

Abstract
The initial event in atherogenesis is the increased transcytosis of low density lipoprotein, and its subsequent deposition, retention and modification in the subendothelium. It is followed by the infiltration of activated inflammatory cells from the coronary circulation into the arterial wall. There they secrete reactive oxygen species (ROS) and produce oxidized lipoproteins capable of inducing endothelial cell apoptosis, and thereby plaque erosion. Activated T lymphocytes, macrophages and mast cells, accumulate in the eroded plaque where they secrete a variety of proteases capable of inducing degradation of extracellular proteins, thereby rendering the plaques more prone to rupture. This review summarizes the recent advancements in the understanding of the roles of ROS and oxidized lipoproteins in the activation of inflammatory cells and inducing signalling pathways related to cell death and apoptosis. In addition, it presents evidence that this vicious circle between oxidative stress and inflammation does not only occur in the diseased arterial wall, but also in adipose tissues. There, oxidative stress and inflammation impair adipocyte maturation resulting in defective insulin action and adipocytokine signalling. The latter is associated with increased infiltration of inflammatory cells, loss of anti-oxidant protection and cell death in the arterial wall.
Keywords: atherosclerosis, metabolic syndrome, diabetes, inflammation, oxidative stress, adipose tissue, vascular tissue, oxidized LDL
Introduction
One of the emerging cardiovascular risk factors is subclinical chronic low-grade inflammation [1]. Population studies showed a strong correlation between pro-inflammatory biomarkers (such as C-reactive protein, interleukin-6 [IL-6] and tumour necrosis factor-α[TNF-α]) and perturbations in glucose homeostasis, obesity and atherosclerosis [2, 3]. Another emerging risk factor is oxidized low-density lipoprotein (ox-LDL) that activates circulating monocytes, thereby increasing their ability to infiltrate the vascular wall. This increased infiltration is a key event in atherogenesis [4].
The metabolic syndrome clusters several cardiovascular risk factors including obesity, dyslipidaemia, hypertension and insulin resistance (IR) [5–8]. Increased inflammation [9, 10] and oxidative stress [11, 12] were found to be associated with the metabolic syndrome. It is a primary risk factor for diabetes and cardiovascular diseases [6, 13–20]. Recent data suggest that increased oxidative stress in adipose tissue is an early instigator of the metabolic syndrome and that the redox state in adipose tissue is a potentially useful therapeutic target for the obesity-associated metabolic syndrome [21]. Oxidative damage of adipose tissues is associated with impaired adipocyte maturation, production of pro-inflammatory adipocytokines by dysfunctional adipocytes and increased infiltration of macrophages into the adipose tissues of obese persons where they produce inflammatory chemokines [22, 23]. This enhanced infiltration is causatively related to the loss of insulin signalling [24]. The goal of this review is to give an overview of the molecular mechanisms in adipose and vascular tissues that can explain the vicious circle between oxidative stress and inflammation, in relation to atherosclerosis and cardiovascular diseases.
Overview of molecular mechanisms explaining the association of inflammation and oxidative stress in the vessel wall
The initial event in atherogenesis is the increased transcytosis of LDL, and its subsequent deposition, retention and modification in the subendothelium within and outside the meshes of the hyperplasic basal lamina where LDL interacts with matrix proteins [25, 26]. It is followed by the recruitment of circulating monocytes into the vascular intima and their subsequent transformation into macrophage/foam cells are key elements of the initiation of atherosclerosis. This infiltration occurs preferentially at sites with disturbed flow dynamics where the endothelium becomes dysfunctional at the cellular and molecular level. Activated by the disturbed flow, the endothelium shows enhanced platelet adhesion, increased endothelial permeability to macromolecules such as LDL, and augmented endothelial gene expression of leucocyte adhesion molecules and chemotactic factors (Fig. 1). Activation of the leucocyte adhesion cascade ultimately leads to transmigration of monocytes into the subendothelial space of the intima and subsequent differentiation into macrophages. Adherence of circulating monocytes and T lymphocytes to the inflamed endothelium is mediated particularly by the vascular adhesion molecule-1 (VCAM-1), the intercellular adhesion molecule-1 (ICAM-1) and E- selectin and fibronectin. The monocyte chemoattractant protein-1 (MCP-1) and IL-8 play an active role as chemoattractants in the infiltration of leucocytes into the arterial wall. There, their activation accelerates the generation of reactive oxygen species (ROS). Activated macrophages express enzymes such as myeloperoxidase (MPO) and NADPH oxidase (NOX-1), which together generate a range of oxidants, including superoxide, hydrogen peroxide and hypochlorous acid [27, 28]. Each of them causes oxidative damage not only to invading micro-organisms, but also to host molecules in surrounding tissues. Among them are phospholipids in cell membranes and circulating lipoproteins which contain polyunsaturated fatty acid chains, such as the abundant phospholipid 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (PAPC) that are particularly susceptible to oxidation by such mediators [29]. Products of PAPC oxidation have been shown to accumulate at sites of inflammation, and in cells treated with inflammatory stimulants such as IL-1β and TNF-α[30]. Oxidation of PAPC leads to the formation of a mixture of products, ranging from epoxyisoprostanes to truncated chain derivatives that are collectively termed ox-PAPC. Oxidation of phospholipids in LDL, that infiltrates into the injured vessel wall, by MPO and NOX-1, results in the accumulation of ox-PAPC in ox-LDL (Fig. 1).
Fig 1.
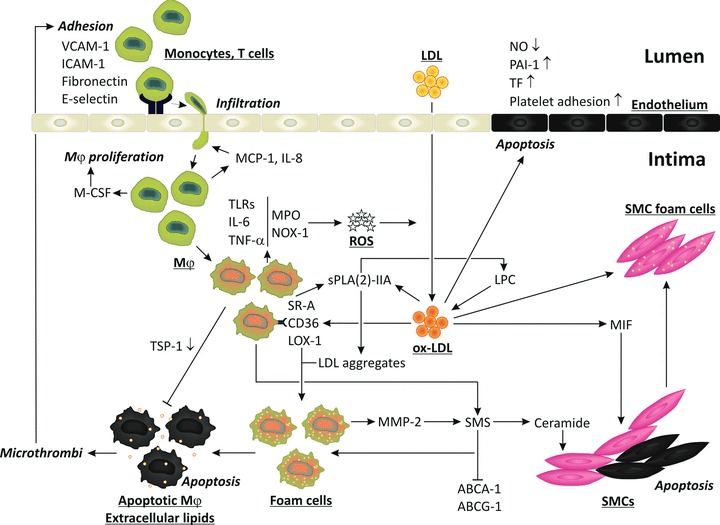
Molecular mechanisms of inflammation and oxidative stress in atherosclerotic plaques. Endothelial dysfunction in relation to hypercholesterolemia, hypertension, type 2 diabetes, and smoking is associated with induction of adhesion molecules for inflammatory cells, ICAM-1, VCAM-1, E-selectin and fibronectin. The infiltration and activation of inflammatory cells are associated with the activation of the oxidant enzymes MPO and NOX-1, resulting in the production of ROS and the oxidation of phospholipids and protein in LDL, resulting in the accumulation of ox-LDL. It stimulates the endothelium to secrete MCP-1 and IL-8, which induce transmigration of leucocytes into the endothelial space. Macrophages secrete M-CSF, thereby stimulating macrophage proliferation and inducing the expression of scavenger receptors CD36, LOX-1 and SR-A. The scavenger receptor mediated uptake of ox-LDL by macrophages leads to massive cholesterol and lipid accumulation and formation of foam cells, finally resulting in apoptotic macrophages and exposure of thrombogenic lipids. Deficient TSP-1 expression is associated with a decreased phagocytosis of dead cells. Foam cells secrete MMPs and SMS resulting in the production of ceramide that induces smooth SMC apoptosis (black cells). Activation of SMS also blunts the action of ABCA-1 and ABCG-1 resulting in impaired cholesterol and lipid efflux from foam cells. Ox-LDL induces TLRs of which the ligands enhance the expression of inflammatory mediators IL-6 and TNF-α. Ox-LDL induces migration inhibitory factor that stimulates SMC migration. The uptake of ox-LDL by SMCs leads to the production of SMC foam cells and secretion of MMPs that degrade the extracellular matrix proteins rendering the plaque more prone to rupture. Ox-LDL stimulates platelet adhesion and aggregation by decreasing endothelial production of nitric oxide, and enhances the pro-coagulant activity of endothelium by inducing the release of tissue factor. Ox-LDL reduces the fibrinolytic activity of endothelium by increasing the release of plasminogen activator inhibitor-1. Finally, ox-LDL induces apoptosis in endothelial cells (black) contributing to plaque erosion and rupture.
The activation of macrophages is also characterized by increased expression of the scavenger receptor-A (SR-A), cluster of differentiation (CD)36, and the lectin-like ox-LDL receptor (LOX-1) which internalize ox-LDL [31]. This scavenger receptor mediated uptake is important for clearing ox-LDL. However, unregulated uptake of ox-LDL leads to production of lipid-loaded foam cells. The unlimited accumulation of lipids, also as a consequence of defective cholesterol and lipid efflux, leads to apoptosis and cell burst resulting in exposure of extracellular lipids. Both apoptotic cells and thrombogenic lipids induce the formation of microthrombi which further stimulate the adhesion and infiltration of inflammatory cells, and the secretion of inflammatory cytokines, and the production of ROS.
Besides its deleterious action in the production of foam cells, CD36 plays also a protective role as a receptor of thrombospondin (TSP-1). This interaction is important for activating TSP-1-mediated phagocytosis, thereby preventing inflammation and macrophage-induced elastin degradation by matrix metalloproteinases (MMPs), and thus vessel wall degeneration and plaque rupture [32]. By binding to TSP-1, ox-LDL does not only impair phagocytosis but also prevents TSP-1-dependent TGF-β activation, thereby increasing inflammation and atherogenesis [33].
Activated macrophages secrete a number of growth factors such as the macrophage colony-stimulating factor (M-CSF) that augments SR-A expression, and induces the production of cytokines and growth factors which stimulate intimal proliferation. Once resident in the arterial wall, the interaction between monocytes/macrophages and T cells results in a broad range of cellular and humoural responses that drive the progression of a relatively simple fatty streak to a more complex lesion.
Modified forms of LDL are immunogenic and activate both cell-mediated and humoural immune responses. Both types of responses are pro-inflammatory and are probably primary players in the perpetuation of the chronic inflammatory reaction characteristic of atherosclerosis. The immunologic response to modified LDL can be directed to major histocompatibilty complex class II (MHC-II)-associated peptides in the case of T helper cells, and to a variety of epitopes-modified lysine groups, modified phospholipids, proteins that become associated with ox-LDL (such as β2 glycoprotein 1) – in the case of B cell responses [34]. At the other hand, the activation of regulatory T cells may be protective. Indeed, recent data showed that alum precipitates, containing antigens derived from ox-LDL, increased regulatory T cells activated by tolerogenic antigen-presenting cells presenting ox-LDL antigens [35]. Similarly macrophages, endothelial cells and smooth muscle cells (SMCs) appear to be activated, demonstrated by increased expression of MHC-II molecules, and of toll-like receptors (TLRs) of which the ligands induce the release of numerous inflammatory products, such as TNF-α, IL-6 and MCP-1. Recently, we found that ox-LDL can also induce inflammation by inducing TLR-2 and -4 and the interferon regulatory factor-1 [36] (Fig. 1).
Moreover, activated macrophages produce pro-inflammatory secretory phospholipase A (2) (sPLA (2))-IIA. This enzyme lipolyzes the phospholipid monolayers of LDL, increasing its affinity to proteoglycans, rendering it more susceptible to aggregation, and enhancing its ability to insert cholesterol into cells [37]. This modification may promote scavenger receptor independent LDL uptake by macrophages leading to the formation of foam cells. In addition to sPLA2IIA that does not hydrolyse phosphatydylcholine (PC) into lyso-PC (LPC) [38], there are at least three sPLA2 that degrade intact PC in human LDL leading to foam cells formation, including PLA2G10 [39] and PLA2GIII [40] and to lesser extent PLA2GV [41]. Moreover there is a specific lipoprotein-associated PLA2 (Lp-PLA2; PLA2G7) that degrades oxidized ceramide found in ox-LDL and that increases its inflammatory action [42, 43]. Interestingly, we showed that ox-LDL induced the phospholipase expression by monocytes and macrophages [42].
Furthermore, activation of inflammatory cells is associated with the activation of sphingomyelinases (SMS) in the trans-Golgi apparatus and in plasma membranes resulting in the synthesis of sphingomyelin (SM), which transfers the phosphorylcholine moiety from phosphatidylcholine onto ceramide [44]. The interaction between SM, cholesterol, and glycosphingolipid drives the formation of SM-enriched lipid rafts resulting in an inhibition of the ATP-binding cassette transporter (ABC) A-1 and ABCG-1-mediated lipid efflux. This is important because macrophages cannot limit the uptake of cholesterol, and therefore depend on cholesterol and lipid efflux pathways to prevent their transformation into foam cells [45]. Ceramide production is associated with enhanced apoptosis of SMCs [46]. This may be beneficial because it prevents plaque growth. However, by preventing fibrous cap formation, it renders plaques more susceptible to rupture, and increases the risk of acute coronary syndromes. Interestingly, enhanced ceramide production was observed in association with the activation of MMP-2 by ox-LDL [47]. The association between ox-LDL, foam cell formation and MMP secretion is, however, not limited to macrophages but does also occur in SMCs [48]. Finally, ox-LDL induces the macrophage migration inhibitory factor that stimulates the migration of SMCs, contributing to intimal hyperplasia (Fig. 1).
Regulatory mechanisms of interactions between oxidative stress and inflammation
Recently, we obtained a mouse model of the metabolic syndrome that allowed the study of molecular mechanisms explaining the relations of the metabolic syndrome components with enhanced inflammation and oxidative stress. Indeed, we found that mice with combined leptin and LDL receptor deficiency (double knockout [DKO] mice) are obese and show severe hypertriglyceridaemia, hypertension and IR and diabetes. This combination of metabolic syndrome factors was associated with accelerated atherosclerosis due to increased accumulation of macrophages in association with endothelial dysfunction demonstrated by increased expression of VCAM-1 and ICAM-1 in the aorta of DKO mice [49]. Increased macrophage accumulation was associated with elevated plaque ox-LDL. The latter could be partly attributed to increased MPO by plaque macrophages. In addition, impaired high-density lipoprotein associated anti-oxidant activity in the blood [50] was associated with more ox-LDL in the plaques. We then investigated the relation between metabolic syndrome components and the oxidation of LDL further by assessing the effect of weight loss. We selected this intervention because it had been demonstrated that the cardiovascular risk of insulin-resistant obese persons is higher than that of insulin-sensitive obese persons, and that weight loss reduces the risk of insulin-resistant obese persons [51].
Weight loss in obese mice was associated with a decrease of metabolic syndrome components, resulting in reduced inflammation and oxidative stress. Ultimately, these changes led to inhibition of atherosclerosis [52] due to decreased accumulation of macrophages and deposition of ox-LDL. We showed that weight loss was not only associated with a decrease of the volume of adipose tissues, but also with improved adipocyte maturation demonstrated by induction of peroxisome proliferator activated receptors (PPARs) resulting in improved glucose uptake and insulin signalling, and fatty acid metabolism. This improved metabolic profile was associated with increased anti-oxidant protection, supported by increased expression of superoxide dismutase (SOD), glutathione peroxidase (GPX) and nitric oxide synthase (NOS). The decreased inflammation was supported by decreased expression of ICAM-1, CD44 and CD68 [52]. Similar effects were obtained with rosuvastatin that is known to increase insulin sensitivity and inhibit the oxidation of LDL [53]. Cell experiments [54] indicated that PPAR-γ can inhibit the accumulation of ox-LDL by increasing the anti-oxidant defence by up-regulating SOD-1 and -3 and GPX (Fig. 2). PPAR-γ also increases CD36-mediated clearance of ox-LDL. The decrease of ox-LDL is associated with a reduction of the TLR-mediated inflammatory response. PPAR-γ also decreases expressions of monocyte-specific adhesion molecules and chemotactic factors, resulting in less macrophage accumulation. In addition, PPAR-γ increases NOS production associated with improved endothelial function, induces the efflux of cholesterol and lipids from foam cells by up-regulating the liver X receptors (LXR)-α and ABCA-1. Finally, PPAR-γ induces the expressions of the glucose transporter-4 and the insulin receptor substrate-2 (IRS-2), which activate glucose uptake and insulin action. The improved IRS-2-mediated insulin signalling is associated with increased SOD-2 expression, resulting in lower mitochondrial oxidative stress and ROS production. Finally, PPAR-γ activation was found to prime monocytes into alternative M2 macrophages with anti-inflammatory properties [55].
Fig 2.
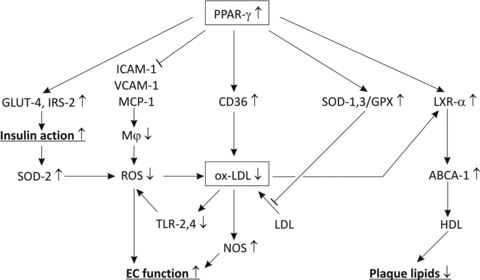
Regulatory mechanisms of interactions between oxidative stress and inflammation. PPAR-γ reduces the expressions of ICAM-1, VCAM-1 and MCP-1, resulting in reduced macrophage accumulation, ROS production and ox-LDL deposit. In addition, PPAR-γ increases expressions of CD36 and anti-oxidant enzymes SOD and GPX, resulting in a further reduction of ox-LDL. Higher PPAR-γ is also associated with increased LXR-α and ABCA-1 expression, resulting in a decrease of cholesterol and lipids. The reduction in ox-LDL is associated with a decrease of TLR-mediated inflammation, and thereby reduced ROS production. In addition this reduction is associated with higher NOS production resulting in improved endothelial vasoreactivity, blood pressure regulation and left ventricle function. Reduction of ox-LDL results in restoring PPAR-γ expression that is associated with increased IRS-2 and glucose transporter-4 expressions, which are important regulators of insulin sensitivity and glucose uptake. Improved insulin action results in decreased mitochondrial oxidative stress, increased SOD-2 expression and reduced ROS production.
Overview of molecular mechanisms explaining the association of inflammation and oxidative stress in adipose tissues
Obesity is associated with reduced adipose tissue oxygenation and hypoxia, without the appropriate angiogenic response, due to defective activation of LXRs [56] and vascular endothelial growth factor (VEGF) [57], but with increased macrophage chemotaxis [23] (Fig. 3). Indeed very recent findings suggest that obese adipose tissue activates CD8+ T cells, which, in turn, promote the recruitment and activation of macrophages in this tissue and support the notion that CD8+ T cells have an essential role in the initiation and propagation of adipose inflammation [58, 59]. Finally, it was found that lean, but not obese, fat is enriched for a unique population of regulatory T cells that protects against the harmful effect of metabolic parameters [60].
Fig 3.
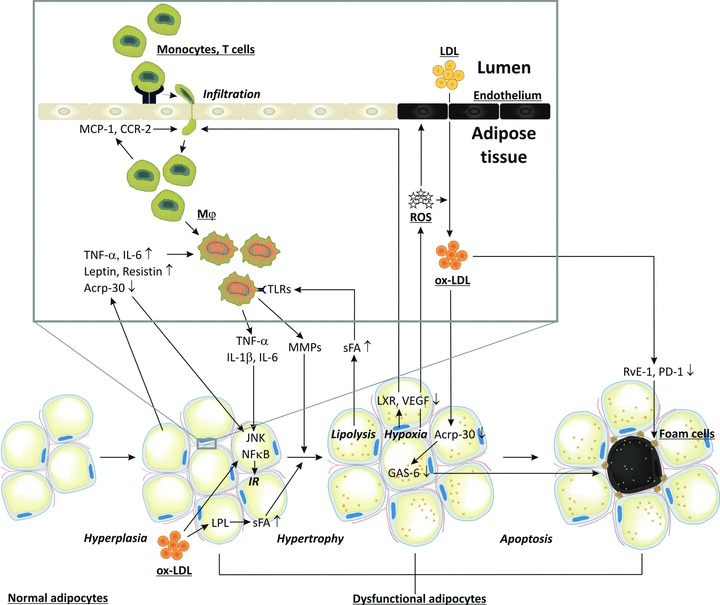
Macrophage infiltration in adipose tissues, oxidative stress and IR. Circulating monocytes adhere to activated endothelial cells. Activated CD8+ T cells and chemokines induce monocyte migration into adipose tissues where they differentiate into macrophages. Interaction of saturated fatty acids with TLRs leads to secretion of inflammatory cytokines/chemokines (IL-6 and TNF-α). Together with the adipocytokines leptin and resistin, they impair c-jun N-terminal kinase and NF-κB signalling, resulting in IR and reduced adiponectin (Acrp-30) secretion, and thereby loss of adipocyte maturation. Together they also induce adipocyte proliferation. Infiltration of inflammatory cells is associated with ROS and ox-LDL production, endothelial cell apoptosis, impaired LXR and VEGF signalling, and decreased blood flow leading to hypoxia and increased oxidative stress. Ox-LDL can further induce adipose tissue hyperplasia, and by inducing lipoprotein lipase it enhances lipid accumulation resulting in adipose tissue hypertrophy. The latter is also facilitated by MMPs secreted by macrophages. Hypoxia and increased oxidative stress induces apoptosis of adipocytes. Increased apoptosis also results from reduced Acrp-30 secretion and growth arrest specific 6 mediated survival pathway. Apoptotic adipocytes attract macrophages which normally remove apoptotic adipocytes. However, ox-LDL impairs phagocytosis of dead adipocytes by inhibiting resolvin E1 and protectin D1 production.
Infiltrated inflammatory cells produce and secrete TNF-α and IL-6 contributing to the pathogenesis of IR, together with factors produced by dysfunctional adipocytes such as leptin and resistin. Conversely, insulin-sensitizing adiponectin is down-regulated during obesity [61]. A range of mouse models with loss-of-function mutations in genes important in macrophage recruitment (MCP-1 and chemokine (C-C motif) receptor 2), inflammatory cytokine production (TNF-α) and pro-inflammatory activation (NF-κB [nuclear factor of κ light polypeptide gene enhancer in B-cells] and IκB kinase [IKK]β) have demonstrated to be protected against high-fat diet-induced IR [62–64]. Interestingly, weight loss was associated with a reduction in the macrophage infiltration of adipose tissues, associated with an improvement of the inflammatory and oxidant profile of adipocytes and monocytes in adipose tissue and the circulation, respectively. Furthermore, weight loss resulted in enhanced insulin sensitivity and a decrease of the cardiovascular risk of insulin-resistant, obese persons [51].
In addition to their roles in inflammation, macrophages also promote vascular remodelling of adipose tissues [65] by producing MMPs. This occurs preferentially at sites of adipocyte death where macrophages form a crown-like structure that envelopes and ingests the moribund adipocyte and its potentially cytotoxic remnant lipid droplets [66]. As a consequence of lipid scavenging, macrophages within crown-like structures become lipid-loaded foam cells that fuse to multinucleate giant cells that have lost their capacity to remove dead adipocytes. During resolution, specific ω-3 polyunsaturated fatty-acid-derived mediators, including resolvin E1 and protectin D1, promote phagocyte removal during acute inflammation by inhibiting leucocyte infiltration and increasing macrophage ingestion of apoptotic cells [67]. This resolution is impaired in obesity leading to defective protection against oxidative stress-initiated inflammation by glutathione conjugates of lipid oxidation-derived aldehydes, such as 4-hydroxy-trans-2-nonenal present in ox-LDL [68].
A possible explanation for the relation between ox-LDL and obesity is that ox-LDL increases the volume of adipose tissues either directly by inducing adipocyte proliferation [69] or indirectly by increasing the infiltration of inflammatory monocytes/macrophages which increase adipogenesis [70]. The increase in adipose tissue mass may also be explained by a cellular hypertrophy due to an increased lipid accumulation in the pre-existing adipocytes rather than an increase in cell number or differentiation. Indeed, ox-LDL increases triglyceride production by inducing the expression of lipoprotein lipase [71], and the accumulation of fatty acids in adipocytes [72]. Interestingly, fatty acids stimulate the accumulation of ceramide that contributes to inflammation that, as discussed above, is associated with adipose hyperplasia. Ox-LDL was also found to decrease the production of adiponectin that in contrast with other adipocytokines is reduced in obese persons, and suppresses excess ROS production under high-glucose conditions, an effect that has implications for vascular protection in diabetes [73]. In addition, the decrease of adiponectin results in a loss of its capacity to protect cells against inactivation of the growth arrest specific 6 mediated survival pathway by TNF-α[74]. The observed relations between obesity and ox-LDL are important to understand the relation of obesity with IR [27] and the metabolic syndrome [75]. All these processes are summarized in Fig. 3.
Conclusions
Experimental, clinical and population studies demonstrated active roles of oxidative stress and inflammation in the development of obesity-associated metabolic syndrome and cardiovascular diseases. Herein, we presented evidence of a vicious circle between inflammation and oxidative stress not only in vascular but also in adipose tissues. Inflammatory cells increase the production of ROS and ox-LDL by secreting oxidant enzymes. They impair the function of endothelial cells and SMCs in vascular, and of endothelial cells and adipocytes in adipose tissues. Especially, inhibiting the IRS-mediated insulin action is associated with a decrease in anti-oxidant enzymes, and an increase of mitochondrial oxidative stress in both tissue types. By inducing the production of foam cells, ox-LDL decreases the phagocytosis of apoptotic cells by macrophages, resulting in impaired insulin action. By attacking endothelial cells, ox-LDL impairs blood flow and causes hypoxia and oxidative stress.
In addition, we showed that PPAR-γ and adiponectin protect against oxidative stress and inflammation. Loss of adiponectin-mediated signalling between adipose and arterial tissues is of particular importance in the development of obesity-associated atherosclerosis and cardiovascular diseases.
Acknowledgments
Funding was provided by the Interuniversitaire Attractiepolen Programma of the Belgian Federal Government (P06/30), the Bijzonder Onderzoeksfonds of the Katholieke Universiteit Leuven (OT/06/56) and by the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (Vascular Biology Network).
References
- 1.Fonseca V, Desouza C, Asnani S, et al. Nontraditional risk factors for cardiovascular disease in diabetes. Endocr Rev. 2004;25:153–75. doi: 10.1210/er.2002-0034. [DOI] [PubMed] [Google Scholar]
- 2.Duncan BB, Schmidt MI, Pankow JS, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes. 2003;52:1799–805. doi: 10.2337/diabetes.52.7.1799. [DOI] [PubMed] [Google Scholar]
- 3.Pradhan AD, Manson JE, Rifai N, et al. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–34. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 4.Cipolletta C, Ryan KE, Hanna EV, et al. Activation of peripheral blood CD14+ monocytes occurs in diabetes. Diabetes. 2005;54:2779–86. doi: 10.2337/diabetes.54.9.2779. [DOI] [PubMed] [Google Scholar]
- 5.Expert Panel on the Detection, Evaluation and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 6.Ford ES. The metabolic syndrome and mortality from cardiovascular disease and all-causes: findings from the National Health and Nutrition Examination Survey II Mortality Study. Atherosclerosis. 2004;173:309–14. doi: 10.1016/j.atherosclerosis.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 7.Grundy SM, Brewer HB, Jr, Cleeman JI, et al. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation. 2004;109:433–8. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 8.Kahn R, Buse J, Ferrannini E, et al. The metabolic syndrome: time for a critical appraisal: joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28:2289–304. doi: 10.2337/diacare.28.9.2289. [DOI] [PubMed] [Google Scholar]
- 9.Conen D, Rexrode KM, Creager MA, et al. Metabolic syndrome, inflammation, and risk of symptomatic peripheral artery disease in women: a prospective study. Circulation. 2009;120:1041–7. doi: 10.1161/CIRCULATIONAHA.109.863092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ridker PM, Buring JE, Cook NR, et al. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation. 2003;107:391–7. doi: 10.1161/01.cir.0000055014.62083.05. [DOI] [PubMed] [Google Scholar]
- 11.Holvoet P, Kritchevsky SB, Tracy RP, et al. The metabolic syndrome, circulating oxidized LDL, and risk of myocardial infarction in well-functioning elderly people in the health, aging, and body composition cohort. Diabetes. 2004;53:1068–73. doi: 10.2337/diabetes.53.4.1068. [DOI] [PubMed] [Google Scholar]
- 12.Holvoet P, Lee DH, Steffes M, et al. Association between circulating oxidized low-density lipoprotein and incidence of the metabolic syndrome. JAMA. 2008;299:2287–93. doi: 10.1001/jama.299.19.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girman CJ, Rhodes T, Mercuri M, et al. The metabolic syndrome and risk of major coronary events in the Scandinavian Simvastatin Survival Study (4S) and the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) Am J Cardiol. 2004;93:136–41. doi: 10.1016/j.amjcard.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 14.Hunt KJ, Resendez RG, Williams K, et al. National Cholesterol Education Program versus World Health Organization metabolic syndrome in relation to all-cause and cardiovascular mortality in the San Antonio Heart Study. Circulation. 2004;110:1251–7. doi: 10.1161/01.CIR.0000140762.04598.F9. [DOI] [PubMed] [Google Scholar]
- 15.Isomaa B, Henricsson M, Almgren P, et al. The metabolic syndrome influences the risk of chronic complications in patients with type II diabetes. Diabetologia. 2001;44:1148–54. doi: 10.1007/s001250100615. [DOI] [PubMed] [Google Scholar]
- 16.Lakka HM, Laaksonen DE, Lakka TA, et al. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA. 2002;288:2709–16. doi: 10.1001/jama.288.21.2709. [DOI] [PubMed] [Google Scholar]
- 17.Lempiainen P, Mykkanen L, Pyorala K, et al. Insulin resistance syndrome predicts coronary heart disease events in elderly nondiabetic men. Circulation. 1999;100:123–8. doi: 10.1161/01.cir.100.2.123. [DOI] [PubMed] [Google Scholar]
- 18.Malik S, Wong ND, Franklin SS, et al. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–50. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 19.Onat A, Ceyhan K, Basar O, et al. Metabolic syndrome: major impact on coronary risk in a population with low cholesterol levels–a prospective and cross-sectional evaluation. Atherosclerosis. 2002;165:285–92. doi: 10.1016/s0021-9150(02)00236-8. [DOI] [PubMed] [Google Scholar]
- 20.Stern MP, Williams K, Hunt KJ. Impact of diabetes/metabolic syndrome in patients with established cardiovascular disease. Atheroscler Suppl. 2005;6:3–6. doi: 10.1016/j.atherosclerosissup.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–61. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14:1225–30. doi: 10.2174/138161208784246153. [DOI] [PubMed] [Google Scholar]
- 23.Reilly MP, Lehrke M, Wolfe ML, et al. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111:932–9. doi: 10.1161/01.CIR.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 24.De Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582:97–105. doi: 10.1016/j.febslet.2007.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holvoet P, Theilmeier G, Shivalkar B, et al. LDL hypercholesterolemia is associated with accumulation of oxidized LDL, atherosclerotic plaque growth, and compensatory vessel enlargement in coronary arteries of miniature pigs. Arterioscler Thromb Vasc Biol. 1998;18:415–22. doi: 10.1161/01.atv.18.3.415. [DOI] [PubMed] [Google Scholar]
- 26.Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:266–74. doi: 10.1161/01.ATV.0000253884.13901.e4. [DOI] [PubMed] [Google Scholar]
- 27.Park K, Gross M, Lee DH, et al. Oxidative stress and insulin resistance: the coronary artery risk development in young adults study. Diabetes Care. 2009;32:1302–7. doi: 10.2337/dc09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronald JA, Chen JW, Chen Y, et al. Enzyme-sensitive magnetic resonance imaging targeting myeloperoxidase identifies active inflammation in experimental rabbit atherosclerotic plaques. Circulation. 2009;120:592–9. doi: 10.1161/CIRCULATIONAHA.108.813998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subbanagounder G, Leitinger N, Schwenke DC, et al. Determinants of bioactivity of oxidized phospholipids. Specific oxidized fatty acyl groups at the sn-2 position. Arterioscler Thromb Vasc Biol. 2000;20:2248–54. doi: 10.1161/01.atv.20.10.2248. [DOI] [PubMed] [Google Scholar]
- 30.Subbanagounder G, Wong JW, Lee H, et al. Epoxyisoprostane and epoxycyclopentenone phospholipids regulate monocyte chemotactic protein-1 and interleukin-8 synthesis. Formation of these oxidized phospholipids in response to interleukin-1beta. J Biol Chem. 2002;277:7271–81. doi: 10.1074/jbc.M107602200. [DOI] [PubMed] [Google Scholar]
- 31.Mukhopadhyay S, Pluddemann A, Gordon S. Macrophage pattern recognition receptors in immunity, homeostasis and self tolerance. Adv Exp Med Biol. 2009;653:1–14. doi: 10.1007/978-1-4419-0901-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moura R, Tjwa M, Vandervoort P, et al. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE-/- mice. Circ Res. 2008;103:1181–9. doi: 10.1161/CIRCRESAHA.108.185645. [DOI] [PubMed] [Google Scholar]
- 33.Sakamoto Y, Miyazaki A, Tamagawa H, et al. Specific interaction of oxidized low-density lipoprotein with thrombospondin-1 inhibits transforming growth factor-beta from its activation. Atherosclerosis. 2005;183:85–93. doi: 10.1016/j.atherosclerosis.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 34.Virella G, Lopes-Virella MF. Atherogenesis and the humoral immune response to modified lipoproteins. Atherosclerosis. 2008;200:239–46. doi: 10.1016/j.atherosclerosis.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wigren M, Bengtsson D, Duner P, et al. Atheroprotective effects of Alum are associated with capture of oxidized LDL antigens and activation of regulatory T cells. Circ Res. 2009;104:e62–e70. doi: 10.1161/CIRCRESAHA.109.196667. [DOI] [PubMed] [Google Scholar]
- 36.Holvoet P, Davey PC, De Keyzer D, et al. Oxidized low-density lipoprotein correlates positively with toll-like receptor 2 and interferon regulatory factor-1 and inversely with superoxide dismutase-1 expression: studies in hypercholesterolemic swine and THP-1 cells. Arterioscler Thromb Vasc Biol. 2006;26:1558–65. doi: 10.1161/01.ATV.0000226553.01555.02. [DOI] [PubMed] [Google Scholar]
- 37.Hakala JK, Oorni K, Pentikainen MO, et al. Lipolysis of LDL by human secretory phospholipase A(2) induces particle fusion and enhances the retention of LDL to human aortic proteoglycans. Arterioscler Thromb Vasc Biol. 2001;21:1053–8. doi: 10.1161/01.atv.21.6.1053. [DOI] [PubMed] [Google Scholar]
- 38.Pruzanski W, Lambeau L, Lazdunsky M, et al. Differential hydrolysis of molecular species of lipoprotein phosphatidylcholine by groups IIA, V and X secretory phospholipases A2. Biochim Biophys Acta. 2005;1736:38–50. doi: 10.1016/j.bbalip.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Karabina SA, Brocheriou I, Le Naour G, et al. Atherogenic properties of LDL particles modified by human group X secreted phospholipase A2 on human endothelial cell function. FASEB J. 2006;20:2547–9. doi: 10.1096/fj.06-6018fje. [DOI] [PubMed] [Google Scholar]
- 40.Sato H, Kato R, Isogai Y, et al. Analyses of group III secreted phospholipase A2 transgenic mice reveal potential participation of this enzyme in plasma lipoprotein modification, macrophage foam cell formation, and atherosclerosis. J Biol Chem. 2008;283:33483–97. doi: 10.1074/jbc.M804628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boyanovsky B, Zack M, Forrest K, et al. The capacity of group V sPLA2 to increase atherogenicity of ApoE-/- and LDLR-/- mouse LDL in vitro predicts its atherogenic role in vivo. Arterioscler Thromb Vasc Biol. 2009;29:532–8. doi: 10.1161/ATVBAHA.108.183038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Keyzer D, Karabina SA, Wei W, et al. Increased PAFAH and oxidized lipids are associated with inflammation and atherosclerosis in hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2009;29:2041–46. doi: 10.1161/ATVBAHA.109.196592. [DOI] [PubMed] [Google Scholar]
- 43.Vickers KC, Maguire CT, Wolfert R, et al. Relationship of lipoprotein-associated phospholipase A2 and oxidized low-density lipoprotein in carotid atherosclerosis. J Lipid Res. 2009;50:1735–43. doi: 10.1194/jlr.M800342-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merrill AH, Jr, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Huan C, Chakraborty M, et al. Macrophage sphingomyelin synthase 2 deficiency decreases atherosclerosis in mice. Circ Res. 2009;105:295–303. doi: 10.1161/CIRCRESAHA.109.194613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kolmakova A, Kwiterovich P, Virgil D, et al. Apolipoprotein C-I induces apoptosis in human aortic smooth muscle cells via recruiting neutral sphingomyelinase. Arterioscler Thromb Vasc Biol. 2004;24:264–9. doi: 10.1161/01.ATV.0000112036.72200.ac. [DOI] [PubMed] [Google Scholar]
- 47.Auge N, Maupas-Schwalm F, Elbaz M, et al. Role for matrix metalloproteinase-2 in oxidized low-density lipoprotein-induced activation of the sphingomyelin/ceramide pathway and smooth muscle cell proliferation. Circulation. 2004;110:571–8. doi: 10.1161/01.CIR.0000136995.83451.1D. [DOI] [PubMed] [Google Scholar]
- 48.Segers D, Helderman F, Cheng C, et al. Gelatinolytic activity in atherosclerotic plaques is highly localized and is associated with both macrophages and smooth muscle cells in vivo. Circulation. 2007;115:609–16. doi: 10.1161/CIRCULATIONAHA.106.636415. [DOI] [PubMed] [Google Scholar]
- 49.Mertens A, Verhamme P, Bielicki JK, et al. Increased low-density lipoprotein oxidation and impaired high-density lipoprotein antioxidant defense are associated with increased macrophage homing and atherosclerosis in dyslipidemic obese mice: LCAT gene transfer decreases atherosclerosis. Circulation. 2003;107:1640–6. doi: 10.1161/01.CIR.0000056523.08033.9F. [DOI] [PubMed] [Google Scholar]
- 50.Mertens A, Holvoet P. Oxidized LDL and HDL: antagonists in atherothrombosis. FASEB J. 2001;15:2073–84. doi: 10.1096/fj.01-0273rev. [DOI] [PubMed] [Google Scholar]
- 51.McLaughlin T, Abbasi F, Kim HS, et al. Relationship between insulin resistance, weight loss, and coronary heart disease risk in healthy, obese women. Metabolism. 2001;50:795–800. doi: 10.1053/meta.2001.24210. [DOI] [PubMed] [Google Scholar]
- 52.Verreth W, De Keyzer D, Pelat M, et al. Weight-loss-associated induction of peroxisome proliferator-activated receptor-alpha and peroxisome proliferator-activated receptor-gamma correlate with reduced atherosclerosis and improved cardiovascular function in obese insulin-resistant mice. Circulation. 2004;110:3259–69. doi: 10.1161/01.CIR.0000147614.85888.7A. [DOI] [PubMed] [Google Scholar]
- 53.Verreth W, De Keyzer D, Davey PC, et al. Rosuvastatin restores superoxide dismutase expression and inhibits accumulation of oxidized LDL in the aortic arch of obese dyslipidemic mice. Br J Pharmacol. 2007;151:347–55. doi: 10.1038/sj.bjp.0707231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Desjardins F, Sekkali B, Verreth W, et al. Rosuvastatin increases vascular endothelial PPARgamma expression and corrects blood pressure variability in obese dyslipidaemic mice. Eur Heart J. 2008;29:128–37. doi: 10.1093/eurheartj/ehm540. [DOI] [PubMed] [Google Scholar]
- 55.Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 56.Walczak R, Joseph SB, Laffitte BA, et al. Transcription of the vascular endothelial growth factor gene in macrophages is regulated by liver X receptors. J Biol Chem. 2004;279:9905–11. doi: 10.1074/jbc.M310587200. [DOI] [PubMed] [Google Scholar]
- 57.Pasarica M, Sereda OR, Redman LM, et al. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes. 2009;58:718–25. doi: 10.2337/db08-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1304–10. doi: 10.1161/ATVBAHA.108.165100. [DOI] [PubMed] [Google Scholar]
- 59.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 60.Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–9. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bastard JP, Maachi M, Lagathu C, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12. [PubMed] [Google Scholar]
- 62.Arkan MC, Hevener AL, Greten FR, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–8. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 63.Uysal KT, Wiesbrock SM, Marino MW, et al. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 64.Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–24. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cho CH, Koh YJ, Han J, et al. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ Res. 2007;100:e47–e57. doi: 10.1161/01.RES.0000259564.92792.93. [DOI] [PubMed] [Google Scholar]
- 66.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 67.Schwab JM, Chiang N, Arita M, et al. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–74. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spite M, Summers L, Porter TF, et al. Resolvin D1 controls inflammation initiated by glutathione-lipid conjugates formed during oxidative stress. Br J Pharmacol. 2009;158:1062–73. doi: 10.1111/j.1476-5381.2009.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Masella R, Vari R, D’Archivio M, et al. Oxidised LDL modulate adipogenesis in 3T3-L1 preadipocytes by affecting the balance between cell proliferation and differentiation. FEBS Lett. 2006;580:2421–9. doi: 10.1016/j.febslet.2006.03.068. [DOI] [PubMed] [Google Scholar]
- 70.Nishimura S, Manabe I, Nagasaki M, et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes. 2007;56:1517–26. doi: 10.2337/db06-1749. [DOI] [PubMed] [Google Scholar]
- 71.Stengel D, Antonucci M, Gaoua W, et al. Inhibition of LPL expression in human monocyte-derived macrophages is dependent on LDL oxidation state: a key role for lysophosphatidylcholine. Arterioscler Thromb Vasc Biol. 1998;18:1172–80. doi: 10.1161/01.atv.18.7.1172. [DOI] [PubMed] [Google Scholar]
- 72.Merkel M, Heeren J, Dudeck W, et al. Inactive lipoprotein lipase (LPL) alone increases selective cholesterol ester uptake in vivo, whereas in the presence of active LPL it also increases triglyceride hydrolysis and whole particle lipoprotein uptake. J Biol Chem. 2002;277:7405–11. doi: 10.1074/jbc.M107914200. [DOI] [PubMed] [Google Scholar]
- 73.Ouedraogo R, Wu X, Xu SQ, et al. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: evidence for involvement of a cAMP signaling pathway. Diabetes. 2006;55:1840–6. doi: 10.2337/db05-1174. [DOI] [PubMed] [Google Scholar]
- 74.Son BK, Akishita M, Iijima K, et al. Adiponectin antagonizes stimulatory effect of tumor necrosis factor-alpha on vascular smooth muscle cell calcification: regulation of growth arrest-specific gene 6-mediated survival pathway by adenosine 5′-monophosphate-activated protein kinase. Endocrinology. 2008;149:1646–53. doi: 10.1210/en.2007-1021. [DOI] [PubMed] [Google Scholar]
- 75.Goodpaster BH, Krishnaswami S, Harris TB, et al. Obesity, regional body fat distribution, and the metabolic syndrome in older men and women. Arch Intern Med. 2005;165:777–83. doi: 10.1001/archinte.165.7.777. [DOI] [PubMed] [Google Scholar]