- Li Y., Tollefsbol T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem. 2010;17:2141–2151. doi: 10.2174/092986710791299966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger, LLC; New York, NY: 2005. LigPrep., version 2.2. [Google Scholar]
- Liu Z.F., Liu S.J., Xie Z.L., Pavlovicz R.E., Wu J., Chen P. Modulation of DNA methylation by a sesquiterpene lactone parthenolide. J. Pharmacol. Exp. Ther. 2009;329:505–514. doi: 10.1124/jpet.108.147934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Vallejo F., Caulfield T., Martínez-Mayorga K., Giulianotti M.A., Nefzi A., Houghten R.A. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Comb. Chem. High Throughput Screen. 2011;14:475–487. doi: 10.2174/138620711795767866. [DOI] [PubMed] [Google Scholar]
- Lyko F., Brown R. DNA methyltransferase inhibitors and the development of epigenetic cancer therapies. J. Natl. Cancer Inst. 2005;97:1498–1506. doi: 10.1093/jnci/dji311. [DOI] [PubMed] [Google Scholar]
- Schrödinger, LLC; New York, NY: 2010. MacroModel, version 9.8. [Google Scholar]
- Medina-Franco J.L., Caulfield T. Advances in the computational development of DNA methyltransferase inhibitors. Drug Discovery Today. 2011;16:418–425. doi: 10.1016/j.drudis.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Medina-Franco J., López-Vallejo F., Kuck D., Lyko F. Natural products as DNA methyltransferase inhibitors: a computer-aided discovery approach. Mol. Diversity. 2011;15:293–304. doi: 10.1007/s11030-010-9262-5. [DOI] [PubMed] [Google Scholar]
- Milani M., Mastrangelo E., Bollati M., Selisko B., Decroly E., Bouvet M. Flaviviral methyltransferase/RNA interaction: structural basis for enzyme inhibition. Antiviral Res. 2009;83:28–34. doi: 10.1016/j.antiviral.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C.A., Gavin C.F., White J.A., Parrish R.R., Honasoge A., Yancey C.R. Cortical DNA methylation maintains remote memory. Nat. Neurosci. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palii S.S., Van Emburgh B.O., Sankpal U.T., Brown K.D., Robertson K.D. DNA methylation inhibitor 5-aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 2008;28:752–771. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podvinec M., Lim S.P., Schmidt T., Scarsi M., Wen D., Sonntag L.-S. Novel inhibitors of dengue virus methyltransferase: discovery by in vitro-driven virtual screening on a desktop computer grid. J. Med. Chem. 2010;53:1483–1495. doi: 10.1021/jm900776m. [DOI] [PubMed] [Google Scholar]
- Portela A., Esteller M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Robertson K.D. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- Salam N.K., Nuti R., Sherman W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009;49:2356–2368. doi: 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- Schermelleh L., Spada F., Easwaran H.P., Zolghadr K., Margot J.B., Cardoso M.C. Trapped in action: direct visualization of DNA methyltransferase activity in living cells. Nat. Methods. 2005;2:751–756. doi: 10.1038/nmeth794. [DOI] [PubMed] [Google Scholar]
- Schrump D.S., Fischette M.R., Nguyen D.M., Zhao M., Li X.M., Kunst T.F. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin. Cancer Res. 2006;12:5777–5785. doi: 10.1158/1078-0432.CCR-06-0669. [DOI] [PubMed] [Google Scholar]
- Siedlecki P., Boy R.G., Musch T., Brueckner B., Suhai S., Lyko F. Discovery of two novel, small-molecule inhibitors of DNA methylation. J. Med. Chem. 2006;49:678–683. doi: 10.1021/jm050844z. [DOI] [PubMed] [Google Scholar]
- Singh N., Dueñas-González A., Lyko F., Medina-Franco J.L. Molecular modeling and dynamics studies of hydralazine with human DNA methyltransferase 1. ChemMedChem. 2009;4:792–799. doi: 10.1002/cmdc.200900017. [DOI] [PubMed] [Google Scholar]
- Sippl W., Jung M. DNA methyltransferase inhibitors. In: Sippl W., Jung M., editors. Epigenetic Targets in Drug Discovery. Wiley-VCH; Weinheim: 2009. pp. 163–183. [Google Scholar]
- Song J., Rechkoblit O., Bestor T.H., Patel D.J. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331:1036–1040. doi: 10.1126/science.1195380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stresemann C., Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607. [DOI] [PubMed] [Google Scholar]
- Vilkaitis G., Merkiene E., Serva S., Weinhold E., Klimasauskas S. The mechanism of DNA cytosine-5 methylation: kinetic and mutational dissection of HhaI methyltransferase. J. Biol. Chem. 2001;276:20924–20934. doi: 10.1074/jbc.M101429200. [DOI] [PubMed] [Google Scholar]
- Yokochi T., Robertson K.D. Preferential methylation of unmethylated DNA by mammalian de novo DNA methyltransferase Dnmt3a. J. Biol. Chem. 2002;277:11735–11745. doi: 10.1074/jbc.M106590200. [DOI] [PubMed] [Google Scholar]
- Yoo J., Medina-Franco J.L. Discovery and optimization of inhibitors of DNA methyltransferase as novel drugs for cancer therapy. In: Chris Dr., Rundfeldt., editor. Drug Development - A Case Study Based Insight into Modern Strategies. InTech; Croatia: 2011. http://www.intechopen.com/books/drug-development-a-case-study-based-insight-into-modern-strategies/discovery-and-optimization-of-inhibitors-of-dna-methyltransferase-as-novel-drugs-for-cancer-therapy 978-953-307-257-9 Available at: [Google Scholar]
- Yoo J., Medina-Franco J.L. Homology modeling, docking, and structure-based pharmacophore of inhibitors of DNA methyltransferase. J. Comput.-Aided Mol. Des. 2011;25:555–567. doi: 10.1007/s10822-011-9441-1. [DOI] [PubMed] [Google Scholar]
- Yoo J., Medina-Franco J.L. Trimethylaurintricarboxylic acid inhibits human DNA methyltransferase 1: insights from enzymatic and molecular modeling studies. J. Mol. Model. 2011;18:1583–1589. doi: 10.1007/s00894-011-1191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawia N.H., Lahiri D.K., Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med. 2009;46:1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]