

Abstract
After bone injuries, several molecular mechanisms establish bone repair from stem/progenitor cells. Inflammation factors attract regenerative cells which expand and differentiate in order to build up a bone highly similar to that before injury. Bone marrow (BM) mesenchymal stem cells (MSCs) as skeletal stem cells and endothelial progenitors (EPCs) are at the origin of such reparation mechanisms. However, discrepancies exist about their identities. Although cultured MSCs are extensively described, their in vivo native forms are poorly known. In addition, recent experiments show that several types of EPC exist. We therefore review up-to-date data on the characterization of such stem/progenitor cells and propose a new point of view of their function in bone regeneration.
Keywords: cultured mesenchymal stem cells, native mesenchymal stem cells, skeletal stem cells, endothelial progenitors, osteoblasts, vasculogenesis, bone healing
Introduction
The repair process in adults closely resembles normal development of the skeleton during embryogenesis, which occurs by intramembranous and endochondral ossification (Fig. 1) [1]. Nonetheless, some aspects are different from the foetal bone-forming process, such as the contribution of inflammation, the scarcity of regenerative cells and the increased prevalence of mechanical forces in adults [2–4]. Of note, the inflammation in fracture healing process is an early event giving rise to signalling of pro-inflammatory cytokines crucial for the wound repair. Interleukin-1 (IL-1) and IL-6 as well as tumour necrosis factor-α (TNF-α) carry out central functions in the induction of downstream responses to injury by having a chemotactic effect on other inflammatory cells, enhancing extracellular matrix synthesis, stimulating angiogenesis and recruiting endogenous fibrogenic cells to the injury site [5–8].
Fig 1.
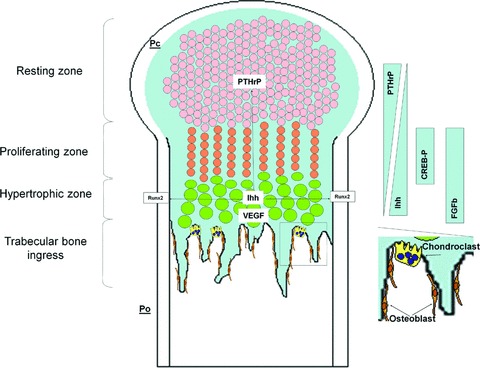
Endochondral bone formation. Long bones are built up by endochondral mechanisms. In the beginning, mesenchymal cells form condensations. At the border of condensations, cells constitute the perichondrium (Pc), and into the inner condensations, cells differentiate into chondrocytes. Chondrocytes from the resting zone start to proliferate, thus generating columns of proliferating chondrocytes. The latter become hypertrophic and by their secretion of VEGF, attract blood vessels and undergo apoptosis, giving rise to a scaffold for osteoblasts accompanying the vascular ingrowth. In addition to VEGF, hypertrophic chondrocytes express Ihh, which regulates positively the expression of PTHrP by chondrocytes from the resting zone. However, PTHrP induces the proliferation of chondrocytes, inhibiting their capacity to become hypertrophic. Ihh-producing hypertrophic chondrocytes are generated when proliferating chondrocytes reach a location where PTHrP is insufficient. Their Ihh secretions also induce Runx2 expression from periosteal cells (Po) differentiating them into osteoblasts from bone collar. Osteoblasts and chondroblasts invading the growth plate through vessels forming primary spongiosa remodel bone to form trabeculation. Crucial proteins for growth plate development are noted according their expression area.
The first step in endochondral ossification is the aggregation of mesenchymal cells into discrete condensations that resemble the shape of the future skeletal element. A similar process occurs during the early stages of fracture repair [9]. After vascular damage, mesenchymal stem cells (MSCs) populate the wound site in hypoxic conditions, where they proliferate and then differentiate along a cartilaginous or an osteogenic lineage in response to growth factors and cytokines released by platelets, inflammatory cells and neighbouring cells and tissues. Indeed, within this process, it can be detected IL-1, IL-6, TNF-α, transforming growth factor-β (TGF-β) as pro-inflammatory molecules, whereas placental growth factor (PlGF) and vascular endothelial growth factor (VEGF) are angiogeneic molecules, and fibroblast growth factor (FGF), bone morphogenetic proteins (BMPs), Indian hedgehog (Ihh) and Wnt are differentiation inducing proteins [2, 10–17]. On the other hand, vascular damage induces angiogenesis or vasculogenesis by recruiting endothelial progenitors (EPCs) locally or after their circulation in blood. The VEGF/PlGF defect impairs their recruitment as well as their proliferation and differentiation [17].
The defects of one of the factors mentioned above yield both delayed union and non-union. However, to date there is not a specific marker of the non-union which could be used as prognostic marker and the molecular mechanisms need to be clarified. This could be resolved by the knowledge of MSCs with capabilities to repair bone fracture in adult. In addition, there are convincing data showing that EPCs are also crucial for this process; however, BM EPCs are also poorly known. We review here recent data on MSC and EPC origins and characterization, and we depict their respective role in bone healing.
Mesenchymal stem cells as skeletal stem cells
Cultured or expanded MSCs
In adult stages, multipotent skeletal stem cells, also referred to as MSCs or multipotential stromal cells (MSCs), contribute to the maintenance of various tissues, particularly bone. MSCs can be isolated from bone marrow (BM) and adipose tissues in adult stages and also from placenta and umbilical cord blood [18–21]. MSCs can be induced in vitro and in vivo to differentiate into various mesenchymal tissues such as bone, cartilage, muscle, tendon, adipose tissue and haematopoiesis-supporting stroma. In addition, human BM-derived MSCs maintain their multipotent capacity and exhibit site-specific differentiation after in utero transplantation in sheep [22]. MSCs are usually selected by their capacity to adhere to plastic culture flasks and then expand via colony forming unit-fibroblasts (CFU-Fs) after several weeks in vitro within basal medium and specific foetal calf serum [18]. However, this type of procedure does not permit the characterization of their native form (nMSCs), whereas extensive works describe cultured MSCs (cMSCs) notably their in vitro-derived phenotype and multipotentiality. Within these conditions, cMSCs are defined as non-haematopoietic cells (CD45–, CD14–, CD34–) expressing some molecules, the combination of which is largely used for their description: CD73+, CD44+, CD105+, CD90+ and CD146+ (Table 1). Cultured MSCs are largely used in experimental bone reconstruction in vitro and in vivo[18, 23–25]. Indeed, several types of animal studies demonstrated their potential to produce bone both in the ectopic position and within the bone environment (e.g. using bone defects) [26, 27]. However, in human beings, there have been few clinical studies thus far. In this context, Quarto et al. reported on different bone defects (loss of a 4.0-cm segment of the mid-diaphysis of the right tibia or distal diaphysis of the right ulna, loss of a 7.0-cm segment of the right humerus), which were filled with scaffolds of macroporous hydroxyapatite loaded with autologous cMSCs. In all three patients, radiographs and computed tomography scans revealed abundant callus formation along the implants and good integration at the interfaces with the host bones by the second month after surgery [28]. In addition, Horwitz et al. performed a clinical study of allogeneic cMSCs infused in children with osteogenesis imperfecta. Five of six patients showed engraftment in bone and BM stroma and accelerated growth velocity during the first 6 months after infusion as compared with the 6 months preceding the infusions [29]. Recently, Le Blanc’s research group injected allogeneic major complex of histocompatibilty mismatched male cMSCs within a human foetus in utero after diagnosis of multiple intrauterine fractures due to severe osteogenesis imperfecta. Data analysis showed engraftment of allogeneic cMSCs within bone notably without immune rejection. The clinical benefits remain to be determined [30]. Finally, a recent published report showed the successful use of a mixture of cMSCs and Ca2+ S-biomaterial for healing refractory non-union bone [31]. Nevertheless, this was only a case report. Therefore, clinical human studies with relevant controls are needed to confirm the potential of cMSCs to be used in bone tissue engineering in the clinical setting, whatever their origin, allogeneic or autologous.
Table 1.
Markers expressed by BM non-haematopoietic stem and progenitor cells
| Markers | Molecules | Native MSCs | Cultured MSCs | CFU-Hill | Late EPCs |
|---|---|---|---|---|---|
| GD2 | Ganglioside | +[56] | +[56] | ||
| Stro-1 | Unknown antigen | +[47] | +[47] | ||
| α-SM actin | α-Smooth muscle actin | +[121] | +[37] | ||
| SSEA4 | Stage-specific embryonic antigen | +[57] | +[57] | ||
| VEGF-R2 | Vascular endothelial growth factor-receptor2 | −[52] | +[108] | +[105, 114, 115] | |
| HlA-II | Human leucocyte antigen-II | −[122] | |||
| CD14 | LPS receptor | −[18] | +[111] | −[115] | |
| CD18 | Integrin β2 chain | −[18] | |||
| CD29 | Integrin β1 chain | +[18] | |||
| CD31 | PECAM-1 (platelet endothelial cell adhesion molecule) | −[59, 60] | −[18] | +[114] | |
| CD34 | Sialoprotein | +[123] | −[18] | dim [114] | +[105] |
| CD36 | Glycoprotein IIIb | +[52] | |||
| CD44 | Hyaluronan receptor | +[18] | |||
| CD45 | Pan-leucocyte antigen | dim [53, 58], −[59] | −[18] | +[115] | −[115] |
| CD49a | Integrin α1 chain | +[49] | +[18] | ||
| CD49b | Integrin α2 chain | +[18] | |||
| CD49c | Integrin α3 chain | +[18] | |||
| CD49d | Integrin α4 chain | −[18] | |||
| CD49e | Integrin α5 chain | +[18] | |||
| CD51 | Integrin aV chain | +[18] | |||
| CD54 | ICAM-1 (inter-cellular adhesion molecule-1) | +[18] | |||
| CD56 | NCAM-1 (neural cell adhesion molecule) | −[60] | −[124] | ||
| CD73 | Ecto 5 nucleotidase | +[125] | +[18] | ||
| CD80 | CD28 ligand | −[52] | |||
| CD90 | Thy-1 | +[58, 126] | +[18] | ||
| CD105 | TGF-βRIII (transforming growth factor-β receptor III) | +[127] | +[18] | +[115] | +[115] |
| CD106 | VCAM-1 (Vascular cell adhesion molecule-1) | +[121] | +[18] | ||
| CD133 | AC133 (prominin) | +[50] | −[50] | +[105] | −[109] |
| CD140b | PDGF-Rβ (platelet-derived growth factor receptor β) | +[128] | +[129] | ||
| CD144 | Vascular endothelial-cadherin | −[18] | +[111] | +[111] | |
| CD146 | Mel-CAM (melanoma-cell adhesion molecule) | +[121] | +[59] | +[61] | +[111, 114] |
| CD166 | ALCAM (activated lymphocyte cell adhesion molecule) | +[72] | +[18] | ||
| CD200 | OX-2 | +[52] | +[52] | ||
| CD271 | NGFR (neural growth factor receptor) | +[53] | −[130] |
See text for abbreviations.
As noted above, another remarkable feature of cMSCs is their immunomodulatory potential. This property, which has been described in vitro as well as in vivo, could allow for their use in allogeneic conditions for tissue regeneration and also render them interesting tools for inducing tolerance in allografting [32]. Notably, clinical studies have been conducted to measure the capacity of cMSCs to inhibit graft-versus-host disease (GvHD) after allogeneic haematopoietic stem-cell transplantation. In their pioneering study, Le Blanc et al. injected allogeneic MSCs in a patient with refractory, acute GvHD. Two productions of cMSCs were infused to finally generate complete suppression of GvHD [33]. A multicenter non-randomized study set up by the European Bone Marrow Transplantation (EBMT) consortium confirmed these data [34]. The implicated mechanisms are under intense investigation and seem to include (i) inhibition of the Th1 pathway, (ii) induction of CD4+ /CD25+ FoxP3+ T-regulatory cells, (iii) inhibition of both differentiation and activation of antigen-presenting cells and (iv) inhibition of cell-lysing properties of both CD8+ and natural killer cells [35]. Several of these mechanisms are thought to be mediated by soluble factors such as prostaglandin E2, TGF-β, IL-10, indole amine 2,3-dioxygenase, nitric oxide and HLA-G5 [36]. Taken together, these in vitro, pre- and clinical data demonstrate the efficiency of cMSCs as putative osteoregenerative cells, even in allograft transplantation.
Albeit these extensive and promising works on regenerative capacities of cMSCs few data were obtained concerning their identities. Cultured MSCs were shown to express the smooth muscle form of actin (αSM-actin) putting them into the compartment of vascular smooth muscle (VSM) like cells although they do not express more mature VSM markers such as SM myosin heavy chain [37]. In addition, when intramedullary injected within compromised non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice the majority of human cMSCs were found in the vicinity of endosteum whereas those, which were not associated with trabecular bone, were often observed in the abluminal position of BM vessels [38]. Moreover, the αSM-actin was detected in more than half of injected cells. These data highlight therefore a link between the VSM-like phenotype of cMSCs and their capacities to engraft BM. Several other VSM markers are observed on cMSCs such as membrane CD166, CD49a, CD105 and CD146 molecules and, by studying their transcriptome, cMSCs were phenotypically closely related to pericytes than fibroblasts or human umbilical vein endothelial cells [39]. This reinforces the link between cMSCs to the VSM system of a tissue as demonstrated for BM stromal cells with long-term haematopoiesis-supporting function [40, 41]. Nevertheless, the main trait of cMSCs is the phenotypic and functional heterogeneities. Friedenstein noted in his pioneering works that stromal cells varied in morphology and also in alkaline phosphatase expression [42]. By selecting different clones, it was shown that only one third were tripotential and half possessed the osteoblastic and chondroblastic differentiation capacities [43]. Therefore, it is possible that there is more than one identity for cMSCs, e.g. VSM like cells and osteoblastic cells. However, such heterogeneity can be explain by the fact that cells clonally derived from CFU-F may express simultaneously several markers specific of different lineages including osteoblasts, chondrocytes, adipocytes and VSM cells [44]. This was also found at single-cell level and, among molecules characteristic of mesenchymal lineages, it could be possible to detect transcripts of unorthodox markers of epithelial, neural and haematopoietic cells [45]. Interestingly, the panel of such markers reduces in favour to the gain of molecules specific of lineage toward cells progress [44]. These interesting observations underline the fact that plasticity is the hallmark of cMSCs [46].
Native MSCs
In contrast to cMSCs, only little information exists regarding the features of the primary precursors, nMSCs, that give rise to these plastic-adherent cells (Fig. 2). Various groups have attempted thus far to purify MSCs by using different strategies. Indeed, using the monoclonal antibody Stro-1 (recognizing an as-yet-unknown antigen), Simmons et al. identified nMSCs as CD34+ cells and found them on the abluminal face of the vessels (Table 1) [47, 48]. Our group used the monoclonal antibody anti-CD49a molecule to enrich these cells and confirmed their stem cell phenotype in human beings and in rodents (CD133+ and Sca-1+, respectively) [49, 50]. CD49a is the α1 subunit of the VLA-1 integrin, a collagen IV and laminin receptor and was primarily described as an early marker of VSM cells in organogenesis [51]. Recently, our group demonstrated that nMSCs could also be enriched within the CD200+ BM fraction [52]. CD200 is a member of the immunoglobulin superfamily and is expressed by various cell types, including myeloid cells, endothelium, ovarian cells, trophoblasts and neurons. Native MSCs are also enriched within the low affinity nerve growth factor (LNGF-R or CD271+ BM fraction [53]. As Stro-1 antigen and CD49a, the CD271 molecule is a reliable marker of nMSCs. In a seminal work, using immunohistochemistry, the LNGF-R was shown to be confined to a subset of BM stromal cells, the reticular cells, with dendrites irradiated toward haematopoietic cells and localized at the abluminal side of sinus. The reticular cells have been described to form a system of lacunae where haematopoietic cells are organized [54]. These CD271+ cells were also positive for alkaline phosphatase, vimentin, CD13 but negative for CD34, CD45 and CD68. Interestingly, CD271+ stromal cells originate from vessels and ingrow within BM [55]. Additional markers for the prospective isolation of nMSCs have been described recently, such as the neural glanglioside GD2 and the embryonic membrane molecule SSEA4 [56, 57]. Unfortunately, for the two latter markers no data exist on the enrichment factor obtained after selection, which does not allow for comparison with previous works. However, in contrast to cells from the GD2+, SSEA4+ or CD200+ fraction, CD49a+ nMSCs and CD271+ nMSCs express at a low level the pan-leucocyte marker CD45, which rapidly disappears when cells are cultured [58]. This discrepancy needs further clarification to verify whether nMSCs do actually express CD45 at a low level, similarly to BM haematopoietic stem cells, or whether this is simply due to an experimental artefact. Finally, the CD146 molecule (an immunoglobulin protein also named MUC18 or S-endo) has been convincingly proposed as a marker of multipotent nMSCs [59]. Immunohistochemical studies ascribed a sub-endothelial localization to CD146+ cells in the BM and, consistent with this finding, CD146+ nMSCs expressed multiple mural cell-specific molecules such as NG2, αSM-actin and calponin 1 and 3. In contrast to CD271+ or CD49a+ cells, CD146+ cells were CD45–. In addition, CD146+ cells were found to express very few osteogenic molecules but acquired these additional markers when induced to differentiate into osteoblasts or when transplanted in SCID mice with hydroxyapatite/βtricalciumphosphate (HA/βTCP) particles used as a scaffold. Interestingly, when CD146+ cells were transplanted, they could self-renew since they could generate multipotent CD146+ perivascular cells in serial transplantations. In contrast, cells in which CD146 was down-regulated (i.e. upon culture with the basic FGF), or silenced, no self-renewal was observed. Therefore, this is the first time that multipotential nMSCs have been defined as a specific population of perivascular cells with self-renewal capacities (Fig. 2). This location can explain the possibilities to obtain CFU-Fs from several tissues such as skeletal muscle, pancreas, adipose tissue, brain, spleen, liver, lung and thymus [37]. Recently, Crisan et al.[60] demonstrated that all nMSCs were of pericytic origin, but not all pericytes were nMSCs. Interestingly, sorted CD146+ perivascular cells, from CD34– and CD45– fractions (to exclude both endothelial and haematopoietic cells) of muscle and non-muscle tissues, displayed myogenic potential in vitro and in vivo. Regardless of their tissue origin, sorted pericytes subsequently cultured in cMSC conditions displayed markers of cMSCs (i.e. positive for CD44, CD73, CD90, CD105, CD166 and SSEA4 but negative for CD45, CD34, CD31 and CD144) and could also differentiate into chondrocytes, adipocytes and osteocytes. Nevertheless, the CD146 molecule was also shown to be expressed by other regenerative cells that have strong importance in bone healing: the EPCs [61]. Therefore, investigating the role of the CD146 molecule in these settings would be of interest.
Fig 2.

Phenotype differences between human native and cMSCs. nMSCs are perivascular cells expressing markers used for their isolation (included within the nMSC box). When cells are cultured, the expression of some native markers is disturbed while it remains unchanged for others. Indeed, CD45 and CD34 molecules are observed on nMSCs but disappear in cMSCs. Other native markers, such as CD271, an antigen recognized by Stro-1 Ab, are down-regulated during their expansion. The expression of CD90 by nMSCs has not been investigated in human beings.
In addition to these recent reports, the perivascular origin of nMSCs is supported by other observations as well. In this context bona fide pericytes were previously shown to be multipotent cells because they were able to differentiate into osteoblasts, chondroblasts and adipocytes [62, 63]. Furthermore, clearly defined smooth muscle cells (αSM-actin+, caldesmon+ and myosin heavy chain+) from bovine aortic media have been described to undergo osteoblastic and chondroblastic differentiation when cultured appropriately [64]. These observations could explain the calcification of vessels during atherosclerosis. The mechanisms of such vascular calcification seem to resemble those taking place during bone formation, because BMP2 and its target genes Runx2, osteocalcin and osteopontin were observed within atherosclerotic lesions [65, 66]. Moreover, inflammatory cytokines in aorta induce BMP2, which then promotes signalling through a muscle segment homeobox homolog (Msx2)/Wnt pathway leading to increased alkaline phosphatase activity and osteogenic differentiation [67]. Interestingly, Msx2 was shown to be a regulatory factor for VSM differentiation in one hand and osteoblastic cell differentiation on the other hand [68, 69]. This latter finding suggests that whatever their tissue origin, nMSCs can retain molecular programs to generate either smooth muscle cells or osteo-chondroblastic cells, and such fine-regulation mechanisms can be disturbed in lesion and pathologic situations. This finding fits well with the description of mural mesodermal progenitors capable of generating either skeletogenic (osteoblasts, chondroblasts and adipocytes) or myogenic cells [70]. The decision to give rise to one or the other differentiation pathway depends on molecular environmental influences.
The pericytic identity of nMSCs further suggests that nMSCs may reach the tissue from invading vessels during early events of bone formation. This process was shown to occur through the CD166 molecules which were found to be highly expressed by nMSCs from the perichondrium in the foetus. Activated leucocyte cell adhesion molecule (ALCAM), or CD166, is a membrane molecule that we and others found to be important for haematopoiesis-supporting stromal cells [71, 72]. Purified ALCAM+ cells could support haematopoiesis, osteoclastogenesis, and angiogenesis. Interestingly, in vitro inhibition of homophilic (ALCAM/ALCAM) and heterophilic (ALCAM/CD6) ALCAM-mediated adhesion prevented the blood vessel invasion into cartilage. It is likely that such an effect may be associated with ALCAM involvement in endothelial cell development probably through immature EPCs [73]. In addition, Arai et al. first characterized nMSCs as cells capable of generating endochondral formation in the human foetus [72].
In contrast, when adult fracture healing is concerned, the origin of nMSCs, contributing to bone regeneration is still controversial since these cells are known to reside in a number of surrounding tissues, such as the periosteum, BM, synovium and trabecular bone [18, 74–77]. Native MSCs were also thought to be derived from surrounding skeletal muscle. This suggestion is a real possibility because MSCs might be obtained from multipotential myoblastic cells such as the well-known C2C12 murine cell line [78]. However, during secondary fracture healing, the primary source of nMSCs giving rise to the callus is thought to be periosteum, notably because (i) callus development after fracture is strongly disturbed when the periosteum is removed and (ii) the periosteum produces BMPs during early events following fracture [79, 80]. Furthermore, to date convincing data support the presence of functional nMSCs within the periosteum that have strong proliferative and osteogenic capabilities in vitro and in vivo[81]. Here also, vascularization is a crucial event for the initiation and propagation of the bone formation deriving from the periosteum. We can therefore suppose that periosteal nMSCs are located in the vessels and are induced to proliferate and differentiate into osteoblasts after fracture after vascular ingrowth into developing callus (Fig. 3).
Fig 3.

Model of bone repair process after fracture. After fracture and inflammation stage, new vessel formed from EPCs ingrow into lesion. Native MSCs are of perivascular origin and accompany new vessels into the cal. The hypertrophic chondrocytes (Hyp. Ch) are crucial elements forming the callus and inducing vascularization notably by their secretion of PlGF and VEGF. Native MSCs within cal or lesion proliferate and differentiate into osteoblastic cells capable to build up new bony structure (O). The bone can be repaired through intramembranous or endochondral ossification. In the intramembranous case, nMSCs condense before to form bone whereas in endochondral ossification nMSCs generate chondrocytes which in turn induce osteoblasts to form bone.
To date, no data exist on the strict purification of nMSCs (i.e. one selected cell generating one multipotent CFU-F). Furthermore, heterogeneity is known to exist between CFU-Fs in terms of multipotentiality, and no observational tests exist to discern differences among them [18, 82, 83]. Finally, crucial questions remain to be elucidated: (i) are nMSCs or cMSCs true multipotent stem cells or are they more committed progenitors? (ii) In a pool of enriched nMSCs or cMSCs, does a hierarchy exist as that found in haematopoiesis (i.e. stem cells versus progenitors versus precursors versus mature cells)? Therefore, the identity of nMSCs remains obscure, and their characterization is undoubtedly crucial for understanding bone biology and its abnormalities (Table 1). Finally, since cMSCs derive from nMSCs, it is important that bone-reconstituting studies comparing both populations should also be performed, to prospectively evaluate their clinical outcome.
The neural crest origin of MSCs
In the skull, bone cartilage and smooth muscle cells are of neural crest (NC) origin [84]. NC cells (NCCs) derive from the dorsal neural tube during the development of the embryo. Therefore, one wonders whether there are several types of nMSCs (i.e. NC or mesodermal MSCs) according to their anatomic situation or whether all nMSCs originate from a unique cell type that develops later into NCCs or mesodermal cells. Until recently, cranial MSCs were thought to derive from NCCs, whereas MSCs from the axis and long bones would derive from the mesoderm [85]. However, to date, several reports have shown that MSCs could derive from NCCs. The first evidence of this derivation was reported by Takashima et al., who observed that murin embryonic stem cells, upon retinoic acid induction, generated Sox1+ MSCs from NCCs endowed with strong proliferation potential, contrary to cells from the mesoderm [86]. However, Sox1 is the most specific marker of neuroepithelial cells. In vitro (and also in embryo) studies demonstrated that Sox1-labelled cells were platelet-derived growth factor-receptorα– (PDGF-Rα) then yielded Sox1–/PDGF-Rα+ cells with MSCs characteristics. Very few PDGF-Rα+ cells was obtained from unlabelled cells (i.e. Sox1– cells) suggesting that all MSCs derived from NCC. The same team stained Sox1+ cells with permanent labelling to follow up post-natal MSCs. They observed a decrease of NC-derived MSCs concomitant with an increase of unknown origin MSCs in adult mice. Therefore, there is a hierarchy in the outcome of the MSC population during development. Some other clues were very recently described in agreement with work described here. In mice, Leucht et al. used NC (Hoxa11–) or mesoderm (Hoxa11+)-derived cells to heal mandibular and tibial bone [87]. Surprisingly, NC-derived MSCs healed both mandibles and tibia, whereas mesoderm-derived MSCs healed only tibia. In addition, Hoxa11– MSCs yielded Hoxa11+ cells on transplantation in tibia or on co-culture with mesodermal cells, but mesodermal Hoxa11+ cells never generated Hoxa11– cells. Thus, these observations strongly suggest that reconstituting MSCs are of NC origin and that mesodermal MSCs derive from them. However, one may alternatively suppose that cranial microenvironment does not permit bone reconstitution by tibial (mesodermal) MSCs. In other words, efficient bone healing by mesodermal MSCs could depend on specific signals, mediated by local microenvironmental factors, that may be absent from the NC-derived MSC microenvironment although the mesodermal MSC-specific factors can be efficient on NC-derived MSCs. In accordance with the data depicted previously, nMSCs demonstrate a consistent expression of the neural marker CD271 also named LNGF-R and p75.
In addition, numerous papers showed that BM MSCs can readily expressed several neural markers and display some properties of neuron-like cells [88]. Several neural specific factors, such as neurotrophic molecules, were also found to be expressed by MSCs and then to have biological activities on neural cells [89, 90]. However, the neural potential of BM MSCs is still controversial since fully differentiated and functional neurons, that are capable of communicating with other through synapses, were not generated. These discrepancies could be due to experimental design or conditions. In vitro protocols for eliciting neural potential of BM MSCs are highly complex requiring several stages and actually even slight modifications in such procedures may change completely the final outcomes [91]. Another fundamental parameter should be taken into account as discussed above: the possible NC origin of BM MSCs. One may argue that the neural potential elicited by experimental design could depend on BM content of NC-derived MSCs; in other words the efficacy of cells to generate neurons is related to the number of multipotent NC cells contained within BM samples. Indeed, when evaluated directly on multipotent NC cells one may observe that they have strong capacities to differentiate into neurons, glial cells, osteocytes and chondrocytes and comprise 7–13% of clonogenic NC cells. In contrast, when MSCs are assessed for their neural capacities, it is observed that such properties are very rare events [92, 93]. This is supported by data from reports of Takashima et al. They showed that neural markers were not observed in cells derived from embryonic cells differentiated by mesodermal inducing conditions. On the other hand, neural markers and MSC differentiation capacities were both detected in cells cultured under neuroepithelial conditions [86]. Therefore, the potential of a population of BM MSCs to give rise to neural cells could depend only on its NC-derived cell content. During development, it would be very interesting to address this assumption directly on nMSCs according their expression of either neural (such as p75) or mesodermal markers (such as PDGF-Rβ).
In conclusion, the relationships between nMSC, NC and VSM cells seem to be tight. However, further studies need to be performed to confirm these data and specifically to define the MSC origin in embryo and post-natally and their respective roles in both bone development and bone regeneration after lesion.
Vascularization by endothelial progenitor cells as prerequisite process before bone repair
Convincing data show that angiogenesis and vasculogenesis, in addition to MSCs, are also crucial for bone repair. This observation is confirmed by the fact that expression of vasculogenic/angiogenic molecules (e.g. VEGF, PlGF, erythropoietin [EPO]) is a prerequisite for the skeletal development and bone healing process, notably during hypoxia events [1, 13, 14, 94–97].
Vascularization and bone healing
In bone defects, there is a breaking in blood supply and the graft (generally constituted by regenerative cells and a scaffold) has to be locally vascularized as soon as possible to circumvent the failure. Indeed, to submit tissue or graft to hypoxic conditions and to the lack of nutrients will ultimately lead to cell death [98]. In rat, the critical bone defect filled with engineered scaffold provided bone formation which was increased when a vascularized periosteal flap was added and this also prevented heterotopic ossification [99]. Therefore, scientists performed new strategies to induce vascularization or to inhibit endothelial cell death. The addition of the anti-apoptotic Bcl2 gene into endothelial cells can increase survival and formation of blood-perfused blood vessels that develop into arteries, veins and capillaries [100]. In addition, immortalization of human dermal microvascular endothelial cells by hTERT results in the development of microvascular structures when implanted subcutaneously [101]. In another way, the use of a mixture consisting of perivascular cell precursors and endothelial cells in engineered constructs leads stable microvessels in vivo, which are fully functional for more than 1 year [102]. These data therefore demonstrate that blood supply of regenerative cells by their vascularization is a prerequisite to improve significantly the reconstruction potential of the transplant filled with MSCs.
Hypoxia accompanying vascular damage after skeletal injury has a strong impact on osteoregenerative cells. Temporary exposure of MSCs to this condition, results in the down-regulation of Cbfa-1/Runx2, osteocalcin and type I collagen, and the up-regulation of osteopontin and VEGF [103]. Low oxygen levels within a milieu induce hypoxia-inducible factor α (HIF-α) to be active. HIFs are transcription factors primarily responsible for changes in gene expression during hypoxia. HIF are members of bHLH-PAS family of proteins that bind to canonical DNA sequences (hypoxia-regulated elements) in the promoter region of target genes. HIF-α is one of these elements and is rapidly degraded in normoxia, whereas below 8% O2, HIF-α becomes increasingly stabilized. Once stabilized, HIF-α proteins bind the constitutively expressed HIF-β then DNA, and activate gene transcription [95]. Notably, HIF-α induces VEGF gene expression in osteoblasts, which generate both angiogenesis and proliferation of osteoregenerative cells. Therefore, hypoxia links vascular biology to bone formation [12]. Very recently, Maes et al. underlined the role of inflammation and vasculogenic/angiogenic cytokines in a model of semi-stabilized bone-fracture healing in mice lacking PlGF, a VEGF-like protein. In such mice, the fracture repair was impaired and characterized by delayed healing or non-union bone [17]. In addition, the authors demonstrated an early action of PlGF in the inflammatory process on attraction of monocytes/macrophages, as well as a necessary role in the vascularization of the fracture wound. This latter role was not clearly defined, because the experiments did not show whether PlGF acted directly or indirectly via induction of the expression of VEGF within the inflammation site. The VEGF/PlGF defect could also impair the recruitment, proliferation/differentiation of EPCs and MSCs (nMSCs and cMSCs since cMSCs were assessed directly by in vitro experiments and nMSCs indirectly by measuring the thickness of the periosteum and by BrdU staining). Finally, the remodelling process was also impaired by the decreased level of matrix metalloproteinase 13 (MMP-13) and MT1-MMP functions, thus giving rise to an abnormal structure of bony callus.
Endothelial progenitor cells
By definition, angiogenesis and vasculogenesis refer to the formation of blood vessels from pre-existing blood vessels or EPCs, respectively. Extensive data support the existence of EPCs, their BM origin, and contribution to the formation of new blood vessels in adults. Moreover, their discovery led to a new concept that vasculogenesis and angiogenesis may occur simultaneously in post-natal life because these cells can differentiate when needed into vascular endothelium through a mechanism recapitulating embryonic vasculogenesis. Most of the circulating EPCs reside in the BM in close association with haematopoietic stem cells and the BM stroma that provides an optimal microenvironment. EPCs can proliferate, migrate and differentiate into endothelial lineage cells but have not characteristics of mature endothelial cells. Urbich and Dimmeler [104] define EPCs as non-endothelial cells that show clonal expression (the ability of a single cell to multiply) and stemness characteristics (proliferative capacity and resistance to stress) and are capable of differentiating into endothelial cells.
In most studies, EPCs are identified and enumerated via flow cytometry identification of cells expressing CD34, CD133, or the VEGF receptor 2 (KDR) [105, 106]. Because these molecules are also expressed on haematopoietic stem/progenitor populations, the presence of haematopoietic contamination of EPCs should be expected. EPCs may be quantitated by the use of a commercially available kit that identifies ‘endothelial cell colony-forming units’ (CFU-ECs; also named CFU-Hill) [107, 108]. Identification of CFU-ECs from peripheral blood has formed the basis for the use of these cells as predictive biomarkers of vascular disease and as a cell source for angiogenic therapies. The diversity in defining the cells that give rise to CFU-ECs has contributed to the controversy in understanding the role these cells may play in neoangiogenesis. Indeed, we and others have identified another type of EPCs from human peripheral blood namely blood outgrowth endothelial cells or late EPCs (also referred to as endothelial colony-forming cells) [109–111]. In fact, three distinctly different populations of EPCs can be identified from peripheral blood mononuclear cells (PBMNCs), based on their in vitro adhesion and morphology: the CFU-Hill, the circulating angiogenic cells and late EPCs [112]. The CFU-Hill are obtained after 5-day culture from non-adherent PBMNCs, and are characterized by a central cluster of round cells surrounded by radiating thin flat cells [108]. These cells are positive for CD45, VEGF-R2, CD146, CD144, and are CD34dim (Table 1). They have weak proliferative and vasculogenic activities. Circulating angiogenic cells, on the other hand, are obtained from adherent PBMNCs, do not form colony in culture and are positive for CD133, CD31, VEGFR-2, CD45 and negative for CD34 and CD144 [113]. Finally, late EPCs derived from adherent PBMNCs and form endothelial colonies after 3–4 weeks of culture. These cells formed a typical cobblestone monolayer, have clonal proliferative capacities and consistent capabilities to regenerate vessels in vitro and in vivo and are positive for CD34, CD144, VEGF-R2, but negative for CD133, CD45 and CD14 (Table 1) [109, 114, 115]. This latter type of EPCs is thought to be the principal actor of revascularization after lesion in adult.
The role of these different types of EPCs could be crucial but has not been yet evaluated in bone repair, despite their crucial role in vascularization. Nevertheless, EPCs (CFU-Hill and late EPCs) express the receptors of VEGF and PlGF and are able to migrate when stimulated by VEGF and EPO [116, 117]. All of these molecules are shown to have a crucial role in bone-fracture healing. Scrutinizing their involvement during bone healing could be of particular interest.
Although the implication of such EPCs in bone repair has been suggested, few experiments have been performed to corroborate this notion. In this context, by using a fracture healing and distraction osteogenesis model in mouse and rat, Lee et al. showed that the number of adherent Dil-Ac-LDL+/Lectin+ or VEGF-R2+ cells, thought to represent EPCs, peaked at day 3 after fracture and returned to basal levels [118]. In the plasma of injured mice, VEGF mRNA level was increased before the EPCs peak. Likewise, the SDF-1 and MCP-1 chemokine levels as well as M-CSF and IL-6 pro-inflammatory cytokines were increased during the healing process. In addition, during distraction in rat, the level of EPCs showed a protracted elevation level even at 3 weeks after distraction osteogenesis, as compared to non-distracted controls. Therefore, EPCs could be mobilized during the whole process of distraction. In human beings, a previous study described mobilization of EPCs in diaphyseal tibial fractures, with EPCs being defined as CD34+ or CD133+ cells [119]. Flow cytometry-assessed counts of circulating CD34+ and CD133+ mononuclear cells increased from day 1 to day 3 and then decreased to reach the basal level at day 7. However, within these two studies, EPCs have been not clearly identified because no experiments have been performed so as to demonstrate that these putative EPCs can indeed generate either endothelial colonies or von Willbrand factor+/CD31+ and CD144+ mature endothelial cells.
In addition to the reports mentioned above, studies of human CD34+ progenitor cells deriving from peripheral blood showed that they expressed mRNA of the osteoblastic marker osteocalcin, and when they were intravenously transplanted after producing non-healing femoral fractures in nude rats, some regenerating osteoblasts and endothelial cells were found to be of human origin [120]. Furthermore, when fractured bones were assessed via radiological, histological and biomechanical studies, they revealed enhanced bone healing in the CD34+ transplanted group as compared with control groups. The CD34+ cells at the fracture site were shown to express various angiogenic factors at the mRNA level and, interestingly, injection of soluble Flt1 (soluble VEGF antagonist) reduced not only angiogenesis/vasculogenesis, but also intrinsic osteogenesis, suggesting that angiogenic factors released by the transplanted CD34+ cells contribute, at least in part, to fracture healing in a paracrine manner. However, as we described above in nMSCs paragraph, there is a possibility that such CD34+ cells were nMSCs.
Therefore, EPCs are crucial regenerative cells for bone healing. However, confusion exists regarding their characterization (Table 1), which implies that further studies are mandatory to define these cells properly and to highlight their role in bone biology.
Conclusion
Bone regeneration is a complex biological process involving a well-coordinated interplay between different local or systemic soluble factors, extracellular matrix, MSCs and EPCs (Fig. 3). While there is compelling evidence that ex vivo expanded MSCs can effectively repair critical bone defects, this has not be proven as yet for nMSCs in either animal or human models, due to the lack of a consensus regarding their phenotypic properties, and also because convincing data about their perivascular anatomic location have only recently started to emerge. Likewise, although several studies have reported the important role of EPCs in bone healing, there is an ongoing controversy regarding the different populations isolated in mice and human beings in terms of their characteristics and their potential functional and/or phenotypic overlap. Therefore, although recent advances on the field favour the potential of MSCs and EPCs in bone regeneration, considerable research needs still to be done to unravel the biology of these cells in bone turnover. The information provided will not only shed light into key mechanisms implicated in fracture healing and bone morphogenesis, but will also have major impact into the eventual introduction of MSCs and EPCs in the clinic.
Acknowledgments
This work was funded by the Agence Nationale de la Recherche Technologies pour la Santé et l’Autonomie, ATOS project N° 024–03 (2007–2010).
References
- 1.Ferguson C, Alpern E, Miclau T, et al. Does adult fracture repair recapitulate embryonic skeletal formation. Mech Dev. 1999;87:57–66. doi: 10.1016/s0925-4773(99)00142-2. [DOI] [PubMed] [Google Scholar]
- 2.Bruder SP, Fink DJ, Caplan AI. Mesenchymal stem cells in bone development, bone repair, and skeletal regeneration therapy. J Cell Biochem. 1994;56:283–94. doi: 10.1002/jcb.240560809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le AX, Miclau T, Hu D, et al. Molecular aspects of healing in stabilized and non-stabilized fractures. J Orthop Res. 2001;19:78–84. doi: 10.1016/S0736-0266(00)00006-1. [DOI] [PubMed] [Google Scholar]
- 4.Simon AM, Manigrasso MB, O’Connor JP. Cyclo-oxygenase 2 function is essential for bone fracture healing. J Bone Miner Res. 2002;17:963–76. doi: 10.1359/jbmr.2002.17.6.963. [DOI] [PubMed] [Google Scholar]
- 5.Gerstenfeld LC, Cho TJ, Kon T, et al. Impaired fracture healing in the absence of TNF-alpha signaling: the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res. 2003;18:1584–92. doi: 10.1359/jbmr.2003.18.9.1584. [DOI] [PubMed] [Google Scholar]
- 6.Lehmann W, Edgar CM, Wang K, et al. Tumor necrosis factor alpha (TNF-alpha) coordinately regulates the expression of specific matrix metalloproteinases (MMPS) and angiogenic factors during fracture healing. Bone. 2005;36:300–10. doi: 10.1016/j.bone.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Kon T, Cho TJ, Aizawa T, et al. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J Bone Miner Res. 2001;16:1004–14. doi: 10.1359/jbmr.2001.16.6.1004. [DOI] [PubMed] [Google Scholar]
- 8.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiltunen A, Vuorio E, Aro HT. A standardized experimental fracture in the mouse tibia. J Orthop Res. 1993;11:305–12. doi: 10.1002/jor.1100110219. [DOI] [PubMed] [Google Scholar]
- 10.St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–86. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–65. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- 12.Emans PJ, Spaapen F, Surtel DA, et al. A novel in vivo model to study endochondral bone formation; HIF-1alpha activation and BMP expression. Bone. 2007;40:409–18. doi: 10.1016/j.bone.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Kloen P, Di Paola M, Borens O, et al. BMP signaling components are expressed in human fracture callus. Bone. 2003;33:362–71. doi: 10.1016/s8756-3282(03)00191-1. [DOI] [PubMed] [Google Scholar]
- 14.Yoshimura Y, Nomura S, Kawasaki S, et al. Colocalization of noggin and bone morphogenetic protein-4 during fracture healing. J Bone Miner Res. 2001;16:876–84. doi: 10.1359/jbmr.2001.16.5.876. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Alman BA. Wnt pathway, an essential role in bone regeneration. J Cell Biochem. 2009;106:353–62. doi: 10.1002/jcb.22020. [DOI] [PubMed] [Google Scholar]
- 16.Ito H, Akiyama H, Shigeno C, et al. Hedgehog signaling molecules in bone marrow cells at the initial stage of fracture repair. Biochem Biophys Res Commun. 1999;262:443–51. doi: 10.1006/bbrc.1999.1197. [DOI] [PubMed] [Google Scholar]
- 17.Maes C, Coenegrachts L, Stockmans I, et al. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest. 2006;116:1230–42. doi: 10.1172/JCI26772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 19.Secco M, Zucconi E, Vieira NM, et al. Multipotent stem cells from umbilical cord: cord is richer than blood. Stem Cells. 2008;26:146–50. doi: 10.1634/stemcells.2007-0381. [DOI] [PubMed] [Google Scholar]
- 20.Zuk PA, Zhu M, Mizuno H, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–28. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]
- 21.Yen BL, Huang HI, Chien CC, et al. Isolation of multipotent cells from human term placenta. Stem Cells. 2005;23:3–9. doi: 10.1634/stemcells.2004-0098. [DOI] [PubMed] [Google Scholar]
- 22.Liechty KW, MacKenzie TC, Shaaban AF, et al. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000;6:1282–6. doi: 10.1038/81395. [DOI] [PubMed] [Google Scholar]
- 23.Meijer GJ, De Bruijn JD, Koole R, et al. Cell-based bone tissue engineering. PLoS Med. 2007;4:260–64. doi: 10.1371/journal.pmed.0040009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heymann D, Touchais S, Bohic S, et al. Heterotopic implantation of mouse bone-marrow cells: an in vivo model allowing analysis of mineral phases during mineralization processes. Connect Tissue Res. 1998;37:219–31. doi: 10.3109/03008209809002441. [DOI] [PubMed] [Google Scholar]
- 25.Bohic S, Rohanizadeh R, Touchais S, et al. Leukemia inhibitory factor and oncostatin M influence the mineral phases formed in a murine heterotopic calcification model: a Fourier transform-infrared microspectroscopic study. J Bone Miner Res. 1998;13:1619–32. doi: 10.1359/jbmr.1998.13.10.1619. [DOI] [PubMed] [Google Scholar]
- 26.Bruder SP, Kraus KH, Goldberg VM, et al. The effect of implants loaded with autologous mesenchymal stem cells on the healing of canine segmental bone defects. J Bone Joint Surg Am. 1998;80:985–96. doi: 10.2106/00004623-199807000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Petite H, Viateau V, Bensaid W, et al. Tissue-engineered bone regeneration. Nat Biotechnol. 2000;18:959–63. doi: 10.1038/79449. [DOI] [PubMed] [Google Scholar]
- 28.Quarto R, Mastrogiacomo M, Cancedda R, et al. Repair of large bone defects with the use of autologous bone marrow stromal cells. N Engl J Med. 2001;344:385–6. doi: 10.1056/NEJM200102013440516. [DOI] [PubMed] [Google Scholar]
- 29.Horwitz EM, Gordon PL, Koo WK, et al. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci USA. 2002;99:8932–7. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Blanc K, Gotherstrom C, Ringden O, et al. Fetal mesenchymal stem-cell engraftment in bone after in utero transplantation in a patient with severe osteogenesis imperfecta. Transplantation. 2005;79:1607–14. doi: 10.1097/01.tp.0000159029.48678.93. [DOI] [PubMed] [Google Scholar]
- 31.Bajada S, Harrison PE, Ashton BA, et al. Successful treatment of refractory tibial nonunion using calcium sulphate and bone marrow stromal cell implantation. J Bone Joint Surg Br. 2007;89:1382–6. doi: 10.1302/0301-620X.89B10.19103. [DOI] [PubMed] [Google Scholar]
- 32.Le Blanc K, Ringden O. Immunobiology of human mesenchymal stem cells and future use in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11:321–34. doi: 10.1016/j.bbmt.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Le Blanc K, Rasmusson I, Sundberg B, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–41. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 34.Le Blanc K, Frassoni F, Ball L, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579–86. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 35.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–22. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 36.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–36. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 37.Da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119:2204–13. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 38.Muguruma Y, Yahata T, Miyatake H, et al. Reconstitution of the functional human hematopoietic microenvironment derived from human mesenchymal stem cells in the murine bone marrow compartment. Blood. 2006;107:1878–87. doi: 10.1182/blood-2005-06-2211. [DOI] [PubMed] [Google Scholar]
- 39.Covas DT, Panepucci RA, Fontes AM, et al. Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Exp Hematol. 2008;36:642–54. doi: 10.1016/j.exphem.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 40.Galmiche MC, Koteliansky VE, Briere J, et al. Stromal cells from human long-term marrow cultures are mesenchymal cells that differentiate following a vascular smooth muscle differentiation pathway. Blood. 1993;82:66–76. [PubMed] [Google Scholar]
- 41.Charbord P, Tavian M, Humeau L, et al. Early ontogeny of the human marrow from long bones: an immunohistochemical study of hematopoiesis and its microenvironment. Blood. 1996;87:4109–19. [PubMed] [Google Scholar]
- 42.Friedenstein AJ, Latzinik NW, Grosheva AG, et al. Marrow microenvironment transfer by heterotopic transplantation of freshly isolated and cultured cells in porous sponges. Exp Hematol. 1982;10:217–27. [PubMed] [Google Scholar]
- 43.Muraglia A, Cancedda R, Quarto R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J Cell Sci. 2000;113:1161–6. doi: 10.1242/jcs.113.7.1161. [DOI] [PubMed] [Google Scholar]
- 44.Delorme B, Ringe J, Pontikoglou C, et al. Specific lineage-priming of bone marrow mesenchymal stem cells provides the molecular framework for their plasticity. Stem Cells. 2009;27:1142–51. doi: 10.1002/stem.34. [DOI] [PubMed] [Google Scholar]
- 45.Seshi B, Kumar S, King D. Multilineage gene expression in human bone marrow stromal cells as evidenced by single-cell microarray analysis. Blood Cells Mol Dis. 2003;31:268–85. doi: 10.1016/s1079-9796(03)00150-5. [DOI] [PubMed] [Google Scholar]
- 46.Zipori D. Mesenchymal stem cells: harnessing cell plasticity to tissue and organ repair. Blood Cells Mol Dis. 2004;33:211–5. doi: 10.1016/j.bcmd.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 47.Simmons PJ, Torok-Storb B. Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood. 1991;78:55–62. [PubMed] [Google Scholar]
- 48.Gronthos S, Zannettino AC, Hay SJ, et al. Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J Cell Sci. 2003;116:1827–35. doi: 10.1242/jcs.00369. [DOI] [PubMed] [Google Scholar]
- 49.Deschaseaux F, Charbord P. Human marrow stromal precursors are alpha 1 integrin subunit-positive. J Cell Physiol. 2000;184:319–25. doi: 10.1002/1097-4652(200009)184:3<319::AID-JCP5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 50.Gindraux F, Selmani Z, Obert L, et al. Human and rodent bone marrow mesenchymal stem cells that express primitive stem cell markers can be directly enriched by using the CD49a molecule. Cell Tissue Res. 2007;327:471–83. doi: 10.1007/s00441-006-0292-3. [DOI] [PubMed] [Google Scholar]
- 51.Belkin VM, Belkin AM, Koteliansky VE. Human smooth muscle VLA-1 integrin: purification, substrate specificity, localization in aorta, and expression during development. J Cell Biol. 1990;111:2159–70. doi: 10.1083/jcb.111.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Delorme B, Ringe J, Gallay N, et al. Specific plasma membrane protein phenotype of culture-amplified and native human bone marrow mesenchymal stem cells. Blood. 2008;111:2631–5. doi: 10.1182/blood-2007-07-099622. [DOI] [PubMed] [Google Scholar]
- 53.Quirici N, Soligo D, Bossolasco P, et al. Isolation of bone marrow mesenchymal stem cells by anti-nerve growth factor receptor antibodies. Exp Hematol. 2002;30:783–91. doi: 10.1016/s0301-472x(02)00812-3. [DOI] [PubMed] [Google Scholar]
- 54.Caneva L, Soligo D, Cattoretti G, et al. Immuno-electron microscopy characterization of human bone marrow stromal cells with anti-NGFR antibodies. Blood Cells Mol Dis. 1995;21:73–85. doi: 10.1006/bcmd.1995.0011. [DOI] [PubMed] [Google Scholar]
- 55.Cattoretti G, Schiro R, Orazi A, et al. Bone marrow stroma in humans: anti-nerve growth factor receptor antibodies selectively stain reticular cells in vivo and in vitro. Blood. 1993;81:1726–38. [PubMed] [Google Scholar]
- 56.Martinez C, Hofmann TJ, Marino R, et al. Human bone marrow mesenchymal stromal cells express the neural ganglioside GD2: a novel surface marker for the identification of MSCs. Blood. 2007;109:4245–8. doi: 10.1182/blood-2006-08-039347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gang EJ, Bosnakovski D, Figueiredo CA, et al. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood. 2007;109:1743–51. doi: 10.1182/blood-2005-11-010504. [DOI] [PubMed] [Google Scholar]
- 58.Deschaseaux F, Gindraux F, Saadi R, et al. Direct selection of human bone marrow mesenchymal stem cells using an anti-CD49a antibody reveals their CD45med,low phenotype. Br J Haematol. 2003;122:506–17. doi: 10.1046/j.1365-2141.2003.04469.x. [DOI] [PubMed] [Google Scholar]
- 59.Sacchetti B, Funari A, Michienzi S, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–36. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 60.Crisan M, Yap S, Casteilla L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–13. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 61.Delorme B, Basire A, Gentile C, et al. Presence of endothelial progenitor cells, distinct from mature endothelial cells, within human CD146+ blood cells. Thromb Haemost. 2005;94:1270–9. doi: 10.1160/TH05-07-0499. [DOI] [PubMed] [Google Scholar]
- 62.Doherty MJ, Ashton BA, Walsh S, et al. Vascular pericytes express osteogenic potential in vitro and in vivo. J Bone Miner Res. 1998;13:828–38. doi: 10.1359/jbmr.1998.13.5.828. [DOI] [PubMed] [Google Scholar]
- 63.Farrington-Rock C, Crofts NJ, Doherty MJ, et al. Chondrogenic and adipogenic potential of microvascular pericytes. Circulation. 2004;110:2226–32. doi: 10.1161/01.CIR.0000144457.55518.E5. [DOI] [PubMed] [Google Scholar]
- 64.Tintut Y, Alfonso Z, Saini T, et al. Multilineage potential of cells from the artery wall. Circulation. 2003;108:2505–10. doi: 10.1161/01.CIR.0000096485.64373.C5. [DOI] [PubMed] [Google Scholar]
- 65.Bostrom K, Watson KE, Horn S, et al. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800–9. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steitz SA, Speer MY, Curinga G, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–54. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 67.Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arterioscler Thromb Vasc Biol. 2006;26:1423–30. doi: 10.1161/01.ATV.0000220441.42041.20. [DOI] [PubMed] [Google Scholar]
- 68.Brunelli S, Tagliafico E, De Angelis FG, et al. Msx2 and necdin combined activities are required for smooth muscle differentiation in mesoangioblast stem cells. Circ Res. 2004;94:1571–8. doi: 10.1161/01.RES.0000132747.12860.10. [DOI] [PubMed] [Google Scholar]
- 69.Towler DA, Rutledge SJ, Rodan GA. Msx-2/Hox 8.1: a transcriptional regulator of the rat osteocalcin promoter. Mol Endocrinol. 1994;8:1484–93. doi: 10.1210/mend.8.11.7877617. [DOI] [PubMed] [Google Scholar]
- 70.Bianco P, Cossu G. Uno, nessuno e centomila: searching for the identity of mesodermal progenitors. Exp Cell Res. 1999;251:257–63. doi: 10.1006/excr.1999.4592. [DOI] [PubMed] [Google Scholar]
- 71.Cortes F, Deschaseaux F, Uchida N, et al. HCA, an immunoglobulin-like adhesion molecule present on the earliest human hematopoietic precursor cells, is also expressed by stromal cells in blood-forming tissues. Blood. 1999;93:826–37. [PubMed] [Google Scholar]
- 72.Arai F, Ohneda O, Miyamoto T, et al. Mesenchymal stem cells in perichondrium express activated leukocyte cell adhesion molecule and participate in bone marrow formation. J Exp Med. 2002;195:1549–63. doi: 10.1084/jem.20011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ohneda O, Ohneda K, Arai F, et al. ALCAM (CD166): its role in hematopoietic and endothelial development. Blood. 2001;98:2134–42. doi: 10.1182/blood.v98.7.2134. [DOI] [PubMed] [Google Scholar]
- 74.De Bari C, Dell’Accio F, Tylzanowski P, et al. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001;44:1928–42. doi: 10.1002/1529-0131(200108)44:8<1928::AID-ART331>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 75.Jones EA, English A, Henshaw K, et al. Enumeration and phenotypic characterization of synovial fluid multipotential mesenchymal progenitor cells in inflammatory and degenerative arthritis. Arthritis Rheum. 2004;50:817–27. doi: 10.1002/art.20203. [DOI] [PubMed] [Google Scholar]
- 76.De Bari C, Dell’Accio F, Vanlauwe J, et al. Mesenchymal multipotency of adult human periosteal cells demonstrated by single-cell lineage analysis. Arthritis Rheum. 2006;54:1209–21. doi: 10.1002/art.21753. [DOI] [PubMed] [Google Scholar]
- 77.Noth U, Osyczka AM, Tuli R, et al. Multilineage mesenchymal differentiation potential of human trabecular bone-derived cells. J Orthop Res. 2002;20:1060–9. doi: 10.1016/S0736-0266(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 78.Katagiri T, Yamaguchi A, Komaki M, et al. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol. 1994;127:1755–66. doi: 10.1083/jcb.127.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakahara H, Bruder SP, Haynesworth SE, et al. Bone and cartilage formation in diffusion chambers by subcultured cells derived from the periosteum. Bone. 1990;11:181–8. doi: 10.1016/8756-3282(90)90212-h. [DOI] [PubMed] [Google Scholar]
- 80.Bostrom MP, Lane JM, Berberian WS, et al. Immunolocalization and expression of bone morphogenetic proteins 2 and 4 in fracture healing. J Orthop Res. 1995;13:357–67. doi: 10.1002/jor.1100130309. [DOI] [PubMed] [Google Scholar]
- 81.De Bari C, Dell’Accio F, Karystinou A, et al. A biomarker-based mathematical model to predict bone-forming potency of human synovial and periosteal mesenchymal stem cells. Arthritis Rheum. 2008;58:240–50. doi: 10.1002/art.23143. [DOI] [PubMed] [Google Scholar]
- 82.Banfi A, Muraglia A, Dozin B, et al. Proliferation kinetics and differentiation potential of ex vivo expanded human bone marrow stromal cells: implications for their use in cell therapy. Exp Hematol. 2000;28:707–15. doi: 10.1016/s0301-472x(00)00160-0. [DOI] [PubMed] [Google Scholar]
- 83.Baksh D, Davies JE, Zandstra PW. Soluble factor cross-talk between human bone marrow-derived hematopoietic and mesenchymal cells enhances in vitro CFU-F and CFU-O growth and reveals heterogeneity in the mesenchymal progenitor cell compartment. Blood. 2005;106:3012–9. doi: 10.1182/blood-2005-01-0433. [DOI] [PubMed] [Google Scholar]
- 84.Couly GF, Coltey PM, Le Douarin NM. The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development. 1993;117:409–29. doi: 10.1242/dev.117.2.409. [DOI] [PubMed] [Google Scholar]
- 85.Le Douarin NM, Creuzet S, Couly G, et al. Neural crest cell plasticity and its limits. Development. 2004;131:4637–50. doi: 10.1242/dev.01350. [DOI] [PubMed] [Google Scholar]
- 86.Takashima Y, Era T, Nakao K, et al. Neuroepithelial cells supply an initial transient wave of MSC differentiation. Cell. 2007;129:1377–88. doi: 10.1016/j.cell.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 87.Leucht P, Kim JB, Amasha R, et al. Embryonic origin and Hox status determine progenitor cell fate during adult bone regeneration. Development. 2008;135:2845–54. doi: 10.1242/dev.023788. [DOI] [PubMed] [Google Scholar]
- 88.Hermann A, Gastl R, Liebau S, et al. Efficient generation of neural stem cell-like cells from adult human bone marrow stromal cells. J Cell Sci. 2004;117:4411–22. doi: 10.1242/jcs.01307. [DOI] [PubMed] [Google Scholar]
- 89.Kan I, Melamed E, Offen D. Autotransplantation of bone marrow-derived stem cells as a therapy for neurodegenerative diseases. Handb Exp Pharmacol. 2007:219–42. doi: 10.1007/978-3-540-68976-8_10. [DOI] [PubMed] [Google Scholar]
- 90.Crigler L, Robey RC, Asawachaicharn A, et al. Human mesenchymal stem cell subpopulations express a variety of neuro-regulatory molecules and promote neuronal cell survival and neuritogenesis. Exp Neurol. 2006;198:54–64. doi: 10.1016/j.expneurol.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 91.Wislet-Gendebien S, Wautier F, Leprince P, et al. Astrocytic and neuronal fate of mesenchymal stem cells expressing nestin. Brain Res Bull. 2005;68:95–102. doi: 10.1016/j.brainresbull.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 92.Li Y, Chen J, Chen XG, et al. Human marrow stromal cell therapy for stroke in rat: neurotrophins and functional recovery. Neurology. 2002;59:514–23. doi: 10.1212/wnl.59.4.514. [DOI] [PubMed] [Google Scholar]
- 93.Fu L, Zhu L, Huang Y, et al. Derivation of neural stem cells from mesenchymal stemcells: evidence for a bipotential stem cell population. Stem Cells Dev. 2008;17:1109–21. doi: 10.1089/scd.2008.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giannoudis PV, Einhorn TA, Marsh D. Fracture healing: the diamond concept. Injury. 2007;38:S3–6. doi: 10.1016/s0020-1383(08)70003-2. [DOI] [PubMed] [Google Scholar]
- 95.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–72. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y, Wan C, Deng L, et al. The hypoxia-inducible factor alpha pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117:1616–26. doi: 10.1172/JCI31581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gerstenfeld LC, Cho TJ, Kon T, et al. Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs. 2001;169:285–94. doi: 10.1159/000047893. [DOI] [PubMed] [Google Scholar]
- 98.Potier E, Ferreira E, Meunier A, et al. Prolonged hypoxia concomitant with serum deprivation induces massive human mesenchymal stem cell death. Tissue Eng. 2007;13:1325–31. doi: 10.1089/ten.2006.0325. [DOI] [PubMed] [Google Scholar]
- 99.Vogelin E, Jones NF, Huang JI, et al. Healing of a critical-sized defect in the rat femur with use of a vascularized periosteal flap, a biodegradable matrix, and bone morphogenetic protein. J Bone Joint Surg Am. 2005;87:1323–31. doi: 10.2106/JBJS.C.00913. [DOI] [PubMed] [Google Scholar]
- 100.Schechner JS, Nath AK, Zheng L, et al. In vivo formation of complex microvessels lined by human endothelial cells in an immunodeficient mouse. Proc Natl Acad Sci USA. 2000;97:9191–6. doi: 10.1073/pnas.150242297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang J, Nagavarapu U, Relloma K, et al. Telomerized human microvasculature is functional in vivo. Nat Biotechnol. 2001;19:219–24. doi: 10.1038/85655. [DOI] [PubMed] [Google Scholar]
- 102.Koike N, Fukumura D, Gralla O, et al. Tissue engineering: creation of long-lasting blood vessels. Nature. 2004;428:138–9. doi: 10.1038/428138a. [DOI] [PubMed] [Google Scholar]
- 103.Potier E, Ferreira E, Andriamanalijaona R, et al. Hypoxia affects mesenchymal stromal cell osteogenic differentiation and angiogenic factor expression. Bone. 2007;40:1078–87. doi: 10.1016/j.bone.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 104.Urbich C, Dimmeler S. Endothelial progenitor cells functional characterization. Trends Cardiovasc Med. 2004;14:318–22. doi: 10.1016/j.tcm.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 105.Peichev M, Naiyer AJ, Pereira D, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–8. [PubMed] [Google Scholar]
- 106.Reyes M, Dudek A, Jahagirdar B, et al. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest. 2002;109:337–46. doi: 10.1172/JCI14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bryder D, Rossi DJ, Weissman IL. Hematopoietic stem cells: the paradigmatic tissue-specific stem cell. Am J Pathol. 2006;169:338–46. doi: 10.2353/ajpath.2006.060312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 109.Lin Y, Weisdorf DJ, Solovey A, et al. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105:71–7. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ingram DA, Mead LE, Tanaka H, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–60. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- 111.Deschaseaux F, Selmani Z, Falcoz PE, et al. Two types of circulating endothelial progenitor cells in patients receiving long term therapy by HMG-CoA reductase inhibitors. Eur J Pharmacol. 2007;562:111–8. doi: 10.1016/j.ejphar.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 112.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1584–95. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vasa M, Fichtlscherer S, Adler K, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–90. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 114.Hur J, Yoon CH, Kim HS, et al. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;24:288–93. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- 115.Yoder MC, Mead LE, Prater D, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–9. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ribatti D. The discovery of endothelial progenitor cells. An historical review. Leuk Res. 2007;31:439–44. doi: 10.1016/j.leukres.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 117.Prunier F, Pfister O, Hadri L, et al. Delayed erythropoietin therapy reduces post-MI cardiac remodeling only at a dose that mobilizes endothelial progenitor cells. Am J Physiol Heart Circ Physiol. 2007;292:522–9. doi: 10.1152/ajpheart.00357.2006. [DOI] [PubMed] [Google Scholar]
- 118.Lee DY, Cho TJ, Kim JA, et al. Mobilization of endothelial progenitor cells in fracture healing and distraction osteogenesis. Bone. 2008;42:932–41. doi: 10.1016/j.bone.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 119.Laing AJ, Dillon JP, Condon ET, et al. Mobilization of endothelial precursor cells: systemic vascular response to musculoskeletal trauma. J Orthop Res. 2007;25:44–50. doi: 10.1002/jor.20228. [DOI] [PubMed] [Google Scholar]
- 120.Matsumoto T, Kawamoto A, Kuroda R, et al. Therapeutic potential of vasculogenesis and osteogenesis promoted by peripheral blood CD34-positive cells for functional bone healing. Am J Pathol. 2006;169:1440–57. doi: 10.2353/ajpath.2006.060064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shi S, Gronthos S. Perivascular niche of postnatal mesenchymal stem cells in human bone marrow and dental pulp. J Bone Miner Res. 2003;18:696–704. doi: 10.1359/jbmr.2003.18.4.696. [DOI] [PubMed] [Google Scholar]
- 122.Le Blanc K, Ringden O. Use of mesenchymal stem cells for the prevention of immune complications of hematopoietic stem cell transplantation. Haematologica. 2005;90:438. [PubMed] [Google Scholar]
- 123.Simmons PJ, Torok-Storb B. CD34 expression by stromal precursors in normal human adult bone marrow. Blood. 1991;78:2848–53. [PubMed] [Google Scholar]
- 124.Conget PA, Minguell JJ. Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J Cell Physiol. 1999;181:67–73. doi: 10.1002/(SICI)1097-4652(199910)181:1<67::AID-JCP7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 125.Barry F, Boynton R, Murphy M, et al. The SH-3 and SH-4 antibodies recognize distinct epitopes on CD73 from human mesenchymal stem cells. Biochem Biophys Res Commun. 2001;289:519–24. doi: 10.1006/bbrc.2001.6013. [DOI] [PubMed] [Google Scholar]
- 126.Ghilzon R, McCulloch CA, Zohar R. Stromal mesenchymal progenitor cells. Leuk Lymphoma. 1999;32:211–21. doi: 10.3109/10428199909167382. [DOI] [PubMed] [Google Scholar]
- 127.Boiret N, Rapatel C, Boisgard S, et al. CD34+CDw90(Thy-1)+ subset colocated with mesenchymal progenitors in human normal bone marrow hematon units is enriched in colony-forming unit megakaryocytes and long-term culture-initiating cells. Exp Hematol. 2003;31:1275–83. doi: 10.1016/j.exphem.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 128.Buhring HJ, Battula VL, Treml S, et al. Novel markers for the prospective isolation of human MSC. Ann N Y Acad Sci. 2007;1106:262–71. doi: 10.1196/annals.1392.000. [DOI] [PubMed] [Google Scholar]
- 129.Vogel JP, Szalay K, Geiger F, et al. Platelet-rich plasma improves expansion of human mesenchymal stem cells and retains differentiation capacity and in vivo bone formation in calcium phosphate ceramics. Platelets. 2006;17:462–9. doi: 10.1080/09537100600758867. [DOI] [PubMed] [Google Scholar]
- 130.Jones E, McGonagle D. Human bone marrow mesenchymal stem cells in vivo. Rheumatology. 2008;47:126–31. doi: 10.1093/rheumatology/kem206. [DOI] [PubMed] [Google Scholar]