

Abstract
Purpose of review
To provide an expert review and expert perspective on important advances related to the genetics of acute and chronic pancreatitis.
Recent findings
Provocative new reports highlight the interplay between genetic, developmental and environmental factors. Key findings include the relationship between pancreas divisum and CFTR mutations, the role of trypsin in acute and recurrent acute pancreatitis and the discovery of a pancreatitis modifier gene on the X chromosome that provides new clues to why the vast majority of patients with alcoholic pancreatitis are men.
Summary
Pancreatic genetics is complex, linked to the multiplicative and modifying effects of multiple interacting genetic, structural and environmental factors. Clinical interpretation will require disease modeling and simulation to understand the combined effect of risk factors that alone are neither sufficient or necessary to cause disease, and to design treatment strategies that prevent the develop of advanced chronic pancreatitis – which by definition is irreversible.
Keywords: Pancreatitis, CFTR, SPINK1, PRSS1, TBN
Introduction
Genetics plays an important role in pancreatitis susceptibility, progression and severity. Autosomal dominate hereditary pancreatitis (HP) and autosomal recessive cystic fibrosis (CF) are examples of classic Mendelian genetic disorders, but these entities are either rare (HP) or are part of a complex syndrome involving multiple organs (CF). In contrast, most recurrent acute and chronic pancreatitis appears to be complex genetic disorder in which every patient has a different combination of factors, and needs a personalized approach (1). Mendelian genetic disorders involve one causative gene mutation whereas a complex genetic disorder involves various gene environment interactions, gene-gene interactions, or more complex interactions that together result in significantly increased or decreased risk of developing organ injury or dysfunction, or modifying the nature or severity of the evolving process.
Five pancreatitis susceptibility genes are well established including variants in the cationic trypsinogen gene (PRSS1), the cystic fibrosis transmembrane conductance regular gene (CFTR), the pancreatic secretory trypsin inhibitor gene (SPINK1), the chymotrypsinogen C gene (CTRC) and the calcium sensing receptor gene (CASR). These have been reviewed in detail elsewhere (2–5). What is emerging is the fact that these genetic factors strongly interact with each other (complex genetics) but not strongly with alcohol or other etiological factors.
The North American Pancreatitis Study 2 (NAPS2) cohort of recurrent acute and chronic pancreatitis patients has reached enrollment targets and is providing major insights into pancreatitis. The primary environmental risk factor in the first 1000 patients was very heavy alcohol drinking, which was present in 38% of men and 11% of women with chronic pancreatitis, and only 17% of men and 6% of women with recurrent acute pancreatitis (6). Smoking was an independent, dose-dependent and additive risk factor to drinking (6). In addition, at lease one of 5 established pancreatitis risk genes were identified in a quarter of the cases, but only 3% of the total had both heavy alcohol and an established pancreatitis risk gene (7). These data, and the meta analysis of Aoun et al (8) on trypsin/SPINK1-dependent pathways, highlight the fact that pancreatic inflammation is not the disorder: it is the consequence of multiple disorders that cause injury and inflammation (see the SAPE hypothesis model (9, 10)).
Further insights into the complex risk of established susceptibility genes occurred through several recent publications. A new breakthrough into understanding the pathophysiology of alcoholic pancreatitis further strengths the multiple pathway / SAPE hypothesis model. This review will end with emphasis on moving new data into the clinic.
A new framework for treating recurrent acute and chronic pancreatitis
Twentieth century medicine was based on the germ theory of disease. This framework suggest that single factors such as a germ, a toxin or a genetic defect is “the” proximal cause of each complex disorders. This framework for the scientific method of null hypothesis significance testing of candidate factors, the system of disease classification, and the curriculum for medical educational was outlined by the Flexner report in 1910 and implemented by government mandate (1). Within this framework chronic pancreatitis is defined as a syndrome of destructive, inflammatory conditions that encompasses the many sequelae of long-standing pancreatic injury. A Pathologic diagnosis of chronic pancreatitis requires evidence of irreversible histological changes from the normal pancreatic architecture include irregular fibrosis, acinar cell loss, islet cell loss, and inflammatory cell infiltrates. A clinical diagnosis requires the identification of specific functional, morphologic, and histological features that characterize the final common pathologic pathway of a variety of pancreatic disorders. While this approach is useful for classification of established and irreversible disease and for billing, it is not useful for early diagnosis (i.e. irreversible damage is required to make the diagnosis), it obscures the true etiology, and it fails to provide guidance to physicians for early intervention and treatments that will minimize progression, complications and cost. Thus, the goal of 20th century medicine is to identify evidence of tissue pathology in a living patient, which, by definition requires that irreversible damage has already occurred.
The power of genetics is that it allows etiologic mechanisms to be identified early in the course of the disease, well before there is irreversible damage. Indeed, this approach has already been implemented at the University of Pittsburgh in the Pancreas Center of Excellence’s multidisciplinary clinic (1). Knowledge of the etiological pathway using modeling of the genetic and environmental data provides the critical insights for true personalized medicine. It allows the underlying problem to be targeted rather than focusing on treating symptoms that are common to all etiologies.
Figure 1 is a simplified view of the etiological pathways leading to chronic pancreatitis. In this example different groups of gene-environmental factors are seen to markedly increase overall risk of developing fibrosis through different mechanism. Risk of developing fibrosis through the alcohol pathway, for example, is only marginally increased by SPINK1 mutations, whereas the risk of chronic pancreatitis is markedly increased by SPINK1 mutations in the trypsin-dependent pathways.
Figure 1.
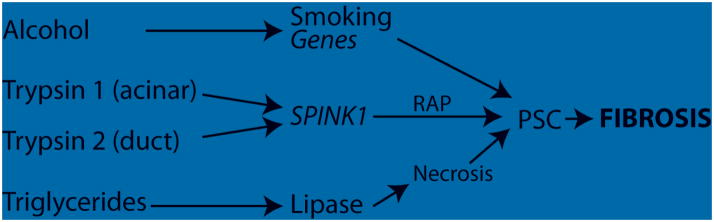
Pathways to fibrosis. Four pathways are illustrated that eventually lead to fibrosis, a hallmark of chronic pancreatitis. The risk of alcoholic pancreatitis progression is enhance by smoking and additional genes, possibly through an interaction with macrophages and pancreatic stellate cells (PSC). Risk from trypsin activation can occur in the acinar cell (e.g. PRSS1, CTRC mutations), or through the duct (CFTR, CASR, pancreas divisum). Mutations that reduce PRSS1 and PRSS2 expression appear to reduce the risk of pancreatitis from both Trypsin Pathways. Hypertryglyceridemia requires leakage of lipase and hydrolysis into fatty acids to case necrosis and eventually fibrosis.
Alcohol Pathway
Alcoholic pancreatitis is the presence of recurrent acute and chronic pancreatitis in a person who is consuming alcohol above a classification threshold and has no other obvious cause. Until more sensitive techniques to identify mild chronic pancreatitis were developed, most clinical-epidemiology studies only identified the most severe cases, and reported that chronic pancreatitis was a disease of alcoholic men. This detection bias may have reflected the more rapid progression of disease in alcoholics who also smoked cigarettes since the combination accelerates disease progression and smoking is strongly associated with calcifications (11) which are easy to detect by CT scan or even by abdominal x-ray. While the risk of chronic pancreatitis with smoking and drinking is nearly 8 fold the general population risk (6), only 3% of alcoholic develop chronic pancreatitis (12), suggesting that other risk factors are also important. Animal studies have demonstrated that the chronic pancreatitis can be replicated in animals that are fed alcohol if they are also given episodes of acute pancreatitis through another mechanism such as repeated injections of cerulean (13–15). Once the inflammatory process has been initiated, then the progression to inflammation and fibrosis is amplified and accelerated in animals that continue to be given alcohol, as outlined by the SAPE hypothesis framework (10). Thus, recurrent attacks of acute pancreatitis in alcoholics represents one mechanism for driving the progression to injury pancreas to chronic pancreatitis in humans (16). However, genetic factors have long been suspected to play a role as well.
Surprisingly the 5 comment susceptibility factors for acute and chronic pancreatitis are less common in alcoholic chronic pancreatitis than expected. In a meta analysis by Aoun, et al. (8)patients were classified according to etiology and according to the presence or absence of SPINK1 mutations. SPINK1 codes for is a specific trypsin inhibitor, pancreatic secretory trypsin inhibitor, and it was hypothesized that patients with an etiological risk factor resulting in recurrent trypsin activation would be much more severe if they also had a SPINK1 mutation. Indeed the risk of pancreatitis was markedly elevated in tropical pancreatitis and idiopathic pancreatitis (which was later demonstrated to be strongly enriched in CFTR mutations (17)). Surprisingly there was a relatively small combined effect of alcohol and SPINK1, suggesting that alcoholic pancreatitis was being driven through a process that was non-trypsin reactivation dependent (Figure 1).
This year, the North American Pancreatic Study Group is completing the first phase of a genome wide association study (GWAS) and initial evaluation suggest an association between pancreatitis and a locus on the X chromosome. Subgroup analysis is suggesting that the risk was especially high in patients who also drink alcohol.
These findings are fascinating from several levels. First it has been observed that men are much more likely to have alcoholic pancreatitis than women. This has been assumed to be related to the fact that the men are more likely to be heavy drinkers than women, but does not explain the magnitude of the sex difference. A risk factor for alcoholic pancreatitis on the X chromosome may partially explain this observation. In women, the high-risk allele acts as a recessive genetic disorder. Thus, regardless of mechanism, the genetic variant in this region plays an important part of the pathophysiology of alcoholic pancreatitis. These are fascinating and potentially important findings and opens up the door to a variety of research avenues in order to understand the biology of complex traits.
Trypsin/SPINK1-dependent pathways
Figure 1 outlines two trypsin-dependent pathways to chronic pancreatitis. The first is linked to the acinar cell while the second is linked to the pancreatic duct. The variable importance of trypsin in different pancreatitis pathways was demonstrated in a rat model in which the most important isoform of trypsinogen (T7) in mice was genetically knocked out (T−/−) (18). In this mouse trypsin activity was significantly reduced, but not eliminated. These mice had protection from severe cerulean-induced acute pancreatitis, but with continued acinar cell hyperstimulation, stress and NFkB activation they still progressed to fibrosis. A similar finding was seen in humans with a common PRSS1 haplotype that reduces PRSS1 and PRSS2 gene expression (19). This variant haplotype appears to provide protection from some forms of pancreatitis. These findings are consistent with Figure 1, in which reduction in trypsin expression protects from the two trypsin pathways, but not the alcohol pathway.
A study on pancreatic divisum was published by Bertin et al (20) reporting on the prevalence of mutations in the SPINK1, PRSS1, or CFTR genes. They found that pancreatic divisum was present in 7% of subjects without pancreatic disease, 7% in patients with alcohol-induced pancreatitis, and 5, 16, 16, and 47% in those with idiopathic, and PRSS1-, SPINK1-, and CFTR-associated pancreatitis, respectively (P<0.0001). Pancreas divisum is a duct problem, which is believed to cause increase resistance to pancreatic fluid flow as the majority of pancreas juice must flowing through the high-resistance minor papilla. The CFTR variants are important because they are key to the mechanism responsible for the secretion of sodium bicarbonate rich fluid into the duct upstream, resulting in the elevated hydrostatic pressure that flushes the zymogens (including trypsinogen) out of the pancreas. The finding that there is a high concordance with pancreatic divisum and known CFTR mutations suggests a multiplicative risk of failed trypsin flushing through a combination of inability to generate a high hydrostatic pressure because of dysfunctional CFTR combined with a high distal resistance because of the flow of the pancreatic juice though the minor papilla. This pathway is illustrated in Figure 1 as the Trypsin Pathway 2 (duct) mechanism, and downstream SPINK1 variants, but not alcohol, further increase risk.
Rosendahl et al (21), recently published a summary of their analysis of CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis in a predominantly German population. On of the interesting observations in their Table 5 are mutations in multiple risk genes within the same patient (trans-heterozygous genotypes). Of note, all cases of pancreatitis in patients with CTRC mutations, for example, were also carriers of a PRSS1, SPINK1 or CFTR mutation (CASR not tested). They also replicated the finding of Schneider et al (17) who noted a very high coincidence of SPINK1 mutations with CFTR mutations. In a review of published studies of CASR mutations LaRusch and Whitcomb (3) noted that CASR loss-of-function mutations had high coincidence with SPINK1 mutations, whereas patients with gain-of-function CASR mutations were mostly alcoholics. Thus, the risk of pancreatitis through the trypsin pathways appears to be multiplied by other trypsin-associated risk factors, as outlined in Table 1, and CTRC and CASR appear to be secondary or dependent risk factors since they are only seen in the presence of other strong pancreatitis risk factors.
Lipase Pathway
Severe hypertriglyceridaemia (HTG) is a condition that is usually associated mutations in genes such as lipoprotein lipase. Hypertriglyceridaemia is a known risk factor for acute pancreatitis. Tryglycerides themselves do not cause pancreatic damage, but in the presence of pancreatic lipase, the tryiglycerides are hydrolyzed to free fatty acids. Navina et al (22) showed that unsaturated fatty acids, but not saturated fatty acids, cause lipotoxicity to many organs during acute pancreatitis. Obese patients are at increased risk because of stress-induced release of triglycerides from adipocytes. The effect is pronounced in the pancreas, where the concentration of both triglycerides and lipase tend to be high.
Severe hypertriglyceridaemia is a risk factor for acute pancreatitis and is usually found in subjects with mutations in genes such as LPL. Surendran et al (23) investigated the molecular basis of severe HTG in patients referred to their lipid disorder center by DNA sequencing. In addition to common variants in LPL and APOA5 they identified multiple rare DNA sequence variants LPL, APOC2, APOA5, GPIHBP1 and LMF1 that accounted for over half of the patients with HTG of unknown cause. The importance of this article for pancreatitis is that most of the focus has been on detecting common genetic variants, whereas a major proportion of idiopathic cases may be due to rare mutations in known genes.
Next-Generation Sequencing
NGS is a powerful and disruptive technology that his at the forefront of personalized medicine. NGS encompasses a wide variety of new and exciting technologies that allow the use of massive parallel sequencing on a patient’s DNA to produce accurate DNA sequences and detection of other variants at a minimal cost. Indeed, the cost of whole exome sequencing for pancreatitis patients is similar to that of sequencing only the CFTR gene (24). Not only are the currently acknowledged pancreatitis susceptibility genes included, but genes related to other relevant disorders (e.g. hyperlipidemia – above) at no extra cost and the anticipatory sequencing of genes that may be of interest in the future. If the NGS is done on patients with the first signs of pancreatitis, then the diagnosis of specific types of pancreatitis are confirmed, others are excluded, and an etiology-based treatment plan can be developed (1). For example, it is often possible to subdivide patients immediately into genetic syndromes such as hereditary pancreatitis, typical or atypical cystic fibrosis, or complex recurrent acute or chronic pancreatitis categories. The reason this is important is that it provides opportunity for targeting therapy. If the problem is associated with a CFTR variant then the recurrent pancreatitis may be related to poor duct clearance because of diminished hydrostatic pressure and measures to increase CFTR function or stimulation could be used to direct therapy. If the problem is with the pancreatic acinar cell where conditions enhance trypsin activation, then therapy is directed as minimizing stimulation of the pancreas or controlling calcium concentration, variations are a possible target. If the problem is autoimmune in nature, then therapies aimed at the immune system may be more effective.
Conclusions
The simplicity of the pancreas and the richness of the genetic information that has been generated through candidate gene studies and GWAS studies is providing a clearer picture of the complex pancreatic risk and disorders in patients complaining of pancreatic disease. As more experience is gained at applying new NGS technologies, it is anticipated that the cost of caring for these patients will be markedly reduced and the quality of life of patients improved by avoiding expensive work-ups and complementing symptomatic treatment with therapies that slow disease progression and possibly prevent an end-stage disorder that cannot be cured.
References
- 1.Whitcomb DC. What is personalized medicine - what does it replace? Nat Rev Gastroenterol Hepatol. 2012 doi: 10.1038/nrgastro.2012.100. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen JM, Ferec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87. doi: 10.1146/annurev-genom-082908-150009. [DOI] [PubMed] [Google Scholar]
- 3.Larusch J, Whitcomb DC. Genetics of pancreatitis. Curr Opin Gastroenterol. 2011 Sep;27(5):467–74. doi: 10.1097/MOG.0b013e328349e2f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teich N, Mossner J. Hereditary chronic pancreatitis. Best Pract Res Clin Gastroenterol. 2008;22(1):115–30. doi: 10.1016/j.bpg.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Whitcomb DC. Genetic aspects of pancreatitis. Annu Rev Med. 2010;61:413–24. doi: 10.1146/annurev.med.041608.121416. [DOI] [PubMed] [Google Scholar]
- 6.Yadav D, Hawes RH, Brand RE, Anderson MA, Money ME, Banks PA, et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch Intern Med. 2009 Jun 8;169(11):1035–45. doi: 10.1001/archinternmed.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitcomb DC. Going MAD: Development of a “Matrix Academic Division” to Facilitate Translating Research to Personalized Medicine. Academic medicine : journal of the Association of American Medical Colleges. 2011 Sep 26;86(11):1353–9. doi: 10.1097/ACM.0b013e3182303d7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aoun E, Chang CC, Greer JB, Papachristou GI, Barmada MM, Whitcomb DC. Pathways to injury in chronic pancreatitis: decoding the role of the high-risk SPINK1 N34S haplotype using meta-analysis. PLoS One. 2008;3(4):e2003. doi: 10.1371/journal.pone.0002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitcomb DC. Hereditary Pancreatitis: New insights into acute and chronic pancreatitis. Gut. 1999;45:317–22. doi: 10.1136/gut.45.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol. 2010 Mar;7(3):131–45. doi: 10.1038/nrgastro.2010.6. [DOI] [PubMed] [Google Scholar]
- 11.Maisonneuve P, Lowenfels AB, Mullhaupt B, Cavallini G, Lankisch PG, Andersen JR, et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut. 2005 Apr;54(4):510–4. doi: 10.1136/gut.2004.039263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yadav D, Eigenbrodt ML, Briggs MJ, Williams DK, Wiseman EJ. Pancreatitis: prevalence and risk factors among male veterans in a detoxification program. Pancreas. 2007 May;34(4):390–8. doi: 10.1097/mpa.0b013e318040b332. [DOI] [PubMed] [Google Scholar]
- 13.Deng X, Wang L, Elm MS, Gabazadeh D, Diorio GJ, Eagon PK, et al. Chronic alcohol consumption accelerates fibrosis in response to cerulein-induced pancreatitis in rats. Am J Pathol. 2005 Jan;166(1):93–106. doi: 10.1016/S0002-9440(10)62235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perides G, Tao X, West N, Sharma A, Steer ML. A mouse model of ethanol dependent pancreatic fibrosis. Gut. 2005 Oct;54(10):1461–7. doi: 10.1136/gut.2004.062919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gukovsky I, Lugea A, Shahsahebi M, Cheng JH, Hong PP, Jung YJ, et al. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2008 Jan;294(1):G68–79. doi: 10.1152/ajpgi.00006.2007. [DOI] [PubMed] [Google Scholar]
- 16.Nordback I, Pelli H, Lappalainen-Lehto R, Jarvinen S, Raty S, Sand J. The recurrence of acute alcohol-associated pancreatitis can be reduced: a randomized controlled trial. Gastroenterology. 2009 Mar;136(3):848–55. doi: 10.1053/j.gastro.2008.11.044. [DOI] [PubMed] [Google Scholar]
- 17.Schneider A, Larusch J, Sun X, Aloe A, Lamb J, Hawes R, et al. Combined Bicarbonate Conductance-Impairing Variants in CFTR and SPINK1 Variants Are Associated With Chronic Pancreatitis in Patients Without Cystic Fibrosis. Gastroenterology. 2011 Jan;140(1):162–71. doi: 10.1053/j.gastro.2010.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dawra R, Sah RP, Dudeja V, Rishi L, Talukdar R, Garg P, et al. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology. 2011 Dec;141(6):2210–7. e2. doi: 10.1053/j.gastro.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LaRusch JA, Orlichenko LS, Singh VK, Whitcomb DC. The second most common PRSS1 haplotype lowers trypsin expression and reduces pancreatitis risk. Pancreas. 2011;40(8):1333. [Google Scholar]
- 20.Bertin C, Pelletier AL, Vullierme MP, Bienvenu T, Rebours V, Hentic O, et al. Pancreas divisum is not a cause of pancreatitis by itself but acts as a partner of genetic mutations. The American journal of gastroenterology. 2012 Feb;107(2):311–7. doi: 10.1038/ajg.2011.424. [DOI] [PubMed] [Google Scholar]
- 21.Rosendahl J, Landt O, Bernadova J, Kovacs P, Teich N, Bodeker H, et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut. 2012 Mar 17; doi: 10.1136/gutjnl-2011-300645. [DOI] [PubMed] [Google Scholar]
- 22.Navina S, Acharya C, Delany JP, Orlichenko LS, Baty CJ, Shiva SS, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med. 2011 Nov 2;3(107):107ra10. doi: 10.1126/scitranslmed.3002573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Surendran RP, Visser ME, Heemelaar S, Wang J, Peter J, Defesche JC, et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012 Jan 12; doi: 10.1111/j.1365-2796.2012.02516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaRusch J, Barmada MM, Solomon S, Whitcomb DC. Whole Exome Sequencing Identifies Multiple, Complex Etiologies in an Idiopathic Hereditary Pancreatitis Kindred. JOP. 2012;13(3):258–62. [PMC free article] [PubMed] [Google Scholar]