

Abstract
Introduction
Intracranial hypertension frequently complicates severe traumatic brain injury (TBI) and may be associated with poor outcomes. TBI induces a neuroinflammatory response by microglial activation and upregulation of proinflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor alpha (TNF-α,) and interleukin-6 (IL-6). To elucidate the effect of elevated intracranial pressure on microglial function, we studied the effects of increased extracellular pressure on primary human microglial cell phagocytosis, proliferation, cytokine secretion and total nitrate production. In addition, since many patients receive propofol during anesthesia or intensive care unit sedation, we evaluated whether propofol alters the effects of pressure.
Methods
Human microglial cells (HMG030) were pretreated with (2.5–20μg/ml) propofol or intralipid as a vehicle control were incubated at ambient atmospheric pressure or at 15 or 30 mmHg increased pressure for 2 hours for phagocytosis assays or 24 hours for proliferation, cytokine secretion and total nitrate production studies. Phagocytosis was determined by incorporation of intracellular fluorescent latex beads. TNF-α, IL1-β and IL-6 were assayed by sandwich ELISA, and total nitrate by Greiss reagent.
Results
Increased extracellular pressure stimulated phagocytosis vs. untreated microglial cells or cells treated with an intralipid vehicle control. In fact, propofol also stimulated microglial phagocytosis at ambient pressure. However, increased pressure reduced phagocytosis in the presence of propofol. Pressure also increased microglial TNF-α and IL-1β secretion and propofol pretreatment blocked the pressure-stimulated effect. However, IL-6 production was not altered either by pressure or propofol. Pressure also induced total nitrate secretion and propofol pretreatment decreased basal as well as pressure-induced microglial nitrate production.
Conclusion
Extracellular pressures consistent with increased intracranial pressure after head injury activate inflammatory signals in human primary microglial cells in vitro, stimulating phagocytosis, proliferation, and TNF-α, IL-1β, and total nitrate secretion but not affecting IL-6. Such inflammatory events may contribute to the worsened prognosis of traumatic brain injury after increased intracranial pressure. Since propofol alleviated these potentially pro-inflammatory effects, these results raise the possibility that the inflammatory cascade activated by intracranial pressure may be targeted by propofol in patients with increased intracranial pressure after TBI.
Keywords: traumatic brain injury, microglia, phagocytosis, proliferation, cytokines, extracellular pressure
Introduction
1.5 million people sustain a head injury in the US each year, producing 50,000 deaths, 80,000 disabling injuries, and $54 billion in hospital care costs.1 Elevated intracranial pressure (ICP) remains a main cause of death and disability in patients with traumatic brain injury (TBI.) Intracranial hypertension leads to cerebral ischemia, brain herniation, and death.2, 3 Indeed, elevated ICP discriminates between TBI patients with good or poor outcomes.4, 5 Clinical management of TBI emphasizes basic intensive care and interventions to control ICP and optimize cerebral perfusion pressure (CPP,) the difference between ICP and the mean arterial pressure (MAP.)6, 7 However, the management of the cellular consequences of increased ICP, if it occurs, is less well understood.
Microglia are resident monocyte-lineage cells in the brain8 that are activated very early after injury.9–11 Activation and proliferation of resident microglia are each typical of inflammatory events in TBI.12 Microglia respond to changes in the brain’s structural integrity independently of blood-brain barrier disruption,13 and changes in neural electrical transmission trigger various degrees of microglial activation.14 Glutamate,15 ATP,16 7-ketocholesterol,15 calcium,17 and zinc18 have each been previously identified as triggers for microglial activation.
Following TBI, microglia can act as scavengers and phagocytize the cell debris employing mechanisms in common with other macrophages.19 The number of microglial cells increases in a wide variety of CNS pathologies, and local proliferation rather than recruitment of blood-derived monocytes has been suspected as a probable mechanism.20 Microglia may be the dominant source of pro-inflammatory molecules, such as TNF-α, IL-1β, IL-6,21, 22 and free oxygen radicals in this setting.23, 24 Activated microglial cells have been proposed to aggravate neuronal damage25 and to be potential targets of immunosuppressive substances such as roscotovine after acute spinal cord or brain injury.26
We have previously reported that exposure to constant low and high extracellular pressure (20–100mmHg) enhances phagocytosis by human peripheral monocytes and PMA-differentiated THP-1 macrophages.27–29 We previously also reported that propofol (2,6-diisopropylphenol) stimulates macrophage phagocytosis but reverses pressure-stimulated macrophage phagocytosis.30 Propofol is known to alter the functions of immunocompetent cells as well as to decrease pro-inflammatory cytokine production and iNOS expression in LPS-stimulated mouse macrophages.31–33. Furthermore, propofol is a commonly used anesthetic agent in surgical patients and is also used for sedation in the intensive care unit setting. Propofol is known to alter the functions of immunocompetent cells, as well as to decrease pro-inflammatory cytokine production and iNOS expression in LPS-stimulated mouse macrophages.31–33. We therefore hypothesized that pressure would modulate phagocytosis, proliferation, and cytokine secretion in the human microglial cells and that propofol would modulate the effects induced by pressure in these cells.
Materials and Methods
Cell Culture
The human microglia cell line (Catalog #HMG 030) was obtained from Clonexpress (Gaithersburg, MD) and maintained at 37°C with 5%CO2 in DMEM Ham’s F-12 (Sigma, St. Louis, MO,) supplemented with 10% fetal bovine serum (FBS) (Sigma, St. Louis, MO,) L-glutamine (200mM,) gentamicin/amphotericin (Invitrogen,) and macrophage colony-stimulating factor (M-CSF) (Sigma.). The waterproof AP74 oxygen meter was obtained from Fisher Thermo scientific (ThermoFisher Scientific Inc, PA).
Microglia cells
The human microglia cell line (Catalog #HMG 030) was developed by Clonexpress (Gaithersburg, MD) using a proprietary process to amplify human brain derived microglial cells for 6–10 population doublings. The microglial cells express HLA class II antigens following treatment with interferon gamma, as well as CD68, CD14, esterase, and other markers as surface markers. The microglial cells do not express GFAP (astrocyte marker) or neuron-specific markers. They do express functions similar to those of primary microglial cells, including the secretion of cytokines in response to pro-inflammatory cytokines. They are commonly used as a research tool to model microglial cell biology34.
Pressure model
Extracellular pressure was manipulated using an apparatus previously described elsewhere.29, 35 Briefly, cells were placed in an airtight Lucite chamber with gas inlet and outlet valves (Lucite International Ltd, Southampton, UK,) thumbscrews, O-ring to achieve an airtight seal, and pressure manometer to monitor inside pressure. The box was pre-warmed to 37°C for 1 hour before each study to prevent fluctuations in internal pressure caused by temperature shifts of the pressurizing gas; the appropriate target pressure of 30 mmHg above ambient pressure was achieved within 1 minute. Temperature and pressure were maintained within ±2°C for temperature and ±1.5 mm Hg for pressure using this method, and the pH level, osmolarity, and pO2 of the culture medium remained essentially the same over the course of the study.36
Assay for phagocytosis
The phagocytosis assay was performed as described previously29. Briefly, fluorescent labeled latex beads (2μm) (Polysciences, Inc. Warrington, PA,) were opsonized with 10% unheated FBS for 60 minutes at 37°C prior to the experiments as described previously (29. 1×106 microglial cells in one milliliter of tissue culture medium supplemented with 10% FBS in a 35mm Petri dish were then mixed with opsonized latex beads at a multiplicity ratio of 1:5, and incubated for 2 hours at 37°C. Microglial cells were pretreated for 30 minutes with propofol (10μg/ml) or with the equivalent amount of intralipid as a vehicle control. One set of dishes was placed in a calibrated pressure box set at 30mmHg, and the pressure box was placed inside an incubator at 37°C and monitored every 15 minutes. The second set of dishes was placed inside the same incubator but under ambient pressure conditions. It should be emphasized that in each experiment, the cells subjected to ambient pressure or increased pressure, with intralipid or propofol, were all seeded simultaneously from the same original culture flask and were studied simultaneously, permitting valid comparison across these groups. After 2 hours, macrophage monolayers were washed vigorously with phosphate buffered saline (PBS) to remove extracellular beads, fixed with methanol for 10 minutes, and then counterstained with methylene blue. The number of intracellular latex particles was determined by counting fluorescent beads within cells under a fluorescence microscope. Five hundred consecutive microglial cells were counted in each culture. A previously described scoring method29 was utilized to calculate the number of latex beads phagocytosed by macrophages or monocytes and the phagocytic index. In brief, cells were separated into the following categories: cells with no beads, cells with 1 bead, cells with 2–5 beads, cells with 6–10 beads, and cells with >10 beads. Percent phagocytosis was calculated as the total number of cells with at least one bead as a percentage of the total number of cells counted.
Proliferation Studies
The proliferation assays were performed using a colorimetric crystal violet staining method as described previously 37. Briefly, primary human microgial cells were seeded at 100,000cells/well six-well culture plates for 24 hours. Subconfluent (30–40%) cells were serum-starved for 24 hours. The serum-starved cells were switched back to normal growth medium supplemented with 10% FBS under ambient pressure or 30 mmHg pressure for 24 hours before staining with crystal violet. Microglial cells were rinsed in PBS and fixed in absolute ethanol-glacial acetic acid (3:1 vol/vol) for 10 minutes at room temperature and left to air dry. The cells were stained with 0.1% crystal violet (wt/vol) for 10 min at room temperature. Excess dye was removed by decantation and washed twice with distilled water. The dye was extracted in 10% acetic acid (vol/vol), and optical density was measured at 550 nm using a Thermomax microplate reader (Molecular Devices, Ramsey, MN.)
TNF-α, IL-1β and IL-6 assays
One milliliter of HMG030 cell suspension (1×106cells/ml) was placed in each well of a 6-well tissue culture plates at 37°C for 24 hours under ambient pressure or 15 or 30mmHg increased pressure conditions. At the end of the culture period, conditioned supernatants were harvested and the production of immunoreactive TNF-α, IL-6, and IL-1β were determined by sandwich enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s recommendation.
Generation of lower oxygen concentration (100 torr pO2)
In some studies cells were incubated at ~100torr pO2 with and without increased pressure for 24 hours and TNF-α production was measured. The lower oxygen tension (~100 torr) was generated by mixing a pure nitrogen gas (100%), with a pre-mixture of nitrogen (74%), oxygen (21%) and CO2 (5%) gas in equal flow using a Y-connector. The oxygen concentration was routinely measured by using a waterproof dissolved oxygen meter (AP74, ThermoFisher Scientific Inc, PA) that had been precalibrated and placed inside the incubator. The same mixed gas was supplemented to the cells incubated in the pressure box at 30mmHg for 24 hours. Since the pressure box was transparent and the incubator used contained an inner door that was transparent, the dissolved oxygen in the fluid could be monitored in real time. At the end of the culture period, conditioned supernatants were harvested and the production of immunoreactive TNF-α was determined by sandwich enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s recommendation.
Nitrite/Nitrate assay
One milliliter of HMG030 cell suspension (1×106 cells/ml) was placed in each well of 6-well tissue culture plates at 37°C for 24 hours under ambient or 30mmHg increased pressure conditions. The total nitrite/nitrate in the supernatant was analyzed by the protocol from the Research and Development Systems nitrite/nitrate assay kit (Minneapolis, MN). Briefly, the reaction is a two-step process with the first step converting NO3− to NO2− using nitrate reductase and the second step utilizing the Griess reagents to chemically convert NO2− to an azo compound. Total NO3−/NO2− then was determined by spectrophotometric absorbance at 540nm.
Statistical Analysis
Each proliferation experiment employed 3 independent replicates, while phagocytosis assays evaluated at least 500 cells per well in at least 3 different wells. ELISAs were done in duplicate from each of 3–6 independent samples from independent wells. The nitrite/nitrate assay was conducted in 9 independent wells. All experiments were then done independently at least three times with similar results, and the mean values obtained by analyzing all of the replicate data from each experiment were analyzed statistically. Data are presented as means ±SE. Statistical analysis of all data sets was conducted using analysis of variance (ANOVA) with Tukey post hoc testing using SYSTAT software (Chicago, IL.)
Results
Extracellular pressure stimulated microglial phagocytosis, but the effect was reversed by propofol pretreatment. [Fig. 1]
Fig 1. Effect of increased extracellular pressure on microglial cell phagocytosis and the role of propofol in the modulation of pressure-induced phagocytosis.
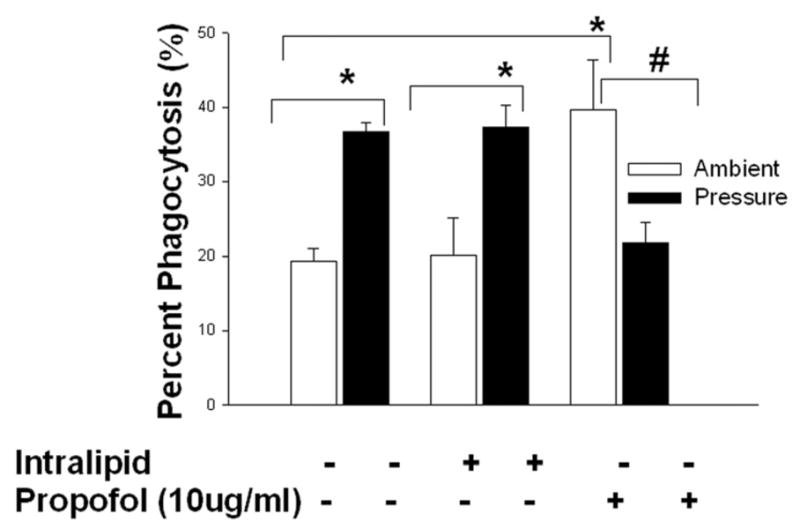

Either untreated or Intralipid-treated microglial cells increased phagocytosis under increased pressure conditions in comparison to cells not exposed to increased pressure (* p<0.01, n=3). Pretreatment with propofol (*p<0.01, n=3) significantly increased phagocytosis in microglial cells compared to cells treated with intralipid at ambient pressure. Propofol reversed the effect of pressure-stimulated phagocytosis in propofol-treated cells (# p<0.01, n=3).
Human microglial (HMG030) cells were untreated or treated with vehicle control (Intralipid) or propofol (10μg/ml) for 30 minute and then cells were either exposed to increased extracellular pressure (30mmHg) or kept at ambient pressure. Exposure to increased pressure significantly increased the percentage of cells exhibiting phagocytosis in comparison to cells incubated at ambient pressure (19.3±1.7% vs. 36.7±1.3%, n=3, p<0.01.). Cells treated with intralipid vehicle control also similarly displayed increased phagocytosis under increased extracellular pressure in comparison to cells pretreated with intralipid but not exposed to pressure (20.0±5.1% vs. 37.4±2.9%, n=3, p<0.01). There was no significant difference between ambient pressure controls and ambient pressure intralipid-treated cells and there was also no significant difference between increased pressure controls and increased pressure intralipid-treated cells. However, propofol pretreatment increased the basal phagocytosis significantly at ambient pressure to 39.7±6.7% in comparison to either ambient pressure control or ambient pressure intralipid-treated cells (n=3, p<0.05). Furthermore, cells pretreated with propofol and exposed to increased pressure significantly reduced the phagocytosis to 21.8±2.6% compared to ambient pressure with propofol (n=3, p<0.05.). We have observed similar results in propofol treated monocytes and THP-1 macrophages stimulated with increased pressure 30. This suggests that propofol increases basal phagocytosis and pressure reverse its effects not only in monocytes, macrophages but also in brain immune cells.
Extracellular pressure stimulated microglial proliferation independently of propofol. [Fig. 2]
Fig 2. Effect of increased extracellular pressure on microglial cell proliferation and the role of propofol in the modulation of pressure-induced proliferation.
Microglial cells were pre-treated with intralipid (vehicle control) or propofol (10μg/ml) for 30 minutes then subjected to ambient or 30mmHg extracellular pressure for 24 hours. Pressure significantly increased proliferation in the vehicle control (intralipid) group (* p<0.05, n=3) and in the propofol pretreatment group (* p<0.01, n=3).
Increased extracellular pressure significantly stimulated the proliferation of microglial cells treated with the intralipid vehicle control (100.0±3.6% vs. 115.2±3.4%, n=3, p<0.05.) Pretreatment with propofol did not alter basal proliferation at ambient pressure (n=3, p=n.s.). Furthermore, propofol pretreatment did not block the pressure-stimulated proliferation (n=3, p<0.05.).
Propofol dose responsive effects on pressure-stimulated TNF-α production
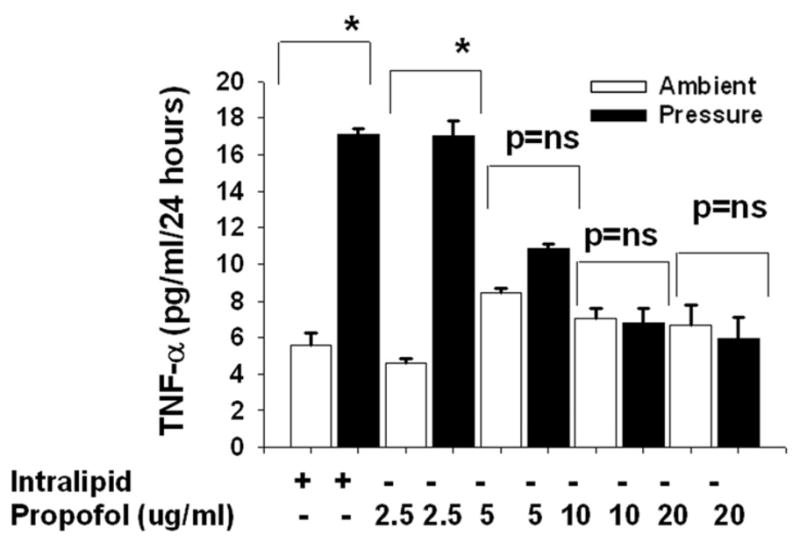
Microglia cells were pretreated with intralipid as vehicle control or propofol (2.5–20 μg/ml) for 30 minutes and then exposed to 30mmHg pressure for 24 hours or incubated at ambient pressure. Microglial cells exposed to extracellular pressure produced significantly more TNF-α in comparison to cells incubated under ambient pressure within the intralipid groups (Fig 3A). The propofol at dose 2.5 μg/ml did not affect pressure-induced TNF-α production. At 5.0 μg/ml, propofol at least attenuated if it did not block the pressure effect. A trend toward increased TNF-α production in response to pressure was still observed in the cells pretreated with 5.0 μg/ml propofol but this did not achieve statistical significance. The propofol doses of 10 and 20 μg/ml completely blocked any stimulation by pressure of TNF-α production in the human microglial cells.
Fig 3. Dose response of propofol, dose response of increased pressure and the effect of lower oxygen concentration (100torr pO2) on TNF- α production.
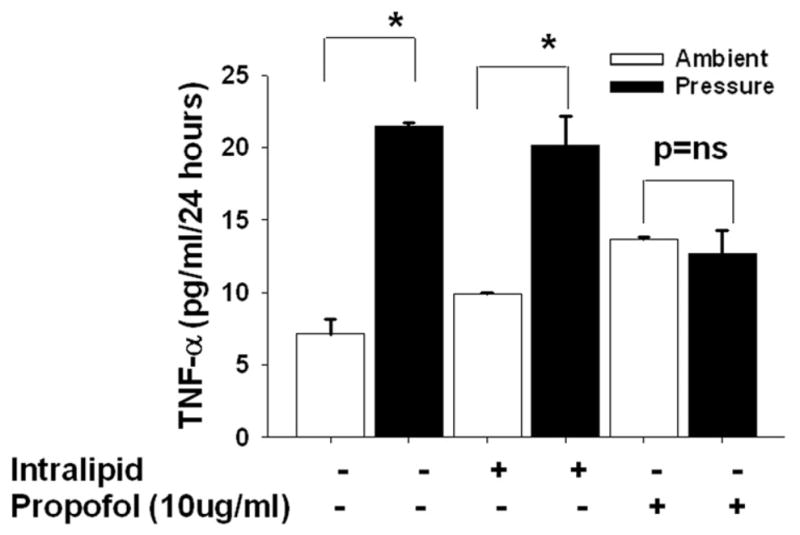
3A) Dose response of propofol on increased pressure stimulated TNF-α production. Microglial cells were pre-treated with intralipid (vehicle control) or propofol (2.5μg/ml-20μg/ml) for 30 minutes and then subjected to ambient or 30mmHg extracellular pressure for 24 hours. Increased extracellular pressure significantly stimulated TNF-α secretion in the vehicle control (intralipid) group as well as in cells pretreated with 2.5μg/ml propofol ((* p<0.05, n=6). Propofol at 5.0 μg/ml attenuated the pressure effect (p=0.07, n=6). The higher dose of (10 and 20μg/ml) propofol completely blocked the pressure effect.
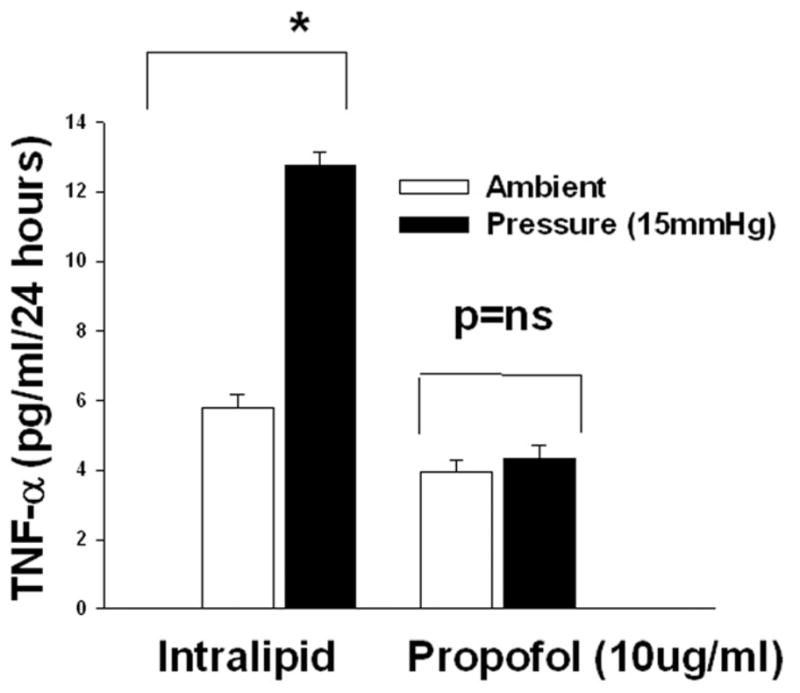
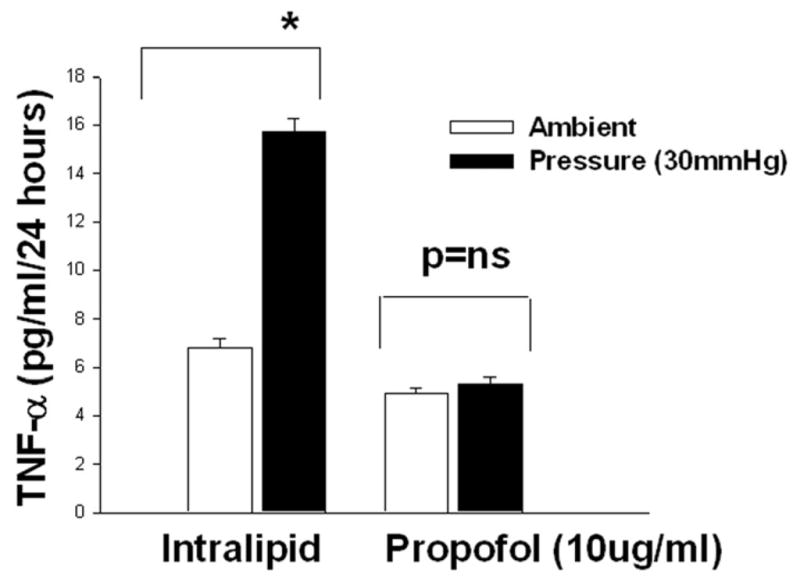
3B). Dose response of increased pressure on the TNF-α production and propofol block the pressure effect. Microglial cells were pre-treated with intralipid (vehicle control) or propofol (10μg/ml) for 30 minutes then subjected to ambient or 15 (B-I) or 30mmHg 9B-II) extracellular pressure for 24 hours as described under “Materials and Method”. Increased extracellular pressures at both 15 and 30mmHg significantly stimulated TNF-α secretion in vehicle control (intralipid) groups (*p<0.05, n=6). Propofol completely inhibited the pressure effect.
3C). Propofol block the pressure-induced TNF-α production at lower oxygen concentration (100torr). Microglial cells were pre-treated with intralipid (vehicle control) or propofol (10μg/ml) for 30 minutes then subjected to continuous flow of lower oxygen concentration(~100 torr) or 30mmHg extracellular pressure for 24 hours as described under “Materials and Method”. Increased extracellular pressure at 30mmHg significantly stimulated TNF-α secretion in vehicle control (intralipid) groups (*p<0.05, n=6). Propofol completely inhibited the pressure effect.
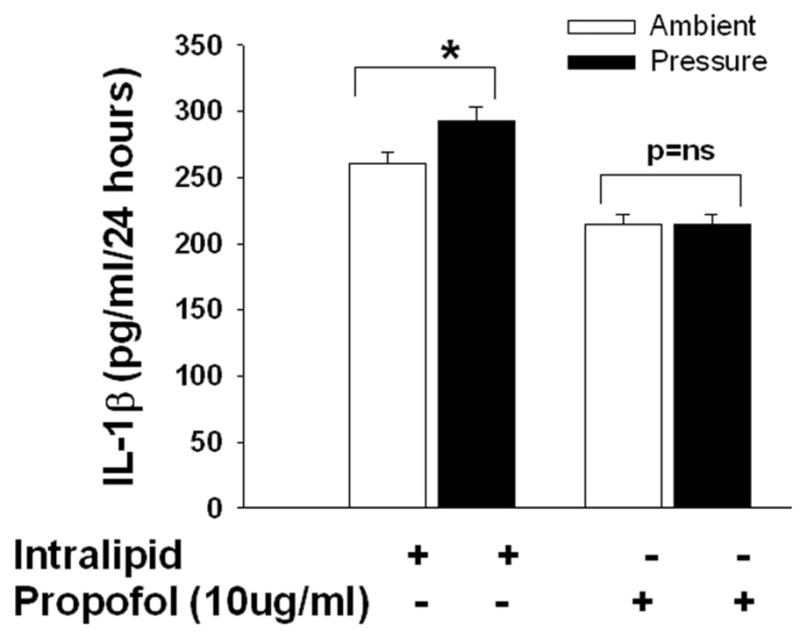
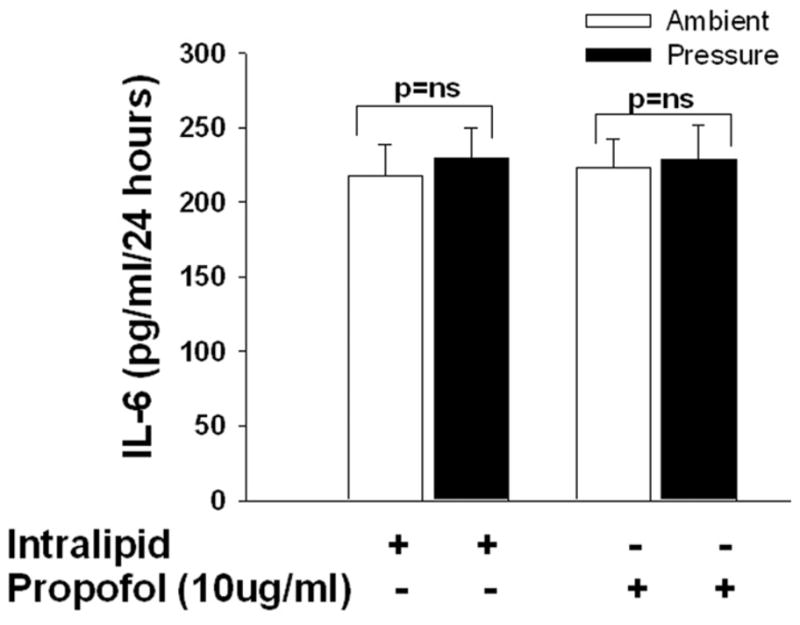
3D and E). Effect of increased extracellular pressure on IL-1beta and Il-6 secretion and the role of propofol in the modulation of pressure-induced effect on. Microglial cells were pre-treated with intralipid (vehicle control) or propofol for 30 minutes then subjected to ambient or 30mmHg extracellular pressure for 24 hours as described under “Materials and Method”. Pressure significantly increased IL-1beta but not IL-6 secretion in the vehicle control (intralipid) group (* p<0.05, n=6, D) and the propofol pretreatment group block the pressure-induced IL- 1beta. Similar to vehicle control, propofol also did not alter the Il-6 (p>0.05, n=6, E) secretion in either increased pressure or ambient pressure conditions.
Effect of increased extracellular pressure (15 and 30mmHg) on TNF-α secretion, and blockade of this effect by propofol: [Fig.3B-I and II]
Microglia cells were pretreated with intralipid as vehicle control or propofol (10μg/ml) for 30 minutes and then either exposed to 15 or 30mmHg pressure for 24 hours or incubated at ambient pressure. Microglial cells exposed to 15mmHg or 30 mmHg extracellular pressures produced significantly more TNF-α in comparison to cells not exposed to pressure within the intralipid groups (Fig 3B-I and II). In cells pretreated with propofol extracellular pressure at either 15 mmHg (Fig. 3B-I) or 30 mmHg ((Fig. 3B-II) mmHg failed to increase TNF- α production.
Effect of extracellular pressure at lower oxygen concentration on TNF-α secretion
Microglia cells were pretreated with intralipid as vehicle control or propofol (10μg/ml) for 30 minutes and then exposed 30mmHg pressure with ~100torr oxygen concentration for 24 hours or incubated in the incubator similarly maintained at the lower oxygen concentration as described in “materials and methods”. At 100torr oxygen concentration, increased extracellular pressure stimulated microglial TNF-α secretion in comparison to cells not exposed to increased pressure but also maintained at 100torr constant oxygen concentration in the incubator (Figure 3C). Furthermore, propofol completely prevented pressure stimulation of TNF-α secretion even at this lower oxygen concentration (Fig. 3C). Thus these results suggest that pressure stimulates TNF-α both at higher (171–181torr) and lower oxygen (100–110torr) concentrations and propofol blocks pressure-induced TNF-α secretion in either condition.
Effect of extracellular pressure on IL-1β and IL-6 secretion
Microglia cells were pretreated with intralipid as vehicle control or propofol (10μg/ml) for 30 minutes and then exposed to 30mmHg pressure for 24 hours or incubated at ambient pressure. Microglial cells exposed to extracellular pressure displayed increased IL-1β secretion in comparison to cells incubated under ambient pressure within the intralipid groups (292.1±2.8pg/ml vs. 260.9±4.1 pg/ml, n=3, p<0.05, Fig 3D). Propofol completely inhibited pressure stimulation of IL-1β secretion (Fig. 3D). Microglial cells exposed to extracellular pressure did not increase IL-6 secretion in comparison to cells incubated under ambient pressure within the intralipid groups (Fig 3E). Propofol pretreatment also did not alter the secretion of IL-6 at either ambient or increased pressure (Fig. 3E). This suggests that IL-16 secretion is not influenced by pressure or propofol in the microglial cells.
Effect of propofol on pressure-mediated total nitrate production
Pressure increased total nitrate production (14.2±0.3 μmol/ml vs. 12.9±0.5μmol/ml, n=9, p<0.05) in microglial cells treated with the intralipid vehicle control. Propofol-treated cells under ambient pressure demonstrated a total nitrate production of 10.5±1.1μmol/ml, which was significantly lower than nitrate release after intralipid treatment under either ambient pressure (n=9, p<0.05) or increased pressure (n=9, p<0.05) Pretreatment with propofol decreased total nitrate production in microglia subjected to extracellular pressure to 8.4±0.8μmol/ml, which was significantly lower than cells under ambient pressure pretreated with propofol (n=9, p<0.05), so propofol not only blocked but reversed the effect of pressure on nitrate production. Indeed, nitrate production by propofol-treated cells under pressure was also lower than that of intralipid-treated cells under increased pressure (n=9, p<0.05).
Effect of Pressure on Solution pH and pO2
Since the 30 mm Hg increase used for most of the studies is relatively small in comparison to ambient atmospheric pressure (approximately 760 mm Hg), this increase in pressure exerts relatively slight effects on the pO2 and pH of the solutions. For instance, in this study, cell culture medium maintained at 30 mm Hg increased pressure at 37°C in room air for 24 hours had a pH of 7.65± 0.01 and a pO2 of 152.58±3.9, whereas medium similarly maintained at ambient pressure had a pH of 7.62±0.02 and a pO2 of 151.90±4.4. Neither of these differences was statistically significant (n=6, n.s.).
Discussion
TBI increases ICP and initiates a neuroinflammatory cascade characterized by astrocyte and microglial activation and cytokine release. This cascade increases brain edema that, in turn, further increases ICP. Microglia, the phagocytic cells of the central nervous system, is the first to initiate cytokine signaling, releasing the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6. Nitric oxide also contributes to inflammation and the immune response in extracranial phagocytic cells, like macrophages and neutrophils38, as well as in microglia.39 Our results are consistent with microglial involvement in the inflammatory and phagocytic cascade, and suggest overall inflammatory activation by extracellular pressure. Propofol, an anesthetic and common intensive care sedative, reversed pressure effects on phagocytosis and TNF-α and nitrate production without affecting the mitogenic effect of pressure. Pressures above 20mmHg (27.19cm H2O) are considered elevated. Our test pressure of 30mmHg thus resembles in vivo ICP after TBI, although our TNF-α studies suggest pressure and propofol can modulate microglial cytokine release even at lower pressures.
We previously reported that propofol inhibits phagocytosis in monocytes from patients anesthetized with propofol and from human monocytes exposed to propofol ex vivo.30 Activation of alveolar macrophages by pressure has also been demonstrated in vitro and in vivo40,41, including a trend toward increased macrophage phagocytosis of Klebsiella pneumonia in response to pressure and significant cytokine activation 40 Fibroblasts and dendritic cells also display increased phagocytosis in response to increased pressure, although propofol was not investigated in those studies.42, 43. Increased extracellular pressure increases phagocytosis in monocytes and macrophages by inhibiting FAK and ERK.29 Microglia, despite their location in the central nervous system, appears to respond to pressure and propofol similarly to monocytes and macrophages. Thus, increased phagocytosis in response to increased pressure may be a generalized response of diverse phagocytic cells, and could similarly represent ERK and FAK inhibition in response to cytoskeletal deformation. Conversely, propofol inhibits pressure effects on macrophage and monocytes phagocytosis through GABAA receptor activation and p130cas inhibition.30 Although the precise mechanism by which microglial cells sense such pressures awaits further study, extrapolation from other cell types exposed to pressure suggests the possibility that the cytoskeleton may act as a mechanosensor that “perceives” mechanical signals from forces such as pressure and deformation and transduces them to the focal adhesion complex 43,44,35.
Previous dose-response studies demonstrated maximal effects on phagocytosis when pressure-stimulated macrophages and monocytes were exposed to propofol at 10μg/ml.30 We focused on 10μg/ml here to improve our ability to detect propofol effects. In that prior study, however, propofol also altered phagocytosis at ambient pressure and increased pressure at 5μg/ml, albeit at a lower magnitude, which demonstrates efficacy at in vivo therapeutic concentrations.30 We found blunting of pressure effects on microglial TNF-α release even with 5μg/ml propofol here.
The concentration of propofol in human brain tissue during prolonged propofol sedation or anesthesia has not been measured. Cerebrospinal fluid concentrations are dramatically lower than blood concentrations because of the blood brain barrier 45. However, propofol levels within the brain of propofol-sedated rats were measured at 5 μg/ml after 15 minutes of sedation, rising to 14 μg/ml after 30 minutes, suggesting that the lipid-soluble propofol molecule accumulates within brain tissue 46. Our present results suggest that propofol can modulate pressure effects on microglial cells at 5–20 μg/ml, within this range. Propofol levels with the much longer infusions used to sedate patients after head injury might result in concentrations at least this high within brain tissue. Ischemia/reperfusion injury, inflammatory syndromes, or other critical illness may impair the blood brain barrier and further facilitate propofol movement across the barrier. Indeed, one recent report suggests chronic impairment of the blood brain barrier in humans even years after TBI 47
Propofol arrests TNF-α production in macrophages at the pretranslational level.48 We previously reported that extracellular pressure increases the release of monocyte cytokines, but did not investigate propofol effects.49 TNF-α and IL-1β expression follow different pathways.48, 50 Despite these different pathways, both TNF-α and IL-1β increased in macrophages under pressure. This corresponds to in vivo observations of these cytokines in TBI.51.
Mitogenesis, increased under pressure, was not inhibited by propofol. This suggests that the effects we have observed are not toxic. Although a literature search revealed no studies of mitogenesis in nerve tissue under pressure, pressure stimulates proliferation in other cells, including cardiac,52 and vascular 53 myocytes, endothelial cells,54, and normal intestinal epithelium55 Pressure stimulates proliferation in some colon cancer cell lines through an unclear pathway that may involve PKC-α and some tyrosine kinase activity.56
Nitric oxide production from activation of inducible nitric oxide synthase (iNOS) impairs the blood-brain barrier, facilitating posttraumatic brain edema. This edema also increases ICP and thus microglial recruitment. Arresting nitric oxide synthase using novel agents has been explored previously.57 However, propofol arrests iNOS production in stimulated macrophages.58, possibly via toll-like receptor protein activation.59
Although the stimulation of microglial phagocytosis by pressure has not previously been described, we and others have previously described the ability of increases in extracellular pressure to stimulate the uptake of serum-opsonized latex beads, IgG complex, tumor cell fragments, and Klebsiella pneumonia 29, 30,27, 28, 40, 42, 43, 60. The mechanisms of this effect, and whether more specific microglial endocytosis is similarly affected by increased extracellular pressure awaits study.
Preclinical data suggest that targeting microglial pro-inflammatory cytokine overproduction may represent an effective new therapeutic intervention for TBI. Microglias are “first responders” to the initial insult of TBI. Modulating their release of pro-inflammatory cytokines may decrease inflammation and edema, permitting improvement in prognosis, outcome, and a closer return to baseline neurologic function. Our present results indicate that extracellular pressure at levels consistent with increased ICP after TBI activates inflammatory signals in microglial cells, stimulating phagocytosis, proliferation, nitrate, TNF-α and IL-1β production without affecting IL-6 and that propofol inhibits some of these critical inflammatory signals in vitro. Although awaiting in vivo confirmation, these results suggest the possibility that propofol may alleviate at least some of the inflammatory effects of increased ICP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Binder S, Corrigan JD, Langlois JA. The public health approach to traumatic brain injury: an overview of CDC’s research and programs. The Journal of Head Trauma and Rehabilitation. 2005 May-Jun;20(3):189–195. doi: 10.1097/00001199-200505000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Marmarou A, Anderson RL, Ward JD, et al. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. Journal of Neruosurgery. 1991 Nov;75(Supplement):s59–s66. [Google Scholar]
- 3.Juul N, Morris GF, Marshall SB, Marshall LF The Executive Committee of the International Selfotel Trial. Intracranial hypertension and cerebral perfusion pressure: influence on neurological deterioration and outcome in severe head injury. Journal of Neurosurgery. 2000 Jan;92(1):1–6. doi: 10.3171/jns.2000.92.1.0001. [DOI] [PubMed] [Google Scholar]
- 4.Czosnyka M, Balestreri M, Steiner L, et al. Age, intracranial pressure, autoregulation, and outcome after brain trauma. Journal of Neurosurgery. 2005 Mar;102(3):450–454. doi: 10.3171/jns.2005.102.3.0450. [DOI] [PubMed] [Google Scholar]
- 5.Hiler M, Czosnyka M, Hutchinson P, et al. Predictive value of initial computerized tomography scan, intracranial pressure, and state of autoregulation in patients with traumatic brain injury. Journal of Neruosurgery. 2006 Mar;104(5):731–737. doi: 10.3171/jns.2006.104.5.731. [DOI] [PubMed] [Google Scholar]
- 6.Steiner LA, Andrews PJ. Monitoring the injured brain: ICP and CBF. British Journal of Anaesthesia. 2006 Jul;97(1):26–38. doi: 10.1093/bja/ael110. [DOI] [PubMed] [Google Scholar]
- 7.Robertson C, Valadka AB, Hannay JH, et al. Prevention of secondary ischemic insults after severe head injury. [Article] Critical Care Medicine. 1999 Oct;27(10):2086–2095. doi: 10.1097/00003246-199910000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Vass K, Hickey WF, Schmidt RE, Lasssmann H. Bone marrow-derived elements in the peripheral nervous system. An immunohistochemical and ultrastructural investigation in chimeric rats. Laboratory Investigations. 1993 Sep;69(3):275–282. [PubMed] [Google Scholar]
- 9.Perry VH, Andersson P-B, Gordon S. Macrophages and inflammation in the central nervous system. Trends in Neurosciences. 1993 Jul;16(7):268–273. doi: 10.1016/0166-2236(93)90180-t. [DOI] [PubMed] [Google Scholar]
- 10.Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: Intrinsic immuneffector cell of the brain. Brain Research Reviews. 1995 Mar;20(3):269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- 11.Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: A review. Glia. 1988 Oct;1(5):301–307. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- 12.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Current Opinion in Critical Care. 2002 Apr;8(2):101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Koshinaga M, Suma T, Fukushima M, Tsuboi I, Aizawa S, Katayama Y. Rapid microglial activation induced by traumatic brain injury is independent of blood brain barrier disruption. Histology and Histopathology. 2007 Feb;22(2):129–135. doi: 10.14670/HH-22.129. [DOI] [PubMed] [Google Scholar]
- 14.Gehrmann J, Mies G, Bonnekoh P, et al. Microglial Reaction in the Rat Cerebral Cortex Induced by Cortical Spreading Depression. Brain Pathology. 1993 Jan;3(1):11–17. doi: 10.1111/j.1750-3639.1993.tb00720.x. [DOI] [PubMed] [Google Scholar]
- 15.Diestel A, Aktas O, Hackel D, et al. Activation of Microglial Poly(ADP-Ribose)-Polymerase-1 by Cholesterol Breakdown Products during Neuroinflammation: a Link between Demyelination and Neuronal Damage. The Journal of Experimental Medicine. 2003 Dec 1;198(11):1729–1740. doi: 10.1084/jem.20030975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Färber K, Kettenmann H. Purinergic signalling and microglia. Pflügers Archiv European Journal of Physiology. 2006;452(5):615–621. doi: 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]
- 17.Whittemore ER, Korotzer AR, Etebari A, Cotman CW. Carbachol increases intracellular free calcium in cultured rat microglia. Brain Research. 1993 Sep;621(1):59–64. doi: 10.1016/0006-8993(93)90297-z. [DOI] [PubMed] [Google Scholar]
- 18.Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc Triggers Microglial Activation. The Journal of Neuroscience. 2008 May 28;28(22):5827–5835. doi: 10.1523/JNEUROSCI.1236-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brockhaus J, Moller T, Kettenmann H. Phagocytozing ameboid microglial cells studied in a mouse corpus callosum slice preparation. Glia. 1996 Dec;16(1):81–90. doi: 10.1002/(SICI)1098-1136(199601)16:1<81::AID-GLIA9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 20.Giordana MT, Attanasio A, Cavalla P, Migheli A, Vigliani MC, Schiffer D. Reactive cell proliferation and microglia following injury to the rat brain. Neuropathology and Applied Neurobiology. 1994;20(2):163–174. doi: 10.1111/j.1365-2990.1994.tb01175.x. [DOI] [PubMed] [Google Scholar]
- 21.Shohami E, Novikov M, Bass R, Yamin A, Gallily R. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. Journal of Cerebral Blood Flow & Metabolism. 1994 Jul;14(4):615–619. doi: 10.1038/jcbfm.1994.76. [DOI] [PubMed] [Google Scholar]
- 22.Woodroofe MN, Sarna GS, Wadhwa M, et al. Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: evidence of a role for microglia in cytokine production. Journal of Neuroimmunology. 1991 Sep;33(3):227–236. doi: 10.1016/0165-5728(91)90110-s. [DOI] [PubMed] [Google Scholar]
- 23.Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Research. 1992 Aug;587(2):250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima K, Kohsaka S. Microglia:Neuroprotective and Neurotrophic Cells in the Central Nervous System. Current Drug Targets - Cardiovascular & Haematological Disorders. 2004 Mar;4(1):65–84. doi: 10.2174/1568006043481284. [DOI] [PubMed] [Google Scholar]
- 25.Byrnes K, Faden A. Role of Cell Cycle Proteins in CNS Injury. Neurochemical Research. 2007;32(10):1799–1807. doi: 10.1007/s11064-007-9312-2. [DOI] [PubMed] [Google Scholar]
- 26.Hilton GD, Stoica BA, Byrnes KR, Faden AI. Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. Journal of Cerebral Blood Flow and Metabolism. 2008 Jul;28(11):1845–1859. doi: 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiratsuchi H, Basson MD. Activation of p38 MAPK{alpha} by extracellular pressure mediates the stimulation of macrophage phagocytosis by pressure. American Journal of Physiology. Cell Physiology. 2005 May 1;288(5):C1083–1093. doi: 10.1152/ajpcell.00543.2004. [DOI] [PubMed] [Google Scholar]
- 28.Shiratsuchi H, Basson MD. Akt2, but not Akt1 or Akt3 mediates pressure-stimulated serum-opsonized latex bead phagocytosis through activating mTOR and p70 S6 kinase. Journal of Cellular Biochemistry. 2007;102(2):353–367. doi: 10.1002/jcb.21295. [DOI] [PubMed] [Google Scholar]
- 29.Shiratsuchi H, Basson MD. Extracellular pressure stimulates macrophage phagocytosis by inhibiting a pathway involving FAK and ERK. American Journal of Physiology. Cell Physiology. 2004 Jun 1;286(6):C1358–1366. doi: 10.1152/ajpcell.00553.2003. [DOI] [PubMed] [Google Scholar]
- 30.Shiratsuchi H, Kouatli Y, Yu GX, Marsh HM, Basson MD. Propofol inhibits pressure-stimulated macrophage phagocytosis via the GABAA receptor and dysregulation of p130cas phosphorylation. American Journal of Physiology. Cell Physiology. 2009 Jun 1;296(6):C1400–1410. doi: 10.1152/ajpcell.00345.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen R-M, Chen T-G, Chen T-L, et al. Anti-Inflammatory and Antioxidative Effects of Propofol on Lipopolysaccharide-Activated Macrophages. Annals of the New York Academy of Sciences. 2005;1042 doi: 10.1196/annals.1338.030. [DOI] [PubMed] [Google Scholar]; The Role of the Mitochondria in Human Aging and Disease: From Genes to Cell Signaling. :262–271. [Google Scholar]
- 32.Chen R-M, Wu C-H, Chang H-C, et al. Propofol Suppresses Macrophage Functions and Modulates Mitochondrial Membrane Potential and Cellular Adenosine Triphosphate Synthesis. Anesthesiology. 2003 May;98(5):1178–1185. doi: 10.1097/00000542-200305000-00021. [DOI] [PubMed] [Google Scholar]
- 33.Mikawa K, Akamatsu H, Nishina K, et al. Propofol inhibits human neutrophil functions. Anesthesia and Analgesia. 1998 Sep 1;87(3):695–700. doi: 10.1097/00000539-199809000-00039. [DOI] [PubMed] [Google Scholar]
- 34.Nagai A, Nakagawa E, Hatori K, et al. Generation and characterization of immortalized human microglial cell lines: expression of cytokines and chemokines. Neurobiol Dis. 2001 Dec;8(6):1057–1068. doi: 10.1006/nbdi.2001.0437. [DOI] [PubMed] [Google Scholar]
- 35.Thamilselvan V, Basson MD. Pressure activates colon cancer cell adhesion by inside-out focal adhesion complex and actin cytoskeletal signaling. Gastroenterology. 2004;126(1):8–18. doi: 10.1053/j.gastro.2003.10.078. [DOI] [PubMed] [Google Scholar]
- 36.Basson MD, Yu CF, Herden-Kirchoff O, et al. Effects of increased ambient pressure on colon cancer cell adhesion. Journal of Cellular Biochemistry. 2000;78(1):47–61. doi: 10.1002/(sici)1097-4644(20000701)78:1<47::aid-jcb5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 37.Chaturvedi LS, Marsh HM, Shang X, Zheng Y, Basson MD. Repetitive deformation activates focal adhesion kinase and ERK mitogenic signals in human Caco-2 intestinal epithelial cells through Src and Rac1. J Biol Chem. 2007 Jan 5;282(1):14–28. doi: 10.1074/jbc.M605817200. [DOI] [PubMed] [Google Scholar]
- 38.Gross SS, Wolin MS. Nitric Oxide: Pathophysiological Mechanisms. Annual Review of Physiology. 1995 Oct;57(1):737–769. doi: 10.1146/annurev.ph.57.030195.003513. [DOI] [PubMed] [Google Scholar]
- 39.Garthwaite J, Boulton CL. Nitric Oxide Signaling in the Central Nervous System. Annual Review of Physiology. 1995;57(1):683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 40.Hall NG, Liu Y, Hickman-Davis JM, et al. Bactericidal function of alveolar macrophages in mechanically ventilated rabbits. Am J Respir Cell Mol Biol. 2006 Jun;34(6):719–726. doi: 10.1165/rcmb.2005-0463OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pugin J, Dunn I, Jolliet P, et al. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol. 1998 Dec;275(6 Pt 1):L1040–1050. doi: 10.1152/ajplung.1998.275.6.L1040. [DOI] [PubMed] [Google Scholar]
- 42.Bhalla S, Shiratsuchi H, Craig DH, Basson MD. [beta]1-integrin mediates pressure-stimulated phagocytosis. The American Journal of Surgery. 2009;198(5):611–616. doi: 10.1016/j.amjsurg.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Craig DH, Schaubert KL, Shiratsuchi H, Kan-Mitchell J, Basson MD. Increased pressure stimulates aberrant dendritic cell maturation. Cell Mol Biol Lett. 2008;13(2):260–270. doi: 10.2478/s11658-007-0054-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Craig DH, Zhang J, Basson MD. Cytoskeletal signaling by way of alpha-actinin-1 mediates ERK1/2 activation by repetitive deformation in human Caco2 intestinal epithelial cells. Am J Surg. 2007 Nov;194(5):618–622. doi: 10.1016/j.amjsurg.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engdahl O, Abrahams M, Bjornsson A, et al. Cerebrospinal fluid concentrations of propofol during anaesthesia in humans. Br J Anaesth. 1998 Dec;81(6):957–959. doi: 10.1093/bja/81.6.957. [DOI] [PubMed] [Google Scholar]
- 46.Shyr M-H, Tsai T-H, Tan PPC, Chen C-F, Chan SHH. Concentration and regional distribution of propofol in brain and spinal cord during propofol anesthesia in the rat. Neuroscience Letters. 1995;184(3):212–215. doi: 10.1016/0304-3940(94)11209-2. [DOI] [PubMed] [Google Scholar]
- 47.Tomkins O, Feintuch A, Benifla M, Cohen A, Friedman A, Shelef I. Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc Psychiatry Neurol. 2011;2011:765923. doi: 10.1155/2011/765923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu G-J, Chen T-L, Chang C-C, Chen R-M. Propofol suppresses tumor necrosis factor-[alpha] biosynthesis in lipopolysaccharide-stimulated macrophages possibly through downregulation of nuclear factor-kappa B-mediated toll-like receptor 4 gene expression. Chemico-Biological Interactions. 2009;180(3):465–471. doi: 10.1016/j.cbi.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Shiratsuch H, Basson MD. Differential regulation of monocyte/macrophage cytokine production by pressure. The American Journal of Surgery. 2005;190(5):757–762. doi: 10.1016/j.amjsurg.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 50.Abdul-Sater AA, Koo E, Häcker G, Ojcius DM. Inflammasome-dependent Caspase-1 Activation in Cervical Epithelial Cells Stimulates Growth of the Intracellular Pathogen Chlamydia trachomatis. Journal of Biological Chemistry. 2009;284(39):26789–26796. doi: 10.1074/jbc.M109.026823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buttram SD, Wisniewski SR, Jackson EK, et al. Multiplex assessment of cytokine and chemokine levels in cerebrospinal fluid following severe pediatric traumatic brain injury: effects of moderate hypothermia. J Neurotrauma. 2007 Nov;24(11):1707–1717. doi: 10.1089/neu.2007.0349. [DOI] [PubMed] [Google Scholar]
- 52.Honsho S, Nishikawa S, Amano K, et al. Pressure-Mediated Hypertrophy and Mechanical Stretch Induces IL-1 Release and Subsequent IGF-1 Generation to Maintain Compensative Hypertrophy by Affecting Akt and JNK Pathways. Circ Res. 2009 Nov 20;105(11):1149–1158. doi: 10.1161/CIRCRESAHA.109.208199. [DOI] [PubMed] [Google Scholar]
- 53.Luo D, Cheng J, Xiong Y, et al. Static pressure drives proliferation of vascular smooth muscle cells via caveolin-1/ERK1/2 pathway. Biochemical and Biophysical Research Communications. 2009 doi: 10.1016/j.bbrc.2009.12.132. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 54.Vouyouka AG, Jiang Y, Basson MD. Pressure alters endothelial effects upon vascular smooth muscle cells by decreasing smooth muscle cell proliferation and increasing smooth muscle cell apoptosis. Surgery. 2004 Aug;136(2):282–290. doi: 10.1016/j.surg.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 55.Flanigan TL, Owen CR, Gayer C, Basson MD. Supraphysiologic extracellular pressure inhibits intestinal epithelial wound healing independently of luminal nutrient flow. The American Journal of Surgery. 2008;196(5):683–689. doi: 10.1016/j.amjsurg.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walsh MF, Woo RK-Y, Gomez R, Basson MD. Extracellular pressure stimulates colon cancer cell proliferation via a mechanism requiring PKC and tyrosine kinase signals. Cell Proliferation. 2004;37(6):427–441. doi: 10.1111/j.1365-2184.2004.00324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Terpolilli NA, Zweckberger K, Trabold R, et al. The novel nitric oxide synthase inhibitor 4-amino-tetrahydro-L-biopterine prevents brain edema formation and intracranial hypertension following traumatic brain injury in mice. Journal of Neurotrauma. 2009 doi: 10.1089/neu.2008.0853. (epub ahead of print);0(ja) [DOI] [PubMed] [Google Scholar]
- 58.Lee C-J, Tai Y-T, Lin Y-L, Chen R-M. Molecular mechanisms of propofol-involved suppression of no biosynthesis and inducible iNOS gene expression in LPS-stimulated macrophage-like Raw 264.7 cells. Shock. 2010;33(1):93–100. doi: 10.1097/SHK.0b013e3181a6eaf5. [DOI] [PubMed] [Google Scholar]
- 59.Chiu W-T, Lin Y-L, Chou C-W, Chen R-M. Propofol inhibits lipoteichoic acid-induced iNOS gene expression in macrophages possibly through downregulation of toll-like receptor 2-mediated activation of Raf-MEK1/2- ERK1/2-IKK-NF[kappa]B. Chemico-Biological Interactions. 2009 Oct 30;181(3):430–439. doi: 10.1016/j.cbi.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 60.Mattana J, Sankaran RT, Singhal PC. Increased applied pressure enhances the uptake of IgG complexes by macrophages. Pathobiology. 1996;64(1):40–45. doi: 10.1159/000164004. [DOI] [PubMed] [Google Scholar]