

Abstract
The ATP binding cassette transporter A-1 (ABCA1) is critical for apolipoprotein-mediated cholesterol efflux, an important mechanism employed by macrophages to avoid becoming lipid-laden foam cells, the hallmark of early atherosclerotic lesions. It has been proposed that lipid-free apolipoprotein A-I (apoA-I) enters the cell and is resecreted as a lipidated particle via a retroendocytosis pathway during ABCA1-mediated cholesterol efflux from macrophages. To determine the functional importance of such a pathway, confocal microscopy was used to characterize the internalization of a fully functional apoA-I cysteine mutant containing a thiol-reactive fluorescent probe in cultured macrophages. ApoA-I was also endogenously labeled with 35S-methionine to quantify cellular uptake and to determine the metabolic fate of the internalized protein. It was found that apoA-I was specifically taken inside macrophages and that a small amount of intact apoA-I was resecreted from the cells. However, a majority of the label that reappeared in the media was degraded. We estimate that the mass of apoA-I retroendocytosed is not sufficient to account for the HDL produced by the cholesterol efflux reaction. Furthermore, we have demonstrated that lipid-free apoA-I-mediated cholesterol efflux from macrophages can be pharmacologically uncoupled from apoA-I internalization into cells. On the basis these findings, we present a model in which the ABCA1-mediated lipid transfer process occurs primarily at the membrane surface in macrophages, but still accounts for the observed specific internalization of apoA-I.
Keywords: ATP binding cassette transporter A-1, apolipoprotein A-I, cell biology, endocytosis, macrophage, confocal microscopy, protein expression
Cardiovascular disease (CVD) is the leading cause of death in the United States (1). Numerous prospective studies have demonstrated an inverse correlation between HDL plasma levels and many manifestations of CVD (2, 3). This may be due, in part, to HDL’s role in the reverse cholesterol transport (RCT) system, the process that packages excess cholesterol into HDL and transports it from the periphery to the liver for excretion as bile salts (4, 5).
Because HDL is a critical part of the RCT system and an important risk factor in CVD, many studies have been performed to better understand the factors controlling its production. It has become clear that HDL biogenesis is dependent on the proper function of the membrane transport protein, ATP binding cassette transporter A-1 (ABCA1) (6–9). The lack of functional ABCA1 can lead to Tangier disease, which is characterized by a near absence of plasma HDL, the accumulation of cholesterol esters in the liver, spleen, lymph nodes, and macrophages, and an increased risk of coronary artery disease (8–10). Numerous studies have demonstrated a dependence on ABCA1 for lipid efflux from peripheral cells (10), particularly macrophages. However, despite compelling evidence of ABCA1’s physiological importance, its molecular role in HDL biogenesis is still not fully understood. It is thought that nascent HDL is formed when ABCA1 facilitates the efflux of phospholipids and cholesterol to lipid-poor apolipoprotein A-I (apoA-I) (11). However, the details of the interaction between apoA-I and ABCA1 are unclear. A better understanding of this interaction would increase our knowledge of the physiological mechanisms of HDL and RCT regulation, and help identify new drug targets to enhance the body’s preexisting protective mechanism against atherosclerotic plaque formation.
The location of apoA-I lipidation and HDL formation is controversial. Evidence exists suggesting that the interaction and lipidation of apoA-I via ABCA1 may occur through a retroendocytosis pathway through which apolipoprotein is taken into the cell, is lipidated, and is then resecreted without degradation (12, 13). Multiple groups have demonstrated that in macrophages, apoA-I and ABCA1 colocalize in endosomal compartments (14, 15). This is congruent with the observations that ABCA1 is rapidly cycled between the cell surface and endosomal or lysosomal compartments, and that these intracellular compartments are the source of a majority of the cholesterol effluxed to apoA-I in an ABCA1-dependent manner (16, 17). However, it is unknown whether this retroendocytic pathway plays a significant role in the efflux of cholesterol from macrophages. If retroendocytosis plays an important role in the lipidation of apoA-I, then the following should be true: a) apoA-I should be specifically internalized into the cell upon activation of ABCA1; b) the mass of apoA-I cycled though the cell should account for most of the nascent HDL particles formed in a given period of time; and c) the degree of apoA-I internalization should correlate with the degree of cholesterol efflux promoted. To test these predictions, both fluorescent and radiolabeled apoA-I was generated so that the movement and possible degradation of apoA-I could be studied throughout the retroendocytosis pathway.
EXPERIMENTAL PROCEDURES
Materials
Enterokinase was purchased from Novagen (Madison, WI). IgA protease was obtained from Mobitec (Marco Island, FL). Isopropyl-β-d-thiogalactoside (IPTG) was from Fisher Scientific (Pittsburgh, PA). POPC was acquired from Avanti Polar lipids (Birmingham, AL). Fatty acid-free BSA was from Calbiochem (San Diego, CA). cAMP was from Sigma (St. Louis, MO). FBS and PBS were from Invitrogen (Carlsbad, CA). DTT was from Amresco (Solon, OH). Tris-(2-carboxyethyl) phosphine (TCEP) was obtained from Molecular Probes (Eugene, OR). All chemical reagents were of the highest quality available.
Methods
Construction, expression and labeling of cysteine mutants of apoA-I. The human apoA-I Cys mutants used in this study were expressed in the bacterial pET30 expression vector (Novagen), which encodes an N-terminal histidine tag sequence that can be cleaved from the protein by enterokinase. The cysteine point mutants shown in Fig. 1 were generated directly in this vector with PCR-based techniques using the Quick Change mutagenesis kit (Stratagene; La Jolla, CA). Human apoA-I ordinarily lacks a cysteine. The sequence of each construct was verified on an Applied Biotechnology System (Foster City, CA) DNA sequencer at the University of Cincinnati DNA Core. The proteins were expressed in BL-21 Escherichia coli cells according to established methods (18). The proteins were then purified over Zn2+ chelating columns (Roche Molecular Biochemicals; Indianapolis, IN) according to manufacturer’s instructions. The use of Zn2+ versus the more commonly used Ni2+ chelation approach was required because DTT, which is necessary to keep the Cys reduced during purification, tended to precipitate out of solution when Ni2+ was used. Fractions containing the pure proteins were dialyzed against standard Tris buffer (STB: 10 mM Tris-HCl, 0.15 M NaCl, 1 mM EDTA, 0.2% NaN3, pH 8.2) with 1 g DTT per liter. Before labeling, the DTT was removed by dialysis against STB. TCEP and sodium-cholate were added to a final concentration of 0.35 μM and 0.1%, respectively. To 7.5 mg of apoA-I mutant, 1 mg of solid Alexa Fluor 546, C5-maleimide, and sodium salt (Molecular Probes) in 1 ml STB, pH 7.1, was added. The label was added to the protein 30 μl at a time at 1 min intervals. The reaction was then incubated at room temperature for 2 h. The labeled protein was reconstituted into HDL particles for cleavage with enterokinase [apoA-I is nonspecifically cleaved by enterokinase if it is lipid-free (19)]. Reconstituted HDL particles were prepared using POPC at a lipid-to-protein molar ratio of 110:1 according to the method of Jonas (20). The histidine tag was cleaved with enterokinase (2.2 U/mg apoA-I) and passed over a Superdex 200 gel filtration column (Amersham-Pharmacia; Piscataway, NJ), to separate the labeled protein from the cleaved histidine tag and unreacted fluorescent label. The fractions containing labeled protein were combined, lyophilized, and subjected to a chloroform-methanol delipidation to remove lipid and lipopolysaccharide (21). The dried protein was dissolved in 3 M guanidine HCl and dialyzed into STB for use.
Fig. 1.

Effect of the introduction of a Cys residue and attachment of an Alexa Fluor probe on the ability of apolipoprotein A-I (apoA-I) to promote apolipoprotein-mediated cholesterol efflux from cultured macrophages. RAW264.7 cells were labeled for 24 h with 3H-cholesterol as described in Methods. After removal of labeling media and three washes, medium containing 10 μg/ml of lipid-free acceptor with 0.3 mM cAMP, to upregulate ATP binding cassette transporter A-1 (ABCA1), was added to the cells for 24 h. All Cys mutants represented as acceptors in this figure have the Alexa Fluor 546 probe attached. The cholesterol efflux data are expressed as percentage of total cell 3H-cholesterol for cells whose lipids were extracted immediately after the washes that followed labeling (t0). Error bars represent 1 SD of triplicate samples from one of three representative experiments. A one-way ANOVA was performed, and the asterisk indicates a significant difference from human plasma apoA-I (P < 0.001) by a Tukey-Kramer multiple comparison test.
Expression and purification of 35S-labeled apoA-I. WT (wild-type) apoA-I cDNA in the pET30 vector containing the IgA protease cleavage site (21) was transfected into a methionine auxotroph line of E. coli (b834-DE3) (Novagen). A single colony was used to generate 100 ml Luria-Bertani cultures, which were pelleted and washed with M9A medium [1.0 g/l ammonium chloride, 5.8 g/l dibasic sodium phosphate, 3.0 g/l monobasic potassium phosphate, 2 mM MgCl2, 0.1 mM CaCl2, 2% glucose (v:v), 0.1 mg/ml Thr, 0.1 mg/ml Leu, 0.2 mg/ml Pro, 0.2 mg/ml Arg, and 0.0001% (v:v) thiamine]. The cells were grown in fresh M9A medium for 1 h at 37°C. Then 35S-Met (Amersham-Pharmacia) and IPTG were added at a concentration of 5 μCi/ml and 0.5 mM, respectively, for 1 h at 37°C. The apoA-I was isolated as described above, with the exception that IgA protease was used to remove the His tag.
Cholesterol efflux studies. The transformed mouse macrophage cell line RAW264.7 (American Type Culture Collection; Manassas, VA) was maintained in DMEM (Invitrogen) with 10% FBS and 50 μg/ml gentamycin. Cells were grown to 75% confluence in a 48-well plate, then this maintenance media was removed and 3H-cholesterol labeling media was added for 24 h [DMEM, 10% FBS, 50 μg/ml gentamycin, 3H-cholesterol, 1.0 μCi/ml (Amersham-Pharmacia); 0.5 ml per well]. After 24 h, the labeling media was removed and cells were washed twice with PBS with 0.2% BSA and once with DMEM containing 0.2% BSA. Efflux media [DMEM, 0.2% BSA, 10 μg/ml acceptor (apoA-I or specified mutant)] was added, with or without 0.3 mM 8-bromo-cAMP. For all instances in this manuscript, the term “cAMP” refers to this analog. After 24 h, a 100 μl sample of efflux media was passed through a 0.45 μm filter to remove any floating cells and then measured by liquid scintillation counting. Percent efflux was calculated by dividing the counts in the media by total counts in the cells at time 0 h (22). In experiments using the endocytosis inhibitors, the cells were preincubated for 1.5 h with the inhibitor alone in DMEM, 0.2% BSA. This was removed, and the cells were incubated with inhibitor and 10 μg/ml apoA-I acceptor in DMEM, 0.2% BSA for 1.5 or 6 h, as indicated in the figure legends. Otherwise, the cholesterol efflux studies were performed as above, and apoA-I uptake studies were performed as below.
Confocal microscopy studies. RAW264.7 macrophages were grown on slide well plates (#138121; Nalge Nunc International, Rochester, NY) in the maintenance media described above. Once the cells were 60% confluent, they were incubated in DMEM containing 0.2% BSA with or without 0.3 mM cAMP for 16 h. Media containing the Alexa Fluor-labeled apoA-I and 0.2% BSA was then added with or without 0.3 mM cAMP for the appropriate length of time. Cells were washed three times with PBS (no BSA). Then 0.75 ml of a 3% paraformaldehyde, 2% sucrose solution was added to the cells. The cells were fixed on ice for 45 min. Fixed cells were washed in PBS with 1% BSA (to block non-specific binding). In some experiments, a rat anti-mouse monoclonal antibody to the macrophage membrane protein CD11b (also known as MAC-1) (Research Diagnostics; Flanders, NJ), 10 μg/ml in PBS with 1% BSA, was added to the cells. The cells were incubated for 40 min on ice and washed in PBS with 1% BSA. An Oregon Green-labeled secondary antibody, which excites at the same wavelength as FITC (Oregon Green 488-conjugated goat anti-rat IgG; Molecular Probes), was added to the cells at a concentration of 20 μg/ml in PBS with 1% BSA. The cells were then incubated for an additional 40 min on ice, washed with PBS with 1% BSA, and sealed with coverslips using melted paraffin. For colocalization experiments, LysoTracker Green (Molecular Probes) was added to cells at a final concentration of 200 nM in the Alexa Fluor-labeled apoA-I media described above. Once the cells were labeled, confocal imaging was performed on a Leica TCS 4D microscope/SCANware system (Heidelberg, Germany) equipped with an Omnichrome krypton-argon laser (Chino, CA). Excitation was performed using the FITC/TRITC channel, which excites at 488 nm and 568 nm simultaneously. Emission was collected using an RSP580 beam splitter and BP530 filter at detector #1 and a BP600 filter at detector #2. The data were analyzed using the image analysis software on the SCANware system. The amount of apoA-I within the cell was calculated by determining the number of pixels in the TRITC channel per cell and subtracting background. For control studies performed using dextran, the dextran conjugate-Alexa Fluor 546 (Molecular Probes) was dissolved in STB to a final concentration of 2 mg/ml. The dextran conjugate was added to the cells in DMEM with 0.2% BSA ± 0.3 mM cAMP at a final dextran concentration of 18 μg/ml for 2 h. The cells were washed and fixed, and the membranes were labeled with CD11b as described above. Confocal studies using Chinese hamster ovary (CHO) cells were performed in a similar manner, but the base medium was Ham’s F-12 (Invitrogen) instead of DMEM. The media for the CHO cells transfected with ABCA1-green fluorescent protein (GFP) additionally contained 3 mg/ml of the antibiotic G-418 sulfate (USB Corp.; Cleveland, OH). Similar to the macrophage experiments, cells were incubated in Ham’s F-12 media with 0.2% BSA for 16 h.
35S-apoA-I uptake, resecretion and degradation. Pulse-chase apoA-I uptake and resecretion experiments were performed in RAW264.7 cells plated in 24-well plates and grown until 85% confluent in DMEM maintenance media. Cells were washed twice with 1 ml PBS with 0.2% BSA and once with 1 ml DMEM with 0.2% BSA, and preincubated for 16 h in DMEM with 0.2% BSA ± 0.3 mM cAMP. Then they were pulsed with 3 μg/ml 35S-radiolabeled apoA-I ± 0.3 mM cAMP in 0.75 ml DMEM with 0.2% BSA at 37°C for 1 h. The cells were washed five times with 0.75 ml cold PBS with 1 mM CaCl2, 0.2% BSA, and 5 μg/ml human HDL to remove adherent apolipoprotein (12). The original media was sampled to determine total counts added to the cells. The chase incubation containing 0.5 ml DMEM, 0.2% BSA, and 50 μg/ml human HDL ± 0.3 mM cAMP was incubated with the cells at 37°C for the indicated time period. The media was collected to find total secreted activity and then subjected to a TCA precipitation in which 50% (w/v) TCA was added in a 1:4 ratio to media, incubated 30 min at 4°C, and centrifuged for 20 min at 15,000 relative centrifugal force. One-hundred micro-liters of supernatant was counted to determine the amount of degraded protein present. The cells were washed once with 1 ml PBS with 0.2% BSA and twice with 1 ml PBS, and dissolved in 0.5 ml 0.2 M NaOH. One-hundred microliters of this solution was counted on the scintillation counter to determine residual cell label. Total cell uptake was calculated as the sum of the medium and residual cell-associated radioactivity. The degraded protein was expressed as the TCA-soluble fraction of the chase medium, and the resecreted (intact) protein was calculated as total counts in the chase medium minus the TCA-soluble counts (12). To determine the rate of particle formation, 35S-apoA-I was added to cells as described above. The media was collected at 3 h and run over a Superdex 200 gel filtration column (Amersham-Pharmacia). These fractions were analyzed by liquid scintillation counting. The same experiment was performed with 3H-cholesterol-labeled cells with or without 10 μg unlabeled apoA-I as the acceptor protein to determine which column fractions contained cholesterol.
Miscellaneous. Human plasma apoA-I was purified as previously described (23). The ABCA1-GFP construct (a gift of Dr. Richard Lawn) in the mammalian expression vector pEGFP-N1 (Clontech; Palo Alto, CA) was stably expressed in CHO cells using the Lipofectamine Plus kit (Invitrogen) following the manufacturer’s instructions.
RESULTS
If a retroendocytosis pathway plays a key role in apolipoprotein-mediated cholesterol efflux, then apoA-I should be detectable within macrophages soon after its introduction to cells expressing ABCA1. To test this, we constructed a panel of apoA-I mutants, each containing a single cysteine to which a thiol-reactive probe was attached. Previous work has shown that the presence of this probe does not have a major effect on the secondary structure content of these proteins, as determined by circular dichroism (18). Absorbance spectroscopy confirmed that each apoA-I molecule contained a single molecule of fluorescent probe. Figure 1 shows that the label introduced at amino acids 9, 93, and 124 did not affect the ability of apoA-I to participate in apolipoprotein-mediated cholesterol efflux in RAW macrophages stimulated with 0.3 mM cAMP. However, when the probe was introduced at position 232 in helix 10, a 40% decrease in ABCA1-mediated cholesterol efflux was observed. This is not surprising, because helix 10 has been shown to be critical to apoA-I’s ability to stimulate ABCA1-mediated cholesterol efflux (24). The V93C mutant was chosen for further studies because it exhibited the highest levels of expression in our recombinant system.
Next, the ability to visualize fluorescent apoA-I (V93C) within RAW264.7 macrophages treated with cAMP to induce expression of ABCA1 was assessed. Panels 1 and 2 of Fig. 2A show an optical slice taken through the center of the macrophage by confocal microscopy after incubation with fluorescent apoA-I (V93C) for 2 h. The red-fluorescing Alexa Fluor probe on apoA-I was clearly identifiable within the confines of the CD11b membrane marker (green) in cells preincubated with cAMP. Within each cell, the apoA-I label was excluded from a large area that appeared to be the cell nucleus. Much less of the label was apparent within the cells in the absence of cAMP pretreatment (cf. panels 1 and 2). This is illustrated quantitatively by computer image analysis, shown in Fig. 2B. The cells shown in Fig. 2 were fixed in paraformaldehyde (see Methods), but similar results were obtained when live cells were imaged (see supplementary Fig. I). We also characterized the time dependence of apoA-I uptake. ApoA-I was detected within the cells 15 min after its addition to the media. However, before 30 min, most of the label was associated with, or just beneath, the cell surface (data not shown). After 30 min, most of the label appeared to be inside the cells, as shown in Fig. 2A.
Fig. 2.

The specificity of apoA-I uptake by RAW264.7 macrophages. RAW264.7 cells were incubated for 16 h ± 0.3 mM cAMP to upregulate ABCA1. Then, either 10 μg/ml Alexa Fluor-labeled apoA-I (V93C) ± 400 μg/ml unlabeled apoA-I competitor, or 18 μg/ml Alexa Fluor-labeled dextran beads were added ± 0.3 mM cAMP for 2 h. A: Representative confocal images (magnification 63×) taken through the approximate center of fixed cells from the various treatments. The apoA-I and dextran labels appear red, whereas the CD11b outer membrane marker is green. The labels on the left edge of the figure refer to treatment with or without cAMP for the entire row of images. The labels across the top identify the internalized component for the entire column of images. B: Computer image analysis in which the red pixels were quantified on a per cell basis from six fields (at least 75 cells) per sample after background subtraction. Bars that have the same symbol are significantly different from each other, $ (P < 0.0001) and * (P < 0.001) by an unpaired two-tailed Student’s t-test. The error bars represent 1 SD of triplicate samples from one of two representative experiments.
A trivial explanation for the cellular internalization of apoA-I is that treatment with cAMP, a second messenger known to exert a multitude of effects on macrophage metabolism, simply stimulates nonspecific endocytosis in macrophages. To evaluate this possibility, the effect of cAMP treatment on the uptake of dextran polymers labeled with the identical Alexa Fluor 546 probe as that present on our apoA-I (V93C) was studied. This method has been used extensively to study phagocytic processes in macrophages (25). Panels 3 and 4 in Fig. 2A show that the uptake of dextran was similar regardless of cAMP treatment. To further determine the specificity of fluorescent apoA-I internalization, a 40-fold excess of unlabeled apoA-I was added to the treatment. Figure 2 shows that the presence of excess unlabeled apoA-I competitively inhibited the internalization of apoA-I label (cf. panels 2 and 6), indicating significant specificity in apoA-I internalization. (In using the term “specific” we do not mean to imply that apoA-I is the only apolipoprotein that can interact with these domains. The other exchangeable apolipoproteins that are capable of promoting apolipoprotein-mediated cholesterol efflux can undoubtedly compete for these same sites.) To gain insight into the identity of the intra-cellular compartments into which the apoA-I label was sequestered, we performed additional confocal studies using the LysoTracker reagent. Figure 3 shows that a substantial fraction of the internalized apoA-I label colocalized with LysoTracker when cAMP was present, indicating that some of the internalized apoA-I is trafficked to lysosomal or late endosomal compartments (26). This colocalization was not observed in the absence of cAMP (data not shown). Finally, we studied the ability of the label to exit the cell after a thorough wash and a chase incubation in clean media. Figure 4 shows a nonsignificant trend toward the release of a small amount of label, hinting that a portion of the label might be resecreted. However, the majority of the fluorescence signal stayed associated with the cells for the duration of the experiment.
Fig. 3.
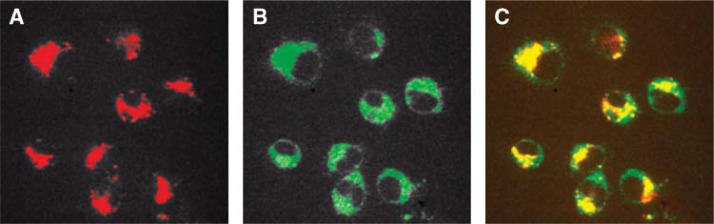
Colocalization of Alexa Fluor apoA-I (V93C) with LysoTracker Green in RAW cells. Fluorescent apoA-I was incubated with RAW cells, as described in Fig. 2, with the addition of LysoTracker Green to the labeling media at a final concentration of 200 nM. The cells were incubated in labeling media for 2 h. A: Representative confocal image (magnification 60×) taken through the approximate center of fixed cells showing Alexa Fluor signal (marking apoA-I location). B: Confocal image identical to that in panel A, viewed through LysoTracker Green channel. C: Merged image of panels A and B. All images are representative of two independent experiments.
Fig. 4.

Pulse-chase incubation of Alexa Fluor apoA-I (V93C) in RAW cells. Fluorescent apoA-I was incubated with cells as described for Fig. 2. However, instead of immediately fixing the cells after the 2 h incubation, the cells were extensively washed. Then, chase medium (0.5 ml DMEM, 0.2% BSA) lacking fluorescent apoA-I was placed on the cells for the indicated period of time. At each time point, the cells were fixed, visualized by confocal microscopy, and then quantified by image analysis as described for Fig. 2. A one-way ANOVA failed to detect a significant difference between uptake at T = 0 and the subsequent time points. The error bars represent 1 SD of triplicate samples from one of two representative experiments.
The confocal microscopy approach, although well suited for measuring fluorescent apoA-I internalization, gives little information on the fate of the label after internalization. Did the protein remain intact or did it undergo immediate lysosomal degradation? To address this question, 35S-methionine was used to endogenously label wild-type apoA-I expressed in bacteria. This method was chosen because studies have shown that radioiodination of apoA-I can have significant effects on its structure and metabolism (27). The purified 35S-apoA-I performed at levels similar to plasma apoA-I in a cholesterol efflux assay using RAW macrophages (data not shown). Figure 5A shows the results of a pulse-chase experiment in which RAW cells were incubated with 35S-apoA-I for 1 h, extensively washed, then chased with clean media for 1.5 h according to the protocol of Takahashi and Smith (12). At the end of the pulse incubation, approximately 66 ng of apoA-I/mg cell protein was associated with the cells when cAMP was present. This represents about 0.9% of the total apoA-I mass initially added to the media. During the 90 min chase period, 70% (or about 46 ng/mg cell protein) of the cellular 35S label appeared in the medium in cAMP-treated cells. However, 29 ng/mg cell protein was found to be degraded. This left 17 ng/mg cell protein (about 37% of the total secreted label) of presumably intact apoA-I. The remaining label stayed associated with the cells. In the absence of cAMP, the amounts of label in each category were substantially smaller, except the amount associated with the cell at the end of the experiment, which was similar to the cAMP treatment level. The effect of the length of the chase incubation on the amount of intact apoA-I resecreted versus that degraded was determined next. For chase times >90 min, it was found that little additional label came out of the cells (data not shown), indicating that the resecretion process was largely complete by 90 min. In addition, the ratio of intact-to-degraded resecreted apoA-I was consistent over time (Fig. 5B).
Fig. 5.

A pulse-chase quantification of uptake, resecretion (intact) and degradation of endogenously labeled 35S-apoA-I from macrophages. A: RAW cells were incubated (pulse) with 3 μg/ml 35S-apoA-I ± 0.3 mM cAMP for 1 h. They were washed thoroughly according to Methods to remove residual surface 35S-apoA-I and then incubated with clean medium containing 50 μg/ml human HDL for 1.5 h (chase) to assess resecretion. Total uptake is defined as the sum of the residual cell label and the label in the chase medium at the end of the experiment. The amount of secreted, degraded label was determined from a TCA precipitation performed on the chase medium. Resecreted (intact) apoA-I was defined as the difference between the total label in the chase medium and the amount that was degraded. Mass values were determined from the specific activity of the initial 35S-apoA-I. B: The pulse-chase experiment was performed exactly as stated above, except that the length of the chase incubation was varied as shown and cAMP was included in all samples. A one-way ANOVA failed to identify a significant difference among the ratios at any time point. The error bars represent 1 SD of triplicate samples from one experiment.
To measure the mass of lipid-free apoA-I incorporated into nascent, discoidal HDL particles, we preincubated macrophages ± cAMP for 8 h, added 35S-apoA-I to the media for an additional 3 h ± cAMP, and then fractionated the media by gel filtration chromatography. The chromatographic profile seen in Fig. 6A shows two major peaks occurring at about 21 and 34 ml, respectively, in samples lacking cAMP. PAGGE analysis identified peak #1 as intact, lipid-free 35S-apoA-I. Peak #2 was completely TCA soluble, indicating that it contained degraded apoA-I.
Fig. 6.

Gel filtration analysis of cholesterol efflux medium. For the experiments shown in panels A and B, RAW264.7 macrophages were preincubated 16 h ± 0.3 mM cAMP to upregulate ABCA1. Medium containing 3 μg/ml 35S-apoA-I ± 0.3 mM cAMP was added to the cells and incubated for 3 h at 37°C. The medium was collected, floating cells were removed, and then it was applied to a Superdex 200 gel filtration column. The fractions were analyzed for 35S-activity. Panel A shows the resulting profile in the absence of cAMP, and panel B shows the result in the presence of cAMP. For the experiment shown in panel C, RAW cells were incubated 16 h with 1 μCi/ml 3H-cholesterol and 0.3 mM cAMP, washed, and incubated in medium containing cAMP ± 10 μg/ml unlabeled wild-type apoA-I. The medium was collected and passed over the sizing column as in A and B. Peak 1: lipid-free 35S-apoA-I; peak 2: degraded 35S-apoA-I; peak 3: nascent HDL particles containing 35S-apoA-I and 3H-cholesterol (see Table 1).
With the addition of cAMP (Fig. 6B), a broad third peak appeared that contained intact 35S-apoA-I in the typical size range of discoidal HDL particles for this column. Figure 6C shows the chromatograph of a similar experiment using unlabeled apoA-I and RAW macrophages prelabeled with 3H-cholesterol and treated with cAMP. Activity measurements identified cholesterol in peak #3, indicating that it represents nascent HDL particles. Table 1 shows the percent distribution of the 35S-apoA-I and the mass of apoA-I associated with each peak shown in Fig. 6. The data in Figs. 5, 6 and Table 1 were used to approximate the flux of intact apoA-I through a retroendocytosis pathway (see Discussion).
TABLE 1.
Gel filtration distribution of 35S-apoA-I label in media after a 3 h incubation with RAW macrophages (see Fig. 4)
| Peak # | Identityb | Distribution of 35S-apoA-I Labela
|
|
|---|---|---|---|
| (−) cAMP | (+) cAMP | ||
| % | |||
| 1 | Intact 35S-apoA-I | 73 (164 ng) | 24 (54 ng) |
| 2 | Degraded (TCA soluble) 35S label | 15 (34 ng) | 27 (61 ng) |
| 3 | Intact 35S-apoA-I in cholesterol-containing particles | 8 (18 ng) | 48 (108 ng) |
| Totalc | 96 | 99 | |
apoA-I, apolipoprotein A-I.
The percent distribution of label from the column runs shown in Fig. 5 was determined by summing the total counts in each fraction. For each experiment, peak 1 was defined as fractions 19–24, peak 2 as fractions 30–36, and peak 3 as fractions 10–18. The percent distribution is shown, along with the approximate mass of apoA-I that would be associated with each peak, given that 2,250 ng of apoA-I was initially used in the incubation (see Discussion)
The state of apoA-I (intact, degraded, lipidated, etc.) was determined by Western blot analysis, TCA precipitation, and cofractionation with cholesterol as described in Results.
The total did not add up to 100%, because there were small amounts of counts present in fractions that were not included in the three major peaks.
To test our third prediction, that the degree of apoA-I internalization should correlate with the degree of cholesterol efflux promoted if a retroendocytosis pathway is the main method of apoA-I lipidation, we studied the effects of various endocytosis inhibitors on apoA-I uptake and cholesterol efflux. We tested cytochalasin D, which blocks cellular endocytosis by disassembling actin micro-filaments and preventing endocytotic vesicle formation; amiloride, a sodium channel inhibitor that disrupts endocytic vesicle formation; and monensin, an ion transport inhibitor that blocks transfer from the endosome to the lysosome by increasing the pH of intracellular vesicles, thus inhibiting trafficking from the Golgi to the plasma membrane (28). Using initial inhibitor concentrations found in the literature, monensin and amiloride could disrupt apoA-I internalization to various extents (see supplementary Fig. II). Although cholesterol efflux decreased slightly from untreated cells, there was no correlation between the degree of apoA-I uptake and cholesterol efflux inhibition. Cytochalasin D did not appear to affect apoA-I uptake or cholesterol efflux under these conditions. We further probed the monensin effect by performing a dose response experiment. Figure 7 shows that with the exception of very low concentrations, the degree of cholesterol efflux did not change substantially with increasing monensin concentration. In contrast, a clear dose-dependent decrease in apoA-I internalization was observed. At the highest monensin concentration, apoA-I label was almost completely excluded from the cell, yet cholesterol was still effluxed at nearly normal levels.
Fig. 7.

Effect of monensin concentration on apolipoprotein-mediated cholesterol efflux and apoA-I uptake. A: For cholesterol efflux studies, RAW264.7 macrophages were labeled with 3H-cholesterol and pre-treated with 0.3 mM cAMP as described in Methods. The cells were incubated with the appropriate concentration of monensin for 1.5 h, and then incubated with medium containing 10 μg/ml apoA-I, 0.3 mM cAMP, and the appropriate concentration of monensin for 6 h. Cholesterol efflux and cellular uptake are displayed as a fraction of activity in the absence of monensin. For the apoA-I uptake studies, the asterisk represents a significant difference from the untreated cells (P < 0.01) by one-way ANOVA followed by a Tukey-Kramer comparison. The error bars represent 1 SD of triplicate samples from one of two representative experiments. B: Representative confocal images taken through the center of the cell. The images were taken within 1 h of each other during the same experiment using identical instrument settings. The outer membrane marker used in Fig. 2 was omitted for these experiments. The concentration of monensin used is indicated under each image.
Finally, using CHO cells, the effect of over-expression of ABCA1 on the internalization of fluorescent apoA-I was determined. CHO cells exhibit higher basal levels of ABCA1 expression than do RAW macrophages and do not require (nor respond to) cAMP pretreatment (29). Expression of an ABCA1 construct containing a GFP tag increased cholesterol efflux in these cells by about 240% (Fig. 8). Despite the increase in cholesterol efflux in the transfected cells, the degree of internalization of the fluorescent apoA-I was actually decreased by about 50%, compared to untransfected cells.
Fig. 8.

Effect of ABCA1 over-expression on apolipoprotein-mediated cholesterol efflux and apoA-I uptake in Chinese hamster ovary (CHO) cells. CHO cells were transfected with an ABCA1 green fluorescent protein construct and compared with untransfected cells in terms of cholesterol efflux and apoA-I uptake over a 6 h incubation, as described in Methods and presented as in Fig. 7. The * and # symbols represent significant (P < 0.0001) differences from untransfected cells for apoA-I uptake and cholesterol efflux, respectively, by unpaired two-tailed Student’s t-test.
DISCUSSION
A retroendocytosis pathway for apolipoprotein-mediated cholesterol efflux is an attractive idea because it would offer numerous sites of regulation for potential pharmacological exploitation. Much of the evidence presented in the Introduction supports the existence of such a pathway, but the significance of this process in apolipoprotein-mediated cholesterol efflux from macrophages is unclear. We reasoned that if such a pathway plays a major role in cholesterol efflux from macrophages, then: a) apoA-I should be specifically internalized upon activation of ABCA1; b) the mass of apoA-I cycled though the cell should account for most of the nascent HDL particles formed in a given period of time; and c) the degree of apoA-I internalization should correlate with the degree of cholesterol efflux promoted. Each of these predictions is discussed below with respect to our data, then we present a model of apolipoprotein-mediated cholesterol efflux in which retroendocytosis plays, at most, a minor role.
To address the cellular uptake of apoA-I, we used a recombinant form of apoA-I that had been labeled with a fluorescent probe at a single and highly targeted site in the molecule. It has been shown previously that the C-terminal helix of apoA-I is critical for the ability of apoA-I to promote cholesterol efflux from these cells (24, 30). Consistent with this, cholesterol efflux capacity was negatively impacted when we tried to place a label in this helix at position 232 (Fig. 1). This demonstrates that a targeted approach to fluorescent labeling of apoA-I is a crucial component of any study designed to follow the cell biology of apoA-I by fluorescent microscopy. Thus, fluorescent labeling techniques that rely on random labeling of lysine residues, such as the Cy5 reagent, should be interpreted with caution.
As predicted, the fluorescent apoA-I was indeed taken into RAW macrophages when ABCA1 was upregulated using a cAMP analog (Fig. 2). This uptake could not be attributed to nonspecific uptake or recognition of the probe because upregulation of ABCA1 with cAMP did not affect the uptake of dextran beads labeled with the same Alexa Fluor probe. In addition, the uptake of fluorescent apoA-I was competitively inhibited by unlabeled apoA-I, indicating that the uptake was specific for the sequence of apoA-I. The uptake occurred rapidly, and the label appeared to be targeted to endosomal/lysosomal compartments within the cell. These observations are consistent with the idea of a retroendocytic pathway for apoA-I lipidation, but are also consistent with a degradation pathway in which apoA-I would also be transported to the lysosome. The fact that a large portion of the fluorescent label could not come back out of the cell during extended chase periods in media lacking labeled apoA-I supports the latter explanation (Fig. 4). Indeed, when we studied the fate of the internalized protein using the endogenously 35S-Met-labeled apoA-I in the presence of cAMP, the total amount of apoA-I associated with the cells was about 0.9% of that present in the medium, and although the majority of the cell-associated label reappeared in the chase medium, much of it had already been degraded. Our results differed from those of Takahashi and Smith (12), who showed that intact apoA-I represented the majority of the resecreted species. These investigators used apoA-I that had been nonspecifically radioiodinated, whereas our studies used endogenously labeled protein that contained no covalent modifications and that was demonstrated to be equally as capable of stimulating cholesterol efflux as WT apoA-I. It is possible that the labeling method may affect the cellular trafficking of apoA-I or its susceptibility to normal degradation processes.
The finding of a relatively small pool of resecreted apoA-I does not preclude the ability of such a pathway to account for the majority of apolipoprotein-mediated cholesterol efflux, especially if this small pool is rapidly turned over. To evaluate this possibility, we devised an estimate of the total flux of apoA-I through the putative retroendocytosis pathway. The estimation uses the degree of apoA-I degradation as a marker for the amount of apoA-I passed through the cells. This estimate depends on the following three assumptions, the merits of which will be discussed below: 1) degradation of apoA-I primarily occurs inside the cell; 2) the ratio of degraded apoA-I to intact apoA-I resecreted from the cell is constant over time, and no further degradation occurs once apoA-I is lipidated (see Fig. 5B); and 3) all apoA-I that goes through the cell is lipidated. From the pulse-chase study shown in Fig. 4, a ratio of intact apoA-I to degraded apoA-I of about 0.60 was measured and was consistent over a wide range of chase incubation lengths. As seen in Table 1, in the presence of cAMP, the macrophages converted 108 ng of apoA-I to a lipidated HDL particle in 3 h, giving an approximate rate of conversion of 90 ng of apoA-I/mg cell protein/h (using an average of 0.4 mg cell protein/well). During that time, 61 ng of apoA-I was degraded. If assumption 2 is correct, then 61 ng * 0.60 (the ratio of intact to degraded apoA-I) = 37 ng of intact apoA-I came out of the cells over 3 h, giving a rate of about 30.5 ng apoA-I/mg cell protein/h. Therefore, we estimate that about one-third of the total lipidated apoA-I product can be accounted for by retroendocytosed apoA-I. If one accounts for the nonspecific degradation of apoA-I evident in the non-cAMP treated macrophages, then only about 11% of the product HDL can be explained by ABCA1-mediated retroendocytosis. It is also likely that a percentage of the “resecreted” apoA-I was never actually internalized. Although much care was taken to wash off the apoA-I present in the pulse incubation, it is highly likely that some of the apoA-I recovered intact in the chase medium was apoA-I that simply stuck to the outside of the cell. Thus, our estimation of the contribution of endocytosed apoA-I to lipidated particle formation is probably an overestimation.
If most of apoA-I is lipidated at the cell surface, as follows from the arguments above, then the cholesterol effluxed from macrophages should not correlate with the amount of apoA-I internalized. From the experiments shown in Fig. 7 and supplementary Fig. II, it is clear that the uptake of apoA-I and the resultant cholesterol efflux can be pharmacologically decoupled. Amiloride and monensin exhibited relatively minor effects on cholesterol efflux, but were more effective at blocking apoA-I uptake. The fact that monensin and amiloride affected cholesterol efflux to some extent was not unexpected, because it has been shown that compounds that affect lysosomal vesicular trafficking can have profound effects on apolipoprotein-mediated lipid efflux (31). In the case of monensin, we were surprised at the relatively modest effect that we observed, compared with the work of others (32). Although the reason for this discrepancy is not immediately clear, perhaps differences in cell type and degree of cholesterol loading may affect monensin-sensitive pathways. The experiments performed in the ABCA1-transfected CHO cells provide further support for the claim that a direct correlation between the degree of internalized apoA-I and the degree of cholesterol efflux does not exist (Fig. 8), at least in these cell types.
On the basis of our data, we propose that although retroendocytosis of intact apoA-I may occur at low levels, the process is probably not critical for the overall efflux of cholesterol from macrophages, i.e., the majority of lipid transfer events probably occur at the cell surface. Based on the work of Vedhachalam et al. (33) and Denis et al. (34), a general model has emerged in which lipid-free apoA-I first binds to ABCA1 and then associates with specialized lipid domains that have been created/modified by ABCA1 translocase activity. Indeed, most of the apoA-I that binds to cells in an ABCA1-dependent manner is bound to lipid rather than to ABCA1 itself (33). Although the time frame of this apoA-I-to-membrane association is not clearly known, we propose that apoA-I retroendocytosis may be less a pathway and more a side effect of macrophage membrane turnover. Because macrophages are professional phagocytes, the high degree of endocytic vesicle formation on the cell surface could be expected to trap some of this membrane-associated apoA-I and drag it into the cell. Depending on the intracellular targeting of the particular vesicle, it is conceivable that much of the trapped apoA-I would be delivered to lysosomal compartments and degraded, whereas a small fraction might find its way back to the membrane without degradation. This idea has been suggested previously by Magnusson, Faerevik, and Berg (35), who concluded that retroendocytosis of certain glycoproteins in liver cells occurs mainly because of incomplete dissociation of ligands from receptors before receptor recycling to the cell surface. They demonstrated a positive relationship between a ligand’s affinity for its receptor and the degree of retroendocytosis. The case of apoA-I in macrophages is similar if one thinks of an ABCA1-generated patch of lipid as the apoA-I “receptor.” It has been shown that ABCA1 is endocytosed during cholesterol efflux (36–38), and it is possible that during this process, apoA-I is occasionally internalized as well. This would explain our observation of the specific nature of the apoA-I uptake and its relationship to ABCA1 activity. The relatively efficient degradation of this internalized apoA-I is consistent with our assertion that the intracellular apoA-I does not account for a significant fraction of the apoA-I that is converted to lipidated HDL. The CHO cells used in the transfection experiment shown in Fig. 8 are probably much less active in terms of endocytosis than are macrophages and probably took up much less apoA-I for that reason. In this same cell line, we have shown that exogenous treatment with ceramide modulates ABCA1 trafficking such that it becomes enriched at the cell surface (39). The fact that this cell surface enrichment of ABCA1 corresponds to a several-fold increase in cholesterol efflux to apoA-I supports the idea that the plasma membrane may be the key site for apolipoprotein-mediated cholesterol efflux.
Finally, although we are proposing that endocytosis of apoA-I plays a minor role in the mechanics of apolipoprotein-mediated cholesterol efflux, we do not preclude the possibility that the internalization of apoA-I may play an important biological role. For example, it is possible that the small amount of apoA-I that is internalized and degraded might function as a signaling agent, perhaps mediating some aspect of macrophage lipid or ABCA1 metabolism such as those elucidated by the elegant studies of Tang, Vaughan, and Oram (40). In addition, this process may be more important in other cell types. For example, Rohrer et al. (13) have demonstrated that lipid-free apoA-I may be transcytosed across endothelial cells. It is clear that more study is required to unravel the complexities of the interaction between apoA-I and peripheral cells.
Supplementary Materials
Supplemental Material can be found at: http://www.jlr.org/content/suppl/2008/03/25/M800048-JLR200.DC1.html
Acknowledgments
The authors thank Dr. Frank McCormack for his insights and experimental suggestions.
Abbreviations:
- ABCA1
ATP binding cassette transporter A-1
- apoA-I
apolipoprotein A-I
- CHO
Chinese hamster ovary
- CVD
cardiovascular disease
- GFP
green fluorescent protein
- IPTG
isopropyl-β-d-thiogalactoside
- RCT
reverse cholesterol transport
- STB
standard Tris buffer
- TCEP
Tris-(2-carboxyethyl) phosphine
Footnotes
This work was supported by RO1 Grants HL-62542 and HL-67093 from the National Heart Lung and Blood Institute (W.S.D.), a summer graduate student fellowship from the University of Cincinnati Research Counsel (L.E.F), and a University of Cincinnati Dean’s Distinguished Dissertation Fellowship (S.E.P.).
REFERENCES
- 1.Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, Zheng ZJ, Flegal K, O'Donnell C, Kittner S, et al. Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:E85–E151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 2.Assmann G, Schulte H, von Eckardstein A, Huang YD. High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis. 1996;124(Suppl):11–20. doi: 10.1016/0021-9150(96)05852-2. [DOI] [PubMed] [Google Scholar]
- 3.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 4.Groen AK, Oude Elferink RP, Verkade HJ, Kuipers F. The ins and outs of reverse cholesterol transport. Ann. Med. 2004;36:135–145. doi: 10.1080/07853890310020635. [DOI] [PubMed] [Google Scholar]
- 5.Tall AR, Wang N. Tangier disease as a test of the reverse cholesterol transport hypothesis. J. Clin. Invest. 2000;106:1205–1207. doi: 10.1172/JCI11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 7.Marcil M, Brooks-Wilson A, Clee SM, Roomp K, Zhang LH, Yu L, Collins JA, van Dam M, Molhuizen HO, Loubster O, et al. Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. Lancet. 1999;354:1341–1346. doi: 10.1016/s0140-6736(99)07026-9. [DOI] [PubMed] [Google Scholar]
- 8.Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, Deleuze JF, Brewer HB, Duverger N, Denefle P, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- 9.Bodzioch M, Orso E, Klucken J, Langmann T, Bottcher A, Diederich W, Drobnik W, Barlage S, Buchler C, Porsch-Ozcurumez M, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 10.Assman G, Eckardstein A, Brewer BJ. Familial An alpha-lipoproteinemia: Tangier Disease. McGraw-Hill; 2001. The online metabolic & molecular bases of inherited disease; NY pp. Chapter 194. [Google Scholar]
- 11.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J. Mol. Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi Y, Smith JD. Cholesterol efflux to apolipoprotein AI involves endocytosis and resecretion in a calcium-dependent pathway. Proc. Natl. Acad. Sci. USA. 1999;96:11358–11363. doi: 10.1073/pnas.96.20.11358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohrer L, Cavelier C, Fuchs S, Schluter MA, Volker W, von Eckardstein A. Binding, internalization and transport of apolipoprotein A-I by vascular endothelial cells. Biochim. Biophys. Acta. 2006;1761:186–194. doi: 10.1016/j.bbalip.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 14.Smith JD, Waelde C, Horwitz A, Zheng P. Evaluation of the role of phosphatidylserine translocase activity in ABCA1-mediated lipid efflux. J. Biol. Chem. 2002;277:17797–17803. doi: 10.1074/jbc.M201594200. [DOI] [PubMed] [Google Scholar]
- 15.Neufeld EB, Stonik JA, Demosky SJ, Knapper CA, Combs CA, Cooney A, Comly M, Dwyer N, Blanchette-Mackie J, Remaley AT, et al. The ABCA1 transporter modulates late endocytic trafficking: insights from the correction of the genetic defect in Tangier disease. J. Biol. Chem. 2004;279:15571–15578. doi: 10.1074/jbc.M314160200. [DOI] [PubMed] [Google Scholar]
- 16.Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, et al. Cellular localization and trafficking of the human ABCA1 transporter. J. Biol. Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- 17.Chen W, Sun Y, Welch C, Gorelik A, Leventhal AR, Tabas I, Tall AR. Preferential ATP-binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux from late endosomes/lysosomes. J. Biol. Chem. 2001;276:43564–43569. doi: 10.1074/jbc.M107938200. [DOI] [PubMed] [Google Scholar]
- 18.Tricerri MA, Behling Agree AK, Sanchez SA, Bronski J, Jonas A. Arrangement of apolipoprotein A-I in reconstituted high-density lipoprotein disks: an alternative model based on fluorescence resonance energy transfer experiments. Biochemistry. 2001;40:5065–5074. doi: 10.1021/bi002815q. [DOI] [PubMed] [Google Scholar]
- 19.Safi W, Maiorano JN, Davidson WS. A proteolytic method for distinguishing between lipid-free and lipid-bound apolipoprotein A-I. J. Lipid Res. 2001;42:864–872. [PubMed] [Google Scholar]
- 20.Jonas A. Reconstitution of high-density lipoproteins. Methods Enzymol. 1986;128:553–582. doi: 10.1016/0076-6879(86)28092-1. [DOI] [PubMed] [Google Scholar]
- 21.Panagotopulos SE, Witting SR, Horace EM, Nicholas MJ, Sean DW. Bacterial expression and characterization of mature apolipoprotein A-I. Protein Expr. Purif. 2002;25:353–361. doi: 10.1016/s1046-5928(02)00020-7. [DOI] [PubMed] [Google Scholar]
- 22.Davidson WS, Lund-Katz S, Johnson WJ, Anantharamaiah GM, Palgunachari MN, Segrest JP, Rothblat GH, Phillips MC. The influence of apolipoprotein structure on the efflux of cellular free cholesterol to high density lipoprotein. J. Biol. Chem. 1994;269:22975–22982. [PubMed] [Google Scholar]
- 23.Lund-Katz S, Phillips MC. Packing of cholesterol molecules in human high-density lipoproteins. Biochemistry. 1984;23:1130–1138. doi: 10.1021/bi00301a015. [DOI] [PubMed] [Google Scholar]
- 24.Panagotopulos SE, Witting SR, Horace EM, Hui DY, Maiorano JN, Davidson WS. The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J. Biol. Chem. 2002;277:39477–39484. doi: 10.1074/jbc.M207005200. [DOI] [PubMed] [Google Scholar]
- 25.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vult von Steyern F, Josefsson JO, Tågerud S. Rhodamine B, a fluorescent probe for acidic organelles in denervated skeletal muscle. J. Histochem. Cytochem. 1996;44:267–274. doi: 10.1177/44.3.8648087. [DOI] [PubMed] [Google Scholar]
- 27.Braschi S, Neville TA, Maugeais C, Ramsamy TA, Seymour R, Sparks DL. Role of the kidney in regulating the metabolism of HDL in rabbits: evidence that iodination alters the catabolism of apolipoprotein A-I by the kidney. Biochemistry. 2000;39:5441–5449. doi: 10.1021/bi9919504. [DOI] [PubMed] [Google Scholar]
- 28.Wileman T, Boshans RL, Schlesinger P, Stahl P. Monensin inhibits recycling of macrophage mannose-glycoprotein receptors and ligand delivery to lysosomes. Biochem. J. 1984;220:665–675. doi: 10.1042/bj2200665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bortnick AE, Rothblat GH, Stoudt G, Hoppe KL, Royer LJ, McNeish J, Francone OL. The correlation of ATP-binding cassette 1 mRNA levels with cholesterol efflux from various cell lines. J. Biol. Chem. 2000;275:28634–28640. doi: 10.1074/jbc.M003407200. [DOI] [PubMed] [Google Scholar]
- 30.Mendez AJ. Cholesterol efflux mediated by apolipoproteins is an active cellular process distinct from efflux mediated by passive diffusion. J. Lipid Res. 1997;38:1807–1821. [PubMed] [Google Scholar]
- 31.Remaley AT, Schumacher UK, Stonik JA, Farsi BD, Nazih H, Brewer HB., Jr Decreased reverse cholesterol transport from Tangier disease fibroblasts. Acceptor specificity and effect of brefeldin on lipid efflux. Arterioscler. Thromb. Vasc. Biol. 1997;17:1813–1821. doi: 10.1161/01.atv.17.9.1813. [DOI] [PubMed] [Google Scholar]
- 32.Mendez AJ, Uint L. Apolipoprotein-mediated cellular cholesterol and phospholipid efflux depend on a functional Golgi apparatus. J. Lipid Res. 1996;37:2510–2524. [PubMed] [Google Scholar]
- 33.Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler. Thromb. Vasc. Biol. 2007;27:1603–1609. doi: 10.1161/ATVBAHA.107.145789. [DOI] [PubMed] [Google Scholar]
- 34.Denis M, Haidar B, Marcil M, Bouvier M, Krimbou L, Genest J., Jr Molecular and cellular physiology of apolipoprotein A-I lipidation by the ATP-binding cassette transporter A1 (ABCA1) J. Biol. Chem. 2004;279:7384–7394. doi: 10.1074/jbc.M306963200. [DOI] [PubMed] [Google Scholar]
- 35.Magnusson S, Faerevik I, Berg T. Characterization of retroendocytosis in rat liver parenchymal cells and sinusoidal endothelial cells. Biochem. J. 1992;287:241–246. doi: 10.1042/bj2870241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen W, Wang N, Tall AR. A PEST deletion mutant of ABCA1 shows impaired internalization and defective cholesterol efflux from late endosomes. J. Biol. Chem. 2005;280:29277–29281. doi: 10.1074/jbc.M505566200. [DOI] [PubMed] [Google Scholar]
- 37.Landry YD, Denis M, Nandi S, Bell S, Vaughan AM, Zha X. ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J. Biol. Chem. 2006;281:36091–36101. doi: 10.1074/jbc.M602247200. [DOI] [PubMed] [Google Scholar]
- 38.Le Goff W, Peng DQ, Settle M, Brubaker G, Morton RE, Smith JD. Cyclosporin A traps ABCA1 at the plasma membrane and inhibits ABCA1-mediated lipid efflux to apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 2004;24:2155–2161. doi: 10.1161/01.ATV.0000144811.94581.52. [DOI] [PubMed] [Google Scholar]
- 39.Witting SR, Maiorano JN, Davidson WS. Ceramide enhances cholesterol efflux to apolipoprotein A-I by increasing the cell surface presence of ATP-binding cassette transporter A1. J. Biol. Chem. 2003;278:40121–40127. doi: 10.1074/jbc.M305193200. [DOI] [PubMed] [Google Scholar]
- 40.Tang C, Vaughan AM, Oram JF. Janus kinase 2 modulates the apolipoprotein interactions with ABCA1 required for removing cellular cholesterol. J. Biol. Chem. 2004;279:7622–7628. doi: 10.1074/jbc.M312571200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material can be found at: http://www.jlr.org/content/suppl/2008/03/25/M800048-JLR200.DC1.html