

Glioblastoma multiform (GBM) is among the deadliest forms of brain cancers, yet few effective advances in treatment have emerged in the last several decades.[1,2] Inefficient delivery of therapeutic drugs across the blood brain barrier and/or the blood brain tumor barrier (BBB and BBTB) and non-specific delivery to brain stroma are in general the major obstacles in brain cancer treatment in the clinic.[3,4] Nanoparticle-drug delivery systems offer various strategies, including the targeting of certain cell surface proteins, to overcome the difficulties of systemic drug administration to treat brain tumors.[5-7] In this work, we examine the hypothesis that EGF-targeted gold nanoparticles will be effective at delivering a noncovalently adsorbed hydrophobic drug molecule into brain tumors.
Colloidal gold nanoparticles (Au NPs) with low toxicity, tuneable size, shape, and wellestablished surface chemistry have been explored as drug delivery systems.[8-11] Rational design of the architecture of the Au NP overcomes the current delivery barriers, such as the BBB and BBTB, for therapeutic drugs with regard to treatment of brain cancers. Previously reported polyethylene glycol (PEG)-coated Au NPs provide an excellent platform to deliver therapeutic drugs into cancer cells.[9,12-14] The PEGylated Au NP surface acts as an amphiphilic reservoir that can adsorb and stabilize hydrophobic molecules, such as hydrophobic drugs, in aqueous media.[9,13] It provides a uniquely efficient drug delivery approach for therapeutics without the need to further modify the molecular drug structure. In addition, these PEGylated Au NPs have long circulation life-times in the blood with reduced uptake by the reticulo-endothelial system.[15] Permeability and retention into solid tumors can be achieved through passive targeting, the so called enhanced permeability and retention (EPR) effect.[15-17] Further, the surface of Au NPs can be modified to improve targeting to cancer cells. Overexpression of cancer biomarkers related to uncontrolled cell proliferation and cell death are attractive targets. Active targeting can be achieved by using moieties, such as peptides and antibodies, that are conjugated to the Au NP surface. The targeted-Au NPs can then identify and target cancer cells and therefore improve the drug efficacy and minimize potential side-effects to the healthy tissue.[14]
Photodynamic therapy (PDT), a non-invasive treatment using light, has the promise of improving treatment for brain cancers by selectively targeting tumor tissue via localized light activation and thereby reducing side effects to the normal brain tissue.[18-20] The most effective PDT drugs are hydrophobic and tend to localize in the lipophilic environment of organelles such as mitochondria and endoplasmic reticulum.[21] However, it is inherently difficult to transport hydrophobic PDT drugs into the brain and selectively target brain tumors by systemic administration, which so far has greatly hampered the broad application of anticancer drugs for brain cancers.[22] A strategy combining both selective delivery of the PDT drug and application of light to the brain tumor provides a great opportunity to improve PDT efficacy.
In the present study, we demonstrate the rational design of a non-covalent Au NP drug delivery platform which can cross the BBB/ BBTB and selectively deliver therapeutic drug to brain glioma tumors for PDT. Scheme 1 shows the design of the EGF-Au NP-Pc 4 conjugates.
Scheme 1.
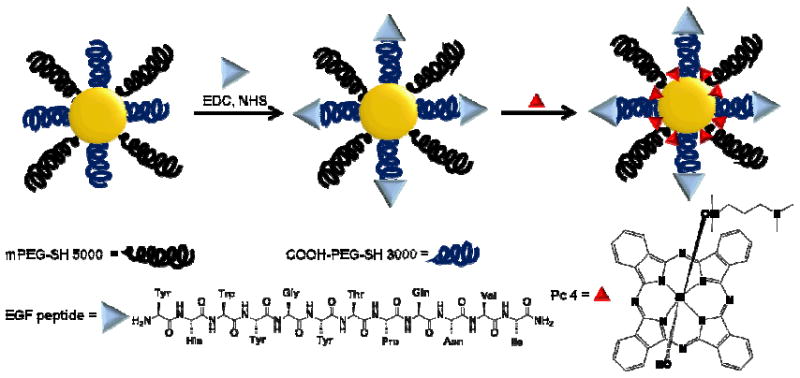
Design of targeted delivery of the PDT drug Pc 4 via EGF-labeled Au NP conjugates.
Without chemical modification of phthalocyanine 4 (Pc 4), a hydrophobic PDT drug which is currently in clinical trials,[23] the active drug is spatially encaged and photophysically quenched through adsorption on the PEGylated Au NPs while being transported to the target tissue. The PEG ligand on the Au NPs creates excellent water miscibility, biocompatibility and long circulation in the blood of the conjugate system.[15] PEG also prevents protein agglomeration on the NP surface. More importantly, the PEG layer provides bifunctionality to conjugate epidermal growth factor (EGF) peptides, which are internalizing and non-mitogenic, to recognize EGFRs on the glioma cancer cell surface. The core of Au NPs is designed to be 5 nm in diameter which is large enough on which to load drug molecules, but small enough to be eventually excreted by renal clearance.[25]
PEGylated Au NPs were synthesized and modified with a mixture of 20% hetero-bifunctional COOH-PEG-SH (MW 3000) and 80% monofunctional mPEG-SH (MW 5000) (Scheme 1). The EGF peptide with the 12 amino acid sequence YHWYGYTPQNVI-amide was linked to the carboxyl group on bifunctional PEG layer via the amide bond. Au NPs with the surface plasmon resonance band at 522 nm were well dispersed and stable in aqueous solution before and after conjugation as shown in Figure 1.
Figure 1.
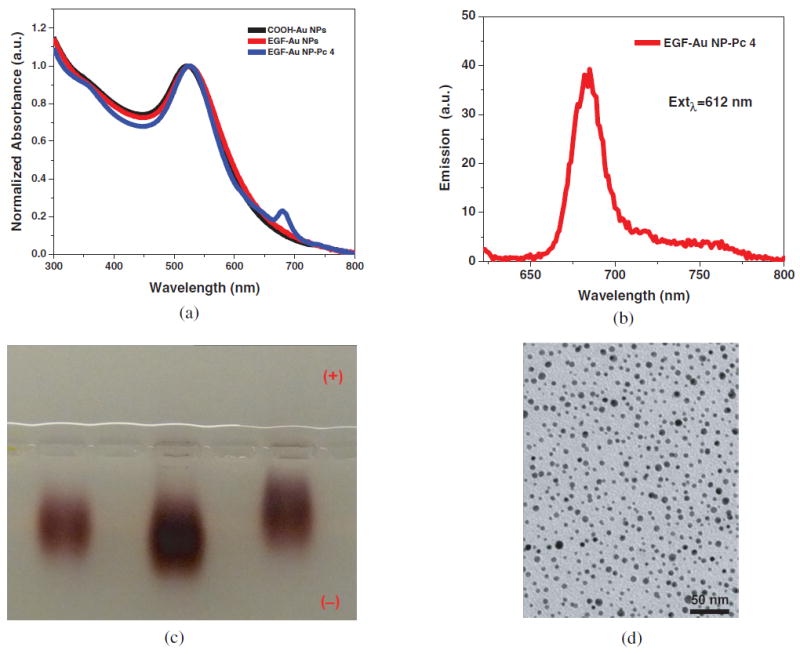
Characterization of EGF-Au NP-Pc 4 conjugates (A-D). (A) Absorbance spectra of COOH-functionalized Au NPs, EGF-Au NPs and EGF-Au NP-Pc 4 conjugates in water. (B) The fluorescence spectrum and (C) 1% Agarose gel at 120 V for 4 hours in TAE buffer. From left to right: COOH-functionalized Au NP, EGF-Au NPs and EGF-Au NP-Pc 4 conjugates. (D) TEM image of the conjugates.
Pc 4 is then adsorbed to the Au NP surface through N-Au bonding by the terminal amine group on the Pc 4 axial ligand. Since PEG does not contain any unconjugated reactive groups to facilitate this amine attachment, it is likely that the Pc 4 is adsorbed to the surface of the Au NP.[9] This adsorption occurs as a result of both hydrophobic and electrostatic interactions in the PEG corona, close to the gold surface. Synthesis and characterization of the particles is described by Cheng et al., 2010.[26] Pc 4 absorption measurements at 679 nm in the UV-vis spectra (Figure 1) determined that 30±3 Pc 4 molecules were adsorbed per Au NP. The conjugates showed emission at 685 nm due to the intrinsic fluorescence property of Pc 4, which allows detection of drug accumulation via in vitro and in vivo fluorescence imaging. According to TEM analysis (Figure 1D), the average size of the Au NP core was ~5 nm in diameter. The hydrodynamic diameter of the EGF-Au NP-Pc 4 conjugates was measured to be 42±2 nm by dynamic light scattering (DLS). The EGF-Au NP-Pc 4 conjugates were found to be effectively neutral as indicated by zeta-potential measurements. Gel electrophoresis studies demonstrated clear changes between the COOH functionalized Au NP, EGF-Au NPs and the conjugates (Figure 1) indicating successful conjugation. Typically, the samples moved toward the negative electrode in an agarose gel electrophoresis experiment. This is mainly due to electro-osmosis under the applied electric field, which we have recently quantified.[27]
Figure 2 shows that the PDT drug on the Au NPs is inactive and no toxicity could be detected without irradiation. The activity of Pc 4 on the Au NPs is quenched as measured by photodecomposition of 1,3-diphenylisobenzofuran (DPBF), a commonly used singlet oxygen trap. However, after Pc 4 releases from the Au NPs, the drug activity can be recovered. It can be excited by using near infrared light to transfer the absorbed energy to molecular oxygen and to generate reactive oxygen species such as singlet oxygen to damage surrounding tissues.[23] The dark toxicity and phototoxicity of the EGF-Au NP-Pc 4 conjugates were evaluated with MTT assays and trypan blue staining (Figure 2C). No toxicity was observed upon incubation with the conjugates in the dark. Upon light exposure, the EGF-Au NP-Pc 4 conjugates showed excellent cytotoxicity. Over 90% of cancer cells were killed with 0.5 or 1 J/cm2 light exposure at 1 μM drug concentration. Also, a PDT effect of the conjugates could be obtained at half the initial drug concentration, although 30% of the cancer cells survived upon 1 J/cm2 light exposure at this lower concentration.
Figure 2.
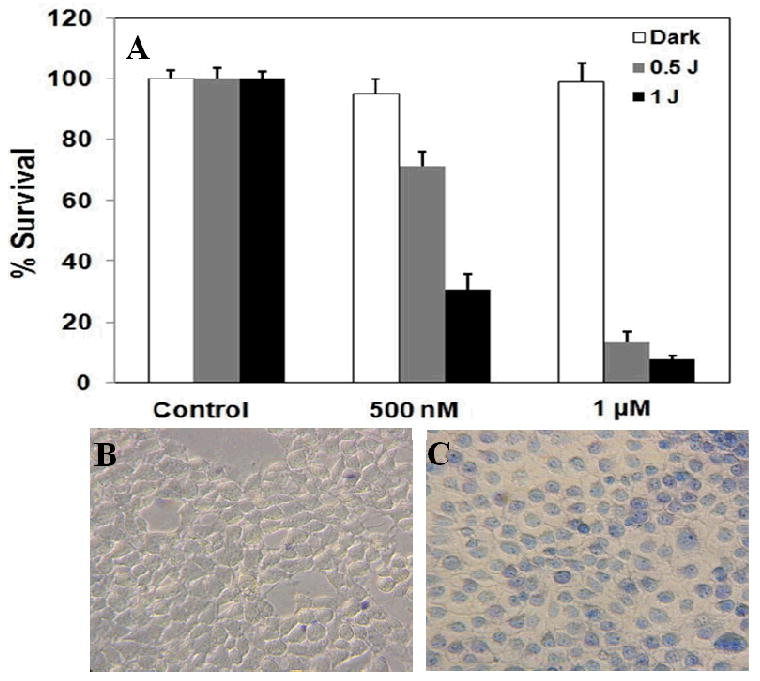
Toxicity study of the EGF-Au NP-Pc 4 conjugates in the dark and under light exposure. (A) PDT effect of the EGF-Au NP-Pc 4 conjugates after 4 hours incubation measured with an MTT assay. (B) Bright field images of the conjugate-incubated cancer cells before and (C) after light exposure. Dead cells are colored blue due to the staining by trypan blue.
Due to the non-covalent attachment, efficient drug release of the EGF-Au NP-Pc 4 conjugate was found in vitro (Figure 3). As shown in previous studies, Pc 4 localized in hydrophobic sites such as the lipid bilayers of mitochondria in cells.[28] The water-toluene two-phase system provides a simplified model to study the drug release and transfer from the aqueous phase into a less polar environment. Significant drug release into the organic phase was detected within 4 hours as shown in Figure 3A, driven by the hydrophobic interaction between the drug and water. The hydrophobic drug was delivered into the toluene rapidly with a time constant of 78±15 min. Similar to the phase transfer studies, the efficient drug uptake into the glioma cancer cells was also observed within 4 hours of incubation with the conjugates, as observed by confocal microscopy (Figure 3B). Pc 4 fluorescence was observed mainly in the cytoplasm and vesicles of the cancer cells.
Figure 3.
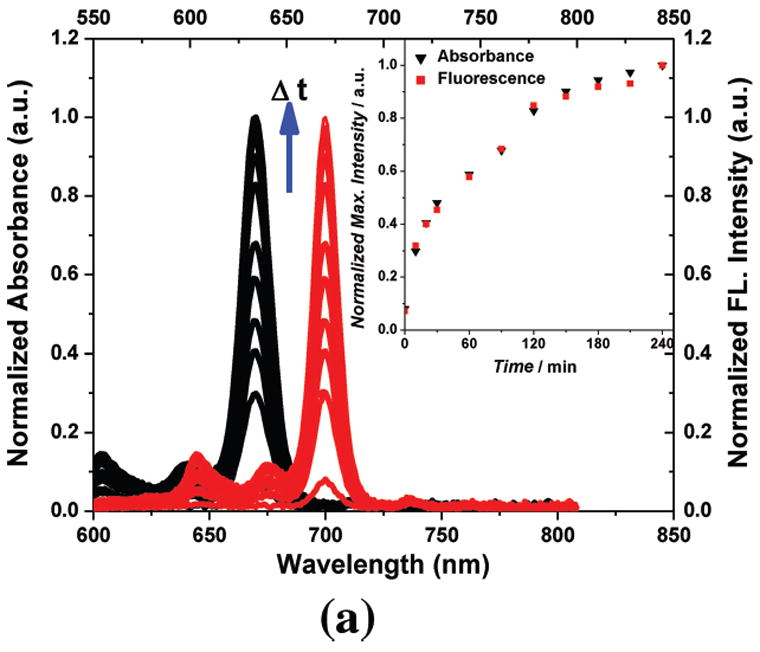
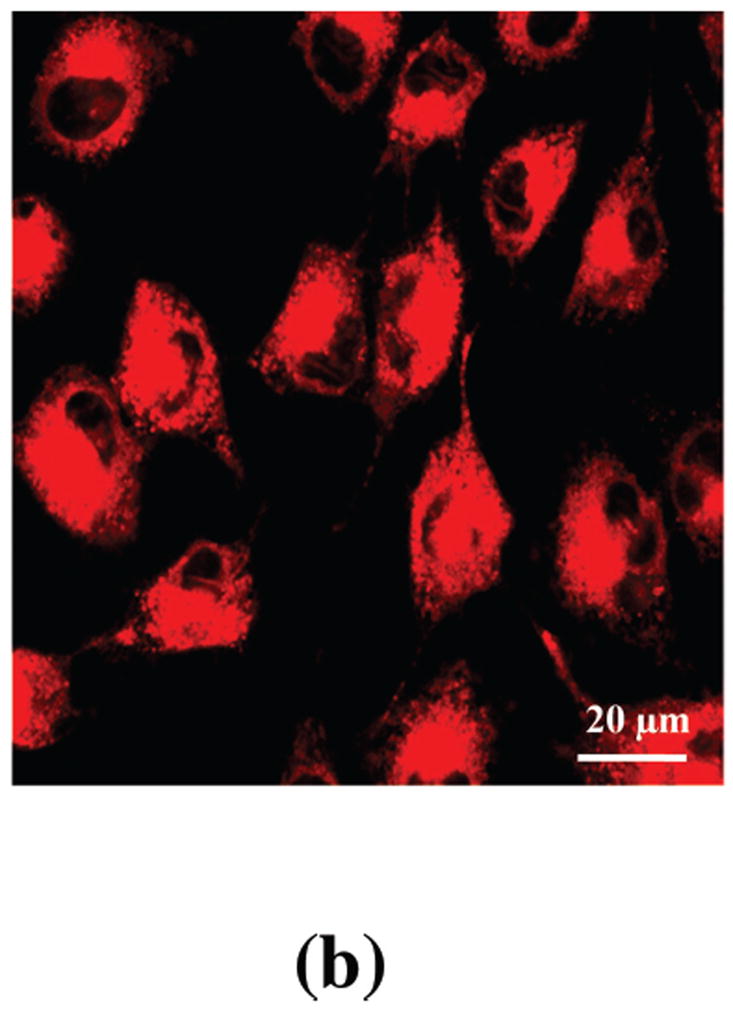
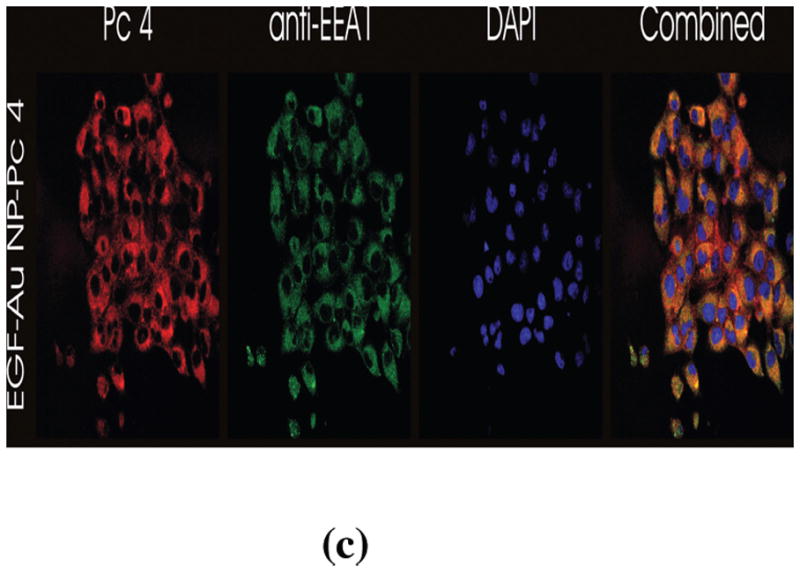
In vitro studies of the PDT drug release from EGF-Au NPs. (A) Time dependent drug release in the water-toluene system. The absorption (in black, left scale) and fluorescence (in red, right scale) of the PDT drug in the toluene phase was monitored within a 4 hour time window. The inset shows the drug release kinetics into the hydrophobic environment. (B) Confocal image of live glioma cancer cells after 4 hours incubation with the conjugates, Pc 4 fluorescence is shown in red color. (C) Confocal fluorescence images of fixed glioma cells after incubation for 24 hours with EGF-Au NP-Pc 4, [Pc 4] = 1 μM. Pc 4 is measured by direct fluorescence of Pc 4 in the Cy5 channel (red). Anti-EEA1 recognizes early endosomes (green). DAPI shows nuclei (blue). The yellow color in the combined image shows the co-localization of Pc 4 in early endosomes.
The free EGF peptide has a specific binding to the EGFR with a dissociation constant of 22 nM.[29] After targeting the Au NPs with the EGF peptide, we found that receptor-mediated endocytosis was the dominant pathway and the drug was taken up into the glioma cancer cells through endocytosis with virtually no uptake of the NPs. The conjugate uptake mechanism was studied by TEM, graphite furnace atomic absorption spectroscopy (GFAAS), UV-vis spectrometry, epi-fluorescence microscopy, and confocal fluorescence microscopy. In contrast to free Pc 4, which is known to locate in the mitochondria,[28] the EGF-targeted Au NP-Pc 4 conjugates altered the drug delivery pathway to early endosomes in the glioma cells (Figure 3C).
TEM imaging showed little Au NP uptake into the cells, in agreement with previous reports of PEGylated Au NP drug delivery systems (Figure S1).[30] In addition, few NPs were found attached or adjacent to the plasma membrane of the cells (Figure S1). The amount of Au NPs in the cancer cells was also quantified by GFAAS, which confirmed the very low uptake of EGF-Au NPs by the cells at 4 and 24 hours after incubation (Figure S2) with increased NP uptake after 24 hours incubation. The amount of Pc 4 in EGF-targeted Au NP cells was quantified by UV-vis spectroscopy (Figure S2). Despite the relatively low uptake of Au NPs, as demonstrated by TEM and GFASS, we found that the Au NPs had different pharmacokinetics as compared to Pc 4 (Figure S2). The Pc 4 to Au NP ratio in the cells was (13.8±1.0)×103:1 after 4 hours incubation and (7.3±1.9)×103:1 after 24 hours incubation times (Figure S2). To confirm this drug uptake mechanism, the cells were preincubated with free EGF peptide, effectively blocking the receptor-mediated pathway. Fluorescence signal from Pc 4 in the cells decreased in comparison to the control without peptide blocking (Figure S3), demonstrating that the drug release takes place when the EGFAu NP-Pc 4 conjugates interact with the EGF receptors on the cell surface.
Animal studies showed that the EGF-Au NP-Pc 4 could pass through the BBB/ BBTB and targeted drug delivery to the brain tumor (Figure 4). The drug biodistribution was monitored via its fluorescence. Human glioma cells were implanted and grown in the right hemisphere of the brain of mice. Four hours after intravenous injection with either the EGF-Au NP-Pc 4 or untargeted Au NP-Pc 4 conjugates, the mice were euthanized and sacrificed, as reported previously.[31] The brains were removed and then serially transected. In contrast to the untargeted Au NPs, a striking accumulation of the drug in the brain tumor was observed in the EGFR-targeted conjugate injected mice as shown in the fluorescence images of the serial transections in Figure 4. A 10-fold increase of the drug accumulated in the tumor as quantified by fluorescence molecular tomography compared to that of the untargeted ones (Figure S4). The EGF peptide-modified conjugates therefore pass through the BBB/BBTB efficiently and transport the drug into the brain. It should also be noted that the conjugates were small enough to leak through the BBTB, which restricts the total Au NP size to 100 nm or less, and selectively accumulated in the tumor region by additional EPR effect and receptor-mediated endocytosis. Only tumor regions showed increased Pc 4 fluorescence with extremely low Pc 4 accumulation in the healthy brain tissue (Figure 4). Biodistribution studies have demonstrated that most tissues, including the tumor, reached maximum Au NP accumulation by 4 hours followed by a gradual decrease over time. An exception to this was observed in the spleen where accumulation of the Au NPs continued over time to reach a maximum at the latest time point measured, 7 days, where liver and spleen together showed a maximum Au NP accumulation of 20 % of the injected dose. Gold content was found in the urine samples which indicated the excretion of Au NPs through renal clearance.
Figure 4.
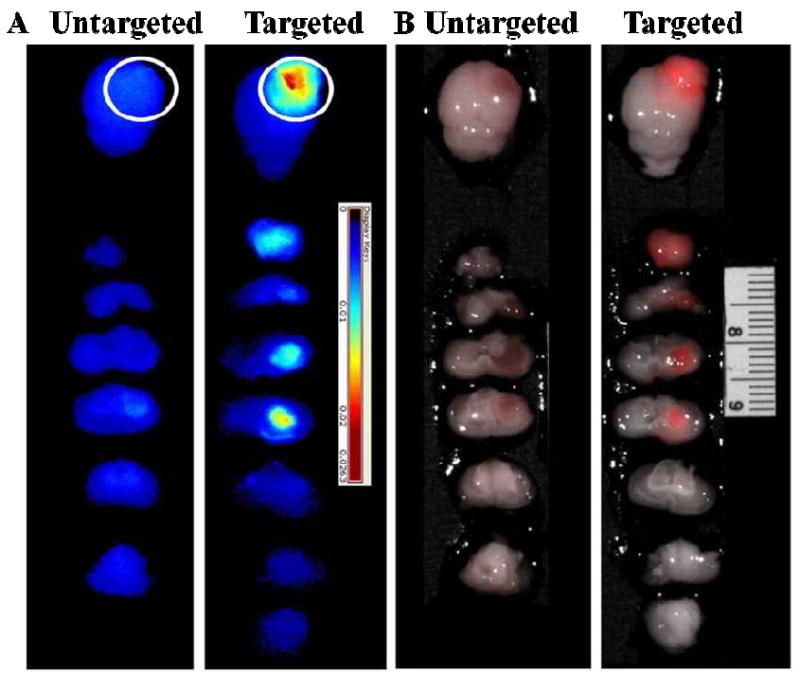
Targeting effect of EGF-Au NP-Pc 4 conjugates compared to untargeted Au NP-Pc 4 conjugates in brain tumor-bearing mice. Fluorescence imaging of extracted brains with tumors (A) and the overlay of fluorescence (shown in red) and monochromatic images (B). The white circle indicates the tumor position in the brain. Slice thickness is 2 mm.
In conclusion, we demonstrate that EGF targeting of Pc 4-loaded Au NPs to the cell surface receptor significantly improves their capacity to deliver drug cargo into brain tumors. These results are interesting and novel in two respects. First, our data suggest that increased drug delivery occurs with significant increases in the uptake of Pc 4 by targeted tissues. The increase in drug delivery occurs via a novel mechanism. Very few Au NPs can be found within targeted cells suggesting that the EGF-peptide receptor interaction likely allows for a prolonged interaction of the particles at the cell surfaces, allowing the hydrophobic Pc 4 to transfer to the cellular membrane. Untargeted Au NPs were 10-fold less effective at delivering the drug to cells in vivo. Second, when conjugated to EGF-targeted Au NPs, therapeutic levels can be realized within 4 hours after administration and the drug is internalized through early endosomal compartments. These studies demonstrate the rational and successful design of a non-covalent drug delivery system to brain tumors using targeted Au NPs.
Experimental Section
Au NPs were synthesized based on a modified Brust-Schiffrin method.[32-33] The untargeted Au NP-Pc 4 conjugates were synthesized as reported and coated with mPEG-SH (MW 5000, Laysan Bio). To synthesize the targeted conjugates, Au NPs were etched and shielded by the mixture of mPEG-SH (MW 5000) and HS-PEG-COOH (MW 3000, Rapp Polymere) with a 4:1 ratio for 48 hours. The carboxyl functionalized Au NPs were purified by centrifugation with 50,000 Dalton molecular weight cutoff filtration membranes. The EGF peptide was attached on the carboxyl functionalized NP surface through the amide bond. COOH- groups on the Au NPs were activated with EDC (1-ethyl-3-[3 dimethylaminopropyl]carbodiimide) and Sulfo-NHS (N-hydroxysulfosuccinimide) in MES (2-[morpholino]ethanesulfonic acid) buffer at pH 4.7 for 15 minutes at room temperature. Then the buffer pH was increased immediately above 7 using concentrated PBS (phosphatebuffered saline). The Sulfo-NHS-activated NPs were well mixed with the EGF peptide at 1:1 ratio for 4 hours at room temperature. Excess reactants were separated from the EGF peptide functionalized Au NPs (EGF Au NPs) by centrifugation with 50,000 Dalton molecular weight cutoff membranes. The purified EGF Au NPs were re-dissolved in chloroform and a 40 fold excess Pc 4 was added into the solution. After 48 hours mixing at room temperature, the solvent was removed under vacuum. The EGF Au NP-Pc 4 conjugates were suspended in aqueous solution and purified by centrifugation and 200 nm pore filters.
The average cell viability in the dark and under light exposure was evaluated by the MTT assay (Cell Proliferation Kit I by Roche). 9L.E29 rat glioma cancer cells, engineered to overexpress EGFR, were added (10,000 per well) in three 96-well plates and incubated for 24 hours at 37°C and 5% CO2. The EGF Au NP-Pc 4 conjugates and free Pc 4 were incubated with the cells in the dark for 4 hours. After three times washing, one plate was placed in the dark. The other two plates were irradiated under light of wavelength >550 nm at 0.5 J/cm2 and 1J/cm2, respectively. The plates were incubated for another 24 hours and 10μL yellow tetrazolium salt MTT labeling reagent was added into each well. After 4 hours incubation, the purple formazan crystals were formed by metabolic active cells. 100 μL of the solubilization solution per well was added to dissolve the cells and placed into the incubator overnight. The absorbance at 550 nm (the formazan salt) and 690 nm as reference wavelength was measured with a Tecan Infinite 200 microplate reader. 8 replicates were used for each condition.
Pc 4 in the cells was extracted with ethyl acetate and quantified with UV-vis spectroscopy. After digesting the cells with 70% HNO3, Au NPs were quantified with graphite furnace atomic absorption spectroscopy (GFAAS). And localization of Au NPs in cells was detected by Transmission Electron Microscopy (TEM). After 24 hours incubation with the conjugates, the cells were fixed in 2.5% glutaraldehyde for 1 hour after incubation with the conjugates. After washes with PBS, the samples were stained with 2% osmium tetroxide and 0.5% uranyl acetate. The samples were gradually dehydrated in ethanol and embedded in Epon-Propylene oxide. Thin sections were obtained with an ultramicrotome and deposited onto TEM grids. The images were taken on a JEOL JEM-1200 EX electron microscope.
Animal experiments were performed according to IACUC policies and guidelines of the animal care and use committee at Case Western Reserve University. Female athymic mice were obtained from the Athymic Animal Core Facility of the Cancer Research Center of Case Western Reserve University. Human glioma (Gli36Δ5) cancer cell lines were implanted on the right parietal lobe of the posterior of the brain using a stereotactic rig designed for mice. Animals were continuously monitored for any signs of discomfort, and tumors were allowed to grow up to 12 days prior to any systemic injection. Animals were fed exclusively on a special rodent diet (Tekland 2018S; Harlan Laboratories, Inc.) to reduce auto-fluorescence. Ex vivo brain images were obtained with the Maestro In Vivo Imaging System (Cambridge Research and Instrumentation, Inc., Woburn, MA), and tomographical fluorescent images were acquired using the VisEn Translational Fluorescence In Vivo Imaging System, the FMT 2500 (VisEn Medical, Inc., Bedford, MA). FMT 2500 analysis began by calibrating the system to the specific fluorescent profile of Pc 4 by imaging a known concentration and volume of Pc 4 in VisEn’s calibration probe to correlate intensity of fluorescence with actual concentration of Pc 4 (in pmol). Mice with tumors were anaesthetized with isoflurane and injected intravenously via the tail with either EGF Au NP-Pc 4 or Au NP-Pc 4 conjugates at a dosage of 1mg kg-1 of Pc 4 per total mouse body weight.
Supplementary Material
Acknowledgments
We thank Dr. Fujioka for helpful discussion. This work was supported by CWRU NFCR and CTSC, and NIH Grant No. 1R01EB012099-01.
Contributor Information
Yu Cheng, Department of Chemistry, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA).
Joseph D. Meyers, Center for Molecular Imaging, NFCR Center for Molecular Imaging at Case, Departments of Biomedical Engineering and Radiology, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA)
Dr. Richard S. Agnes, Center for Molecular Imaging, NFCR Center for Molecular Imaging at Case, Departments of Biomedical Engineering and Radiology, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA)
Tennyson L. Doane, Department of Chemistry, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA)
Prof. Dr. Malcolm E. Kenney, Department of Chemistry, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA)
Prof. Dr. Ann-Marie Broome, Prof. Dr., Email: axb110@case.edu, Center for Molecular Imaging, NFCR Center for Molecular Imaging at Case, Departments of Biomedical Engineering and Radiology, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA).
Prof. Dr. Clemens Burda, Email: burda@case.edu, Department of Chemistry, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA), Fax: (+1) 216-368-3006.
Prof. Dr. James P. Basilion, Email: jxb206@case.edu, Center for Molecular Imaging, NFCR Center for Molecular Imaging at Case, Departments of Biomedical Engineering and Radiology, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH, 44106 (USA).
References
- 1.Huse JT, Holland EC. Nat Rev Cancer. 2010;10:319. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 2.Jansen M, de Witt Hamer PC, Witmer AN, Troost D, van Noorden CJ. Brain Res Rev. 2004;45:143. doi: 10.1016/j.brainresrev.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Pardridge WM. Nat Rev Drug Discovery. 2002;1:131. doi: 10.1038/nrd725. [DOI] [PubMed] [Google Scholar]
- 4.Huynh GH, Deen DF, Jr, Szoka FC. J Controlled Release. 2006;110:236. doi: 10.1016/j.jconrel.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 5.Shi J, Votruba AR, Farokhzad OC, Langer R. Nano Lett. 2010;10:3223. doi: 10.1021/nl102184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding H, Inoue S, Ljubimov AV, Patil R, Portilla-Arias J, Hu J, Konda B, Wawrowsky KA, Fujita M, Karabalin N, Sasaki T, Black KL, Holler E, Ljubimova JY. Proc Natl Acad Sci U S A. 2010;107:18143. doi: 10.1073/pnas.1003919107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veiseh O, Sun C, Fang C, Bhattarai N, Gunn J, Kievit F, Du K, Pullar B, Lee D, Ellenbogen RG, Olson J, Zhang M. Cancer Res. 2009;69:6200. doi: 10.1158/0008-5472.CAN-09-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paciotti GF, Kingston DGI, Tamarkin L. Drug Dev Res. 2006;67:47. [Google Scholar]
- 9.Cheng Y, Samia AC, Meyers JD, Panagopoulos I, Fei BW, Burda C. J Am Chem Soc. 2008;130:10643. doi: 10.1021/ja801631c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh P, Han G, De M, Kim CK, Rotello VM. Adv Drug Deliv Rev. 2008;60:1307. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L. Drug Deliv. 2004;11:169. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 13.Kim CK, Ghosh P, Pagliuca C, Zhu Z, Menichetti S, Rotello VM. J Am Chem Soc. 2009;131:1360. doi: 10.1021/ja808137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi CHJ, Alabi CA, Webster P, Davis ME. Proc Natl Acad Sci U S A. 2010;107:1235. doi: 10.1073/pnas.0914140107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW. Nano Lett. 2009;9:1909. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 16.Matsumura Y, Maeda H. Cancer Res. 1986;12:6387. [PubMed] [Google Scholar]
- 17.Maeda H, Fang J, Inutsuka T, Kitamoto Y. Int Immunopharmacol. 2003;3:319. doi: 10.1016/S1567-5769(02)00271-0. [DOI] [PubMed] [Google Scholar]
- 18.Dougherty TJ. J Clin Laser Med Surg. 2002;20:3. doi: 10.1089/104454702753474931. [DOI] [PubMed] [Google Scholar]
- 19.Popovic E, Kaye A, Hill J. J Clin Laser Med Surg. 1996;14:251. doi: 10.1089/clm.1996.14.251. [DOI] [PubMed] [Google Scholar]
- 20.Stylli SS, Kaye AH, Macgregor L, Howes M, Rajendra P. J Clin Neurosci. 2005;12:389. doi: 10.1016/j.jocn.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Konan YN, Gurny R, Allemann E. J Photochem Photobiol B:Biol. 2002;66:89. doi: 10.1016/s1011-1344(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 22.Koo YEL, Reddy GR, Bhojani M, Schneider R, Philbert MA, Rehemtulla A, Ross BD, Kopelman R. Adv Drug Delivery Rev. 2006;58:1556. doi: 10.1016/j.addr.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Miller JD, Baron ED, Scull H, Hsia A, Berlin JC, McConnick T, Colussi V, Kenney ME, Cooper KD, Oleinick NL. Toxicol Appl Pharmacol. 2007;224:290. doi: 10.1016/j.taap.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salomon DS, Brandt R, Ciardiello F, Normanno N. Crit Rev Oncol Hemat. 1995;19:183. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 25.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Nat Biotechnol. 2007;25:1165. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Y, Samia AC, Li J, Kenney ME, Resnick A, Burda C. Langmuir. 2010;26:2248. doi: 10.1021/la902390d. [DOI] [PubMed] [Google Scholar]
- 27.Doane TL, Cheng Y, Babar A, Hill RJ, Burda C. J Am Chem Soc. 2010;132:15624. doi: 10.1021/ja1049093. [DOI] [PubMed] [Google Scholar]
- 28.Morris RL, Azizuddin K, Lam M, Berlin J, Nieminen AL, Kenney ME, Samia ACS, Burda C, Oleinick NL. Cancer Res. 2003;63:5194. [PubMed] [Google Scholar]
- 29.Li Z, Zhao R, Wu X, Sun Y, Yao M, Li J, Xu Y, Gu J. FASEB J. 2005;19:1978. doi: 10.1096/fj.05-4058com. [DOI] [PubMed] [Google Scholar]
- 30.Nativo P, Prior IA, Brust M. ACS Nano. 2008;2:1639. doi: 10.1021/nn800330a. [DOI] [PubMed] [Google Scholar]
- 31.Cheng Y, Meyers JD, Broome AM, Kenney ME, Basilion JP, Burda C. J Am Chem Soc. 2011;133:2583. doi: 10.1021/ja108846h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. J Chem Soc, Chem Comm. 1994;7:801. [Google Scholar]
- 33.Duan H, Nie S. J Am Chem Soc. 2007;129:2412. doi: 10.1021/ja067727t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.