

Synopsis
Malignant melanoma is the most aggressive form of skin cancer and its incidence has increased dramatically in the past two decades. Even with a high rate of success in the treatment of early stages of this malignancy, currently there are no effective strategies for the treatment of advanced metastatic melanoma. Much effort has been put into the use of different target-specific drugs amongst which BRAF kinase-specific small molecule inhibitors have rendered promising results as therapeutic agents in metastatic melanoma. Nonetheless, some side effects, such as development of squamous cell carcinoma, as well as tumor resistance and recurrence are common limitations of this therapeutic strategy. The use of combination treatments in which different regulatory pathways or the immunological response are targeted seems promising in the future of melanoma therapeutics.
Keywords: BRAF inhibitors, Combination therapy, MAP kinases, Melanoma, Tumorigenesis
INTRODUCTION
Melanoma is a major form of skin cancer and arises from malignant transformation of melanocytes, specialized black-pigmented cells producing melanins [1, 2]. In general, skin cancer is the most common cancer in the United States and melanoma, which accounts for about 5% of all the cancers of the skin, is the most aggressive form. Its incidence is increasing faster than that of any other cancer and has more than doubled in the past 20 years. The lifetime risk for melanoma is one in 68. The American Cancer Society predicted 68,130 new cases of melanoma and 8,700 consequential deaths in 2010 while no more than 0.1% of the more common non-melanoma skin cancers will result in death. Risk factors for melanoma include sun exposure, tanning salon use, pale skin, red-hair, inability to tan, tendency to sunburn, increased numbers of freckles and moles, and the presence of dysplastic nevi [2–4]. If detected and surgically removed at an early stage, the cure rate for melanoma is approximately 90%. However, there is no effective treatment for melanoma metastases, and patients with this metastatic disease have a short life expectancy with a 5-year survival rate of 11% and a median survival of 6 to 12 months [5]. The high propensity to metastasize in addition to the rapidly rising incidence and morbidity rate of melanoma underscores the urgency to better understand its pathogenesis and identify effective therapeutic targets and strategies.
BRAF KINASE: A POTENTIAL THERAPEUTIC TARGET IN MELANOMA
The RAS-RAF-MEK-ERK MAP kinases pathway has a central role in the biology of various cell types, including melanocytes, the cell type from which a melanoma arises. Stimulation of different membrane-bound receptors, mainly tyrosine kinases and G-protein coupled receptors, promotes activation of RAS and this activates RAF kinases (ARAF, BRAF and CRAF). Activated RAF then sequentially activates MEK and phosphorylates ERK, which in turn targets different cytoplasmic and nuclear molecules involved in proliferation, differentiation and cell survival [6–8]. Davies and coworkers [9] reported a high frequency of BRAF somatic missense mutation within the kinase domain in melanoma tumor cells. A single amino acid substitution, V600E, accounts for at least 80% of those BRAF mutations [9–11] and results in a protein with RAS-independent elevated kinase activity by mimicking BRAF phosphorylation in the activation segment [12]. This mutation also confers on BRAF the ability to transform cells, which are tumorigenic in vivo [9]. In addition to Val to Glu substitution, other mutations at this position such as, V600K and V600E have also been reported with variable frequency [11, 13, 14]. In addition to melanoma, mutations in BRAF are also quite frequent in thyroid (40–70 %) and colorectal cancers (5–20%) [9, 15, 16].
Melanoma cells harboring BRAF mutations depend on activated BRAF for their growth and maintenance. It has been shown that silencing BRAF activity by RNA interference blocks ERK activity and inhibits DNA synthesis causing reduced growth and increased apoptosis of melanoma cells in vitro [17–20]. Moreover, this siRNA-mediated block of BRAFV600E inhibits tumor development in xenograft models [20]. In addition, silencing of mutant BRAF inhibits melanoma cell extravasations in an in vitro flow migration model and the development of lung metastases in vivo [19].
The high frequency of BRAF mutations in melanoma as well as the critical role of BRAF in tumor proliferation, survival and malignancy suggested that BRAF is a potentially valuable molecular target and has lead to the development of BRAF kinase inhibitors for targeted therapy particularly in the treatment of metastatic melanoma.
PRECLINICAL STUDIES ON USING BRAF-SPECIFIC INHIBITORS IN MELANOMA
One of the first attempts targeting the serine-threonine protein kinase BRAF pathway as a therapeutic intervention in melanoma was the development of the small molecule multikinase inhibitor sorafenib which inhibits ERK activation, cell proliferation and induces apoptosis in cultured cells [18, 21]. This drug was originally designed as a C-RAF kinase inhibitor; however it was demonstrated that it also inhibits the B-RAF kinase as well as VEGFR-2, PDGFR-β and c-Kit receptor tyrosine kinases (RTK) among others [21, 22]. When tested in vitro in an extensive panel of melanoma cell lines, no correlation was observed between sensitivity to sorafenib and BRAF mutation status [23]. Besides, it has been unequivocally demonstrated that its antitumor effects are not due to specific inhibition of oncogenic BRAF [24], suggesting that the down regulation of the RAF/MEK/ERK pathway and the anti-tumoral effects are probably due to inhibition of various RTK targets or CRAF [21–23].
Based on the above-described high frequency of activating V600E mutations in the BRAF kinase and the so-called “BRAF addiction” in melanoma, different small molecule BRAF-specific inhibitors have been developed based on co-crystallography and chemical scaffolding technology which seems especially well-suited for kinase inhibitor design due to the conserved conformation of the kinase domain [25]. Among these small molecule BRAF-kinase specific inhibitors, PLX4720 and its homologue PLX4032 (also known as RG7204) as well as GDC-0879, GSK2118436 and AZ628 are specific inhibitors of BRAFV600E kinase activity at significantly lower concentrations than their inhibitory effect in wild-type (WT) BRAF [26–30]. Treatment of an extensive collection of melanoma cell lines with these BRAF inhibitors has shown a consistent inhibition of cell viability and cell growth with selectivity for the BRAFV600E mutant exceeding 100-fold over the WT BRAF, suggesting that these drugs have anti-melanoma activity only against cells that harbor BRAFV600E [26, 28–33]. Upon treatment with PLX4720, PLX4032 or GDC-0879, BRAFV600E mutant cells show a decrease in phosphorylation of ERK [29, 31, 34–36] and MEK [33, 36, 37] that indicates inactivation of the MAPK pathway [26, 28, 32]. The effect of GDC-0879 on global gene expression in A375 cells, particularly on those involved in cell proliferation, has been shown to be very similar to that observed with BRAF blockade by siRNA [29]. PLX4720/PLX4032 treated BRAF mutant melanoma cells undergo cell cycle arrest in G1 phase with a reduction in cyclin D1 expression and increase in p27 expression. These changes do not occur in WT BRAF or NRAS mutated melanoma cells [32, 35, 36], regardless of zygosity [37]. Furthermore, cells more sensitive to PLX4032 growth inhibitory effects are affected in a cytotoxic manner as demonstrated by an increase in apoptosis and cleavage of PARP after treatment with this drug [34–36]. Interestingly, PLX4032 treatment was shown to induce the expression of melanocyte-specific genes (TYR, TYRP1 and MITF among others) as well as genes associated with melanosome function in BRAF-mutated cell lines, such as RAB27A, MYO5 and RILP [37]. Therefore PLX4032 not only inhibits proliferation and survival but also may lead to resumed melanin production by counteracting the mutant BRAF-induced melanocytic differentiation arrest. Thus the inhibition of pERK may relieve the inhibition on melanogenesis and explain why differentiation markers specific to melanin production and transport are increased after treatment with PLX4032 [37].
Although small molecule BRAF-kinase specific inhibitors were promising as a therapeutic alternative for metastatic melanoma in BRAFV600E bearing melanoma cell lines, it was unexpectedly found that PLX4720, PLX4032 and GDC-0879 cause an increase in viable cell numbers in WT BRAF or NRAS mutant cell lines that is associated with a markedly increased MAPK pathway activation [28, 31, 34, 38, 39]. The molecular mechanisms involved in this increased activation of the MAPK pathway in WT BRAF or NRAS mutant melanoma cell lines remain to be fully elucidated. Nonetheless, it has been suggested that selective BRAF inhibition induces RAS-dependent dimerization of CRAF with WT BRAF, but not BRAFV600E, as well as the formation of CRAF homodimers, leading to subsequent activation of CRAF and downstream MEK-ERK signaling (Figure 1) [8, 28, 38, 39].
Figure 1. Tumor cells harboring BRAFV600E mutations and non-mutant BRAF cells respond differently to BRAF inhibitors.
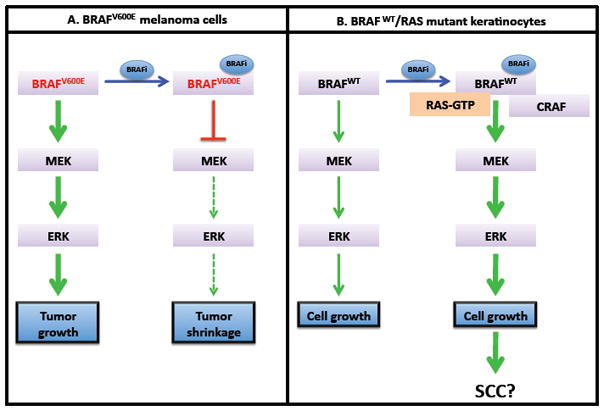
A. BRAFV600E promotes tumor growth through hyperactivation of the MEK-ERK pathway. Treatment with BRAF inhibitors (BRAFi) such as PLX4032 blocks this signaling pathway, leading to tumor shrinkage. B. When cells bearing BRAFWT are treated with BRAF inhibitors, a BRAF/CRAF heterodimerization induces activation of the MEK-ERK pathway. This results in increased cell proliferation, which may contribute to the development of squamous cell carcinoma (SCC).
While characterizing the biological responses associated to these completely opposed biochemical and proliferative responses to PLX compounds between BRAFV600E bearing cell lines and those with a WT gene, Halaban et al. [31], demonstrated that PLX4032 induces proliferation in WT BRAF melanoma cells treated after culture in suboptimal conditions. Furthermore, WT melanoma cells show a time-dependent enhanced detachment, reduced cell adhesion and enhanced motility after treatment with PLX4032 for up to 24h hours (with 99% of viability), effects that correlate with increased phosphorylation of focal adhesion kinase at the ERK phosphorylation site S910 [31]. This suggests that PLX4032 actually may confer an advantage for proliferation and enhanced metastatic capability to cells bearing non-mutated BRAF.
In different animal models of melanoma, mostly based on orthologous growth of human malignant melanoma cell lines in immunocompromised mice (xenografts), PLX4720, PLX4032 or GDC-0879 treatment have resulted in a significant reduction in tumor growth and in all cases this correlated with a high percentage of inhibition of ERK phosphorylation [26, 28, 29, 33]. At higher doses, PLX4720 induces regression of these grafted melanoma tumors. Conversely, WT BRAF bearing tumors are unaffected by the treatment or even exhibit accelerated tumor growth [26, 28]. Importantly, in these xenograft models using highly aggressive melanoma cells, the oral administration of a crystalline form of PLX4032 has proven to be of low efficacy while a microprecipitated-bulk powder (MBP), formulation that increases drug exposure, has a dose-dependent positive effect as an anti-tumor drug [36]. In addition, no toxic effects were seen in these animal models. Low doses induce regression but recurrence, an effect that was not seen when a higher dose was used, suggesting that the maximum tolerated dose in the clinical settings should be used to achieve the most beneficial result [36, 40].
THE CLINICAL TRIALS WITH BRAF-SPECIFIC INHIBITORS IN METASTATIC MELANOMA: THE PROMISE
Based on the successful results derived from preclinical studies using BRAF kinase specific inhibitors, several clinical trials have been carried out with these potential therapeutics. Among them, PLX4032 and GSK2118436 have shown strong promise in the early stages of clinical development [30, 41–45].
Despite the very similar preclinical results between PLX4032 and its analogue PLX4720, the former has been chosen due to more favorable pharmacokinetics properties [40]. In agreement with the preclinical data [36], in a dose-escalation study using an MBP formulation, PLX4032 proved to be more effective and had a much higher bioavailability than the original formulation [40, 43, 45]. About 69% and 81% of the patients with BRAFV600E bearing melanomas treated with PLX4032 showed at least partial objective responses (based on RECIST, Response Evaluation Criteria in Solid Tumors) in the Phase I dose-escalation and extension phases, respectively, with latency in a range between 2 to more than 18 months. The estimated median progression-free survival among these patients is estimated to be at least 7 months [43, 45]. In agreement with the preclinical studies, this positive response correlates with inhibition of the MAPK kinase pathway that induces a decrease in cyclin D1 levels and ultimately decreased proliferative responses within the tumors. Moreover, since the inhibition of cytoplasmic pERK is greater than 80% in patients with tumor regression, it seems that near-complete inhibition of ERK signaling may be needed to achieve a significant anti-tumor response [40, 46]. Together, these in vitro and in vivo mechanistic data predict that BRAF kinase inhibitors will not inhibit ERK signaling in normal cells and therefore the toxicity associated to MAPK inhibition would be low. Thus, it seems possible to reach greater clinical efficacy based on the possibility of the use of doses that are high enough to reach more complete ERK signaling inhibition with lower toxic effects [34]. Furthermore, these studies demonstrated again the high specificity of PLX4032 since patients with metastatic melanoma without BRAF mutations did not respond to the treatment, and even had progression of this disease during the treatment [40, 43, 45]. Currently, a phase III clinical study of PLX4032 is ongoing to assess the overall survival (OS) and progression-free survival (PFS) in melanoma patients with BRAF mutations, as compared to patients treated with the current standard of care.
GSK2118436 is another BRAF inhibitor in active clinical development. Recent data from a phase I clinical trial of GSK2118436 revealed a > 20% tumor decrease by RECIST at 8 to 9 weeks in about 60% of patients with BRAFV600E melanomas [30, 41]. Preliminary data from an ongoing phase I/II trial of this drug suggests that GSK2118436 may be effective against brain metastases in patients with tumors bearing mutant BRAF, rendering a promising therapy for metastatic melanoma patients [42].
Despite the positive responses and therefore encouraging results of the treatment with PLX4032, some adverse effects, which were proportional to the dose and exposure to the drug, were observed at high doses [43, 45]. The most common being arthralgia, rash, nausea, phosphosensitivity, fatigue, pruritus and palmo-plantar dysesthesia. Similar adverse effects were also commonly seen during the clinical trial with GSK2118436 [41]. Nevertheless, none of these adverse effects prompted the discontinuation of treatment [43, 45]. Importantly, 31% of the patients treated with PLX4032 at higher yet well-tolerated doses, developed cutaneous squamous cell carcinoma (SCC) mainly of the keratoacanthoma type within a median time of 8 weeks of treatment initiation [43, 45, 47]. The molecular mechanisms underlying the development of SCC in these PLX4032-treated melanoma patients are under active investigations. It has been speculated that the paradoxical activation of RAF-MEK-ERK signaling pathway by PLX4032 in the WT BRAF cells might be involved [15, 43]. Activating RAS mutations and hyperactivation of the ERK signaling pathway are known to play a very important role in the initiation of SCC [48–50]. As discussed above, when WT BRAF cells bearing activating RAS mutations are treated with BRAF-specific inhibitors, a BRAF/CRAF heterodimerization induces activation of the MAPK pathway via CRAF [8, 28, 38, 39] (Figure 1). This could result in an increased proliferative response in the epidermis leading to the development of SCC. Therefore it seems possible that some pre-existing oncogenic mutations in keratinocytes of the skin, such as those in RAS proteins, can potentiate the effect of BRAF inhibitors leading to SCC development [15, 51]. Cutaneous SCC as a result of melanoma therapy with BRAF inhibitors are considered innocuous and are surgically removable. They are confined to the skin, mainly in areas that are subjected to sun exposure, and no metastatic evolution has been reported [15, 43]. However, the potential for SCC development in other locations or perhaps other cancer types during long-term treatment must be considered. Therefore, it is necessary to further investigate this toxicity to fully understand the mechanisms involved in its development and ultimately to identify therapeutic strategies to prevent it.
ACQUIRED AND INTRINSIC DRUG RESISTANCE TO BRAF INHIBITIORS: A MAJOR DRAWBACK
As discussed, a high percent of patients with melanomas carrying activating BRAFV600E mutations treated with PLX4032, currently the most promising BRAF kinase inhibitor under clinical trials, respond to treatment at least partially at all sites of metastasis with as yet unknown durability of the response [40, 45]. Even when these clinical results are very encouraging, one must remember the high specificity of PLX4032 and other BRAF inhibitors for BRAFV600E bearing tumors. In addition to WT BRAF bearing tumors, there is a fraction (around 20%) of patients with BRAF mutated melanoma tumors that are not responsive at all due to an intrinsic resistance, and another population in which tumors reappear due to the generation of acquired resistance during the course of treatment [35, 36, 45]. The mechanisms for intrinsic and acquired resistance to BRAF inhibitors therapy are currently under intense investigation.
In order to understand the intrinsic resistance to BRAF-specific inhibitors in mutant BRAF cells and tumors, one must consider that tumor cells are heterogeneous entities and particular attention should be paid to the fact that there may be different genetic alterations in cell proliferation pathways that can bypass BRAF inhibition. Genomic alterations in the PI3K/Akt pathway, including deletions in PTEN and increase of Akt3 activity have been described in resistant cell lines and tumors [35, 52, 53]. Moreover, it has been proposed that overexpression of cyclin D1, probably due to CCND1 amplification, in combination with activating mutations in CDK4 may contribute to resistance to BRAF–specific inhibitors in BRAFV600E bearing melanoma cells [54].
Acquired resistance to BRAF-specific inhibitors is one of the greatest challenges for these targeted therapies. Emerging evidence point toward the reactivation of the RAF-MEK-ERK pathway in an “oncogene bypass” manner as a major mechanism, with no secondary BRAF mutations involved [32, 55–57]. However, it is likely that there are a number of mechanisms involved in the recovery of the MEK-ERK activation during the development of resistance to BRAF inhibitors (Figure 2). Amongst these mechanisms, it has been shown that in some PLX4032-resistant melanoma cell lines as well as in cells derived from a subset of resistant tumors from treated patients which resist PLX4032-induced downregulation of the MEK-ERK pathway, this resistance is dependent on activating NRAS mutations [56]. On the other hand, resistance to BRAF inhibitors may be associated with elevated ARAF and CRAF protein levels and pERK activity in some cases. When this occurs, the BRAF dependence is lost and tumor cells switch their oncogenic addiction to another RAF kinase [8, 58, 59]. This is indeed in line with the hypothesis that cells only require one active RAF isoform to activate the MAPK pathway [59]. Lastly, acquired gain-of-function mutations in MEK1 have been reported as a result of the treatment with AZD6244 and also confer cross-resistance to PLX4720 [60].
Figure 2. Targeting the BRAF Signaling pathway in melanoma.
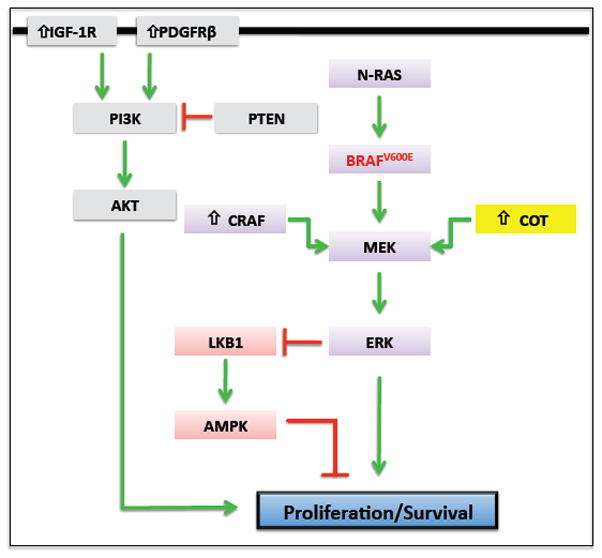
BRAF-specific inhibitors block the RAS-RAF-MEK-ERK signaling pathway in BRAFV600E mutant cells. However, tumor cells acquire resistance to this therapy during the course of treatment. Several mechanisms that involve reactivation of ERK signaling through amplification or mutation of proteins in the RAS-RAF-MEK-ERK pathway (i.e. NRAS, BRAF, CRAF and COT1) have been recently described for the development of acquired resistance. In addition, hyper-activation of the PI3K-Akt signaling pathway (i.e. through upregulation of IGF-1R or PDGFRβ) can also be involved in resistance by promoting survival or proliferation. BRAFV600E suppresses the LKB1 tumor suppressor and AMPK through the BRAF-MEK-ERK signaling cascade, and this regulation is critical for melanoma cell proliferation.
Negative regulators of the RAF-MEK-ERK pathways might be also involved in the development of acquired resistance to BRAF inhibitors. For example, the genetic signature in several cell lines showing acquired resistance revealed inhibition of expression of the MAPK phosphatases (DUSP) 4, 5 and 6 and Sprouty proteins (SPRY) 2 and 4 [37]. These proteins are important negative regulators of the RAS/RAF signaling pathway. Thus MAPK activity in melanoma cell lines that are resistant to BRAF inhibitors may be insensitive to the physiologic negative feedback inhibition provided by DUSP and SPRY [37, 61].
More recently, yet another potential mechanism to reactivate the RAF-MEK-ERK pathway for acquired resistance to BRAF inhibitors in melanoma cell lines has been described. This mechanism involves COT protein kinase (also known as MAP3K8 or Tpl2) [62], which is a MAPK agonist that activates ERK in a MEK-dependent, but RAF-independent manner [63] (Figure 2). Therefore it is possible that BRAF inhibition potentiates the outgrowth of COT–expressing cells during the course of treatment [62]. In fact, there is a correlation between COT expression and acquired resistance to BRAF inhibitors in tissue from patients with relapsing tumors [62].
In addition to re-activation of RAF-MEK-ERK signaling pathway, it is likely that resistance acquisition is dependent on other signaling pathways that are involved in the regulation of cancer cell proliferation and survival. It has been proposed that acquired resistance to BRAF-specific inhibitors may be partially associated with the activation of the PI3K-PTEN-Akt pathway [37] (Figure 2). In this regard, it was shown that mutations in PTEN can affect responses to BRAF inhibitors. Elevated levels of Akt phosphorylation due to loss of PTEN are seen in melanoma cells resistant to PLX4720 whereas inhibition of class I PI3K enhances responses to BRAF inhibitors [37, 52, 59, 64]. Moreover, Receptor Tyrosine Kinase (RTK)-mediated activation of alternative survival pathways has been described as another important mechanism of acquired resistance to BRAF inhibitors [56, 59]. In this regard, it was shown that resistance to the BRAF inhibitor PLX4032, in cell lines and patient-derived tumor cells that do not exhibit reactivation of the RAF-MEK-ERK pathway, can instead be acquired through up-regulation of PDGFRβ [56]. In fact, induction of PDGFRβ RNA, as well as protein and tyrosine phosphorylation with no reactivation of the MAPK pathway is a dominant feature of these resistant cells [56]. In a different study examining the levels of various RTKs in BRAF inhibitor-resistant melanoma cells, it was described that these cells up-regulate IGF-1R surface expression and phosphorylation at the posttranscriptional level. Interestingly, pharmacological inhibition of IGF-1R abrogated viability in these melanoma cells that were resistant to BRAF inhibitors. Furthermore, persistent IGF-1R signaling induces PI3K/Akt activation in these resistant cells whereas treatment with an IGF1R inhibitor blocked Akt phosphorylation with no inhibition of ERK [59]. Of note, it was found that increased expression on IGF-1R and pAkt correlated with resistance to BRAF inhibitors in one of five tissue samples from relapsed patients [52, 59]. Finally, as mentioned before, activating mutations in RAS may be at least partially responsible for resistance to BRAF inhibitors in melanoma cells [56]. However, the RAS-dependent reactivation of the MAPK pathway [56] may not be the only mechanism of resistance. Actually, RAS also signals through activation of PI3K [51, 65], an event that can be responsible for acquired resistance to BRAF specific inhibitors [51, 52, 59, 64].
RATIONAL COMBINATORIAL THERAPY: THE FUTURE
Target-specific therapeutics for cancer represents a very useful weapon against different forms of malignancy. As described throughout this review, both preclinical and clinical studies targeting mutated BRAF have rendered encouraging information toward the future of melanoma treatment, especially the metastatic form for which there is no current effective therapeutic strategy. However, the toxicities associated with BRAF inhibitors therapy, such as the appearance of cutaneous SCC and, both intrinsic and secondary to exposure resistance, which seem likely to be mechanistically related, need to be overcome. Increased proliferative responses in WT BRAF melanoma and non-melanoma cells as well as resistance to the inhibitory effects of BRAF inhibitors are likely to be a multifactorial process in which different biochemical pathways may be involved. Rational combination strategies that target oncogenic pathways along with BRAF have been proposed to overcome limitations associated with BRAF inhibitor single agent therapy [5, 43]. For example, when acquired resistance to PLX4032 is at least partially dependent on restoration of the MAPK signaling, treatment with the MEK inhibitor U0126 together with PLX4032 will not only counteract this resistance but also prevents the development of resistance [29, 32, 61, 66]. In fact, a phase I clinical trial combining the BRAF inhibitor GSK2118436 and the MEK inhibitor GSK1120212 is underway (http://clinicaltrials.gov/ct2/show/NTC01072175). Similarly, because of the potential involvement of the PI3K-PTEN-AKT pathway in developing acquired resistance, it has been suggested that the PI3K-PTEN-AKT pathway could be one of the additional targets to be included in combination therapies [29, 31, 64]. It has been reported that simultaneous MEK and PI3K inhibition leads to cytotoxicity in certain melanoma cells that are resistant to BRAF inhibitors [59], providing a rational basis for this combination therapeutic strategy.
We have recently discovered an intriguing molecular linkage between BRAF and the LKB1-AMPK (AMP-activated protein kinase) signaling pathway [67]. We found that BRAFV600E mutant suppresses LKB1 and AMPK through the BRAF-MEK-ERK signaling cascade, and that this regulation is critical for melanoma cell proliferation and anchorage-independent growth. The tumor suppressor LKB1 is a serine-threonine protein kinase mutated in Peutz-Jeghers syndrome and several sporadic cancers, including melanoma [68]. Its downstream kinase AMPK is an evolutionarily conserved energy sensor that regulates energy homeostasis by monitoring changes in the intracellular AMP and ATP concentrations [69]. Recent studies have shown that the LKB1-AMPK signaling pathway plays an important role in suppressing cell growth, proliferation and survival under energy stress [69]. These studies also raise interesting possibilities for pharmaceutical intervention to suppress tumor growth through activation of this pathway. Drugs that activate the LKB1-AMPK pathway, such as metformin and its analog phenformin, are being used clinically to treat type II diabetes and could be quickly adapted for cancer treatment. Indeed, recent preclinical studies have demonstrated the anti-tumor activities of metformin and phenformin, and metformin is being evaluated for the treatment of breast and prostate cancers as a single agent in several clinical trials [70–72]. The cross-talk between the LKB1-AMPK and BRAF signaling pathways suggests that targeting the LKB1-AMPK pathway with metformin or other AMPK activators together with BRAF inhibitor could be another rational combinatorial strategy for melanoma therapy (Figure 2).
Finally, regulation of the immunological responses as a part of the anti-melanoma therapy is an attractive possibility [73]. In fact, activating BRAFV600E mutations increase inflammatory responses that ultimately contribute to an increased metastatic potential [19]. In addition, there is evidence of increased expression of melanocyte differentiation antigens upon BRAF inhibition, therefore increasing immune recognition and elimination of tumors [74]. Furthermore, it has been demonstrated that the BRAF inhibitor PLX4032 does not have negative effects in the viability or function of T cells and that peripheral blood mononuclear cells activated with anti-CD3/IL-2 are highly resistant to this inhibitor [75]. Thus it is possible that inhibition of BRAF signaling may increase the efficacy of immunotherapy in melanoma. Taken together, combination therapy with BRAF-specific inhibitors and drugs targeting other oncogenic pathways or with immunotherapy may be a great opportunity for the success of the next generation therapeutics of melanoma.
Acknowledgments
Work in the Zheng laboratory is supported by NIH grant R00-CA133245 to B.Z. We thank Ms. Kelly Hogan and Drs. Julide Celebi and Lauren Mordasky Markell for critical reading of the manuscript.
Abbreviations used
- AMPK
AMP-activated protein kinase
- DUSP
dual-specificity protein phosphatase
- ERK
extracellular-signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- MBP
microprecipitated-bulk powder
- MEK (MKK)
MAPK kinase
- RECIST
Response Evaluation Criteria in Solid Tumors
- RTK
receptor tyrosine kinase
- SCC
squamous cell carcinoma
References
- 1.Marks JG, Miller JJ. Lookingbill & Marks’ Principles of dermatology. Elsevier; Philadelphia: 2006. [Google Scholar]
- 2.Williams ML, Sagebiel RW. Melanoma risk factors and atypical moles. West J Med. 1994;160:343–350. [PMC free article] [PubMed] [Google Scholar]
- 3.Bandarchi B, Ma L, Navab R, Seth A, Rasty G. From Melanocyte to Metastatic Malignant Melanoma. Dermatol Res Pract. 2010;2010:1–9. doi: 10.1155/2010/583748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siskind V, Hughes MCB, Palmer JM, Symmons JM, Aitken JF, Martin NG, Hayward NK, Whiteman DC. Nevi, Family History, and Fair Skin Increase the Risk of Second Primary Melanoma. J Invest Dermatol. 2010;131:461–467. doi: 10.1038/jid.2010.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hersey P, Bastholt L, Chiarion-Sileni V, Cinat G, Dummer R, Eggermont AMM, Espinosa E, Hauschild A, Quirt I, Robert C, Schadendorf D. Small molecules and targeted therapies in distant metastatic disease. Ann Oncol. 2009;20:vi35–vi40. doi: 10.1093/annonc/mdp254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wellbrock C, Hurlstone A. BRAF as therapeutic target in melanoma. Biochem Pharmacol. 2010;80:561–567. doi: 10.1016/j.bcp.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 7.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 8.Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-Type and Mutant B-RAF Activate C-RAF through Distinct Mechanisms Involving Heterodimerization. Mol Cell. 2005;20:963–969. doi: 10.1016/j.molcel.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 9.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 10.Uribe P, Wistuba II, Gonzalez S. BRAF mutation: a frequent event in benign, atypical, and malignant melanocytic lesions of the skin. Am J Dermatopathol. 2003;25:365–370. doi: 10.1097/00000372-200310000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Willmorepayne C, Holden J, Tripp S, Layfield L. Human malignant melanoma: detection of BRAF- and c-kit–activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005;36:486–493. doi: 10.1016/j.humpath.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 12.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 13.Poynter JN, Elder JT, Fullen DR, Nair RP, Soengas MS, Johnson TM, Redman B, Thomas NE, Gruber SB. BRAF and NRAS mutations in melanoma and melanocytic nevi. Melanoma Res. 2006;16:267–273. doi: 10.1097/01.cmr.0000222600.73179.f3. [DOI] [PubMed] [Google Scholar]
- 14.Rubinstein JC, Sznol M, Pavlick AC, Ariyan S, Cheng E, Bacchiocchi A, Kluger HM, Narayan D, Halaban R. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med. 2010;8:67. doi: 10.1186/1479-5876-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert C, Arnault JP, Mateus C. RAF inhibition and induction of cutaneous squamous cell carcinoma. Curr Opin Oncol. 2011;23:177–182. doi: 10.1097/CCO.0b013e3283436e8c. [DOI] [PubMed] [Google Scholar]
- 16.Spittle C, Ward MR, Nathanson KL, Gimotty PA, Rappaport E, Brose MS, Medina A, Letrero R, Herlyn M, Edwards RH. Application of a BRAF Pyrosequencing Assay for Mutation Detection and Copy Number Analysis in Malignant Melanoma. J Mol Diagn. 2007;9:464–471. doi: 10.2353/jmoldx.2007.060191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garnett MJ, Marais R. Guilty as chargedB-RAF is a human oncogene. Cancer Cell. 2004;6:313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 18.Karasarides M, Chiloeches A, Hayward R, Niculescu-Duvaz D, Scanlon I, Friedlos F, Ogilvie L, Hedley D, Martin J, Marshall CJ, Springer CJ, Marais R. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;23:6292–6298. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- 19.Liang S, Sharma A, Peng HH, Robertson G, Dong C. Targeting Mutant (V600E) B-Raf in Melanoma Interrupts Immunoediting of Leukocyte Functions and Melanoma Extravasation. Cancer Res. 2007;67:5814–5820. doi: 10.1158/0008-5472.CAN-06-4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma A. Mutant V599EB-Raf Regulates Growth and Vascular Development of Malignant Melanoma Tumors. Cancer Res. 2005;65:2412–2421. doi: 10.1158/0008-5472.CAN-04-2423. [DOI] [PubMed] [Google Scholar]
- 21.Panka DJ. The Raf Inhibitor BAY 43-9006 (Sorafenib) Induces Caspase-Independent Apoptosis in Melanoma Cells. Cancer Res. 2006;66:1611–1619. doi: 10.1158/0008-5472.CAN-05-0808. [DOI] [PubMed] [Google Scholar]
- 22.Wilhelm SM. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 23.Augustine CK, Toshimitsu H, Jung SH, Zipfel PA, Yoo JS, Yoshimoto Y, Selim MA, Burchette J, Beasley GM, McMahon N, Padussis J, Pruitt SK, Ali-Osman F, Tyler DS. Sorafenib, a multikinase inhibitor, enhances the response of melanoma to regional chemotherapy. Mol Cancer Ther. 2010;9:2090–2101. doi: 10.1158/1535-7163.MCT-10-0073. [DOI] [PubMed] [Google Scholar]
- 24.Whittaker S, Kirk R, Hayward R, Zambon A, Viros A, Cantarino N, Affolter A, Nourry A, Niculescu-Duvaz D, Springer C, Marais R. Gatekeeper Mutations Mediate Resistance to BRAF-Targeted Therapies. Sci Transl Med. 2010;2:35ra41–35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- 25.Zhang C, Bollag G. Scaffold-based design of kinase inhibitors for cancer therapy. Curr Opin Genet Dev. 2010;20:79–86. doi: 10.1016/j.gde.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 26.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KSM, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KYJ, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci, U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen JD, Grina J, Newhouse B, Welch M, Topalov G, Littman N, Callejo M, Gloor S, Martinson M, Laird E. Potent and selective pyrazole-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett. 2008;18:4692–4695. doi: 10.1016/j.bmcl.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 29.Hoeflich KP, Herter S, Tien J, Wong L, Berry L, Chan J, O’Brien C, Modrusan Z, Seshagiri S, Lackner M, Stern H, Choo E, Murray L, Friedman LS, Belvin M. Antitumor Efficacy of the Novel RAF Inhibitor GDC-0879 Is Predicted by BRAFV600E Mutational Status and Sustained Extracellular Signal-Regulated Kinase/Mitogen-Activated Protein Kinase Pathway Suppression. Cancer Res. 2009;69:3042–3051. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- 30.Kefford R, Long G, Arkenau H-T, Brown MP, Millward M, O’Day S, Infante J, Pavlick A, Ouellet D, Curtis M, Ma B, Erhardt J, Lebowitz P, Kurzrock R, Falchook G. Selective inhibition of oncogenic BRAF V600E/K/D by GSK2118436: Evidence of clinical activity in subjects with metastatic melanoma. Pigmen Cell Melanoma Res; Proceedings of the 7th international melanoma research congress; 2010. p. abtr 100. [Google Scholar]
- 31.Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, Krauthammer M, McCusker JP, Kluger Y, Sznol M. PLX4032, a Selective BRAFV600EKinase Inhibitor, Activates the ERK Pathway and Enhances Cell Migration and Proliferation of BRAFWTMelanoma Cells. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paraiso KHT, Fedorenko IV, Cantini LP, Munko AC, Hall M, Sondak VK, Messina JL, Flaherty KT, Smalley KSM. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724–1730. doi: 10.1038/sj.bjc.6605714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong H, Belvin M, Herter S, Hoeflich KP, Murray LJ, Wong L, Choo EF. Pharmacodynamics of 2- ethan-1-ol (GDC-0879), a Potent and Selective B-Raf Kinase Inhibitor: Understanding Relationships between Systemic Concentrations, Phosphorylated Mitogen-Activated Protein Kinase Kinase 1 Inhibition, and Efficacy. J Pharmacol Exp Ther. 2009;329:360–367. doi: 10.1124/jpet.108.148189. [DOI] [PubMed] [Google Scholar]
- 34.Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, Halilovic E, Persaud Y, Xing F, Viale A, Tsai J, Chapman PB, Bollag G, Solit DB, Rosen N. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci, U S A. 2010;107:14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Søndergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, Sazegar H, MacConaill LE, Barretina JG, Kehoe SM, Attar N, von Euw E, Zuckerman JE, Chmielowski B, Comin-Anduix B, Koya RC, Mischel PS, Lo RS, Ribas A. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, Kolis S, Zhao S, Lee R, Grippo JF, Schostack K, Simcox ME, Heimbrook D, Bollag G, Su F. RG7204 (PLX4032), a Selective BRAFV600E Inhibitor, Displays Potent Antitumor Activity in Preclinical Melanoma Models. Cancer Res. 2010;70:5518–5527. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 37.Tap WD, Gong KW, Dering J, Tseng Y, Ginther C, Pauletti G, Glaspy JA, Essner R, Bollag G, Hirth P, Zhang C, Slamon DJ. Pharmacodynamic characterization of the efficacy signals due to selective BRAF inhibition with PLX4032 in malignant melanoma. Neoplasia. 2010;12:637–649. doi: 10.1593/neo.10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KYJ, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D’Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu X, Nathanson KL, Nolop K. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kefford R, Arkenau H, Brown MP, Millward M, Infante JR, Long GV, Ouellet D, Curtis M, Lebowitz PF, Falchook GS. Phase I/II study of GSK2118436, a selective inhibitor of oncogenic mutnat BRAF kinase, in patients with metastatic melanoma and other solid tumors. J Clin Oncol. 2010;28:abstr 8503. [Google Scholar]
- 42.Long GV, Kefford RF, Carr PJA, Brown MP, Curtis M, Ma B, Lebowitz P, Kim KB, Kurzrock R, Flachook G. Phase 1/2 study of GSK2118436, a selective inhibitor of V600 mutant (mut) BRAF kinase: evidence of activity in melanoma brain metastases (mets) Ann Oncol. 2010;21:viii12. [Google Scholar]
- 43.Flaherty KT, McArthur G. BRAF, a target in melanoma: implications for solid tumor drug development. Cancer. 2010;116:4902–4913. doi: 10.1002/cncr.25261. [DOI] [PubMed] [Google Scholar]
- 44.Sosman J, Kim K, Schuchter L, Gonzalez R, Pavlick A, Weber J, McArthur G, Hutson T, Lawrence D, Moschos S, Flaherty K, Hersey P, Kefford R, Chmielowski B, Amaravadi R, Puzanov I, Li J, Bhattacharya S, Nolop K, Lee R, Joe A, Riba A. An open-label, multicenter Phase II study of continuous oral dosing of RG7204 (PLX4032) in previously treated patients with BRAF V600E mutation-positive metastatic melanoma. Proceedings of the 7th international melanoma research congress. Pigmen Cell Melanoma Res. 2010;23:abstr 101. [Google Scholar]
- 45.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puzanov I, Nathanson KL, Chapman PB, Xu X, Sosman JA, McArthur GA, Ribas A, Kim KB, Grippo JF, Flaherty KT. PLX4032, a highly selective V600EBRAF kinase inhibitor: Clinical correlation of activity with pharmacokinetic and pharmacodynamic parameters in a phase I trial. J Clin Oncol. 2009;27:abstr 9021. [Google Scholar]
- 47.Lacouture ME, McArthur GA, Chapman PB, Ribas A, Flaherty KT, Lee RJ, Nolop KB, Kim KB, Duvic M, Sosman JA, group Dw. PLX4032 (RG7204), a selective mutant RAF inhibitor: Clinical and histologic characterisitics of therapy-associated cutaneous neoplasms in a phase I trial. J Clin Oncol. 2010;28:abstr 8592. [Google Scholar]
- 48.Bourcier C. p44 Mitogen-Activated Protein Kinase (Extracellular Signal-Regulated Kinase 1)-Dependent Signaling Contributes to Epithelial Skin Carcinogenesis. Cancer Res. 2006;66:2700–2707. doi: 10.1158/0008-5472.CAN-05-3129. [DOI] [PubMed] [Google Scholar]
- 49.Warmka JK. Mitogen-activated Protein Kinase Phosphatase-3 Is a Tumor Promoter Target in Initiated Cells That Express Oncogenic Ras. J Biol Chem. 2004;279:33085–33092. doi: 10.1074/jbc.M403120200. [DOI] [PubMed] [Google Scholar]
- 50.Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26:1657–1667. doi: 10.1093/carcin/bgi123. [DOI] [PubMed] [Google Scholar]
- 51.Arkenau HT, Kefford R, Long GV. Targeting BRAF for patients with melanoma. Br J Cancer. 2011;104:392–398. doi: 10.1038/sj.bjc.6606030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shao Y, Aplin AE. Akt3-Mediated Resistance to Apoptosis in B-RAF-Targeted Melanoma Cells. Cancer Res. 2010;70:6670–6681. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paraiso KHT, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, Wood E, Fedorenko IV, Sondak VK, Anderson ARA, Ribas A, Dalla Palma M, Nathanson KL, Koomen JM, Messina JL, Smalley KSM. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smalley KSM, Lioni M, Palma MD, Xiao M, Desai B, Egyhazi S, Hansson J, Wu H, King AJ, Van Belle P, Elder DE, Flaherty KT, Herlyn M, Nathanson KL. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7:2876–2883. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Solit D, Sawyers CL. Drug discovery: How melanomas bypass new therapy. Nature. 2010;468:902–903. doi: 10.1038/468902a. [DOI] [PubMed] [Google Scholar]
- 56.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poulikakos PI, Rosen N. Mutant BRAF melanomas--dependence and resistance. Cancer Cell. 2011;19:11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 58.Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J. Elevated CRAF as a Potential Mechanism of Acquired Resistance to BRAF Inhibition in Melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D. Acquired Resistance to BRAF Inhibitors Mediated by a RAF Kinase Switch in Melanoma Can Be Overcome by Cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, MacConaill LE, Zhang J, Gray NS, Sellers WR, Dummer R, Garraway LA. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci, USA. 2009;106:20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pratilas CA, Solit DB. Targeting the Mitogen-Activated Protein Kinase Pathway: Physiological Feedback and Drug Response. Clin Cancer Res. 2010;16:3329–3334. doi: 10.1158/1078-0432.CCR-09-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, Salehi-Ashtiani K, Hill DE, Vidal M, Zhao JJ, Yang X, Alkan O, Kim S, Harris JL, Wilson CJ, Myer VE, Finan PM, Root DE, Roberts TM, Golub T, Flaherty KT, Dummer R, Weber BL, Sellers WR, Schlegel R, Wargo JA, Hahn WC, Garraway LA. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hagemann D, Troppmair J, Rapp UR. Cot protooncoprotein activates the dual specificity kinases MEK-1 and SEK-1 and induces differentiation of PC12 cells. Oncogene. 1999;18:1391–1400. doi: 10.1038/sj.onc.1202431. [DOI] [PubMed] [Google Scholar]
- 64.Mordant P, Loriot Y, Leteur C, Calderaro J, Bourhis J, Wislez M, Soria JC, Deutsch E. Dependence on Phosphoinositide 3-Kinase and RAS-RAF Pathways Drive the Activity of RAF265, a Novel RAF/VEGFR2 Inhibitor, and RAD001 (Everolimus) in Combination. Mol Cancer Ther. 2010;9:358–368. doi: 10.1158/1535-7163.MCT-09-1014. [DOI] [PubMed] [Google Scholar]
- 65.Vakiani E, Solit DB. KRAS and BRAF: drug targets and predictive biomarkers. J Pathol. 2011;223:219–229. doi: 10.1002/path.2796. [DOI] [PubMed] [Google Scholar]
- 66.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. V600EBRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci, U S A. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC. Oncogenic B-RAF Negatively Regulates the Tumor Suppressor LKB1 to Promote Melanoma Cell Proliferation. Mol Cell. 2009;33:237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 69.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 70.Gonzalez-Angulo AM, Meric-Bernstam F. Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res. 2010;16:1695–1700. doi: 10.1158/1078-0432.CCR-09-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9:1092–1099. doi: 10.1158/1535-7163.MCT-09-1186. [DOI] [PubMed] [Google Scholar]
- 72.Jalving M, Gietema JA, Lefrandt JD, de Jong S, Reyners AK, Gans RO, de Vries EG. Metformin: taking away the candy for cancer? Eur J Cancer. 2010;46:2369–2380. doi: 10.1016/j.ejca.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 73.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CNJ, Sloss CM, Ferrone CR, Flaherty KT, Lawrence DP, Fisher DE, Tsao H, Wargo JA. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma without Affecting Lymphocyte Function. Cancer Res. 2010;70:5213–5219. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 75.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, Escuin-Ordinas H, Chmielowski B, Koya RC, Ribas A. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16:6040–6048. doi: 10.1158/1078-0432.CCR-10-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]