

Summary
Mutations in methyl CpG binding protein 2 (MECP2) cause Rett Syndrome, the most severe autism spectrum disorder. Re-expressing Mecp2 in symptomatic Mecp2 null mice dramatically improves function and longevity, providing hope that therapeutic intervention is possible in humans. To identify pathways in disease pathology for therapeutic intervention, a dominant ENU mutagenesis suppressor screen was carried out in Mecp2 null mice. Five suppressors that ameliorate symptoms of Mecp2 loss were isolated. Here we show that a stop codon mutation in squalene epoxidase (Sqle), a rate-limiting enzyme in cholesterol biosynthesis underlies suppression in one line. Subsequently, we show that lipid metabolism is perturbed in the brain and liver of Mecp2 null males. Consistently, statin drugs improve systemic perturbations of lipid metabolism, alleviate motor symptoms and confer increased longevity in Mecp2 mutant mice. The genetic screen therefore points to cholesterol homeostasis as a potential target for the treatment of Rett patients.
Rett Syndrome (RTT: OMIM 312750) is an X-linked neurological disorder presenting with autistic features that afflicts approximately 1 in 10,000 females. Within a few months or years of apparently normal postnatal development, progressive neurological manifestations of disease occur, including loss of speech and motor skills, stereotypic hand movements, difficulty walking, irregular breathing, and seizures. Mutations in the X-linked gene, methyl CpG binding protein 2 (MECP2), are the primary cause of RTT1. Hemizygous males with truncating or loss-of-function mutations typically die of encephalopathy, whereas mild mutations in either sex are associated with a variety of intellectual disabilities and autism2.
Mouse models recapitulate many of the symptoms of RTT and their study has provided insight into the physiological basis of disease3–5. Although female Mecp2/+ mice show phenotypic variance in part due to random X-chromosome inactivation, male Mecp2/Y mice have a fully penetrant phenotype. Null males are normal at birth and weaning, but develop limb clasping, tremors, lethargy and abnormal breathing, symptoms that progressively worsen until death at 6 – 16 weeks of age. Restoration of Mecp2 expression in mutant mice after the onset of symptoms rescues the neurological deficits, including motor function, and significantly prolongs survival6. Therefore, MECP2 plays roles in the postnatal maturation and/or maintenance of neuronal properties and circuits4. These findings suggest that the disease may be ameliorated or even reversed by genetic or pharmacological means after symptom onset2,7. Mechanistically, MECP2 binds to methylated DNA to regulate gene transcription through repression or activation8. When MECP2 represses gene transcription, it associates with chromatin-remodeling complexes that contain Type I histone deacetylases (HDACs)2,9. As an epigenetic factor, MECP2 levels are therefore critical within a relatively narrow range, making gene therapy a difficult approach for symptom rescue.
We reasoned that a genetic screen for suppressors of symptoms using a Mecp2 mouse model could reveal pathways responsible for disease pathology and thereby help to identify therapeutic targets. Genetic modifier screens are commonly applied to identify genes that act in developmental or biochemical pathways in fruit flies, worms, and bacteria10; however, this approach is rare in the mouse11,12. Here, we take advantage of advances in mutagenesis, sequencing and genotyping methods13 to identify five loci that suppress disease phenotypes in Mecp2 null mice. One suppressing mutation occurs in a gene that encodes squalene epoxidase (Sqle), a rate-limiting enzyme in cholesterol synthesis; its study suggests that downregulation of the pathway can ameliorate symptoms. Ensuing pharmacologic studies show that administration of statins, which inhibit the cholesterol synthesis pathway, improves motor performance and increases longevity in Mecp2 mutant mice. Cholesterol homeostasis may therefore be a therapeutic target for treating specific features of RTT pathology.
Results
ENU screen identifies five suppressors of Mecp2
C57BL/6J males were treated with N-ethyl-N-nitrosourea (ENU) and mated to 129.Mecp2tm1.1Bird/+ females (Fig. 1a). First generation (G1) Mecp2 null males were screened for rescue of neurological defects using a health scoring system that included assessments of limb clasping, tremors, body size, cage activity, and development of skin/eye inflammation or malocclusion. Of 2963 G1 animals weaned, 1522 were males, and 679 were Mecp2tm1.1Bird/Y. Most G1 Mecp2tm1.1Bird/Y animals had mild neurological abnormalities by four weeks, and died or had to be killed by 6–16 weeks of age; however, some showed amelioration of one or more of the health assessment traits. These 25 males were placed in mating cages to assess heritability and longevity caused by a putative second site mutation; litters were obtained from seven. Five lines showed inherited suppressor loci, which were named Suppressor of Mecp2 mutations 1 – 5 (Sum1m1Jus; MGI:5489912, Sum2m1Jus; MGI;5489913, Sum3m1Jus; MGI;5489914, Sum4m1Jus; MGI;5489915, Sum5m1Jus; MGI;5489916; Fig. 1b). Each suppressor showed different degrees of health trait rescue (Table 1; Supplementary Fig. 1); however, all suppressors increased the lifespan of Mecp2/Y mice by at least 3 weeks from the average 50% lethality (L50) time point. Each survival curve showed a characteristic pattern, reflecting the manifestation of health traits (Fig. 2). Two males, 352 (Sum1m1Jus) and 1395 (Sum4m1Jus), displayed rescue of multiple traits, even though they did not live particularly long lives (163 and 94 days, respectively). Both died suddenly without displaying Mecp2 null symptoms, but each founded lines in which the offspring lived much longer, highlighting the predictive nature of neurological and health assessments on longevity. Unexpected phenotypes appeared in some of the lines, which compromised the health of the animals. Long-lived Mecp2/Y from four of the lines, excluding only Sum1m1Jus, developed varying degrees of dermatitis, eye inflammation or malocclusions at different ages (Table 1). Inflammation is a key manifestation of Mecp2 mutation in non-neural cells14. The rescue of different traits and the varying penetrance of phenotypes in rescue animals suggests that the mutations are suppressing through multiple mechanisms.
Figure 1. A dominant suppressor screen shows inheritance of longevity in five lines.

a) ENU-treated C57BL/6J males were mated to 129.Mecp2tm1.1Bird/+ females. G1 Mecp2tm1.1Bird/Y males were aged and assessed for fore- and hindlimb clasping, tremors, body size, cage activity, and longevity. b) N2 animals from five lines (352 (Sum1m1Jus; blue), 856 (Sum2m1Jus; green), 895 (Sum3m1Jus; red), 1395 (Sum4m1Jus; brown) and 1527 (Sum5m1Jus; purple)) produced N3 offspring that exhibited increased longevity. A closed circle represents offspring of male N2 parents, and an open circle represents offspring of female N2 parents. The longevity of the G1 founder of each line is indicated by the colored square.
Table 1.
Subjective health scores are averaged for offspring assessed in the line. Shown are the average health scores for each parameter at 8 and 20 weeks of age. Health scores are 0=not present, 1=normal similar to wild type, increasing to 5=near death, warranting sacrifice. Inflammation is D=dermatitis, E=eye inflammation. 129.Mecp2tm1.1Bird/Y; grey
|
Mecp2tm1.1Bird/Y Sum1m1Jus/+ |
Mecp2tm1.1Bird/Y Sum2m1Jus/+ |
Mecp2tm1.1Bird/Y Sum3m1Jus/+ |
Mecp2tm1.1Bird/Y Sum4m1Jus/+ |
Mecp2tm1.1Bird/Y Sum5m1Jus/+ |
Mecp2tm1.1Bird/Y +/+ |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (weeks) | 8 | 20 | 8 | 20 | 8 | 20 | 8 | 20 | 8 | 20 | 8 | 20 |
| Clasping | 0 | 2 | 1 | 3 | 1 | 3 | 2 | 2 | 1 | 2 | 4 | N/A |
| Tremors | 0 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 1 | 2 | 3 | N/A |
| Body | 0 | 0 | 0 | 2 | 1 | 1 | 0 | 0 | 1 | 2 | 3 | N/A |
| Lethargy | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 2 | 4 | N/A |
| Inflammation | 0 | 0 | D E | D E | 0 | D | D | D E | E | E | 0 | N/A |
| Malocclusion | 0 | -- | 0 | -- | 1 | -- | 1 | -- | 0 | -- | 1 | -- |
Figure 2.

Survival curves for each line with a confirmed modifier map location are shown assessed at the N3 generation. Survival of Mecp2tm1.1Bird/Y mice is significantly increased by the presence of each suppressing mutation: a) Line 352 (Sum1m1Jus); p=.001. b) Line 856 (Sum2m1Jus); p=.005. c) Line 895 (Sum3m1Jus); p=.002. d) Line 1395 (Sum4m1Jus); p=.016.
To identify a chromosomal location for the suppressing traits, DNA from 7–12 long-lived Mecp2tm1.1Bird/Y M/+ from each founder line was genotyped along with DNA from short-lived Mecp2tm1.1Bird/Y +/+ N3 littermates using an Illumina medium density single nucleotide polymorphism panel. The resulting genotypes were analyzed for heterozygous C57BL/6J linkage to the rescue phenotype according to haplotype assessment at the N3 generation15,16. These data showed significant linkage for Sum1m1Jus on mouse chromosome 16 (LOD score=4.82), Sum2m1Jus on chromosome 3 (LOD score=3.61), Sum3m1Jus on chromosome 15 (LOD score=3.03), and Sum4m1Jus on chromosome 7 (LOD score=3.40; Supplementary Fig. 2a–d). Linkage was confirmed and fine mapping was achieved by assessing markers within the putative map location on additional animals in the line (Supplementary Fig. 3a–d). The maximum LOD score for Sum5m1Jus was 2.86; a map location was not confirmed.
Whole exome sequencing reveals a stop codon mutation that confers rescue
Genomic DNA from two third-generation Mecp2tm1.1Bird/Y Sum3m1Jus/+ long-lived animal “cousins” was subjected to exome capture and massively parallel sequencing17. Seven heterozygous, protein-altering variants were observed in both animals but not in 15 control mouse exomes (a set including C57BL/6J and 129S6/SvEvTac DNAs; Supplementary Fig. 4a). Only one of these variants ranked highly with respect to both overlap with Sum3m1Jus/+ mapping data (chr15:34260887–95144876; Supplementary Fig. 4b) and predicted functional impact – a nonsense mutation (c.1195C>T, p.Arg399Ter) in squalene epoxidase (Sqle; ENSMUSG00000022351), also known as squalene monooxygenase (Fig. 3a).
Figure 3. A stop codon mutation in Sqle confers rescue at Sum3m1Jus.
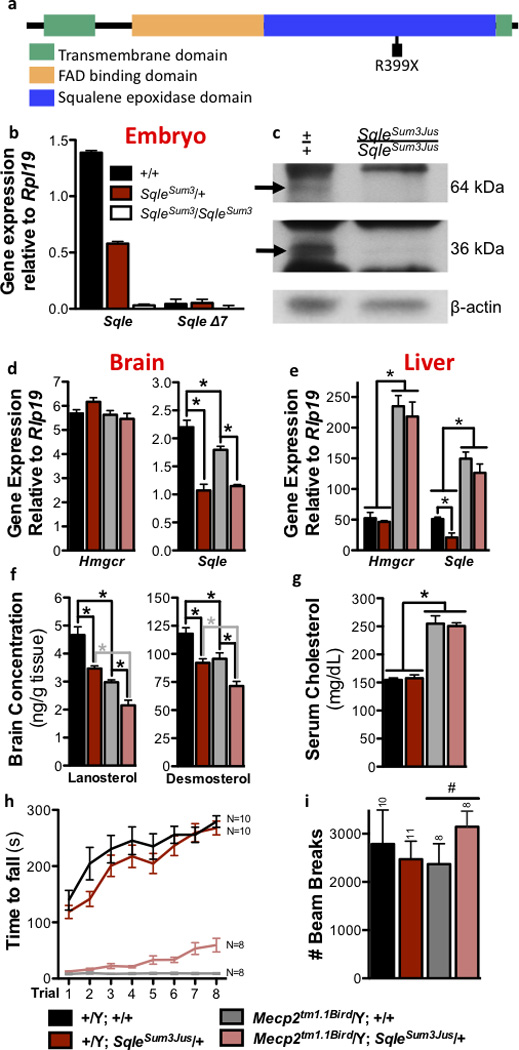
a) The ENU-induced Sqle R399× mutation occurs in exon 7 in the squalene epoxidase domain. b) Sqle is not expressed in homozygous E8.0 mutant embryos. SqleΔ7 is the short predicted transcript lacking exon 7. c) Western blot on stage-matched E8.0 embryos shows that the expected 64kDa protein and 36kDa degradation product are absent in homozygous mutant embryos. d) Expression of Sqle but not Hmgcr is decreased in SqleSum3/+ brain; e) gene expression is unchanged by SqleSum3/+ in liver. i) Brain concentration of cholesterol precursors lanosterol and desmosterol is also decreased by SqleSum3/+. f) Serum cholesterol concentration is unchanged by SqleSum3/+. Tissue (d–g) analysis performed at P70; N=6 per group. Mecp2tm1.1Bird/Y SqleSum3/+ animals at backcross generation N7 to 129S6/SvEvTac show g) significantly improved rotarod performance at P56 (p=.0001), h) improved open field activity at P70. All error bars represent s.e.m.
SQLE catalyzes the first oxygenation reaction in the committed production of cholesterol, an essential lipid either supplied by diet or synthesized from acetate by a complex pathway18,19. Cholesterol homeostasis is maintained by negative feedback regulation of genes and their encoded enzymes in the pathway, including the rate limiting enzymes HMG CoA reductase (HMGCR) and SQLE20–22. SQLE’s enzymatic reaction requires a mitochondrial electron donor to produce 2,3-oxidosqualene, a transient intermediate that is cyclized to lanosterol by lanosterol synthase (LSS)23. SQLE is expressed in many tissues24 and is conserved throughout evolution; the mouse and human proteins are 84% identical at the amino acid level. Mice homozygous for SqleSum3Jus die prior to birth at embryonic day (E)8.5, a phenotype consistent with that found in other mice with mutations affecting cholesterol synthesis25 (Supplementary Table 1). Two isoforms are annotated in mice: the SqleSum3Jus stop mutation identified here occurs in a highly conserved exon present in the long isoform, which unlike the short isoform, is consistently translated to protein in mammalian species. The long isoform is absent in SqleSum3Jus/SqleSum3Jus embryos, suggesting nonsense-mediated RNA decay, and the predicted short form is not upregulated in SqleSum3Jus/SqleSum3Jus embryos or SqleSum3Jus/+ mice (Fig. 3b; Supplementary Fig. 5). Western blot analysis of E8.0 SqleSum3Jus homozygous embryos shows that the expected 64kDa protein is absent as is a 36kDa presumed degradation product, consistent with a null mutation (Fig. 3c). Only one other null eukaryotic mutation of Sqle has been identified, resulting in an inability for plants to tolerate drought stress26,27.
Cholesterol synthesis takes place through complex pathways, all of which use the rate-limiting enzymes HMGCR and SQLE28. Prior analysis suggested that expression of Hmgcr and Sqle were predictive of the behavior of genes encoding intermediate enzymes in the pathways (data not shown). To determine the effect of the mutation on cholesterol biosynthesis, expression of Hmgcr and Sqle was assessed in the brain and liver of SqleSum3Jus/+ mice. Sqle expression was reduced by nearly 50% in SqleSum3Jus/+ brains at P70, with no compensatory change in Hmgcr (Fig. 3d; Supplementary Table 2). Sqle expression was also reduced in the brains of 129.Mecp2tm1.1Bird/Y, regardless of SqleSum3Jus mutation status. Consistently, concentrations of the cholesterol precursors desmosterol and lanosterol were decreased in SqleSum3Jus/+ brains at P70 (Fig. 3f; Supplementary Table 3a). Livers of SqleSum3Jus/+ mice at P70 showed little decrease in Sqle or Hmgcr expression, though 129.Mecp2tm1.1Bird/Y mice have increased expression of both genes, regardless of SqleSum3Jus mutation status (Fig. 3e). Likewise, these changes in expression are reflected in serum cholesterol (Fig. 3g). Together, these data suggest that SqleSum3Jus/+ mice carry a loss of function allele of Sqle.
To determine which RTT-like traits were ameliorated in 129.Mecp2tm1.1Bird/Y SqleSum3Jus/+ animals, a series of behavioral and breathing assessments were carried out at the N=7 backcross generation. In addition to increased longevity, mice showed improved motor activity on a rotarod (Fig. 3h), and increased activity in the open field (Fig. 3i). However, the mice showed little improvement of irregular respiration as measured by plethysmography, and no change in the acoustic startle response (Supplementary Fig. 6a–d). Thus, heterozygous loss of Sqle does not improve all symptoms associated with Mecp2 mutation.
Mecp2 null males have abnormal cholesterol metabolism
We hypothesized that the heterozygous Sqle mutation ameliorates a previously unrecognized dysregulation of cholesterol metabolism in Mecp2 null mice. Cholesterol is a major and essential component of the brain, which must be produced there exclusively via synthesis as cholesterol-rich lipoproteins cannot cross the blood brain barrier (BBB)29. To facilitate proper neurotransmitter release and dendrite remodeling, neuronal membranes constantly require turnover to eliminate cholesterol that has been adulterated by reactive oxygen species, byproducts of their high rate of metabolism30. When neurons require cholesterol turnover or accumulate too much cholesterol or its intermediates, the cytochrome P450 oxidase Cyp46a1 hydroxylates cholesterol to produce 24S-hydroxycholesterol (24SOHC), allowing for egress by one-way diffusion into the circulation across the BBB (Fig. 4a)31. Two alleles of Mecp2 are primarily used as mouse models of RTT: Mecp2tm1.1Bird, a null mutation3, and Mecp2tm1.1Jae, which expresses low levels of a truncated protein32. At postnatal day 28 (P28), when Mecp2/Y mice display mild symptoms, Cyp46a1 expression was increased 38 percent over wild type in the 129.Mecp2tm1.1Bird null brain (p=.02) with a similar trend in B6.Mecp2tm1.1Jae, indicating a heightened need for cholesterol turnover by neurons in early stages of disease (Fig. 4b). Notably, brains from the two different Mecp2 alleles also showed a higher concentration of total cholesterol at P56 (Bird: p=.05, Jae: p=.21; Fig. 4c). However, by P56 when mutant males are severely symptomatic, cholesterol biosynthesis and turnover genes Hmgcr, Sqle and Cyp46a1 were downregulated in both alleles (Fig. 4b). Desmosterol is the most prevalent intermediate produced by the primary cholesterol synthesis pathway in the adult brain33. Interestingly, desmosterol was also decreased at P56 in Mecp2tm1.1Bird/Y brain (p=.01), and both lanosterol and desmosterol were decreased by P70. Furthermore, by P70 brain cholesterol concentration had returned to wild type levels, reflecting cholesterol biosynthesis pathway downregulation in the adult Mecp2tm1.1Bird/Y brain (Fig. 4c; Supplementary Table 3a,b). Similarly, de novo cholesterol synthesis assayed by the in vivo incorporation of 3H2O into sterols34 confirmed that sterol synthesis decreased in the adult brain of B6.Mecp2tm1.1Jae/Y (Fig. 4d; Supplementary Table 4a,b). Therefore, brain cholesterol metabolism is perturbed in two Mecp2 alleles on two different genetic backgrounds. Either increased or decreased amounts of sterols can impair neurological function, demanding a tight regulation of brain sterol synthesis35. In Mecp2 mutant mice, our data suggest that over production of cholesterol in the brain likely feeds back to then decrease synthesis.
Figure 4. Cholesterol metabolism is disrupted in Mecp2 null male mice.

a) A simplified schematic of the enzymes and products in cholesterol biosynthesis via desmosterol is shown. b) Expression of Hmgcr, Sqle and Cyp46a1 in Mecp2tm1.1Bird/Y and Mecp2tm1.1Jae/Y are similar in brain. c) Lanosterol (Lan), desmosterol (Des) and total cholesterol (TC) concentrations are displayed per gram of brain tissue at P56 (N=8 per group) and P70 (N=4 per group). d) Cholesterol synthesis is decreased in Mecp2tm1.1Jae/Y brain at P56 (wild type N=4; null N=5). e) Expression of Hmgcr and Sqle in Mecp2tm1.1Bird/Y and Mecp2tm1.1Jae/Y differ in liver. f) Triacylglyceride (TAG) and TC concentrations are displayed per gram of liver tissue at P56 (N=6 per group). g) Cholesterol synthesis is slightly increased in Mecp2tm1.1Jae/Y liver per gram of tissue at P56 (wild type N=4; null N=5). Serum h) total cholesterol, i) LDL-cholesterol and j) triglyceride levels are significantly higher in Mecp2tm1.1Bird/Y mice by P56 (N=8–11 per group), but unchanged in Mecp2tm1.1Jae/Y mice (N=6 per group). For gene expression data (b,e) Bird: N=6 per genotype at P28, and 12 per genotype at P56; Jae: N=4 per genotype at P28, and 6 per genotype at P56. Tissue data (b–g) represent percentage change from wild type levels. *p≤0.05; All error bars represent s.e.m.
Although RTT is primarily a neurological disease, most cholesterol synthesis studies have been carried out in the liver, which regulates systemic release of cholesterol in conjunction with dietary consumption. Expression of Hmgcr and Sqle was unchanged in P28 and significantly higher in P56 129.Mecp2tm1.1Bird/Y livers compared to wild type (p≤.05; Fig. 4e). Although total liver cholesterol and phospholipid concentrations were unchanged, reflecting the transient nature and rapid packaging of newly synthesized cholesterol into lipoproteins for secretion, the concentration of primary liver storage lipids, triglycerides (TAG), was greatly increased in 129.Mecp2tm1.1Bird/Y mice by P56 (p=.003; Fig. 4f; Supplementary Fig. 7). In contrast, B6.Mecp2tm1.1Jae showed no significant perturbations in gene expression at P28 or P56, a modest increase in de novo liver cholesterol synthesis, and a modest increase in liver TAG at P56 (Fig. 4e–g; Supplementary Table 4a,b). Consistently, total serum cholesterol, LDL-cholesterol and triglycerides were significantly elevated in 129.Mecp2tm1.1Bird/Y mice (p≤.01), but not in B6.Mecp2tm1.1Jae/Y mice at P56 (Fig. 4h–j). The C57BL/6J inbred strain and the 129/Sv substrains inherently manage peripheral cholesterol metabolism differently36. Together, these data suggest that perturbations in brain cholesterol synthesis are common in the two alleles on two different strain backgrounds; however, severe peripheral dysregulation of the pathway occurs only in 129.Mecp2tm1.1Bird.
Statin administration ameliorates behavioral and metabolic symptoms in Mecp2 mice
Aberrant cholesterol metabolism may therefore contribute to Mecp2 null symptoms. We reasoned that a pharmacologic inhibitor of cholesterol synthesis might produce a comparable attenuation of symptoms to the genetic inhibitor SqleSum3Jus/+ in Mecp2 null mice. Wild type and 129.Mecp2tm1.1Bird/Y mutant mice were treated with statin drugs, which are commonly prescribed to reduce systemic cholesterol by interfering with cholesterol synthesis through competitive inhibition of HMG CoA-reductase. As a preliminary trial, age-matched littermates were treated with 3 mg/kg of fluvastatin weekly starting at five weeks of age. This treatment lowered serum cholesterol and increased cage activity during a three-week trial period (data not shown). Starting at week eight, administration of the 3 mg/kg fluvastatin dose was increased to three times per week (Supplementary Table 5). Fluvastatin treatment lowered cholesterol, improved subjective health scores, rotarod performance, open field activity and increased lifespan when compared with control mice receiving a sham dose (Fig. 5a–d), and in fact was more effective at improving these parameters than the Sqle mutation. Similar to the Sqle mutation, fluvastatin treatment did not improve baseline breathing irregularity or the acoustic startle response (Supplementary Fig. 8a–d).
Figure 5. Statin treatment improves health in 129.Mecp2tm1.1Bird/Y males.

Total animals assessed were 37 Mecp2tm1.1Bird/Y fluvastatin-treated, 12 Mecp2tm1.1Bird/Y lovastatin-treated, 31 Mecp2tm1.1Bird/Y vehicle-treated, 29 wild type +/Y fluvastatin-treated, 8 +/Y lovastatin-treated, and 29 wild type +/Y vehicle-treated mice for the following tests. a) Fluvastatin treatment of 129.Mecp2tm1.1Bird/Y confers increased longevity: median 122 days compared to 87 days with 57% survival beyond 120 days (p<.0001). Three animals were sacrificed due to dermatitis (boxes) while active and otherwise healthy. b) Rotarod performance improves in P56 treated null males (fluvastatin p=.015; lovastatin p=.009), c) Open field activity is increased in P70 treated null males as assessed by beam breaks (fluvastatin: p=.011, lovastatin: p=.049). d) Statin treatment lowers plasma cholesterol by P70 (fluvastatin: p=.001, lovastatin: p=.001). e) Statin treatment ameliorates elevated lipid concentration in 129.Mecp2tm1.1Bird/Y livers at P70 (fluvastatin p=.020; lovastatin: p=.386). The concentration of f) lanosterol slightly increases and g) desmosterol significantly increases in the brains of fluvastatin-treated 129.Mecp2tm1.1Bird/Y mice at P70 (N=4 per group; p=.042). All error bars represent s.e.m.
Markedly, brains of fluvastatin treated 129.Mecp2tm1.1Bird/Y mice showed an increase in cholesterol intermediates, particularly desmosterol, towards wild type levels at P70 (Fig. 5f,g; Supplementary Table 3a). The cholesterol biosynthesis pathway is inhibited in brains of null mice likely because aberrant turnover results in cholesterol accumulation33. Treatment starting at an early age may therefore maintain some level of pathway activity in the brain to increase lifespan and ameliorate motor symptoms in Mecp2 mutant mice.
To determine whether the effects of fluvastatin were shared by other statin drugs, 129.Mecp2tm1.1Bird/Y mice were treated with lovastatin, which differs from fluvastatin in its hydrophobicity, pharmacokinetics, transport, and metabolism37,38. Lovastatin was of particular interest because it is highly lipophilic, increasing the likelihood it will reach the brain; it ameliorates neurological symptoms in mouse models of Fragile X39 and is being used in clinical trials to ameliorate cognitive problems in children with neurofibromatosis Type I40. Like the fluvastatin dosing regimen, lovastatin treatment of 129.Mecp2tm1.1Bird/Y mutant mice began at five weeks of age, but mice were administered a 2× weekly dose of 1.5 mg/kg throughout the trial (Supplementary Table 5). Lovastatin was also successful at ameliorating motor symptoms, as assessed by rotarod performance and open field activity (Fig. 5b,c; Supplementary Video). Treatment with either statin drug at 10× of the effective doses, however, was detrimental to the animals (Supplementary Fig. 9).
Statin drugs regulate systemic cholesterol through two mechanisms: by inhibiting HMGCR and by modifying liver uptake of serum LDL and triglycerides38. Notably, livers of 129.Mecp2tm1.1Bird/Y mice had 39% higher lipid content per gram of tissue compared to wild type littermates at P70 (Fig. 5e). These data suggest that systemic dyslipidemia resulted in an increased accumulation of fat in the liver during the course of disease. Statin treatment not only decreased serum cholesterol in 129.Mecp2tm1.1Bird/Y mice (Fig. 5d), but also successfully ameliorated the accumulation of lipids in the liver (Fig. 5e). Decreased fat accumulation likely occurs through the established effect of statins on liver LDL uptake38.
To evaluate statin effects in mice that more closely model predominantly female RTT patients, cohorts of 129.Mecp2tm1.1Bird/+ female mice were treated with a single dose of 3 mg/kg fluvastatin weekly from P42 to 8 months of age (Supplementary Table 5). The milder treatment regimen for female mice reflects their delayed onset of behavioral symptoms, as well as their lack of high serum cholesterol early in life (Supplementary Fig. 10). Motor performance assessed by rotarod at 5 months improved significantly and all treated mice lived for the length of the trial (Fig. 6a–c). Notably, livers of female 129.Mecp2tm1.1Bird/+ mice had 164% higher lipid content per gram of tissue than wild type littermates at 8 months of age, a drastic increase (Fig. 6e). Strikingly, although serum cholesterol was unchanged, fluvastatin treatment reduced liver lipid accumulation to wild type levels in female mice assessed at the end of the 8-month trial (Fig. 6d,e).
Figure 6. Fluvastatin treatment improves health in 129.Mecp2tm1.1Bird/+ females.
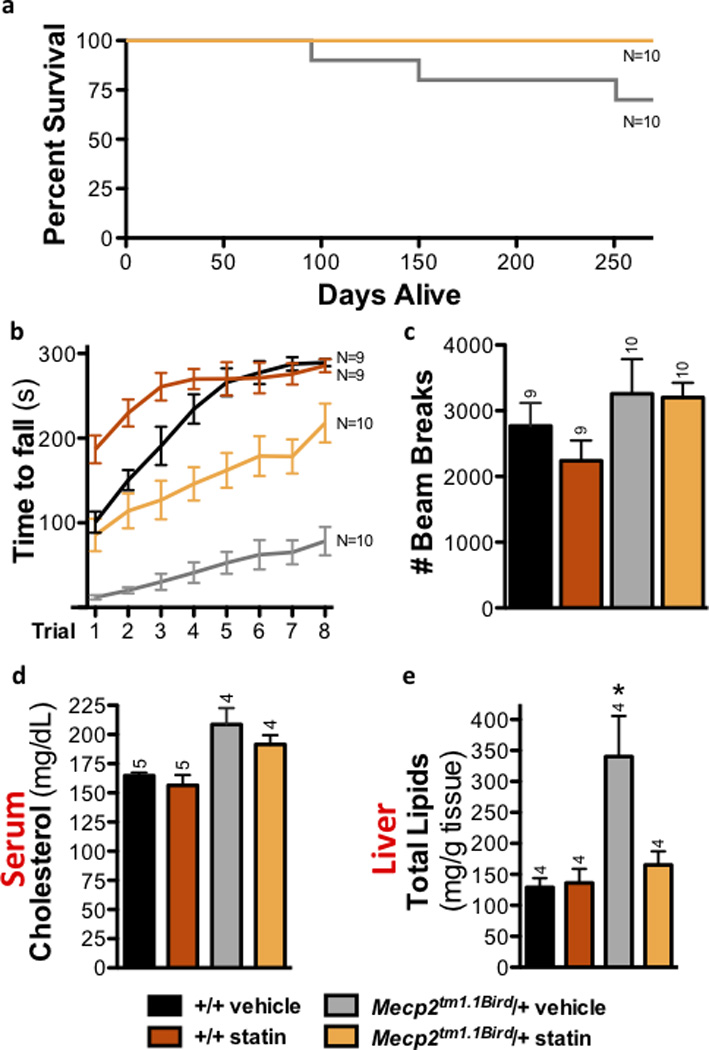
a) No fluvastatin-treated 129.Mecp2tm1.1Bird/+ females died prior to eight months, but three vehicle-treated females died. b) Rotarod performance improves in five-month-old fluvastatin-treated 129.Mecp2tm1.1Bird/+ females (p=.001). c) Open field activity assessed at four months shows no significant differences in fluvastatin- or vehicle-treated groups. d) Fluvastatin treatment does not significantly change serum cholesterol levels at eight months. e) Fluvastatin treatment ameliorates elevated lipid concentration in 129.Mecp2tm1.1Bird/+ livers assessed at eight months (p=.045). All error bars represent s.e.m.
Discussion
Our data show that cholesterol metabolism is abnormal in mouse models of Rett Syndrome and that genetic and pharmacologic interventions to lower cholesterol synthesis lead to improvements in symptoms. Given the diverse roles played by cholesterol in the nervous system, including membrane trafficking, signal transduction, myelin formation, dendrite remodeling, neuropeptide formation and synaptogenesis33, abnormal brain cholesterol metabolism may be a critical link between loss of MECP2 function and neuronal and glial dysfunction. Even small perturbations of cholesterol metabolism can have large effects on neuronal function, disrupting normal development41,42, and contributing to aging disorders including Huntington and Alzheimer disease43. Within a fixed window of time, cholesterol synthesis in the brain is likely to vary among cell types, including neuronal subtypes in various states of differentiation and activity, as well as among non-neuronal cells, including astrocytes, oligodendrocytes and microglia. Whole brain cholesterol synthesis was decreased in Mecp2 null mice by 23% per gram of tissue, a striking difference considering the variety of cell types. The only other mouse mutant to show decreased brain cholesterol synthesis is Cyp46a1, which lacks 24SOHC, required for brain cholesterol turnover44. The discovery that re-expression of Mecp2 in astrocytes contributes to the mitigation of RTT symptoms in a non-cell autonomous manner may be linked to cholesterol turnover, since astrocytes supplement neuronal cholesterol through lipoprotein transfer45,46. Cholesterol turnover is also required to produce geranylgeraniol, a product of HMGCR upstream of SQLE that is essential for learning and synaptic plasticity31, and is important for the interaction between neurons and astrocytes at the synapse28. Furthermore, microglia, which function as brain macrophages, recycle neuronal cholesterol during dendrite pruning39, and replacing mutant with wild type microglia via bone marrow transfer ameliorated RTT symptoms in Mecp2 null mice13. Modulating cholesterol homeostasis and turnover in the brain also ameliorated motor symptoms in Mecp2 null mice: fluvastatin treatment affected the abundance of desmosterol, which is produced primarily by astrocytes in the adult brain33,47.
Perturbed lipid metabolism is evident in liver as well as the brain of 129.Mecp2tm1.1Bird mice, and is ameliorated by statin treatment. The precise mechanisms by which statin drugs alter brain cholesterol are unknown, but likely include indirect effects of altered systemic lipid metabolism48,49. The SqleSum3Jus/+ mutation in the 129.Mecp2tm1.1Bird/Y genetic background rescued similar behavioral abnormalities as statin treatment, although it did not change peripheral cholesterol levels. Furthermore, 129.Mecp2tm1.1Bird/Y SqleSum3Jus/+ brains displayed different patterns of sterol changes than did statin treated mice. Statins affect pathways upstream of cholesterol synthesis and are indiscriminate of cell type, whereas SQLE’s effect is limited to cholesterol synthesis in a subset of neurons and glial cells. Interestingly, statin treatment improved behavioral parameters better than the mutation on this genetic background, raising the idea that modifiers may point to a targetable pathway without providing the most effective rescue.
Dysregulation of cholesterol homeostasis in the brain occurs concomitantly with symptom onset in Mecp2 null mice; however, male null and female heterozygous Mecp2 mutant mice eventually accumulate fat in their livers. This fat accumulation is a sign of perturbed metabolism that can lead to fatty liver disease (FLD) when hepatic lipid accumulation is followed by an increase in oxidative and metabolic stress50. Thus, metabolic changes could exacerbate symptoms in Mecp2 mutant mice, although pinpointing the cause of peripheral dyslipidemia is a future endeavor, since conditional mutation of Mecp2 by a Sim1-cre results in a subset of Mecp2 null symptoms that affect metabolism through the dysregulation of neuroendocrine pathways51. B6.Mecp2tm1.1Jae/Y mice showed perturbations in brain lipid metabolism, even though they did not display systemic evidence of high cholesterol. Further, 129.Mecp2tm1.1Bird/+ females accumulated huge amounts of lipids in the liver, yet showed only mildly elevated serum cholesterol and triglycerides. Lipid homeostasis is controlled by an interplay of multiple organ systems at many levels of feedback: therefore, it is likely that allele and genetic background play a role in the full phenotypic outcome of disrupted lipid homeostasis that was uncovered by this genetic screen. However, our data suggest that altering brain cholesterol metabolism in Mecp2 mutants is essential to the improvements in motor function and longevity. A full understanding of lipid homeostasis in all organs of Mecp2 null mice, including muscle and the gastrointestinal systems, remains to be uncovered. Sterols are the precursors of steroid hormones, bile acids and vitamin D; thus, perturbations in lipid homeostasis may influence both neurological and non-neurological RTT symptoms, including abnormal responses to stress, high anxiety levels, and bone anomalies52. The genetic and pharmacologic amelioration of brain and liver cholesterol perturbation suggests that downstream consequences of loss of Mecp2 contribute to progressively worsening stages in disease pathology, and should be considered when evaluating patients. Even so, our data would suggest that peripheral dyslipidemia could be a biomarker in only a subset of RTT patients.
A subset of MECP2 missense mutations abolish interactions with a transcriptional co-repressor complex containing HDAC3, NCoR1, SMRT, and TBLR1. Such mutations recapitulate Rett Syndrome phenotypes in mice, and suggest for the first time that RTT dysfunction is caused by a failure of MECP2 to anchor the HDAC3/NCoR/SMRT complex to chromatin53,54. FLD in 129.Mecp2tm1.1Bird mice is similar to that in a liver-specific knockout of HDAC355,56. HDAC3 regulates lipid homeostasis, and its loss in liver cells leads to metabolic syndrome and FLD56,57. Interestingly, the promoter regions of lipid/cholesterol homeostasis genes, including Sqle, appear to be targets of HDAC3 in the liver55. Our data support the idea that a repressor complex such as the one containing HDAC3 may perform additional roles in regulating lipid homeostasis genes such as Sqle in the brain.
Screens for disease suppression have the potential to transform translational biology by identifying unrecognized pathways of pathogenesis, facilitating rational design of novel therapeutic strategies. Here, we carried out a suppressor screen in mice that identified an unanticipated role for cholesterol biosynthesis in RTT disease pathology, a finding supported by the amelioration of symptoms in Mecp2 null mice with statin treatment. These data suggest that a subset of RTT patients may be aided by pharmacological interventions designed to modulate lipid metabolism. Notably, moderate amounts of statin drugs were effective, whereas large doses were detrimental, demonstrating the importance of further studies to optimize treatment protocols. Although these findings do not comprise a pre-clinical trial, they point to mechanism-based strategies for treatment that may ameliorate a subset of symptoms and thereby improve quality of life for some RTT patients. None of the suppressors eliminated symptoms entirely, supporting the idea that combination therapy will be required for thorough alleviation of RTT symptoms. The identification of additional suppressor loci may point to other potential targets.
Online Methods
Mouse Strains and Genetic Screen
All animal experiments were conducted under protocols approved by local Animal Care and Use Committees in AALAC-accredited animal facilities at BCM and UTSW. Congenic 129.Mecp2tm1.1Bird/+ female mice were maintained by backcrossing females to males of the 129S6/SvEvTac strain. C57BL/6J males were imported from The Jackson Laboratory at six weeks of age, and injected with three weekly doses of 100 mg/kg ENU at 8 weeks as described58. After recovery of fertility, 60 ENU-treated males were mated to 129.Mecp2tm1.1Bird/+ females, and their G1 male offspring were genotyped for Mecp2 according to The Jackson Laboratory standard protocol. B6.Mecp2tm1.1Jae was made in hybrid 129/Ola C57BL/6 ES cell line32, and backcrossed to C57BL/6J for over 10 generations. The line is maintained by additional backcrosses to C57BL/6J mice.
For each founder line, long-lived G1 males were mated to 129S6/SvEvTac females; G2 offspring were mated to 129.Mecp2tm1.1Bird/+ females or 129S6/SvEvTac males to produce N3 animals for mapping. Fine structure mapping was achieved with informative MIT markers identified on the Mouse Genome Informatics website (informatics.jax.org). PCR (ABI) followed by gel electrophoresis with MetaPhor agarose (Lonza) on relevant primer pairs determined whether a given mouse carried heterozygous B6/129S6 or 129S6 homozygous DNA at each locus of interest. Primers are available upon request.
Lipid and sterol analysis
129.Mecp2tm1.1Bird/Y and age matched +/Y littermate controls housed in plastic Lab Products cages with corncob bedding in rooms alternating 13-hr and 11-hr periods of light and dark were provided acidified water, and a Harlan Teklad 2920× diet ad libitum (19.1% protein, 6.5% fat; 0% cholesterol). Gene expression, serum cholesterol (Cobas Mira clinical chemistry analyzer), and tissue lipids were assessed within a two hour afternoon window following a 4 – 6 hour fast at P28 and P56. Brain analyses were performed on the subcortical region, which contains the corpus callosum, striatum, thalamus, hypothalamus, and hippocampus. Cholesterol intermediates were measured after extraction from tissue from mice treated the same as above by tandem mass spectrometry following a previously published protocol59. Prior to gas-liquid chromatography, lipids were isolated from tissue using CHCl3:CH3OH extraction, followed by drying of the organic phase under N2 pressure. Tissue cholesterol concentrations were assessed by gas-liquid chromatography, and cholesterol synthesis was assessed from saponified tissue after the incorporation of 100 mCi 3H2O after intraperitoneal injection as published34. For the in vivo cholesterol synthesis study, samples were obtained from mice in a fed state the late dark phase of a 12-hour on/off light dark cycle. These mice were adapted to individual housing and a Harlan Teklad 7001 rodent chow (low cholesterol 0.02% w/w, low fat 4% w/w) starting at P38 prior to analysis. The age of mice at sampling was P54–56.
Real time RT-PCR
Brain RNA was isolated using RNAeasy Lipid Tissue Mini Kit (Qiagen) and liver RNA using Trizol (Invitrogen), according to manufacturer’s instructions. Liver RNA was treated with 1IU DNAse (Ambion Inc.) at 37°C for 1h per manufacturer’s instructions. First strand complementary DNA (cDNA) was synthesized from 5000ng of total RNA using SuperScript III First Strand Synthesis System (Invitrogen) per manufacturer’s instructions. RT-PCR was performed in triplicate for each sample on an ABI 7900 (Applied Biosystems CA, USA). Gene primers for QRT-PCR were designed against published mRNA sequences using Primer3 software and synthesized by Integrated DNA Technologies (Iowa, USA). Primer sequences will we provided upon request. QRT-PCR was performed in triplicate on an ABI 7900 (Applied Biosystems CA, USA). Reactions contained cDNA from 10ng total RNA, 0.1µL forward and reverse primers, 5µL Power SYBR®Green Master Mix, and water to a final volume of 10µL. PCR conditions: 95°C for 10 min, 40 cycles of 95°C for 15 sec, 60°C for 60 sec. Single product amplification was confirmed by disassociation curves and agarose gel electrophoresis. Gene expression was normalized to an RpL19 (L19) internal loading control, and analyzed using the 2−(ΔΔCT) method expressed either as raw 2−(ΔΔCT) or as Mecp2/Y expression relative to WT60.
Exome sequencing
Genomic DNA was isolated from two N3 cousin offspring of two N2 animals (female 5 and female 8) from Line 895 (Sum3m1Jus), and from C57BL/6J and 129S6/SvEvTac by standard methods. Mouse exome capture reagents were designed to a 54.3 MB target including 203,225 exonic regions (C57BL/6J, NCBI37/mm9)17. Sequencing of post-enrichment shotgun libraries was performed on an Illumina GA2× (paired-end 76 bp reads) or Illumina HiSeq 2000 (paired-end 100 bp reads). Sequence reads were mapped to the mm9 reference genome with bwa61, and variants were called with samtools62 requiring a minimum SNP quality of 20. Custom scripts were used to annotate variants with respect to their predicted impact on protein sequence. To remove inbred strain polymorphisms as well as systematic sequencing artifacts, variants were removed from consideration if identified in any of 15 other mouse strains (including the parental strains, resequenced alongside the mutants), other lines from this study, or other phenotypes.
Drug Administration
Fluvastatin (Selleckchem) was dissolved in sterile ultrapure water such that the desired dose for a 20g mouse was given in 100ul and administered subcutaneously. Male mice were given a single 3mg/kg weekly dose at five, six and seven weeks, then were given 3× weekly (M,W,F) 3 mg/kg doses beginning at 8 weeks of age. Female mice also received 3 mg/kg doses, but were treated only once per week, beginning at 6 weeks of age. Lovastatin (Tocris Bioscience) preparation required activation in ethanol followed by adjustment to pH7.2, per product information guidelines. The activated stock solution was diluted with ethanol to 20× the injected dose and kept at −20°C for up to one month. The day of injection a 1× working solution was prepared by diluting the stock solution in sterile saline such that the desired dose for a 20g mouse was given in 100ul. Male mice were injected subcutaneously with a twice-weekly 1.5 mg/kg dose, beginning at five weeks of age. Precise timing of injections and assays can be found in Supplemental Table 5.
Assessments of behavior and breathing
Open field locomotor activity was assessed using Versamax Animal Activity Monitors. Recordings were taken in a secluded room with dim light (20–25 lux) and artificial white noise (55–60 dB). Each mouse was placed in the center of the open field chamber and activity was recorded for 30 minutes. An aspect of motor performance was measured using the accelerating rotating rod (rotarod) (Stoelting). At 8 weeks (males) or 12 weeks (females), mice were placed on a grooved rod, rotating at a speed of four revolutions per minute. Over the course of a five-minute trial, the revolution rate increases steadily to a maximum of forty revolutions per minute. The time each mouse is able to stay on the rod is recorded for eight trials, four each over two consecutive days, with a minimum of thirty-minutes between trials. A trial ends when the mouse falls off the rod, spins with the rod for two consecutive revolutions, or successfully completes five minutes. Pre-pulse inhibition was measured using SR-Lab Startle Chambers (San Diego Instruments). Unrestrained, whole-body plethysmography was carried out using the Buxco FinePointe system. Breathing parameters were recorded for one hour, following a 30-minute acclimation period. Mice were monitored for activity. Data represent breathing during periods of stillness.
Statistics
LOD scores were generated using the R/qtl statistics package63. Survival curves were compared using SPSS by Kaplan-Meier analysis followed by log rank comparison. Statistical comparisons between two groups (wild type compared to Mecp2 mutant) were performed in GraphPad Prism 5 using an unpaired, two-tailed student’s t-test; equal variances were not assumed, as the Mecp2 mutant group typically showed increased variability compared to wild type. Statistical tests requiring multiple comparisons were analyzed in SPSS. Excepting rotarod data, comparisons between multiple groups were analyzed by one-way ANOVA, sphericity not assumed; the Bonferroni adjustment was applied when comparing more than two genotypes, the Dunnett post hoc test was used to compare statin treated groups with the vehicle control. Rotarod data was analyzed using repeated measures ANOVA.
Supplementary Material
Acknowledgements
The Genetics Analysis Facility (Tara Patton and Chris Marshall) at the Centre for Applied Genomics, Toronto Hospital for Sick Kids, Toronto, ON, Canada performed the Illumina Goldengate SNP analysis. We thank Jill Crowe, Marijke Schrock, Jennifer Borkey, Misty Hill, Anna Willis, Justine Shaw (Justice laboratory), Choli Lee, Alexandra MacKenzie, and Lindsay Felker (Shendure laboratory), Ian Adams (Katz laboratory), Bonne Thompson (McDonald/Russell laboratory), Kenneth S Posey and Adam M Lopez (Turley laboratory) for technical assistance. We thank Drs. Corinne Spencer and Richard Paylor for advice on assessing mouse behavior, which was carried out in the BCM Mouse Neurobehavior Core, and Dr. Corey Reynolds for advice on plethysmography, which was carried out in the BCM Mouse Phenotyping Core. We thank Drs. Huda Zoghbi and Jeff Neul for Mecp2 mutant mice. We also thank Drs. Huda Zoghbi and Richard Behringer for valuable discussions during revision of the manuscript. Monica Coenraads of the RSRT provided crucial moral and uninterrupted financial support, while she aided intellectually through literature searches and advice.
The work was supported by grants from the Rett Syndrome Research Trust, the Rett Syndrome Research Foundation, the International Rett Syndrome Foundation (ANGEL award 2608 to MJJ and ANGEL award 2583 to DMK), Autism Science Foundation predoctoral fellowship #11–1015 and NIH T32 GM08307 to CMB, NIH GM69338 to DWR, NIH R01 HL09610 to SDT, NIH R01 CA115503 to MJJ and the National Institute of Neurologic Diseases and Stroke, including funding from the American Recovery and Reinvestment Act (DMK). Grants to the BCM Diabetes and Endocrinology Research Center 2P30DK079638-05, and to the BCM Intellectual and Developmental Disabilities Research Center 5P30HD024064-23 from the National Institutes of Health Eunice Kennedy Shriver National Institute Of Child Health & Human Development also supported this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute Of Child Health & Human Development or the National Institutes of Health.
Footnotes
Author Contributions: MJJ conceived of the work, carried out the genetic screen and dissected embryos, JS and WH carried out the capture sequencing and analysis, CB confirmed map locations and lesions, performed statin injections, carried out behavior and plethysmography testing and QRT-PCR, SK performed Western blotting and liver histopathology, HB performed preliminary QRT-PCR, JGM, BL, and SDT analyzed sterols and performed synthesis studies and SDT evaluated liver cholesterol and triglycerides. AP and DK provided Jaenisch mice and laboratory facilities, and DK helped to analyze plethysmography data. MJJ, DWR, DK, SDT, SK and CB wrote the manuscript with input from the other co-authors.
References
- 1.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature genetics. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 2.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nature reviews. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 3.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature genetics. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 4.Shepherd GM, Katz DM. Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: focus on Mecp2 and Met. Current opinion in neurobiology. 2011;21:827–833. doi: 10.1016/j.conb.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kavalali ET, Nelson ED, Monteggia LM. Role of MeCP2, DNA methylation, and HDACs in regulating synapse function. J Neurodev Disord. 2011;3:250–256. doi: 10.1007/s11689-011-9078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science (New York, N.Y. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins AL, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human molecular genetics. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 8.Chahrour M, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science (New York, N.Y. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stancheva I, Collins AL, Van den Veyver IB, Zoghbi H, Meehan RR. A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Molecular cell. 2003;12:425–435. doi: 10.1016/s1097-2765(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 10.St Johnston D. The art and design of genetic screens: Drosophila melanogaster. Nature reviews. 2002;3:176–188. doi: 10.1038/nrg751. [DOI] [PubMed] [Google Scholar]
- 11.Carpinelli MR, et al. Suppressor screen in Mpl−/− mice: c-Myb mutation causes supraphysiological production of platelets in the absence of thrombopoietin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:6553–6558. doi: 10.1073/pnas.0401496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matera I, et al. A sensitized mutagenesis screen identifies Gli3 as a modifier of Sox10 neurocristopathy. Human molecular genetics. 2008;17:2118–2131. doi: 10.1093/hmg/ddn110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Justice MJ, Siracusa LD, Stewart AF. Technical approaches for Mouse Models of Human Disease. Disease Models and Mechanisms. 2011;4:305–310. doi: 10.1242/dmm.000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derecki NC, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neuhaus IM, Beier DR. Efficient localization of mutations by interval haplotype analysis. Mamm Genome. 1998;9:150–154. doi: 10.1007/s003359900706. [DOI] [PubMed] [Google Scholar]
- 16.Moran JL, et al. Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome research. 2006;16:436–440. doi: 10.1101/gr.4563306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fairfield H, et al. Mutation discovery in mice by whole exome sequencing. Genome biology. 2011;12:R86. doi: 10.1186/gb-2011-12-9-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurevics HA, Kidwai FZ, Morell P. Sources of cholesterol during development of the rat fetus and fetal organs. J Lipid Res. 1997;38:723–733. [PubMed] [Google Scholar]
- 19.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Gill S, Stevenson J, Kristiana I, Brown AJ. Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell metabolism. 2011;13:260–273. doi: 10.1016/j.cmet.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Cory EJ, Russey WE, Ortiz de Montellano PR. 2,3-oxidosqualene, an intermediate in the biological synthesis of sterols from squalene. J Am Chem Soc. 1966;88:4750–4751. doi: 10.1021/ja00972a056. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto S, Bloch K. Studies on squalene epoxidase of rat liver. J Biol Chem. 1970;245:1670–1674. [PubMed] [Google Scholar]
- 23.Shibata N, et al. Supernatant protein factor, which stimulates the conversion of squalene to lanosterol, is a cytosolic squalene transfer protein and enhances cholesterol biosynthesis. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2244–2249. doi: 10.1073/pnas.041620398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Astruc M, Tabacik C, Descomps B, de Paulet AC. Squalene epoxidase and oxidosqualene lanosterol-cyclase activities in cholesterogenic and non-cholesterogenic tissues. Biochim Biophys Acta. 1977;487:204–211. doi: 10.1016/0005-2760(77)90056-x. [DOI] [PubMed] [Google Scholar]
- 25.Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nature reviews. 2011;12:393–406. doi: 10.1038/nrg2984. [DOI] [PubMed] [Google Scholar]
- 26.Pose D, Botella MA. Analysis of the arabidopsis dry2/sqe1-5 mutant suggests a role for sterols in signaling. Plant signaling & behavior. 2009;4:873–874. doi: 10.4161/psb.4.9.9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pose D, et al. Identification of the Arabidopsis dry2/sqe1–5 mutant reveals a central role for sterols in drought tolerance and regulation of reactive oxygen species. The Plant journal : for cell and molecular biology. 2009;59:63–76. doi: 10.1111/j.1365-313X.2009.03849.x. [DOI] [PubMed] [Google Scholar]
- 28.Nieweg K, Schaller H, Pfrieger FW. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem. 2009;109:125–134. doi: 10.1111/j.1471-4159.2009.05917.x. [DOI] [PubMed] [Google Scholar]
- 29.Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res. 1993;34:1637–1659. [PubMed] [Google Scholar]
- 30.Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biological chemistry. 2009;390:287–293. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell DW, Halford RW, Ramirez DM, Shah R, Kotti T. Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annual review of biochemistry. 2009;78:1017–1040. doi: 10.1146/annurev.biochem.78.072407.103859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nature genetics. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 33.Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Progress in lipid research. 2011;50:357–371. doi: 10.1016/j.plipres.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Xie C, Lund EG, Turley SD, Russell DW, Dietschy JM. Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J Lipid Res. 2003;44:1780–1789. doi: 10.1194/jlr.M300164-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Ko M, et al. Cholesterol-mediated neurite outgrowth is differently regulated between cortical and hippocampal neurons. The Journal of biological chemistry. 2005;280:42759–42765. doi: 10.1074/jbc.M509164200. [DOI] [PubMed] [Google Scholar]
- 36.Jolley CD, Dietschy JM, Turley SD. Genetic differences in cholesterol absorption in 129/Sv and C57BL/6 mice: effect on cholesterol responsiveness. The American journal of physiology. 1999;276:G1117–G1124. doi: 10.1152/ajpgi.1999.276.5.G1117. [DOI] [PubMed] [Google Scholar]
- 37.Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109:III50–III57. doi: 10.1161/01.CIR.0000131519.15067.1f. [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Sabina A, Gulin-Davila J, Sempere-Serrano P, Gonzalez-Juanatey C, Martinez-Pacheco R. Specific considerations on the prescription and therapeutic interchange of statins. Farmacia hospitalaria: organo oficial de expresion cientifica de la Sociedad Espanola de Farmacia Hospitalaria. 2012;36:97–108. doi: 10.1016/j.farma.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 39.Osterweil EK, et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile×syndrome. Neuron. 2013;77:243–250. doi: 10.1016/j.neuron.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ardern-Holmes SL, North KN. Therapeutics for childhood neurofibromatosis type 1 and type 2. Current treatment options in neurology. 2011;13:529–543. doi: 10.1007/s11940-011-0142-9. [DOI] [PubMed] [Google Scholar]
- 41.Keber R, et al. Mouse knockout of the cholesterogenic cytochrome P450 lanosterol 14alpha-demethylase (Cyp51) resembles Antley-Bixler syndrome. The Journal of biological chemistry. 2011;286:29086–29097. doi: 10.1074/jbc.M111.253245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waterham HR. Defects of cholesterol biosynthesis. FEBS letters. 2006;580:5442–5449. doi: 10.1016/j.febslet.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 43.Bjorkhem I, Hansson M. Cerebrotendinous xanthomatosis: an inborn error in bile acid synthesis with defined mutations but still a challenge. Biochem Biophys Res Commun. 2010;396:46–49. doi: 10.1016/j.bbrc.2010.02.140. [DOI] [PubMed] [Google Scholar]
- 44.Lund EG, et al. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. The Journal of biological chemistry. 2003;278:22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- 45.Lioy DT, et al. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vance JE. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Disease models & mechanisms. 2012;5:746–755. doi: 10.1242/dmm.010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vance JE, Karten B, Hayashi H. Lipid dynamics in neurons. Biochem Soc Trans. 2006;34:399–403. doi: 10.1042/BST0340399. [DOI] [PubMed] [Google Scholar]
- 48.Cibickova L. Statins and their influence on brain cholesterol. Journal of clinical lipidology. 2011;5:373–379. doi: 10.1016/j.jacl.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Stranahan AM, Cutler RG, Button C, Telljohann R, Mattson MP. Diet-induced elevations in serum cholesterol are associated with alterations in hippocampal lipid metabolism and increased oxidative stress. J Neurochem. 2011;118:611–615. doi: 10.1111/j.1471-4159.2011.07351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 51.Fyffe SL, et al. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Percy AK. Rett syndrome: exploring the autism link. Archives of neurology. 2011;68:985–989. doi: 10.1001/archneurol.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyst MJ, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT transcriptional co-repressor. Nature Neuroscience. 2013 doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ebert DH, et al. Activity-dependent phosphorylation of MECP2 threonine 308 regulates interaction with NcoR. Nature. 2013 doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knutson SK, et al. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. The EMBO journal. 2008;27:1017–1028. doi: 10.1038/emboj.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Z, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nature medicine. 2012;18:934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feng D, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–1319. doi: 10.1126/science.1198125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kile BT, et al. Functional genetic analysis of mouse chromosome 11. Nature. 2003;425:81–86. doi: 10.1038/nature01865. [DOI] [PubMed] [Google Scholar]
- 59.McDonald JG, Smith DD, Stiles AR, Russell DW. A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J Lipid Res. 2012;53:1399–1409. doi: 10.1194/jlr.D022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 61.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.