

Abstract
Ultraviolet radiation (UVR) phototherapy is a promising new treatment for inflammatory airway diseases. However, the potential carcinogenic risks associated with this treatment are not well understood. UV-specific DNA photoproducts were used as biomarkers to address this issue. Radioimmunoassay was used to quantify cyclobutane pyrimidine dimers (CPDs) and (6–4) photoproducts in DNA purified from two milieus: nasal mucosa samples from subjects exposed to intranasal phototherapy and human airway (EpiAirway™) and human skin (EpiDerm™) tissue models. Immunohistochemistry was used to detect CPD formation and persistence in human nasal biopsies and human tissue models. In subjects exposed to broadband ultraviolet radiation, DNA damage frequencies were determined prior to as well as immediately after treatment and at increasing times post-treatment. We observed significant levels of DNA damage immediately after treatment and efficient removal of the damage within a few days. No residual damage was observed in human subjects exposed to multiple UVB treatments several weeks after the last treatment. To better understand the molecular response of the nasal epithelium to DNA damage, parallel experiments were conducted in EpiAirway and EpiDerm model systems. Repair rates in these two tissues were very similar and comparable to that observed in human skin. The data suggest that the UV-induced DNA damage response of respiratory epithelia is very similar to that of the human epidermis and that nasal mucosa is able to efficiently repair UVB induced DNA damage.
Keywords: nucleotide excision repair, ultraviolet radiation, cyclobutane pyrimidine dimer, pyrimidine(6–4)pyrimidone photoproduct, reconstructed 3-D human respiratory epithelium, reconstructed 3-D human epidermis, rhinophototherapy
Introduction
Phototherapy of inflammatory and immune-mediated skin diseases such as psoriasis vulgaris, atopic dermatitis and vitiligo is a well-recognized treatment option in routine dermatological practice; its efficacy has been demonstrated in several studies and therefore is recommended by the guidelines of dermatological societies all over the world [1–3]. Recently, phototherapy has been applied to treat immune-mediated mucosal diseases (oral lichen planus, oral manifestations of graft versus host disease and allergic rhinitis) and therefore may represent a promising new therapy for several manifestations of inflammatory airway disease [4–6].
Evidence strongly suggests that UV-B (285–315 nm) induced DNA photoproducts are the predominant premutagenic events responsible for the initiation of human carcinomas [7]. The two major photoproducts induced in DNA by UVB occur at sites of adjacent pyrimidine bases and include the cyclobutane pyrimidine dimer (CPD) and pyrimidine-pyrimidone (6–4) dimer [(6–4)PD]. Transition mutations arising chiefly at the 3′ base of a T-C dipyrimidine have been found in the p53 tumour suppressor gene of ∼50% of human basal cell carcinomas [8] and in the ras proto-oncogene at a much lower rate [9]. Along with CC→TT tandem double mutations, C→T transitions are considered the ‘signature’ mutations of UVB light. Numerous studies have shown that UV B-induced DNA damage can increase the expression of p53 protein, a key element for cell cycle regulation [10]. The accumulation of p53 protein induces cell cycle arrest at the G1/S boundary and increases repair of DNA damage prior to replication. If the amount of damage is excessive and cannot be completely removed, cells may enter apoptosis. Apoptosis is partly determined by DNA damage and p53 and partly by other mechanisms, such as cross-linking of surface receptors [10].
Nucleotide excision repair (NER) is the predominant DNA repair pathway used by cells to remove bulky lesions such as pyrimidine dimers [11]. NER consists of two subpathways, global genome repair, which removes lesions from the entire genome and transcription coupled repair, which preferentially repairs damage in actively transcribing regions of DNA. Failure to properly repair DNA damage results in mutations that impair cell function and eventually lead to cancer. Several mutation hotspots have been identified in the p53 gene of non-melanoma skin cancers [12] and all have been correlated with either UVB-induced DNA damage hotspots [13] or CPD excision coldspots [14]. It is thought that chronic UVR confers a selective advantage on cells with a dysfunctional p53 gene and that reduced apoptosis and increased clonal expansion of these damaged cells leads to formation of mutant p53 clusters, actinic keratoses and, ultimately carcinomas [7, 8, 15, 16].
DNA photoproducts caused by chronic exposure to sunlight are responsible for initiating photocarcinogenesis in the skin (e.g. basal and squamous cell carcinomas). Previous studies demonstrate that skin cells exposed to UV slowly over a long period of time, conditions that mimic those associated with many occupational and recreational behaviours, respond in quite unexpected ways. We have identified cells at the dermal-epidermal boundary that accumulate significant levels of DNA damage in chronically irradiated mice as well as in sun-exposed human skin [17, 18]. These cells appear to be stem cells and may indeed represent pre-initiation events in photocarcinogenesis. The persistence of DNA damage-retaining stem cells in chronically irradiated tissues is, thus, an indicator of the carcinogenic potential of a phototherapeutic treatment protocol.
Because phototherapy of inflammatory airway diseases requires several exposures to UV light, it is important to determine if rhinophototherapy (RPT) poses any risk for carcinogenesis. As respiratory tissues are not routinely exposed to UV radiation, risk assessment necessarily involves comparing the biological and molecular response of these tissues to those in a system where UV-induced carcinogenesis is well characterized, namely human epidermis. Recent publications have indicated that controlled delivery of UV B phototherapy for inflammatory skin diseases is safe and is not accompanied by a significant increase of skin carcinoma [19, 20]. With this in mind, our risk assessment focused on the type of DNA damage associated with initiation in squamous and basal cell carcinoma, that is the production and resolution of CPDs. The formation and persistence of CPDs was monitored in the nasal epithelia from subjects receiving single and multiple UV exposures and compared to previously published data for human epidermis. In addition, we quantified and compared the induction and repair of the two major photoproducts formed after exposure to UV, the CPD and (6–4) photoproduct, in reconstructed normal human three-dimensional respiratory epithelium (EpiAirway) and skin (EpiDerm) (MatTek Corp, Ashland, MA, USA).
Materials and methods
DNA damage and repair after a single UVR exposure of human nasal mucosa
Thirty adult subjects participated in the study, which was approved by the Central Ethics Committee in Hungary and by the Local Ethics Committee of the University of Szeged, Hungary. All subjects gave written informed consent before participating in the study. All subjects underwent baseline nasal mucosa sampling using a plastic curette (Rhino-probe™, ASI, Arlington, TX, USA) from the antero-medial aspect of the inferior turbinate. Two weeks after baseline sampling, all subjects were exposed to one intranasal UV treatment in both nostrils using a broadband ultraviolet (BB-UV) light source (Allux Medical Inc., Menlo Park, CA, USA). The light source uses a mercury xenon bulb and a series of filters to create the light spectrum that is a combination of UVC (2.4 mW), UVB (8.2 mW) and UVA (23.8 mW) as well as some visible light. Each nostril was treated for 3 min. using a similar technique as used for treating allergic rhinitis [4]. Immediately after exposure, nasal mucosa samples were collected from one nostril. A second nasal cytology sample was collected from the contralateral nostril 24 hrs, 48 hrs and 72 hrs after UV exposure from 10 subjects at each time-point (subjects were randomly assigned to one of the 3 time-points listed above). From the nasal cytology samples DNA was extracted for RIA analysis.
DNA damage and repair after multiple UV light exposures of human nasal mucosa
Twenty-six allergic rhinitis subjects were included in the study. The study was approved by the Central Ethics Committee in Hungary and by the Local Ethics Committee of the University of Szeged, Hungary. As above, all subjects gave written informed consent before participating in the study. The study was performed during the 2006 ragweed pollen season in Szeged, Hungary. Subjects were randomized to either receive intranasal phototherapy using the same BB-UV light source (Allux Medical Inc.) or low-dose visible light placebo therapy for 3 weeks (3 times per week). Nasal mucosa samples were collected from the antero-medial surface of the inferior turbinate at the following time-points: before any exposure, immediately after last exposure, 1 week after the last exposure and 1 month after the last exposure. DNA was extracted from the nasal mucosal samples and shipped to Texas for DNA damage analysis.
It has been shown that CPDs can accumulate in basal epidermal cells after chronic irradiation of mouse and human epidermis and that this damage may play an important role in carcinogenesis. With this in mind, we examined the persistence of DNA damage in individual cells of subjects that received multiple UV exposures. Biopsies were collected from 13 subjects who participated in the multiple exposure experiment (9 from the UVR-treated group and 4 from the placebo group). The history of prior RPT in this group was also determined. Tissue collection was performed 2 months after finishing the treatment regimen and a biopsy of the inferior turbinate was performed under local anaesthesia. Tissue was fixed in 10% buffered formalin, embedded in paraffin blocks and processed for routine staining (H&E stain) and for detection of CPDs with immunohistochemical techniques. A core laboratory with extensive experience in airway pathology examined the slides and the pathologist was blinded to the identity of the tissue samples.
DNA damage and repair in EpiDerm and EpiAirway exposed to UVR
The EpiAirway™ tissue (MatTek Corporation, Ashland, MA, USA) consists of normal, human-derived respiratory epithelial cells that were cultured to form a pseudo-stratified, highly differentiated 3-dimensional model that closely resembles the epithelial tissue of the upper respiratory tract. The EpiDerm™ tissue (MatTek Corporation, Ashland, MA, USA) consists of normal, human-derived epidermal keratinocytes, which have been cultured to form a multilayer, highly differentiated three-dimensional model of human epidermis; the tissue possesses all the cell layers and the ultrastructural characteristics of human epidermis.
After receipt, the tissues were transferred to 6-well culture plates and fed with either AIR-100-ASY or EPI-100-ASY supplied culture medium (MatTek Corporation, Ashland, MA, USA). Tissues were re-fed the following day. After 2 days, when growth was at an optimum, UV exposure experiments were performed. Two different types of experiments were performed: one set of experiments was designed to address the repair of UV-specific photoproducts in the two tissues and the other set to explore the depth of penetration of DNA damage induced by different light sources in the two tissue types.
For the DNA repair experiments EpiAirway tissues were exposed to UV light (100 J/m2 and 1000 J/m2). EpiDerm tissues were exposed to 3 doses: 100 J/m2, an adjusted dose of 258 J/m2 (to account for shielding by stratum corneum) and 1000 J/m2 dose. The tissue was harvested at 0, 3, 6, 12 and 24 hrs after exposure. The entire tissue was separated from the support material and harvested for DNA isolation using standard techniques. Each experiment was performed in duplicate. An unirradiated control was harvested also for each dose.
DNA damage penetration in EpiDerm and EpiAirway tissues exposed to UVR
EpiAirway and EpiDerm tissues were exposed to three different doses from each of 4 different light sources; including (1) a narrow-band germicidal UVC source consisting of 5 Philips Sterilamp G8T5 emitting predominantly 254 nm radiation (NB-UVC), (2) a UVB source consisting of 4 Philips TL-01 100W lamps filtered through cellulose acetate (Kodacel, Kodak) emitting narrow band 313 nm radiation (NB-UVB), (3) the broad-band UV light source provided by Allux Medical (BB-UV) and (4) the BB-UV light source from which the UVB had been filtered out, resulting in a broad-band UVC spectrum (BB-UVC). Dose rates were measured using an IL1400A radiometer coupled to appropriate probes for each light source (International Light, Place). Probes included NS 254 #11306 for UVC, SCS 280 #11174 for UVB and WBS320 #19118/QNDS1 #27218/T #25347 for the BB-UV light source. Doses were adjusted to reflect CPD induction frequencies equivalent to those produced by the unmodified BB-UV light source determined from preliminary MatTek experiments. For example, the CPD induction rate in DNA by UVC is 5.9-fold greater than of the BB-UV light source; hence the UVC doses were adjusted by this factor. Tissues were harvested immediately after exposure. The tissue ‘disc’ was bisected using scissors and half was ‘snap frozen’ in 0.5 mL TE buffer in liquid nitrogen. The other half was inserted into a histology cassette, placed in 10% formalin overnight and transferred to TE buffer the following day. DNA for both the repair and penetration experiments was isolated for RIA analysis and slides were prepared for immunohistochemistry of CPDs.
DNA isolation and damage analysis
DNA was extracted from human and MatTek tissues using standard techniques (Gentra Systems Inc, Minneapolis MN, USA). DNA concentrations and purity were determined by reading the absorbance at 230, 260, 280 and 320 nm. DNA damage was quantified using radioimmunoassay (RIA). Briefly, RIA is a competitive binding assay between radiolabelled DNA and sample DNA for antisera raised against UV-irradiated DNA. For the RIA, 50 ng of heat-denatured sample DNA was incubated with 5–10 pg of poly(dA):poly(dT) (labeled to >5 × 108 cpm/μg by nick translation with 32P-dTTP) in a total volume of 1 mL 10 mM Tris, pH 7.8, 150 mM NaCl, 1 mM EDTA and 0.15% gelatin (Sigma). Antiserum was added at a dilution that yielded ∼35% binding to labeled ligand and after incubation overnight at 4°C the immune complex was precipitated with goat anti-rabbit immunoglobulin (Calbiochem, San Diego, CA, USA) and carrier serum from non-immunized rabbits (UTMDACC, Science Park/Veterinary Division, Bastrop, TX, USA). After centrifugation, the pellet was dissolved in tissue solubilizer (NCS, Amersham), mixed with ScintiSafe (Fisher) containing 0.1% glacial acetic acid, and the 32P quantified by liquid scintillation spectrometry. Under these conditions, antibody binding to an unlabelled competitor inhibits antibody binding to the radiolabeled ligand. DNA damage frequencies in samples used for the standard curve were determined using HPLC-MS/MS (Thierry Douki, CEA, Grenoble). These details, as well as those concerning the specificities of the RIAs and standards used for quantification, are described in Mitchell [21, 22].
Immunohistochemistry
Human nasal biopsies were fixed in 10% formalin, paraffin-embedded, and 4 μm sections were cut from each. Sections were extracted from paraffin blocks by incubation at 60°C for 30 min. followed by 2 immersions in xylene for 5 min. each, re-hydrated by 5 min. incubations in 100%, 95% and 70% EtOH and rinsed in ddH20. The slides were then placed immediately into 3.0% H2O2 for 10 min. at 23°C then washed in running ddH20. Following denaturation in 0.1 N NaOH/70% EtOH for 3 min. at 23°C the slides were dehydrated for 1 min. each in 70%, 90% and 100% EtOH. The slides were washed 3 times in phosphate-buffered saline (PBS) for 5 min. each and incubated with proteinase K (10 μg/ml in PBS) at 37°C for 10 min. After 3 washes for 5 min. each in PBS sections were incubated with 5% horse (blocking) serum for 30 min. at 23°C and incubated overnight at 4°C with monoclonal antibody specific for CPDs (clone KTM53, Kamiya Biomedical, Seattle, WA, USA) diluted 1:10,000 in PBS. After primary antibody incubation, the slides were washed 3 times for 3 min. in PBS at 23°C and incubated with biotin-conjugated horse anti-mouse IgG (Vector Laboratories, Burlingame, CA, USA) diluted 1:500. The slides were then washed 3 times for 3 min. each in PBS, incubated in Vector Elite ABC™ peroxidase for 30 min. and washed 3 times for 3 min. in PBS at 23°C. The slides were incubated in a diaminobenzidine (DAB) solution prepared from a DNA Substrate Kit for Peroxidase (Vector Cat. Nr. SK-4100) until optimal staining in the positive control was observed (30–40 sec.). After rinsing in ddH20 the slides were counterstained in haematoxylin for 10–15 sec., washed in ddH20, rinsed in PBS for 30 sec., rinsed again in ddH20 and then dehydrated through graded EtOH as above (i.e. 70%, 95% and 100%).
Statistical analyses
Two-sample independent t-tests using the R Statistical Package ver. 2.7.1 (http://www.r-project.org/) were used to determine significant differences between data. Exponential decay curves and linear regressions were generated using SigmaPlot ver. 10 (Systat, San Jose, CA, USA).
Results
DNA damage and repair after single exposure of the nasal mucosa to UV light
In order to understand the nucleotide repair efficiency in human respiratory epithelia exposed to UVR, nasal cytology samples were taken from UV exposed tissues (single exposure) prior to, immediately after the treatment (T0) and at 24, 48 and 72 hrs later (Fig. 1). As expected, baseline CPD and (6–4)PD levels were very similar for all of the subjects and were not significantly above the RIA background levels (i.e. measured in the absence of sample DNA). Immediately after UV exposure (T0), CPD and (6–4)PD frequencies were measured in all subjects and were significantly greater than baseline levels (P < 0.0001). The large standard deviations immediately after exposure reflect the significant inter-individual variation for damage induction. It is evident that both CPD and (6–4)PD repair is efficient in the respiratory epithelium with 60–70% of the original damage (T0) removed by 24 hrs post-treatment (P < 0.001) and near background levels 72 hrs (P < 0.0003) after UV exposure. The 24, 48 and 72 hrs CPD and (6–4)PD frequencies showed no significant deviation from baseline.
Fig 1.
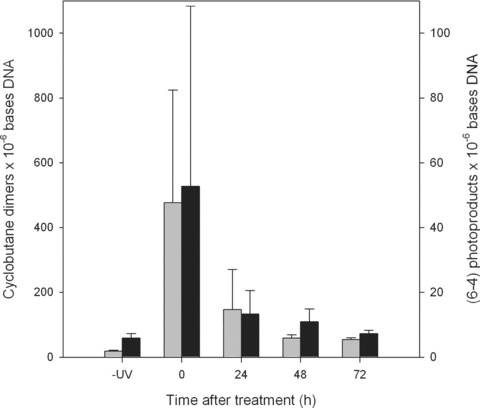
Formation and resolution of DNA damage in human nasal epithelia from patients with seasonal allergic rhinitis receiving a single treatment of rhinophototherapy (RPT). Sample nasal epithelial tissue was collected from RPT patients that had received no RPT (-UV), immediately after the RPT treatment (T0) and at 24 (T24), 48 (T48) and 72 (T72) hrs after exposure. Cyclobutane dimers (grey bars) and (6–4) photoproducts (black bars) were determined using radioimmunoassay. Standard deviations were calculated from 4 data points from the RIAs of DNA extracted from tissue from various patients at different time points; including 30 patients prior to treatment, 30 immediately after treatment (T0) and 10 patients at T24, T48 and T72. Of the 30 original patients, the same patients were not necessarily available for tissue collection at all of the various time points.
DNA damage and repair after multiple UV light exposures of human nasal mucosa
The current phototherapy treatment protocol for allergic rhinitis requires 9 UV treatments of the nasal mucosa during 3 consecutive weeks (3 exposures per week). DNA damage and repair of nasal epithelium was evaluated in nasal cytology samples from symptomatic allergic rhinitis patients at baseline (no UVR exposure), immediately after the final (9th) exposure and then 1 week and 4 weeks later (Fig. 2). As in the single exposure experiments, baseline CPD levels were very similar for all of the subjects and were not significantly above background levels in either the placebo (Panel A) or treated groups (Panel B). However, immediately after the final treatment significant CPD frequencies were measured in patients receiving the UV irradiation treatment (P < 0.0001). As above, significant variation was observed in the amount of DNA damage immediately after treatment and is reflected in the large error bar associated with this group when averaged together. Whereas about half of the treated patients (i.e. 7 of 13) displayed damage levels ranging from approximately 700 to 1300 CPDs × 106 bases DNA, the remainder showed significantly lower levels ranging from 300 to less than 100 CPDs × 106 bases DNA, CPD frequencies were below the limit of sensitivity of the RIA in placebo-treated patients. At 1 and 4 weeks after the last treatment, DNA damage frequencies had returned to the original baseline level in both the treated and untreated groups. A pairwise comparison of all of the data showed no significant differences except for T0.
Fig 2.
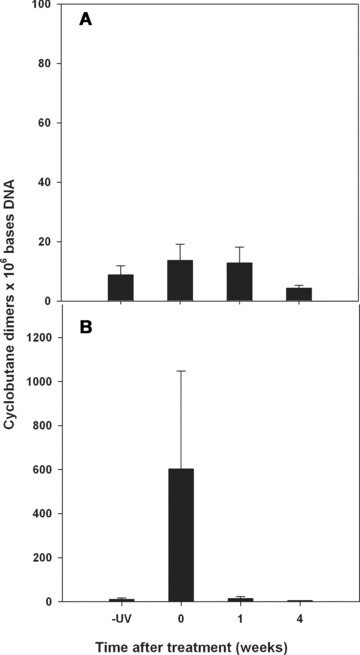
Long-term resolution of DNA damage in human nasal epithelia from patients receiving a 3-week course of rhinophototherapy for the treatment of seasonal allergies. Patients received either RPT (B) or a placebo treatment containing visible light with no UV radiation (A). Sample nasal epithelial tissue was collected either prior to treatment (-UV) or immediately after the 9th treatment (T0) and then at 1 week (T1) and 4 week (T4) intervals after the final treatment. The standard deviations were calculated from 4 data points from the RIAs of DNA extracted from tissues from 13 placebo and 13 treated patients.
Samples biopsied 2 months after the last treatment showed no morphological changes that could be attributed to the UV exposure (not shown). Both placebo-treated and UVR-treated subjects exhibited similar histological changes, most probably associated with the chronic inflammatory process associated with allergic rhinitis of the nasal mucosa. CPD positive cells were not detected in the tissue samples from subjects exposed to multiple UVR treatments or in placebo-treated subjects (Fig. 3B and C). As a positive control, EpiAirway tissue exposed to UVB and processed immediately after irradiation was used. A strong positive staining was detected (Fig. 3A).
Fig 3.
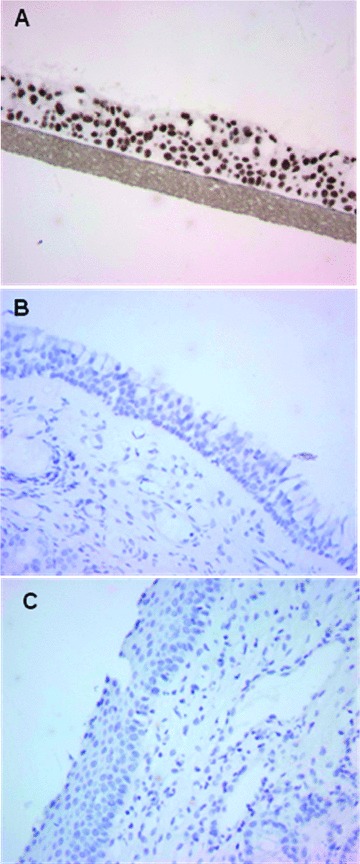
Immunohistochemical analysis of cyclobutane pyrimidine dimers in nasal epithelial tissues from patients subsequent to treatment with rhinophototherapy. Monoclonal antibodies specific for CPDs were used to visualize DNA damage in the nasal epithelium of patients 2 months after the final exposure to a RPT treatment for seasonal allergies (i.e. 9 treatments over a 3-week period). Tissue sections are shown for RPT-treated patients (C), sham-treated (visible light only) patients (B) and a positive control of UV irradiated 3D reconstructed normal human respiratory epithelium (EpiAirway) (A).
Biologic response of EpiDerm and Ep-Airway to UV radiation
We conducted a comparative analysis of 3-dimensional reconstructed respiratory epithelium (EpiAirway) and epidermis (EpiDerm) to better understand similarities and differences between these tissues with regard to their biological response to UVR, particularly DNA damage induction and repair. An experiment was performed to quantify the rates of CPD induction in these tissues using various UV radiation sources (data not shown). Tissues were exposed to increasing doses from the NB-UVC and NB-UVB radiation sources and from the BB-UV source with and without the UVB component removed (BB-UVC). Doses were adjusted to yield approximately equivalent levels of DNA damage from each light source in purified DNA. In all cases significantly more damage was induced in the EpiAirway compared to the EpiDerm by the same dose of UVR due to shielding by the stratum corneum in the EpiDerm tissues.
The EpiAirway tissues were also used to investigate the depth of penetration of different UV sources. The unfiltered, full-spectrum BB-UV light (Allux Medical) closely resembled the NB-UVB pattern in its depth of penetration (Fig. 4). Likewise, the NB-UVC closely resembled the BB-UVC light source (from which the UVB emission had been blocked). It is evident that the UVB component of the BB-UV light source is responsible for inducing DNA damage in all cell layers of the tissue. UVC radiation from either the germicidal light source or emitted exclusively from the BB-UV light source damaged only those cells on the surface. The UVA component of the BB-UV light source does not induce measurable CPD levels in tissues [23, 24]. These data are consistent with previous results showing that longer wavelength UV radiation (i.e. UVB and UVA) penetrates deeper and damages human skin DNA greater than UVC.
Fig 4.
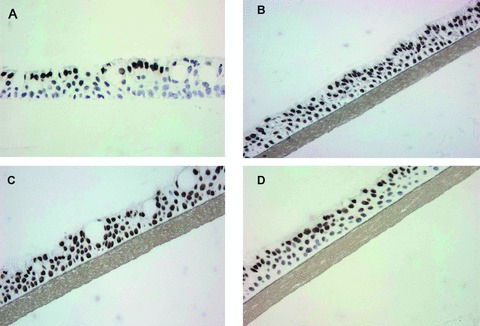
DNA damage distribution in 3D reconstructed normal human respiratory epithelia (EpiAirway) exposed to different light sources. Immunohistochemistry using monoclonal antibodies specific for CPDs is shown immediately after irradiation with a broad-band UVC/UVB lamp used for RPT (Allux Medical) (C), a narrow-band Philips TL01 UVB lamp (B), a narrow-band UVC germicidal lamp (A) and the broadband UVC/UVB lamp shown in Panel A from which the UVB component had been removed (D).
The data reported here using samples obtained from subjects exposed to single or multiple UV treatments suggest that the human respiratory epithelium can efficiently remove lesions in DNA resulting from exposure to UV radiation. To further examine this question, the rates of NER in EpiAirway and EpiDerm were compared. Doses were adjusted to account for shielding by the stratum corneum. In Fig. 5, the kinetics for CPD and (6–4)PD repair are shown in EpiDerm (closed circles) and EpiAirway tissues (open circles). The number of CPDs and (6–4)PDs per megabase DNA are shown in Fig. 5A and 5B, respectively. These values normalized to the T0 value for each photoproduct in each tissue are shown in Fig. 5C (CPDs) and 5D [(6–4)PDs]. It is evident that both photoproducts are efficiently repaired in both engineered tissue systems. There appears to be a slight lag in NER of CPDs in the EpiAirway, which may reflect the greater amount of damage induced. Exponential decay curves were generated for each of the normalized curves. The curves for the (6–4)PD (Fig. 5D) were nearly identical for the two tissues with correlations of determination (r2) of 0.9717 and 0.9780 for Epi-Airway and EpiDerm, respectively. A slight difference in the CPD repair curves (Fig. 5C) was observed. The r2 values for the Epi-Airway and EpiDerm curves were 0.9110 and 0.8943, respectively; the 95% confidence intervals for the curves showed significant overlap. Thus we conclude that NER of CPDs and (6–4)PDs was comparable in the EpiAirway and the EpiDerm systems.
Fig 5.
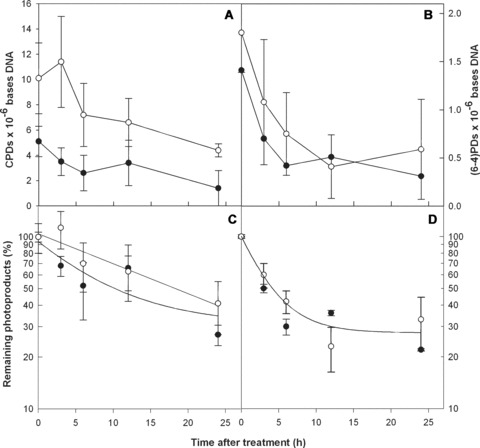
Nucleotide excision repair kinetics in 3D reconstructed normal human nasal epithelial and epidermal tissues. CPD and (6–4)PD repair are shown in EpiDerm (•–•) and EpiAirway (○–○) artificial tissues. The upper panels show the frequencies of CPDs (A) and (6–4)PDs (B) at increasing times post-irradiation as lesions per megabase DNA. Directly below each panel the frequencies have been normalized to the amount of damage measured at T0 and are expressed as the percentage of CPDs (C) and (6–4)PDs (D) remaining at increasing times post-irradiation with a single sublethal dose of UVB. Exponential decay curves for each data set are shown. Standard errors of the mean were calculated from standard deviations using a total of 8 data points from duplicate tissues.
Discussion
Although UV light has been applied in other locations besides the skin (nasal mucosa, oral mucosa), limited data exist about the capacity of tissues other than the epidermis to repair UV-induced DNA damage. Studies performed in bronchial fibroblasts and epithelial cells showed similar DNA repair as in human skin fibroblasts, suggesting that DNA repair mechanisms are equally efficient in all cell types [25]. As demonstrated in a recent pilot study, nasal epithelial cells are capable of repairing UV-induced DNA damage in allergic rhinitis patients receiving intranasal phototherapy [26]. A limitation of this latter study was the use of the Comet assay to assess DNA damage, a technique that cannot distinguish between strand breaks associated with CPDs (and their removal) and those associated with the oxidative stress that is a characteristic of inflamed nasal mucosa in allergic rhinitis.
The goal of the present study was to assess the capacity of airway mucosa to repair UV-induced DNA damage and compare the biological response of a tissue that is not normally exposed to UV radiation (i.e. the human respiratory epithelium) with a well-studied system that is exposed to high levels of UV radiation (i.e. human skin). We focused on the early events in the carcinogenic process, namely DNA damage induction and repair, for this comparison. We believe that if the DNA damage and repair responses in the respiratory epithelium and human epidermis are similar, then the extensive knowledge base available for assessing the risks of UV light in human skin can be used to evaluate the risks associated with phototherapy of the nasal epithelium.
The data show very similar repair kinetics of both photoproducts (CPDs and 6–4 PDs) in human skin and respiratory airway using three-dimensional reconstructed normal human tissue models. EpiAirway and EpiDerm have been previously used in genotoxicity experiments and are considered good and reliable models to predict in vivo behaviour of these tissues after different insults or therapeutic interventions [27, 28]. In order to compare our data with other systems (e.g. human skin and cell culture), we calculated the half-lives (T1/2) for each type of damage in each tissue using the exponential decay curves shown in Fig. 5C and D. We determined that the T1/2 for (6–4)PD repair was 4 hrs for both tissues and 10.3 and 17.9 hrs for CPD repair in the EpiDerm and EpiAirway tissues, respectively. This difference in the T1/2 for CPDs was not statistically significant. By comparison, the half-life for CPD and (6–4)PD removal in human skin in situ is 33.3 and 2.3 hrs, respectively, with significant levels remaining 7 days post-irradiation [29]; in cultured human keratinocytes (HaCaT) the T1/2 for CPDs is 11–13 hrs at doses similar to those used in the current study [30] and < 3 hrs for the (6–4)PD (unpublished observation); in normal human fibroblasts (i.e. GM637 and HS27) the T1/2 values for the excision of CPDs and (6–4)PDs are ∼2 and 6 hrs, respectively [31]. These repair rates closely approximate those measured in the EpiDerm and EpiAirway tissues. Hence, the rate of NER of the two major photoproducts highly implicated as the mutagenic precursors of the UVB signature mutations associated with basal and squamous cell carcinoma [i.e. the CPD and (6–4) PD] are essentially identical in human respiratory epithelia and epidermal tissues. Significant levels of DNA damage were not evident in respiratory epithelia at 7 days and 1 month after a 9-dose regimen of UV light delivered to the nasal mucosa.
Significant amounts of DNA damage accumulate in a small number of epidermal cells after chronic exposure to carcinogens (i.e. UVR and benzo[a]pyrene diolepoxide) [17, 18, 32–34]. The observation that certain basal cells retain high levels of DNA damage for several weeks after UV exposure indicates that these cells are non-cycling. Indeed, the behaviour of these cells is very similar to the slowly cycling, carcinogen-retaining basal cells found in the central regions of the epidermal proliferative unit, which Morris and Potten suggest are stem cells [33, 34]. It is probable that these DNA damage-retaining cells play a significant role in generating the pre-neoplastic clones that ultimately lead to tumour formation.
The limitation of the RIA technique, which was used in the single and multiple exposure experiments, is that it measures damage in DNA extracted from a pool of cells containing varying levels of damage. Although the technique is very specific with a high sensitivity, it may not detect CPD-retaining single cells occurring at very low density in the epithelium. Therefore, nasal mucosa samples were also stained for CPDs, a technique which can detect this type of DNA damage in single cells and allow us to determine if damage is accumulating in certain cells as shown in human epidermis. No CPD-retaining cells were observed in human respiratory epithelial biopsies from patients that had undergone phototherapy of the nasal mucosa (multiple exposures). Moreover, because RPT is commercially available in Hungary (Rhinolight Ltd, Szeged, Hungary), five of the subjects had received a total of two or more courses of intranasal phototherapy. The light source used to treat this condition has an emission spectrum comparable to the BB-UVB light source used in the current study. Therefore, the absence of CPD-retaining cells in nasal mucosa suggests that nasal epithelium is capable of efficient repair in all of the exposed cells after chronic exposure to UVR.
In conclusion, our results show that human nasal mucosa is capable of efficient repair of UV-induced DNA damage and suggest that treatment regimens similar in duration with those used for the treatment of allergic rhinitis may be useful for other immune-mediated or inflammatory diseases of the airways, such as nasal polyps or chronic rhinosinusitis.
References
- 1.Callen JP, Krueger GG, Lebwohl M, et al. AAD consensus statement on psoriasis therapies. J Am Acad Dermatol. 2003;49:897–9. doi: 10.1016/s0190-9622(03)01870-x. [DOI] [PubMed] [Google Scholar]
- 2.Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for vitiligo. American Academy of Dermatology. J Am Acad Dermatol. 1996;35:620–6. doi: 10.1016/s0190-9622(96)90691-x. [DOI] [PubMed] [Google Scholar]
- 3.Ibbotson SH, Bilsland D, Cox NH, et al. An update and guidance on narrowband ultraviolet B phototherapy: a British Photodermatology Group Workshop Report. Br J Dermatol. 2004;151:283–97. doi: 10.1111/j.1365-2133.2004.06128.x. [DOI] [PubMed] [Google Scholar]
- 4.Koreck AI, Csoma Z, Bodai L, et al. Rhinophototherapy: a new therapeutic tool for the management of allergic rhinitis. J Allergy Clin Immunol. 2005;115:541–7. doi: 10.1016/j.jaci.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Redding SW, Callander NS, Haveman CW, et al. Treatment of oral chronic graft-versus-host disease with PUVA therapy: case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;86:183–7. doi: 10.1016/s1079-2104(98)90123-8. [DOI] [PubMed] [Google Scholar]
- 6.Trehan M, Taylor CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol. 2004;140:415–20. doi: 10.1001/archderm.140.4.415. [DOI] [PubMed] [Google Scholar]
- 7.Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–8. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ziegler A, Leffell DJ, Kunala S, et al. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc Natl Acad Sci USA. 1993;90:4216–20. doi: 10.1073/pnas.90.9.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ananthaswamy HN, Pierceall WE. Molecular mechanisms of ultraviolet radiation carcinogenesis. Photochem Photobiol. 1990;52:1119–36. doi: 10.1111/j.1751-1097.1990.tb08452.x. [DOI] [PubMed] [Google Scholar]
- 10.Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res. 2005;571:91–106. doi: 10.1016/j.mrfmmm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 11.De Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–85. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 12.Daya-Grosjean L, Robert C, Drougard C, et al. High mutation frequency in ras genes of skin tumors isolated from DNA repair deficient xeroderma pigmentosum patients. Cancer Res. 1993;53:1625–9. [PubMed] [Google Scholar]
- 13.Drouin R, Therrien JP. UVB-induced cyclobutane pyrimidine dimer frequency correlates with skin cancer mutational hotspots in p53. Photochem Photobiol. 1997;66:719–26. doi: 10.1111/j.1751-1097.1997.tb03213.x. [DOI] [PubMed] [Google Scholar]
- 14.Tornaletti S, Rozek D, Pfeifer GP. The distribution of UV photoproducts along the human p53 gene and its relation to mutations in skin cancer. Oncogene. 1993;8:2051–7. [PubMed] [Google Scholar]
- 15.Berg RJ, Van Kranen HJ, Rebel HG, et al. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc Natl Acad Sci USA. 1996;93:274–8. doi: 10.1073/pnas.93.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated keratinocytes in normal human skin [see comments] Proc Natl Acad Sci USA. 1996;93:14025–9. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell DL, Volkmer B, Breitbart EW, et al. Identification of a non-dividing subpopulation of mouse and human epidermal cerlls exhibiting high levels of persistent UV photodamage. J Invest Dermatol. 2001;117:590–5. doi: 10.1046/j.0022-202x.2001.01442.x. [DOI] [PubMed] [Google Scholar]
- 18.Nijhof JG, Van Pelt C, Mulder AA, et al. Epidermal stem and progenitor cells in murine epidermis accumulate UV damage despite NER proficiency. Carcinogenesis. 2007;28:792–800. doi: 10.1093/carcin/bgl213. [DOI] [PubMed] [Google Scholar]
- 19.Lee E, Koo J, Berger T. UVB phototherapy and skin cancer risk: a review of the literature. Int J Dermatol. 2005;44:355–60. doi: 10.1111/j.1365-4632.2004.02186.x. [DOI] [PubMed] [Google Scholar]
- 20.Weischer M, Blum A, Eberhard F, et al. No evidence for increased skin cancer risk in psoriasis patients treated with broadband or narrowband UVB phototherapy: a first retrospective study. Acta Derm Venereol. 2004;84:370–4. doi: 10.1080/00015550410026948. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell DL. Radioimmunoassay of DNA damaged by ultraviolet light. In: Pfeifer G, editor. Technologies for detection of DNA damage and mutations. New York: Plenum Publishing Corp; 1996. pp. 73–85. [Google Scholar]
- 22.Mitchell DL. Quantification of photoproducts in mammalian cell DNA using radioimmunoassay. Methods Mol Biol. 2006;314:239–49. doi: 10.1385/1-59259-973-7:239. [DOI] [PubMed] [Google Scholar]
- 23.Mouret S, Favier A, Beani JC, et al. Differential p53-mediated responses to solar-simulated radiation in human papillomavirus type 16-infected keratinocytes. Exp Dermatol. 2007;16:476–84. doi: 10.1111/j.1600-0625.2007.00560.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Rosenstein BS, Wang Y, et al. Induction of 8-oxo-7,8-dihydro-2′-deoxyguanosine by ultraviolet radiation in calf thymus DNA and HeLa cells. Photochem Photobiol. 1997;65:119–24. doi: 10.1111/j.1751-1097.1997.tb01886.x. [DOI] [PubMed] [Google Scholar]
- 25.Fornace AJ, Jr, Lechner JF, Grafstrom RC, et al. DNA repair in human bronchial epithelial cells. Carcinogenesis. 1982;3:1373–7. doi: 10.1093/carcin/3.12.1373. [DOI] [PubMed] [Google Scholar]
- 26.Koreck A, Szechenyi A, Morocz M, et al. Effects of intranasal phototherapy on nasal mucosa in patients with allergic rhinitis. J Photochem Photobiol B. 2007;89:163–9. doi: 10.1016/j.jphotobiol.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 27.Belyakov OV, Mitchell SA, Parikh D, et al. Biological effects in unirradiated human tissue induced by radiation damage up to 1 mm away. Proc Natl Acad Sci USA. 2005;102:14203–8. doi: 10.1073/pnas.0505020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curren RD, Mun GC, Gibson DP, et al. Development of a method for assessing micronucleus induction in a 3D human skin model (EpiDerm) Mutat Res. 2006;607:192–204. doi: 10.1016/j.mrgentox.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Young AR, Chadwick CA, Harrison GI, et al. The in situ repair kinetics of epidermal thymine dimers and 6–4 photoproducts in human skin types I and II. J Invest Dermatol. 1996;106:1307–13. doi: 10.1111/1523-1747.ep12349031. [DOI] [PubMed] [Google Scholar]
- 30.Greinert R, Boguhn O, Harder D, et al. The dose dependence of cyclobutane dimer induction and repair in UVB-irradiated human keratinocytes. Photochem Photobiol. 2000;72:701–8. doi: 10.1562/0031-8655(2000)072<0701:tddocd>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 31.Cleaver JE, Cortes F, Karentz D, et al. The relative biological importance of cyclobutane dimer and (6–4) pyrimidine-pyrimidone dimer photoproducts in human cells: evidence from a Xeroderma pigmentosum revertant. Photochem Photobiol. 1988;48:41–9. doi: 10.1111/j.1751-1097.1988.tb02784.x. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell DL, Greinert R, De Gruijl FR, et al. Effects of chronic low-dose ultraviolet B radiation on DNA damage and repair in mouse skin. Cancer Res. 1999;59:2875–84. [PubMed] [Google Scholar]
- 33.Morris RJ, Coulter K, Tryson K, et al. Evidence that cutaneous carcinogen-initiated epithelial cells from mice are quiescent rather than actively cycling. Cancer Res. 1997;57:3436–43. [PubMed] [Google Scholar]
- 34.Morris RJ, Fischer SM, Slaga TJ. Evidence that a slowly cycling subpopulation of adult murine epidermal cells retains carcinogen. Cancer Res. 1986;46:3061–6. [PubMed] [Google Scholar]