

Abstract
Objectives
The concept of inflammation-induced sensitization is emerging in the field of perinatal brain injury, stroke, Alzheimer disease and multiple sclerosis. However, mechanisms underpinning this process remain unidentified.
Methods
We combined in vivo systemic lipopolysaccharide (LPS) or Interleukin-1β (IL-1β) induced sensitization of neonatal and adult rodent cortical neurons to excitotoxic neurodegeneration with in vitro IL-1β sensitization of human and rodent neurons to excitotoxic neurodegeneration. Within these inflammation-induced sensitization models we assessed metabotropic glutamatergic receptor (mGluR) signaling and regulation.
Results
We demonstrate for the first time that group I mGluRs mediate inflammation-induced sensitization to neuronal excitotoxicity in neonatal and adult neurons across species. Inflammation induced G protein–coupled receptor kinase 2 (GRK2) down-regulation and genetic deletion of GRK2 mimicked the sensitizing effect of inflammation on excitotoxic neurodegeneration. Thus, we identify GRK2 as a potential molecular link between inflammation and mGluR-mediated sensitization.
Interpretation
Collectively, our findings indicate that inflammation-induced sensitization is universal across species and ages and that group I mGluRs and GRK2 represent new avenues for neuroprotection in perinatal and adult neurological disorders.
Introduction
Excessive activation of glutamate receptors leads to excitotoxicity1, and the latter plays a key role both in acute brain disorders such as stroke, traumatic brain injury, and perinatal brain lesions, and in chronic neurodegenerative disorders such as Alzheimer disease, Parkinson disease, or amyotrophic lateral sclerosis.2
Numerous clinical and preclinical studies have highlighted that systemic inflammation has a sensitizing effect on perinatal brain lesions.3 In particular, it has been shown that systemic exposure to pro-inflammatory cytokines such as interleukin-1β (IL-1β) or to systemic lipopolysaccharide (LPS) prior to an excitotoxic or hypoxic-ischemic challenge leads to a major exacerbation of neuronal damage in newborn rodents.4 More recently, a similar sensitizing mechanism of systemic inflammation has been suggested in a rodent model of adult stroke involving specifically IL-1β.5 Moreover, clinical evidence supports the existence of such a potential mechanism in human adult stroke.6
In this context, deciphering the as yet poorly understood molecular mechanisms linking systemic inflammation and neuronal excitotoxicity is paramount and could unravel novel targets for neuroprotection relevant for many brain diseases. To address this, we have developed a rodent model in which systemic administration of pro-inflammatory cytokines exacerbates excitotoxic brain lesions induced by intracerebral ibotenate, a glutamate analog acting on both N-methyl-D-aspartate (NMDA) and group I metabotropic (mGlu1 and mGlu5) receptors.4, 7
Activity of mGluRs is regulated by G protein–coupled receptor kinase 2 (GRK2) through phosphorylation and subsequent desensitization.8 Uncoupling and internalization of G protein–coupled receptors (GPCRs) are crucial regulatory processes to ensure attenuation of signaling, preventing overstimulation and damage of cells.9 Both in vivo and in vitro studies have shown that inflammation regulates the expression of GRK2 in multiple cell types including neurons.10
In the present study, we test the hypothesis that group I mGluRs and GRK2 mediate the inflammation-induced sensitization to neuronal excitotoxicity, using an extensive combination of in vivo and in vitro approaches.
Methods
Animals
Experiments on rodents were carried out in compliance with the European Community Commission guidelines (86/609/EEC) and were approved by the institutional review board (Bichat-Robert Debré ethical committee, Paris, France). GRK2+/+ (wild type), GRK2+/-, GRK2flox/+CamK2aCre/+, GRK2+/+CamK2aCre/+, GRK2flox/+GFAPCre/+ and GRK2+/+GFAPCre/+ pups (C57BL/6 background, The Jackson Laboratory, Bar Harbor, MI, USA) were used (homozygous deletion of GRK2 are not viable). Time-lines of the in vivo and the in vitro rodent experiments are presented in Supplementary Figure 1.
Systemic inflammation
In a first set of experiments, mouse pups were injected intraperitoneally (ip) with recombinant mouse IL-1β (R&D systems, Oxon, UK) during 5 days (P1 to P5). In a second set of experiments, adult mice (P45) were injected ip with purified lipopolysaccharide (LPS, Escherichia coli, serotype 055:B5; Sigma, St Louis, MO) during 3 days (P45 to P47). In the last set of experiments, pregnant rats were injected intraperitoneally (ip) with LPS on embryonic days 19 and 20 (E19 and E20).
Excitotoxic lesions
Two hours after the last injection of IL-1β or LPS for P5 and P47 mice, and at P5 for the rat pups, intracerebral injection of glutamate analogues was performed. Glutamate analogue administration was performed as previously described.11
Statistical analysis
Quantitative data are expressed as mean±SEM for each treatment group. Results were compared using Student's t-test (2-tailed) or ANOVA with Bonferroni's correction for multiple comparisons of means test. A p<0.05 was considered as significant (GraphPad Prism, GraphPad Software, La Jolla, CA). In addition, to determine any possible effect of the number of experimental replicates (number of cultures for in vitro data and number of litters for in vivo data), we have performed a two-way ANOVA with “replicate” and “sample” as variables (Supplementary Table 1).
See Supplementary Methods for a description of the systemic inflammation models, intracerebrally administrated drugs, lesion size determination, in vitro drugs, mouse primary neuronal cultures, Human SK-N-MC neuronal culture, quantification of cell viability and of cell death, quantitative RT-PCR, western blotting, immunohistochemistry, and calcium imaging.
Results
Inflammation sensitizes rodent and human neurons to excitotoxic neurodegeneration
As previously shown4, daily intraperitoneal (ip) administration of IL-1β to mouse pups between postnatal day (P) 1 and P5, when compared to ip injection of PBS, exacerbated cortical grey matter lesion size induced by intracerebral ibotenate injection at P5 (Figure 1A-B). Similarly, daily ip injection of Escherichia coli lipopolysaccharide (LPS) to pregnant rats between embryonic day (E) 19 and E2012 exacerbated cortical grey matter lesion size induced by intracerebral ibotenate injection at P5 (Figure 1C). In addition, daily ip injection of LPS to adult mice between P45 and P47 exacerbated cortical grey matter lesion size induced by intracerebral ibotenate injection at P47 (Figure 1D). These data demonstrate that systemic inflammation produced by LPS or IL-1β sensitizes the newborn and adult rodent brain to excitotoxicity.
Figure 1. Inflammation sensitizes neurons to excitotoxic neurodegeneration in vivo and in vitro.
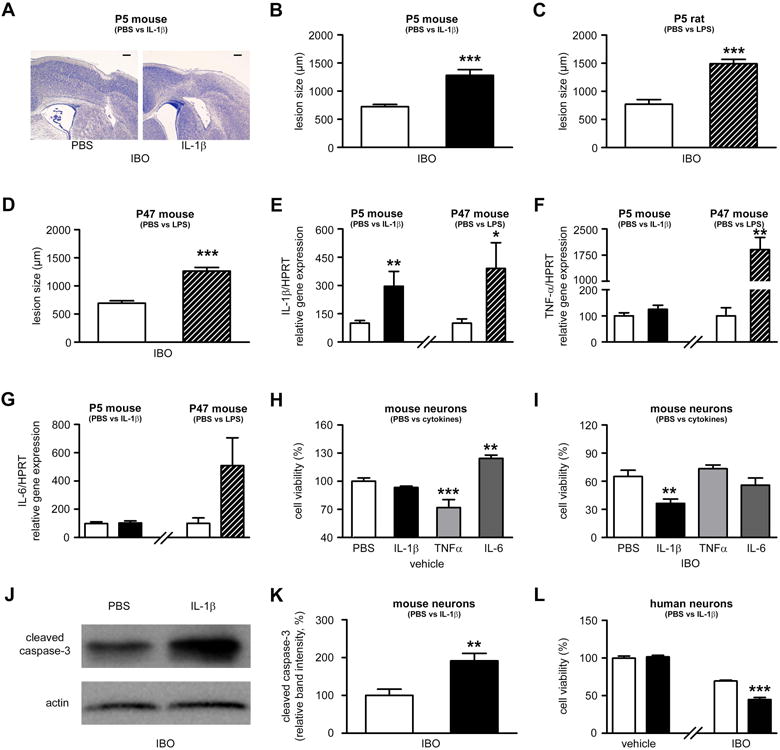
A. Cresyl violet-stained sections showing lesions induced by ibotenate (IBO) injected on P5 following PBS or IL-1β injection between P1 and P5. Bar=80µm. B. Quantification of cortical plate lesions induced by ibotenate in P5 mice following PBS (white bar) or IL-1β (black bar) injections between P1 and P5 (n=16-28/group). C. Quantification of cortical plate lesions induced by ibotenate in P5 rats following PBS (white bar) or LPS (hatched bar) injections between E19 and E20 (n=12-16/group). D. Quantification of cortical plate lesions induced by ibotenate in P47 mice following PBS (white bar) or LPS (hatched bar) injections between P45 and P47 (n=9-15/group). E-G. qRT-PCR of the cortical mRNA levels of IL-1β (E), TNF-α (F), and IL-6 (G) in mice treated between P1 and P5 with PBS (white bar) or IL-1β (black bar) (n=14-22/group), and in mice treated between P45 and P47 with PBS (white bar) or LPS (hatched bar) (n=6/group). H. Cell viability quantification in mouse neurons cultured with PBS, IL-1β, TNFα, or IL-6 (n=30-40 wells/group). I. Cell viability quantification in mouse neurons cultured with PBS, IL-1β, TNFα, or IL-6 and exposed to ibotenate on the fourth day (n=12 wells/group). J-K. Western blot for cleaved caspase-3 in mouse neurons cultured with PBS (white bar) or IL-1β (black bar) and exposed to ibotenate on the fourth day (n=6 wells/group). L. Cell viability quantification in human neurons cultured with PBS (white bar) or IL-1β (black bar) and exposed to vehicle or ibotenate on the fifth day (n=12 wells/group). Bars represent mean + SEM. * p<0.05, ** p<0.01, *** p<0.001 in Student's t-test (B-G, K-L) or ANOVA with Bonferroni's multiple comparison tests (H-I). PBS: Phosphate buffered saline; IL-1β: interleukin-1β; IBO: ibotenate; LPS: lipopolysaccharide.
Sensitization by systemic inflammation is associated with increased production of pro-inflammatory molecules within the brain.13-15 Accordingly, quantitative RT-PCR (qRT-PCR) analysis of pro-inflammatory cytokine expression performed in neocortices showed that ip injection of IL-1β between P1 and P5 induced an increased expression of IL-1β but not of TNFα or IL-6 while ip injection of LPS between P45 and P47 induced an increased expression of both IL-1β and TNFα (Figure 1E-G).
In order to decipher the molecular mechanisms underlying the inflammation-induced sensitization to excitotoxicity, we developed an in vitro model. Mimicking the in vivo conditions of increased intra-cerebral expression of IL-1b seen in the above models of systemic inflammation we tested the effects of IL-1b on neuronal survival in vitro, using mouse primary cortical neurons. We also assessed the effects in this system of exposure to TNFα and IL-6. A four-day exposure to IL-1β had no detectable effect on neuronal viability when compared to PBS, while a four-day treatment with TNFα decreased neuronal viability and a four-day treatment with IL-6 increased neuronal viability (Figure 1H). When mouse cortical neurons were exposed to ibotenate following 4 days of exposure to cytokines (IL-1β, IL-6 or TNFα) or PBS, IL-1β exacerbated neuronal cell death (Figure 1I-K) while TNFα and IL-6 did not have a sensitizing effect (Figure 1I). Supporting the concept of sensitization across species, IL-1β-induced sensitization was also observed when human SK-N-MC neurons were exposed to IL-1β for 5 days prior to ibotenate administration (Figure 1L). Based on the above in vivo and in vitro data, we selected IL-1β as the inflammatory stimulus in all subsequent in vitro experiments.
Sensitization by inflammation to excitotoxicity requires the activation of group I mGluRs
In both P5 mouse pups pre-exposed to ip IL-1β and in P47 mice pre-exposed to ip LPS, an exacerbation of excitotoxic brain damage was observed when these animals were injected intracerebrally with ibotenate (Figure 2A-B). This effect was not elicited when mice were similarly injected intracerebrally with NMDA (Figure 2A-B). Exacerbation of excitotoxicity was also observed in animals injected with a combination of NMDA and the group I mGluR agonist 3,5-dihydroxyphenylglycine (DHPG), mimicking the targets of ibotenate. Conversely exacerbation of excitotoxicity was abolished when IL-1β- or LPS-treated animals were injected intracerebrally with a combination of ibotenate and 1-aminoindan-1,5-dicarboxylic acid (AIDA), a selective mGlu1 antagonist (Figure 2A). The sensitizing effect of IL-1β in the P5 mice was also abolished when ibotenate was co-administered with the selective negative allosteric modulator (NAM) of mGlu1, JNJ16259685 (3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl)-methanone, or co-administered with the selective NAM of mGlu5, MTEP (3-(2-Methyl-1,3-thiazol-4-yl) ethynyl) pyridine hydrochloride). (Figure 2A). P5 pups injected with DHPG alone did not display detectable brain lesion (data not shown).
Figure 2. Sensitization requires the activation of group I mGluR in vivo and in vitro.
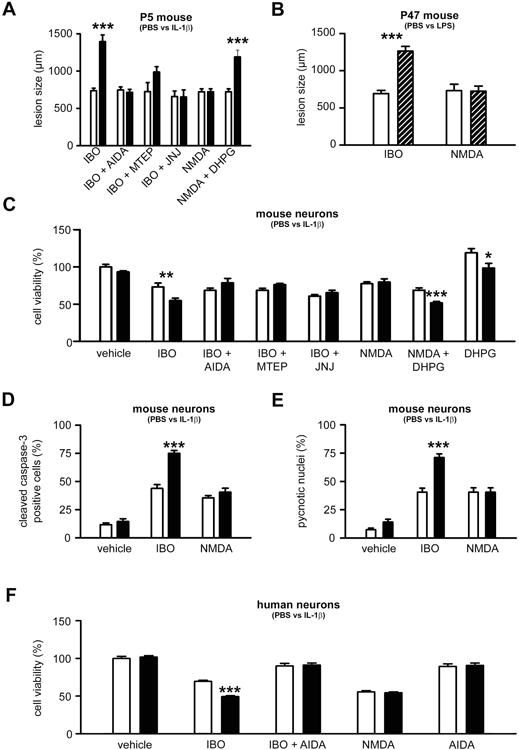
A. Quantification of cortical plate lesions induced by glutamate analogues (indicated on the X axis) in P5 mice following PBS (white bar) or IL-1β (black bar) injections between P1 and P5 (n=12-28/group). B. Quantification of cortical plate lesions induced by glutamate analogues (indicated on the X axis) in P47 mice following PBS (white bar) or LPS (hatched bar) injections between P45 and P47 (n=10-12/group). C-E. Quantification of cell viability (C), western blot for cleaved caspase-3 (D) and pycnotic nuclei (E) in mouse neurons cultured in the presence of PBS (white bars) or IL-1β (black bars) and exposed to glutamate analogues (indicated on the X axis) on the fourth day (n=10-18 well/group). F. Quantification of cell viability in human neurons cultured in the presence of PBS (white bar) or IL-1β (black bar) and exposed to glutamate analogues (indicated on the X axis) on the fifth day (n=8 wells/group). Bars represent mean + SEM. * p<0.05, ** p<0.01, *** p<0.001 in ANOVA with Bonferroni's multiple comparison tests vs. PBS-injected mice (A-B) and vs PBS-treated neurons (C-F). PBS: Phosphate buffered saline; IL-1β: interleukin-1β; LPS: lipopolysaccharide; IBO: ibotenate; AIDA: 1-aminoindan-1,5-dicarboxylic acid (group I mGluR antagonist); MTEP: 3-((2-Methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (mGlu5 negative allosteric modulator); JNJ: JNJ16259685, 3,4-dihydro-2H-pyrano[2,3-b]quinolin-7-yl)-(cis-4-methoxycyclohexyl)-methanone, (mGlu1 negative allosteric modulator); NMDA: N-methyl-D-aspartate, DHPG: 3,5-dihydroxyphenylglycin, (group I mGluR agonist).
Similar to the in vivo situation, in cultured human SK-N-MC neurons and mouse primary cortical neurons, the sensitizing effect of IL-1β only occurred after activation of both NMDA receptors and group I mGluRs (Figure 2C-F). In mouse primary neurons, exposure to DHPG alone induced an increase in cell survival, which was abolished by pre-exposure to IL-1β (Figure 2C).
In P5 mouse pups and in mouse primary neurons, no sensitizing effect of IL-1β was observed when S-bromo-willardiine (alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid -AMPA- and kainate receptor agonist) was used alone or in combination with DHPG (Supplementary Figure 2).
Inflammation does not lead to detectable changes in expression of glutamate receptors
To test the hypothesis that IL-1β-induced sensitization to excitotoxicity is linked to changes in the expression of mGluRs, the expression of mRNA for mGlu1-8 was quantified by qRT-PCR in mouse primary cortical neurons exposed to IL-1β or PBS for 4 days (Figure 3A). In addition, group I mGluR (mGlu1 and mGlu5) protein expression was determined by western blot analysis in the same experimental conditions (Figure 3B-C). IL-1β did not induce any detectable change in the expression of metabotropic glutamate receptors.
Figure 3. Effect of inflammation on the metabotropic glutamatergic receptor expression.
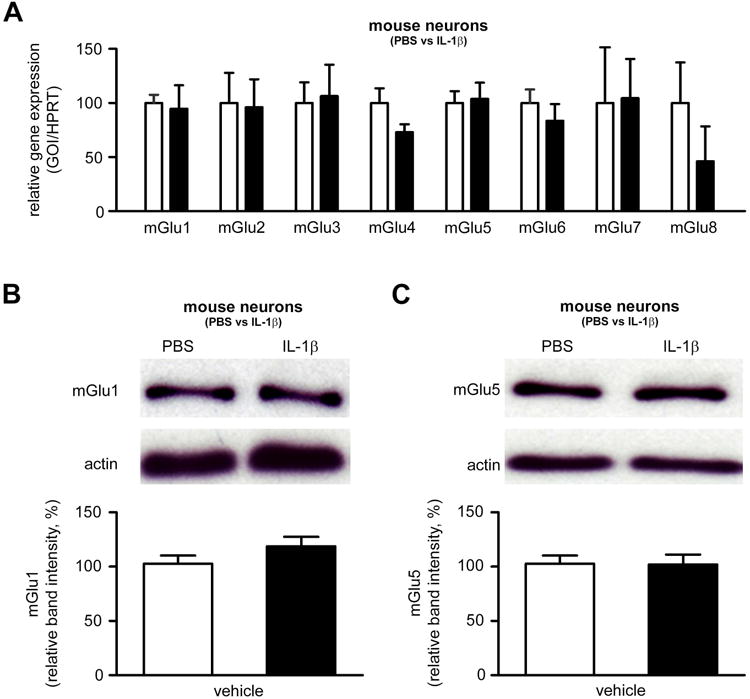
A. qRT-PCR of mGluRs in mouse neurons cultured in the presence of PBS (white bars) or IL-1β (black bars; 50ng/ml once a day for 4 days) and exposed to ibotenate on the fourth day (n=10 wells/group). B-C. Quantification of western blot for mGlu1α (C) and mGlu5 (D) in mouse neurons cultured in the presence of PBS (white bars) or IL-1β (black bars; 50 ng/ml once a day for 4 days) and exposed to ibotenate on the fourth day (n=12 wells/group). Bars represent mean + SEM. PBS: Phosphate buffered saline; IL-1β: interleukin-1β.
Inflammation exacerbates ibotenate-induced calcium mobilization in neurons
In the absence of detectable IL-1β-induced changes in the expression of mGluRs, we hypothesized that inflammation induces a functional change of group I mGluRs. Group I mGluRs are GPCRs. Binding of glutamate to mGluRs leads to the activation of phospholipase Cβ1 (PLCβ1). Activation of PLCβ1 results in formation of diacylglycerol (DAG) which causes the activation of protein kinase C (PKC), and also induces the formation of IP3, which leads to calcium mobilization from endogenous stores.
In P5 mouse pups sensitized by IL-1β and in P47 mice sensitized by LPS, intracerebral injection of the PLCβ inhibitor U73122 reduced ibotenate-induced neuronal loss (Figure 4A-B). Similarly, in mouse primary cortical neurons sensitized by IL-1β, U73122 protected neurons against ibotenate-induced cell death (Figure 4C). In contrast, treatment with one of the two PKC inhibitors bis-indolylmaleimide and chelerythrine had no protective effect (Figure 4C). IL-1β administration did not modify the expression of PLCβ1 in cultured neurons, as determined by qRT-PCR (data not shown) or western blot analysis (Figure 4D). Altogether, these data support a role of the mGluR-PLCβ1 pathway in the increased ibotenate-induced neuronal loss after IL-1β sensitization. However, the DAG-PKC-pathway does not seem to play a significant role.
Figure 4. Inflammation exacerbates ibotenate-induced calcium mobilization in neurons.
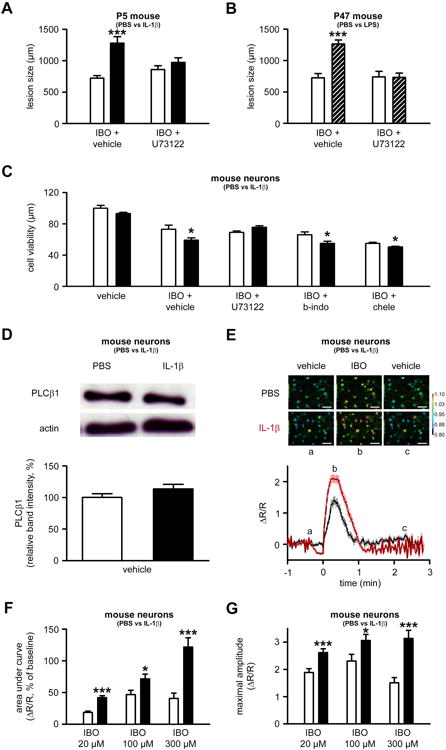
A. Quantification of cortical plate lesions induced by ibotenate+vehicle or ibotenate+U73122 (PLCβ1 inhibitor) on P5 following PBS (white bar) or IL-1β (black bar) between P1 and P5 (n=8-10/group). B. Quantification of cortical plate lesions induced by ibotenate+vehicle or ibotenate+U73122 on P47 following PBS (white bar) or LPS (hatched bar) between P45 and P47 (n=12/group). C. Quantification of cell viability in mouse neurons cultured in the presence of PBS (white bar) or IL-1β (black bar) and exposed to vehicle, ibotenate+vehicle or ibotenate+transduction inhibitors (b-indo, bis-indolylmaleimide; chele, chelerythrine) on the fourth day (n=12-18 wells/group). D. Quantification of western blot for PLCβ1 in mouse neurons cultured with PBS (white bars) or IL-1β (black bars) (n=8 wells/group). E. Representative images and traces of calcium levels in mouse neurons cultured with PBS (black trace) or IL-1β (red trace) and exposed on the fourth day to vehicle (a) prior to 100μM ibotenate (b), followed by vehicle wash (c). Bar=50μm. F-G. Quantification of the area under the curve (F) and maximal amplitude (G) of calcium levels in mouse neurons cultured with PBS (white bar) or IL-1β (black bar) and exposed to different concentrations of ibotenate (n=35-50 wells/group). The scale indicates Ca2+ level, expressed as ΔR/R values, where R is the ratio (R) between fluorescence signals at 340 and 380 nm obtained before the addition of any agent, and ΔR the difference between the ratios measured during a response and R. Bars represent mean + SEM. * p<0.05, *** p<0.001 in ANOVA with Bonferroni's multiple comparison tests vs. PBS-injected mice (A-B) and vs PBS-treated neurons (C-G). PBS: Phosphate buffered saline; IL-1β: interleukin-1β; LPS: lipopolysaccharide; IBO: ibotenate; PLCβ1: Phospholipase Cβ1.
To test the potential role of ibotenate-induced calcium mobilization in the sensitizing effect of inflammation, the impact of IL-1β on ibotenate-induced increases in calcium levels in mouse primary cortical neurons was determined in vitro. Exposure of primary cortical neurons to IL-1β significantly increased ibotenate-induced calcium mobilization, as determined by the area under the curve and the maximum amplitude of calcium concentration (Figure 4E-G), as well as by the proportion of non-responsive neurons (Supplementary Figure 3).
GRK2 is a potential molecular link between sensitizing inflammation and excitotoxicity
GRK2 regulates phosphorylation and subsequent desensitization of different GPCRs, including group I mGluRs.16 In addition, we have shown that inflammation is associated with a decrease in GRK2 protein levels in neurons and other cell types.10 Moreover, newborn mice with reduced GRK2 levels in neurons have been shown to be more sensitive to hypoxic-ischemic insults.17 Based on these findings, we hypothesized that inflammation will reduce GRK2 levels in neurons and that this will contribute to the group I mGluR-mediated exacerbation of excitotoxicity.
In support of our hypothesis, western blot analysis showed that human SK-N-MC neurons exposed to IL-1β for 5 days had a reduced GRK2 content (Figure 5A). Similarly, exposure of primary cortical neurons to IL-1β decreased the GRK2 immunostaining signal (Supplementary Figure 4). Overexpression of GRK2 in human SK-N-MC neurons protected these cells against the combined activation of NMDA receptors + mGluRs but had no effect when only NMDA receptors were activated (Figure 5B). Conversely, GRK2+/- mouse primary cortical neurons that have a ∼50% reduction in GRK2 protein18 were more sensitive to ibotenate excitotoxicity, but not to NMDA alone, when compared to GRK2+/+ control neurons (Figure 5C). These data were further confirmed in vivo. P5 mouse pups with a GRK2 deficiency in all cells (GRK2+/- mice) were more sensitive to ibotenate, but not to NMDA alone or to ibotenate + AIDA, when compared to GRK2+/+ control pups (Figure 5D). Further pointing to neurons as a primary target, P5 mouse pups with a GRK2 deficiency restricted to neurons (GRK2flox/+CamK2aCre/+ mice) also exhibited a higher sensitivity to ibotenate when compared with control pups (GRK2+/+CamK2aCre/+ mice) (Figure 5E). In contrast, P5 mouse pups with a GRK2 deficiency restricted to astrocytes (GRK2flox/+GFAPCre/+ mice) exhibited a reduced sensitivity to ibotenate when compared with control pups (GRK2+/+GFAPCre/+ mice) (Supplementary Figure 5).
Figure 5. Role of GRK2 in the sensitizing effect of inflammation.
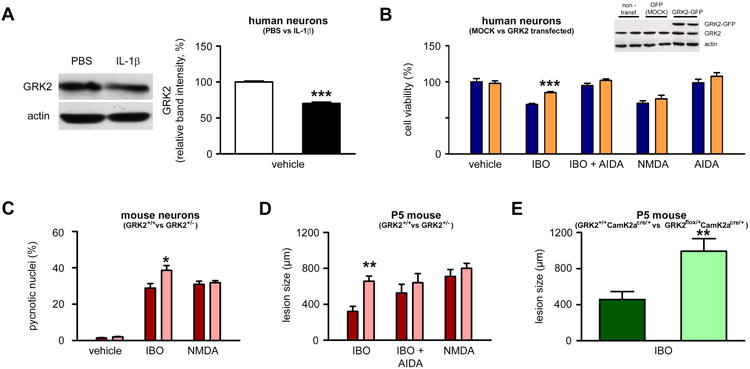
A. Quantification of western blot for GRK2 in human neurons cultured in the presence of PBS (white bar) or IL-1β (black bar) (n=24 wells/group). B. Quantification of cell viability in human neurons transfected with MOCK vector (blue bar) or with GRK2-expressing vector (orange bar) and exposed to glutamate analogues (indicated on the X axis) on the fifth day (n=8 wells/group). C. Quantification of cell viability in mouse neurons from GRK2+/+ (red bars) and GRK2+/- (pink bars) mice exposed to vehicle, ibotenate or NMDA (n=15-18 wells/group). D. Quantification of the cortical plate size of brain lesions induced by glutamate analogues (indicated on the X axis) on P5 and studied on P10 in GRK2+/+ (red bars) and GRK2+/- (pink bars) mice (n=7-9/group). E. Quantification of the cortical plate size of brain lesions induced by ibotenate on P5 and studied on P10 in GRK2+/+CamK2aCre/+(dark green bar) and GRK2flox/+CamK2aCre/+(light green bar) mice (n=7/group). Bars represent mean + SEM. * p<0.05, ** p<0.01, *** p<0.001 in Student's t-test (A, E) or in ANOVA with Bonferroni's multiple comparison tests vs. MOCK-transferred neurons (B) and vs. GRK2+/+ mice (C-D) . PBS: Phosphate buffered saline; IL-1β: interleukin-1β; IBO: ibotenate; AIDA: 1-aminoindan-1,5-dicarboxylic acid (group I mGluR antagonist); NMDA: N-methyl-D-aspartate; DHPG: 3,5-dihydroxyphenylglycine.
Discussion
We have shown that inflammation sensitizes human and rodent neurons to excitotoxic neurodegeneration. We further demonstrate that this sensitization process involves GRK2 down-regulation, leading to over activation of group I mGluRs and subsequent sustained calcium mobilization. Figure 6 provides a schematic view of the potential molecular mechanisms underlying this sensitization process.
Figure 6. Schematic representation of the potential mechanisms by which group 1 mGluR and GRK2 mediate IL-1β-induced sensitization.
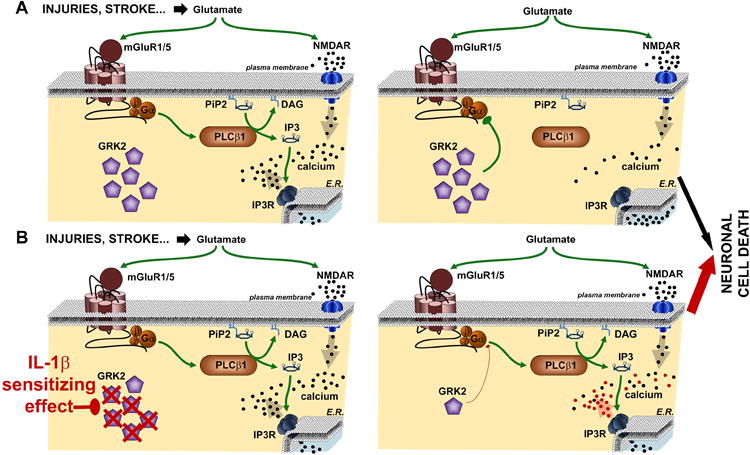
The diagram depicts the molecular mechanisms leading to excitotoxic neuronal cell death in the absence (A) or presence (B) of IL-1β sensitizing effect. In the absence of IL-1β, GRK2 leads to a rapid desensitization of mGlu1/5, limiting the PLCβ1-mediated calcium release from endogenous stores. Exposure to IL-1β leads to a reduced content of GRK2, preventing the complete desensitization of mGlu1/5 and allowing a more prolonged calcium release from endogenous stores. This enhanced calcium mobilization exacerbates neuronal cell death. E.R., endoplasmic reticulum.
Role of group I mGluRs in the sensitization process: a “universal” process in neurodegeneration?
The key role of group I mGluRs in inflammation-induced sensitization to excitotoxicity is strongly supported by the pharmacological data from the present study. In addition, a requirement for mGluRs for sensitization was observed i) both in vivo and in vitro, ii) with IL-1β and LPS, iii) with human and rodent neurons, and iv) in neonates and adults. Together these lines of evidence suggest that this mGluR-dependent mechanism might be involved a large array of neurological disorders where inflammation has been demonstrated or suggested to play a sensitizing role4, 12, 19-23. The role of mGlu1 and mGlu5 in mechanisms of neurodegeneration/neuroprotection has been extensively studied both in vitro and in vivo24, 25. Amplification or reduction of injury is reported following activation of mGlu1 and mGlu5 with DHPG or other orthosteric agonists, depending on the model of neurodegeneration, the nature of the insult, and the functional state of the two receptor subtypes.24 At most synapses, mGlu1 and mGlu5 are localized in the extrasynaptic portion of dendritic spines. A bi-directional cross-talk exists between group-I mGluRs and NMDA receptors26-28, possibly related to interactions between them via scaffolding protein.
mGlu1a and mGlu5 receptors bear a long C-terminus domain that allows the interaction with Homer proteins. 29-31 In the postsynaptic elements, long isoforms of Homer proteins allow a physical interaction between group-I mGlu receptors and the NR2 subunit of NMDA receptors via a chain of interacting proteins, which include PSD-95 and Shank, and Homer.32 Several lines of evidence indicate that activation of group-I mGlu receptors (in particular, mGlu5 receptors) enhances NMDA receptor function26, 33-35, and the dual mGlu1/5 receptor agonist, DHPG, amplifies NMDA toxicity in cultured cortical cells. 36 To our knowledge, there is no evidence for a direct interaction between group-I mGlu receptors and AMPA receptors, although in one report pharmacological activation of mGlu5 receptors with 2-chloro-5-hydroxyphenylglycine (mGluR5 agonist) has been found to enhance AMPA (and NMDA) responses in the spinal cord.37
As reported above, activation of group-I mGluRs can become neurotoxic when combined with the activation of NMDA receptors. For example, isolated activation of mGlu1a stimulates both inositol phospholipid hydrolysis and the phosphatodylinositol-3-kinase (PI3K) pathway. Simultaneous activation of NMDA receptors leads to a calpain-mediated cleavage of the C-terminus domain of mGlu1a, thereby preventing the receptors from activating the neuroprotective PI3K pathway while leaving the stimulation of inositol phospholipid hydrolysis intact and causing neuronal death27. Our data show that inflammation amplifies the neurotoxic component mediated by group-I mGluRs through a mechanism that involves the down-regulation of GRK2, which makes mGlu1 and mGlu5 resistant to homologous desensitization (see below). This amplifies PLCβ1 activation and the ensuing increase in intracellular Ca2+ release, eventually leading to neuronal cell death (Figure 6). All together, this contributes to explain the inflammation-induced sensitization to ibotenate, which is a dual group-I mGluR and NMDA receptor agonist, or with the combination of NMDA with DHPG, but not NMDA alone.
Role of GRK2 in the sensitization process: a key link between inflammation and mGluR
Several lines of evidence support the hypothesis that GRK2 plays a role in mediating the effects of inflammation on group I mGluRs: i) it has been previously shown that inflammation down-regulates the expression of GRK2 in multiple cell types including neuronal cells in vivo and in vitro10; ii) accordingly, in the present study, IL-1β reduced GRK2 content in human SK-N-MC neurons; iii) neurons with reduced GRK2 levels were more sensitive to excitotoxic insult (present study), and newborn mice with reduced GRK2 levels in neurons have been reported to be more sensitive to hypoxic-ischemic38 and excitotoxic (present study) insults; iv) overexpression of GRK2 has been shown to reduce glutamate signaling via group I mGluRs8; and, v) accordingly, in the present study, human SK-N-MC neurons with increased GRK2 levels were more resistant to an excitotoxic insult.
The present data do not allow excluding alternate GRK2-independent pathways by which inflammation could impact on mGluRs. For example, the JAK-STAT pathway induced by IL-1β can potentially regulate the expression or function of multiple receptors in neurons, including GABA, muscarinic, NMDA and AMPA receptors.39 Additional studies will be necessary to decipher the specific contribution of the various mechanisms linking inflammation to mGluR activity.
Role of IL-1β and other cytokines in the sensitization process
In the present study, we considered brain IL-1β as a potential molecular link between systemic inflammation and exacerbated sensitivity of neurons to excitotoxicity, based on a series of evidence: i) in vivo, both systemic LPS and IL-1β induced an increased brain production of IL-1β; ii) in vitro, IL-1β exacerbated neuronal death induced by ibotenate; and iii) in vitro, IL-1β exacerbated calcium mobilization induced by ibotenate in neurons. Constitutive expression of IL-1β in normal brain tissue is low, but expression is induced following systemic stress or brain insults, both in rodents and humans.15, 40, 41 In this mechanism of sensitization, the precise cellular source of brain IL-1β remains to be determined but could be the resident microglia or perivascular macrophages.42 Although our in vitro data point to a neuronal mechanism by which IL-1β induces sensitization, other cell types including microglia, astrocytes, endothelial cells, and systemic immune cells may play an important role in the in vivo sensitization process.
Recently, Gardoni et al. have shown that there is a dynamic interaction between NMDA receptors and IL-1 receptor type I, providing evidence that IL-1β can act as a modulator in pathological events relying on glutamate receptor activation.43Although the implication of brain IL-1β44 in the sensitization process is well supported, one cannot exclude the role of other inflammatory molecules such as other cytokines, including TNF-α45, IL-646 or IL-1847, as well as chemokines48, and matrix metalloproteinases49, which have been implicated in inflammation-mediated brain damage.
Implications for neuroprotective strategies
As highlighted above, group I mGluR agonists have shown both neuroprotective and neurotoxic effects in vitro and in vivo models of neurodegeneration.24, 25 However, it is clear from our data that neurotoxicity prevails in a context of inflammation-induced sensitization. One interesting question is whether mGlu1, mGlu5 or both mediate neuronal death during inflammation. We have found that inflammation-induced sensitization of excitotoxic death was abolished by pharmacological blockade of either mGlu1 or mGlu5. A recent report demonstrates that, at least in recombinant cells, mGlu1 and mGlu5 can form functional heterodimers50, and it is known that one molecule of a NAM is sufficient to fully block mGluR dimers.51 Thus, an attractive hypothesis is that native mGlu1/mGlu5 heterodimers become sensitized by inflammatory cytokines, and that either mGlu1 or mGlu5 NAMs are fully protective against excitototoxic death occurring during neuroinflammation.
Altogether, our data support the concept that, while group I mGluR agonists might be neuroprotective in the absence of systemic inflammation, they could become toxic in the context of inflammation-induced sensitization. As discussed in the Introduction, growing evidence suggests that systemic inflammation might play a role in a large variety of neurological disorders.4, 5, 12, 20-22 Interestingly, in a model of amyotrophic lateral sclerosis, it was recently shown that blockade of mGlu5 (one of the two group I mGluRs) was protective against excitotoxic neuronal cell death.52 Interestingly, mGlu5 NAMs are in the late stage of clinical development53. Thus, these drugs will soon be available to examine the precise role of group-I mGluRs in neuronal degeneration associated with neuroinflammation in humans. In addition to blocking group I mGluRs, targeting GRK2 directly could be another promising avenue for protecting the brain in the context of inflammation-induced sensitization. Strategies aiming at increasing GRK2 production or rather stabilizing the molecule should be experimentally tested in the future.
In conclusion the present study provides experimental support that group I mGluRs and GRK2 are involved in the mechanisms underlying inflammation-mediated sensitization to excitotoxic neurodegeneration. This study therefore offers new avenues for neuroprotection in a large array of neurological disorders across the perinatal period and throughout adulthood that we increasingly recognize to be sensitized by inflammation.
Supplementary Material
Acknowledgments
The authors thank Bobbi Fleiss for discussions and critical reading of the manuscript, Vincent Lelièvre and Sophie Lebon for the design of the primers, and Leslie Schwendimann and Tifenn Le Charpentier for invaluable technical support. This study was supported by grants from Inserm, Université Paris 7, APHP (Contrat Hospitalier de Recherche Translationnelle to Dr Pierre Gressens), PremUP, Sixth Framework Program of the European Commission (contract no LSHM-CT-2006-036534/NEOBRAIN), Seventh Framework Program of the European Union (grant agreement no. HEALTH-F2-2009-241778/NEUROBID), Fondation Roger de Spoelberch, Fondation Grace de Monaco, Fondation Leducq, Fondation pour la Recherche Médicale, Institut pour la Recherche sur la Moelle épinière et l'Encéphale (IRME), Fondation des Gueules Cassées, Fondation Areva, Société Française d'Anesthésie Réanimation (SFAR), and Medical Research Council (UK). Graham Collingridge is WCU International Scholar and supported by WCU program through the KOSEF funded by the MEST (R31-10089).
References
- 1.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 2.Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–808. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- 3.Degos V, Favrais G, Kaindl AM, et al. Inflammation processes in perinatal brain damage. J Neural Transm. 2010;117:1009–1017. doi: 10.1007/s00702-010-0411-x. [DOI] [PubMed] [Google Scholar]
- 4.Dommergues MA, Patkai J, Renauld JC, et al. Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann Neurol. 2000;47:54–63. [PubMed] [Google Scholar]
- 5.McColl BW, Allan SM, Rothwell NJ. Systemic infection, inflammation and acute ischemic stroke. Neuroscience. 2009;158:1049–1061. doi: 10.1016/j.neuroscience.2008.08.019. [DOI] [PubMed] [Google Scholar]
- 6.Smith CJ, Emsley HC, Gavin CM, et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004;4:2. doi: 10.1186/1471-2377-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aden U, Favrais G, Plaisant F, et al. Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: key role of TNFalpha pathway and protection by etanercept. Brain Behav Immun. 2010;24:747–758. doi: 10.1016/j.bbi.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Dale LB, Bhattacharya M, Anborgh PH, et al. G protein-coupled receptor kinase-mediated desensitization of metabotropic glutamate receptor 1A protects against cell death. J Biol Chem. 2000;275:38213–38220. doi: 10.1074/jbc.M006075200. [DOI] [PubMed] [Google Scholar]
- 9.Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Alves-Filho JC, Sonego F, Souto FO, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. 2010;16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 11.Marret S, Mukendi R, Gadisseux JF, et al. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol. 1995;54:358–370. doi: 10.1097/00005072-199505000-00009. [DOI] [PubMed] [Google Scholar]
- 12.Rousset CI, Kassem J, Olivier P, et al. Antenatal bacterial endotoxin sensitizes the immature rat brain to postnatal excitotoxic injury. J Neuropathol Exp Neurol. 2008;67:994–1000. doi: 10.1097/NEN.0b013e31818894a1. [DOI] [PubMed] [Google Scholar]
- 13.Favrais G, Schwendimann L, Gressens P, Lelievre V. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol Dis. 2007;25:496–505. doi: 10.1016/j.nbd.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Stridh L, Li W, et al. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009;183:7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- 15.Dantzer R, O'Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribeiro FM, Ferreira LT, Paquet M, et al. Phosphorylation-independent regulation of metabotropic glutamate receptor 5 desensitization and internalization by G protein-coupled receptor kinase 2 in neurons. J Biol Chem. 2009;284:23444–23453. doi: 10.1074/jbc.M109.000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nijboer CH, Kavelaars A, Vroon A, et al. Low endogenous G-protein-coupled receptor kinase 2 sensitizes the immature brain to hypoxia-ischemia-induced gray and white matter damage. J Neurosci. 2008;28:3324–3332. doi: 10.1523/JNEUROSCI.4769-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nijboer CH, Heijnen CJ, Willemen HL, et al. Cell-specific roles of GRK2 in onset and severity of hypoxic-ischemic brain damage in neonatal mice. Brain Behav Immun. 2010;24:420–426. doi: 10.1016/j.bbi.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eklind S, Mallard C, Leverin AL, et al. Bacterial endotoxin sensitizes the immature brain to hypoxic--ischaemic injury. Eur J Neurosci. 2001;13:1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- 21.Parry-Jones AR, Liimatainen T, Kauppinen RA, et al. Interleukin-1 exacerbates focal cerebral ischemia and reduces ischemic brain temperature in the rat. Magn Reson Med. 2008;59:1239–1249. doi: 10.1002/mrm.21531. [DOI] [PubMed] [Google Scholar]
- 22.Holmes C, Cunningham C, Zotova E, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreno B, Jukes JP, Vergara-Irigaray N, et al. Systemic inflammation induces axon injury during brain inflammation. Ann Neurol. 2011;70:932–942. doi: 10.1002/ana.22550. [DOI] [PubMed] [Google Scholar]
- 24.Nicoletti F, Bruno V, Catania MV, et al. Group-I metabotropic glutamate receptors: hypotheses to explain their dual role in neurotoxicity and neuroprotection. Neuropharmacology. 1999;38:1477–1484. doi: 10.1016/s0028-3908(99)00102-1. [DOI] [PubMed] [Google Scholar]
- 25.Caraci F, Battaglia G, Sortino MA, et al. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: Still a hot topic? Neurochem Int. 2012 doi: 10.1016/j.neuint.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Pisani A, Gubellini P, Bonsi P, et al. Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience. 2001;106:579–587. doi: 10.1016/s0306-4522(01)00297-4. [DOI] [PubMed] [Google Scholar]
- 27.Xu W, Wong TP, Chery N, et al. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 28.Alagarsamy S, Saugstad J, Warren L, et al. NMDA-induced potentiation of mGluR5 is mediated by activation of protein phosphatase 2B/calcineurin. Neuropharmacology. 2005;49(1):135–145. doi: 10.1016/j.neuropharm.2005.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brakeman PR, Lanahan AA, O'Brien R, et al. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 1997;386:284–288. doi: 10.1038/386284a0. [DOI] [PubMed] [Google Scholar]
- 30.Kato A, Ozawa F, Saitoh Y, et al. Novel members of the Vesl/Homer family of PDZ proteins that bind metabotropic glutamate receptors. J Biol Chem. 1998;273:23969–23975. doi: 10.1074/jbc.273.37.23969. [DOI] [PubMed] [Google Scholar]
- 31.Xiao B, Tu JC, Petralia RS, et al. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron. 1998;21:707–716. doi: 10.1016/s0896-6273(00)80588-7. [DOI] [PubMed] [Google Scholar]
- 32.Tu JC, Xiao B, Naisbitt S, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 33.Doherty AJ, Palmer MJ, Henley JM, et al. (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) activates mGlu5, but no mGlu1, receptors expressed in CHO cells and potentiates NMDA responses in the hippocampus. Neuropharmacology. 1997;36:265–267. doi: 10.1016/s0028-3908(97)00001-4. [DOI] [PubMed] [Google Scholar]
- 34.Awad H, Hubert GW, Smith Y, et al. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20:7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collett VJ, Collingridge GL. Interactions between NMDA receptors and mGlu5 receptors expressed in HEK293 cells. Br J Pharmacol. 2004;142:991–1001. doi: 10.1038/sj.bjp.0705861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruno V, Copani A, Knopfel T, et al. Activation of metabotropic glutamate receptors coupled to inositol phospholipid hydrolysis amplifies NMDA-induced neuronal degeneration in cultured cortical cells. Neuropharmacology. 1995;34:1089–1098. doi: 10.1016/0028-3908(95)00077-j. [DOI] [PubMed] [Google Scholar]
- 37.Ugolini A, Corsi M, Bordi F. Potentiation of NMDA and AMPA responses by the specific mGluR5 agonist CHPG in spinal cord motoneurons. Neuropharmacology. 1999;38:1569–1576. doi: 10.1016/s0028-3908(99)00095-7. [DOI] [PubMed] [Google Scholar]
- 38.Nijboer CH, van der Kooij MA, van Bel F, et al. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav Immun. 2010;24:812–821. doi: 10.1016/j.bbi.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Nicolas CS, Peineau S, Amici M, et al. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brochu ME, Girard S, Lavoie K, Sebire G. Developmental regulation of the neuroinflammatory responses to LPS and/or hypoxia-ischemia between preterm and term neonates: An experimental study. J Neuroinflammation. 2011;8:55. doi: 10.1186/1742-2094-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McClain CJ, Cohen D, Ott L, et al. Ventricular fluid interleukin-1 activity in patients with head injury. J Lab Clin Med. 1987;110:48–54. [PubMed] [Google Scholar]
- 42.Serrats J, Schiltz JC, Garcia-Bueno B, et al. Dual roles for perivascular macrophages in immune-to-brain signaling. Neuron. 2010;65:94–106. doi: 10.1016/j.neuron.2009.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gardoni F, Boraso M, Zianni E, et al. Distribution of interleukin-1 receptor complex at the synaptic membrane driven by interleukin-1beta and NMDA stimulation. J Neuroinflammation. 2011;8:14. doi: 10.1186/1742-2094-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- 45.Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 46.Raivich G, Bohatschek M, Kloss CU, et al. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- 47.Hedtjarn M, Leverin AL, Eriksson K, et al. Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci. 2002;22:5910–5919. doi: 10.1523/JNEUROSCI.22-14-05910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mirabelli-Badenier M, Braunersreuther V, Viviani GL, et al. CC and CXC chemokines are pivotal mediators of cerebral injury in ischaemic stroke. Thromb Haemost. 2011;105:409–420. doi: 10.1160/TH10-10-0662. [DOI] [PubMed] [Google Scholar]
- 49.Bednarek N, Clement Y, Lelievre V, et al. Ontogeny of MMPs and TIMPs in the murine neocortex. Pediatr Res. 2009;65:296–300. doi: 10.1203/PDR.0b013e3181973aee. [DOI] [PubMed] [Google Scholar]
- 50.Doumazane E, Scholler P, Zwier JM, et al. A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 2011;25:66–77. doi: 10.1096/fj.10-163147. [DOI] [PubMed] [Google Scholar]
- 51.Kniazeff J, Bessis AS, Maurel D, et al. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol. 2004;11:706–713. doi: 10.1038/nsmb794. [DOI] [PubMed] [Google Scholar]
- 52.D'Antoni S, Berretta A, Seminara G, et al. A prolonged pharmacological blockade of type-5 metabotropic glutamate receptors protects cultured spinal cord motor neurons against excitotoxic death. Neurobiol Dis. 2011;42:252–264. doi: 10.1016/j.nbd.2011.01.013. [DOI] [PubMed] [Google Scholar]
- 53.Nicoletti F, Bockaert J, Collingridge GL, et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology. 2011;60:1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.