

Abstract
More than two decades ago, dysregulation of the intracellular Ca2+ homeostasis was suggested to underlie the development of Alzheimer’s disease (AD). This hypothesis was tested in numerous in vitro studies, which revealed multiple Ca2+ signalling pathways able to contribute to AD pathology. It remained, however, unclear whether these pathways are also activated in vivo, in cells involved in signal processing in the living brain. Here we review recent data analysing intracellular Ca2+ signalling in vivo in the context of previous in vitro findings. We particularly focus on the processes taking place in the immediate vicinity of amyloid plaques and on their possible role for AD-mediated brain dysfunction.
Keywords: hyperactivity, plaque vicinity, calcium dyshomeostasis, seizure, in vivo calcium imaging, two-photon microscopy
Introduction
Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder, characterized by distinct neuropathological lesions. These include extracellular deposits of β amyloid (Aβ) in senile plaques, accumulation of intraneuronal neurofibrillary tangles and profound neuronal death (reviewed in [1–3]). Clinical symptoms include the inability to encode new memories as well as cognitive and behavioural impairments [4]. Most cases of the disease are sporadic, with advancing age being the major risk factor for developing AD. The prevalence of AD rises exponentially with age from approximately 1% at 65 years to 40% after the age of 90 [5–7]. Furthermore, individuals harbouring the 4 allele of apolipoprotein E have an increased risk for developing sporadic, late-onset AD [8, 9]. A small fraction of AD patients, however, have an inherited autosomal dominant form of the disease. These hereditary AD forms are characterized by an earlier onset and are typically caused by mutations in genes encoding human amyloid precursor protein (APP) or presenilin 1 (PS1) and presenilin 2 (PS2) [1, 3, 10]. The presenilins are the part of the γ-secretase complex involved in the synthesis of Aβ, which is derived from APP by sequential enzymatic cleavage by β-APP cleaving enzyme and γ-secretase complex [10, 11].
Expressed in transgenic mice, APP and presenilins with familial mutations allow various aspects of AD neuropathology to be modelled. The mutant mice develop senile plaques and neurofibrillary tangles, exhibit dysregulation of the intracellular Ca2+ homeostasis, brain inflammatory response and memory impairment. However, they do not recapitulate the widespread neuronal loss seen in humans [12].
Accumulation of Aβ plays a crucial role in the genesis of AD [2, 3, 13]. Among the three forms of Aβ (Aβ38, Aβ40, Aβ42), Aβ42 seems to be the most important for the pathogenesis of the disease because it more easily aggregates into oligomers and amyloid fibrils [14]. Mounting evidence suggests that the soluble oligomers (presumably dimers and trimers) are the neurotoxic species in AD [13]. Indeed, naturally secreted small Aβ oligomers have been shown to inhibit long-term potentiation (LTP, [15]), the electrophysiological correlate of learning and memory [13, 16] and to induce a loss of hippocampal synapses [17–19]. Moreover, similar effects were caused by oligomers extracted from the cerebral cortex of AD patients. In wild-type rodents, human oligomers inhibited LTP, enhanced long-term depression, reduced dendritic spine density and interfered with the memory of a learned behaviour [20]. Interestingly, the soluble Aβ dodecamer (Aβ*56, [21]) also seems to impair memory. Thus, young rats injected intracranially with Aβ*56 purified from the brains of old AD mouse mutants showed a reduced performance in the Morris water maze test (a common test for spatial learning).
As a matter of course, formation of Aβ oligomers is abetted by Aβ accumulation within the brain. Interestingly, mutations associated with inherited forms of AD promote both accumulation and oligomerization of Aβ. Thus, familial APP mutations that flank or occur within the Aβ region alter the amount or aggregation properties of Aβ, whereas mutations within presenilins were found to increase the Aβ42/Aβ40 ratio [10, 13]. Another source of Aβ accumulation within the brain is the imbalance between its production and clearance caused by impaired degradation of Aβ (reviewed in [22, 23]). Aβ is cleaved by several proteases, including neprilysin [24, 25], insulin-degrading enzyme [25], endothelin-converting enzyme [26], plasmin [27] and cathepsin B [28]. Neprilysin [25, 29] and cathepsin B [28] overexpression in transgenic mice reduces total Aβ levels and plaque deposition, whereas their pharmacological blockade or genetic ablation increases Aβ load [28, 30, 31]. The activity/expression of neprilysin is down-regulated with aging and at the early stage of AD [28, 32–34], suggesting that decreased activity of Aβ-degrading enzymes may contribute to the sporadic form of the disease.
In this paper we are going to review recent findings regarding the mechanisms of AD. In particular, we will focus on the role of calcium (Ca2+) signalling as well as the regulation of the intracellular Ca2+ homeostasis in AD, discussing data obtained in various AD mouse models, both in vitro and in vivo.
Dysregulation of Ca2+ homeostasis in AD
Numerous studies suggest that besides Aβ accumulation, dysregulation of the intracellular Ca2+ homeostasis might act as an important progenitor of AD. The Ca2+ hypothesis of AD is supported by the fact that many AD-related genes (e.g. those encoding APP, PS1 and PS2) are also involved in Ca2+ signalling [35]. The role of disturbed Ca2+ homeostasis as a proximal cause of brain aging and neurodegenerative processes like AD was postulated by Khachaturian more than 20 years ago [36, 37]. However, it is still debated whether the disturbed intracellular Ca2+ homeostasis is the cause or the result of altered Aβ and tau production [38]. Recent data reviewed below suggest a complex mutually potentiating interaction between Aβ accumulation and Ca2+ dyshomeostasis.
Aβ accumulation causes Ca2+ dyshomeostasis
In vitro studies have revealed several mechanisms by which Aβ can increase intracellular free Ca2+ concentration ([Ca2+]i). In lipid bilayers, for example Aβ causes the formation of cation-selective ion pores through which Ca2+ passes into the cytosol [39, 40]. The formation of the pores by Aβ oligomers is enhanced by the presence of phosphatidylserine, one of the earliest signs of apoptosis, on the cell surface [41]. Thus, an already suffering neuron with increased phosphatidylserine on the cell surface might show an enhanced vulnerability towards Aβ oligomers. Interestingly, specific blockage of ion-conducting Aβ-channels with the peptide NA-7 eliminates any signs of Aβ-induced apoptosis in cell culture models [42].
Studies in human neuroblastoma cells have revealed that oligomeric Aβ can non-selectively increase Ca2+ permeability of cellular membranes, thus increasing both Ca2+ influx from the extracellular space and Ca2+ leakage from the intracellular Ca2+ stores [43]. Furthermore, Aβ can interact with endogenous Ca2+-permeable channels in the cell membrane, thus increasing NMDA receptor-dependent Ca2+ influx [44] or causing free radical-mediated potentiation of Ca2+ entry through voltage-gated Ca2+ channels [45]. The finding that the blocker of L-type Ca2+ channels nimodipine reduces Aβ-mediated cell death [45] has suggested that neuronal death is directly related to Aβ-induced Ca2+ influx through voltage-gated Ca2+ channels. Other Ca2+-permeable channels, however, are inhibited by Aβ. It blocks, for example presynaptic P/Q-type voltage-gated Ca2+ channels [46] as well as Ca2+-permeable α7-containing nicotinic acetylcholine receptor-channels [47].
Membrane-associated oxidative stress represents another mechanism by which Aβ can impair intracellular Ca2+ homeostasis. Reactive oxygen species (H2O2 and hydroxyl radicals), generated during formation of Aβ oligomers, could attack cell membranes and initiate lipoperoxidation [48]. Membrane lipid peroxidation results in the generation of neurotoxic lipid aldehydes (such as 4-hydroxynonenal), which impair the function of membrane proteins involved in ion transport. The latter include ATPases (e.g. Na+/K+-ATPase and Ca2+-ATPase) and glutamate and glucose transporters [49, 50]. The Aβ-mediated impairment of ion-motive ATPases was observed in both primary neuron cultures and synaptosomes from adult postmortem hippocampus [49]. It results in membrane depolarization and opening of NMDA receptor-channels as well as voltage-gated Ca2+ channels (see above), while impaired Ca2+-ATPase activity reduces the ability of the cell to extrude Ca2+[49]. Impairment of glutamate transport results in increased extracellular glutamate and overstimulation of glutamate receptors, whereas the impairment of glucose transport causes ATP depletion and decreased activity of ion-motive ATPases [50]. Both processes might further increase intracellular Ca2+ levels (reviewed in [51]).
Not only Aβ itself but also other products of APP metabolism may affect the intracellular Ca2+ homeostasis. During APP processing, the amyloidogenic carboxy-terminal fragment is cleaved by γ-secretase, liberating the APP intracellular domain (AICD). AICD, too, was shown to influence intracellular Ca2+ signalling [52]. In mouse embryonic fibroblasts, inositol-triphosphate (IP3)-mediated Ca2+ release from the intracellular Ca2+ stores was significantly reduced both in the absence of presenilins (PS1−/−PS2−/− mice, γ-secretase inactive) and in the absence of APP (APP−/− mice). Importantly, this functional deficit was rescued only by cotransfecting fibroblasts with cDNA encoding either AICD itself, or AICD-containing parts of APP. Based on the finding that AICD can form a transcriptionally active complex [53], the authors suggested that AICD may affect Ca2+ signalling by regulating the expression of genes involved in Ca2+ homeostasis. This latter statement, however, caused controversial discussions within the AD community (for details see [54, 55]). On the other hand, the activation of the non-amyloidogenic secretory pathway (e.g. cutting APP by α-secretase) results in the generation of sAPPα. This protein is neuroprotective because it activates K+ channels via cGMP, thus causing membrane hyperpolarization and reducing Ca2+ influx [56, 57].
Ca2+ dyshomeostasis increases Aβ production
Several in vitro studies have shown that increased intracellular Ca2+ levels can trigger Aβ formation and aggregation to protofibrils, implicating Ca2+ dyshomeostasis as a possible causal factor in sporadic forms of AD (reviewed in [38]).
Initial evidence that APP processing is regulated by intracellular Ca2+ came from studies in non-neural cells (HEK-293) overexpressing human APP [58, 59]. These early studies have shown that a global increase in the cytosolic free Ca2+ concentration caused by Ca2+ ionophore A23187 enhances Aβ production. Aβ levels were also increased when stimulating ryanodine receptors (Ca2+ release channels of endoplasmic reticulum [ER]) by caffeine [59]. Thus, Ca2+ release from intracellular Ca2+ stores can also contribute to the genesis of Aβ. Thapsigargin, a compound that inhibits uptake of Ca2+ into ER by sarco-/endoplasmic reticulum calcium ATPases (SERCAs), thereby causing an increase in [Ca2+]i, augmented the caffeine-stimulated release of Aβ[59]. In CHO cells overexpressing human APP, Buxbaum et al.[60] observed a more complex effect of thapsigargin: the formation of Aβ was stimulated at lower concentrations (10 nM) only and was inhibited at higher concentrations (20 nM). Later studies on neuronal cell cultures supported the finding that Ca2+ dyshomeostasis can influence APP processing. A depolarization-induced increase in [Ca2+]i, for example specifically induced the production of large amounts of intraneuronal Aβ42, causing neuronal death [61]. In contrast to non-neural cells [59], however, it was found that Ca2+ release from the intracellular Ca2+ stores was not sufficient to induce generation of Aβ in neurons [61].
Very recently, Dreses-Werringloer et al.[62] identified a novel transmembrane glycoprotein with Ca2+ channel properties, which they called calcium homeostasis modulator 1 (CALHM1). Surprisingly, Ca2+ influx via CALHM1 decreased the total amount of extracellular Aβ. On the contrary, the CALHM1 polymorphism P86L increases Aβ levels by interfering with CALHM1-mediated Ca2+ permeability. These results are in contrast to the previous studies, which have shown that increased transmembrane Ca2+ influx enhances Aβ production (see above). The authors have also suggested that the CALHM1 polymorphism P86L is associated with an increased risk for late-onset AD [62]. This observation, however, was not confirmed in several recent studies analysing the potential association between AD risk and CALHM1 polymorphism in independent datasets of AD patients and control individuals [63, 64]. Thus, the role of CALHM1 as a risk factor in AD remains unclear.
Taken together, the in vitro data strongly suggest that Aβ accumulation causes Ca2+ dyshomeostasis and, vice versa, Ca2+ dysregulation causes Aβ overproduction. Furthermore, increases in [Ca2+]i may trigger Ca2+-activated kinases, which mediate the phosphorylation of tau [65, 66] thereby facilitating the development of neurofibrillary tangles. These neuropathological changes may worsen disease symptoms and ultimately may lead to neuronal death. The ‘chicken or the egg’ conundrum (i.e. whether Ca2+ acts upstream or downstream of Aβ), however, is very difficult to resolve, since altered Ca2+ homeostasis affects the metabolism of the AD-related pathological proteins (Aβ and tau) and, conversely, the accumulation of these proteins further disturbs the Ca2+ metabolism.
Presenilins and Ca2+ homeostasis
Presenilins play a double role in the pathogenesis of AD. First, presenilins are the part of the γ-secretase complex, which generates Aβ through APP cleavage (see above). Familial AD-linked presenilin mutations were shown to elevate the concentration of the aggregation-prone form of Aβ (Aβ42) in expense of Aβ40 (reviewed in [10]). As a consequence, the presenilin-mediated elevation of the Aβ42/Aβ40-ratio activates Aβ-dependent mechanisms of Ca2+ dyshomeostasis (see section ‘Aβ accumulation causes Ca2+ dyshomeostasis’). Secondly, presenilins can directly alter intracellular Ca2+ homeostasis. They interact with three key components of the Ca2+ signalling cascade: IP3 receptors (reviewed in [67, 68]), ryanodine receptors [69–71] and SERCA pumps (Fig. 1, [72]). Already early studies in fibroblasts from AD patients [73, 74] have revealed the altered properties of IP3 receptor-mediated Ca2+ release from the intracellular Ca2+ stores. Subsequently, potentiation of IP3-mediated Ca2+ signals by presenilin mutations has been documented in different experimental systems ranging from Xenopus oocytes to cells from transgenic animals [75, 76]. While the above studies have suggested that exaggerated [Ca2+]i responses in cells expressing mutant presenilins are caused by overfilling of intracellular Ca2+ stores (discussed in [35, 67]), Cheung et al.[68] recently discovered a mechanism that can account for potentiated IP3-mediated Ca2+ signalling in the absence of elevated Ca2+ within the ER. The authors have shown that presenilins can physically interact with the IP3 receptor-channel and thereby stimulate its gating activity (Fig. 1). Expression of two different presenilins with familial mutations (PS1 (M146L) and PS2 (N141I)) sensitized the IP3 receptor-channel to IP3 and enhanced IP3-mediated Ca2+ release from the intracellular Ca2+ stores. In addition to potentiating agonist-induced Ca2+ transients, this sensitization enabled Ca2+ release even at very low resting concentrations of IP3, causing continuous IP3 receptor-mediated ‘leak’ of Ca2+ from the ER.
Fig 1.
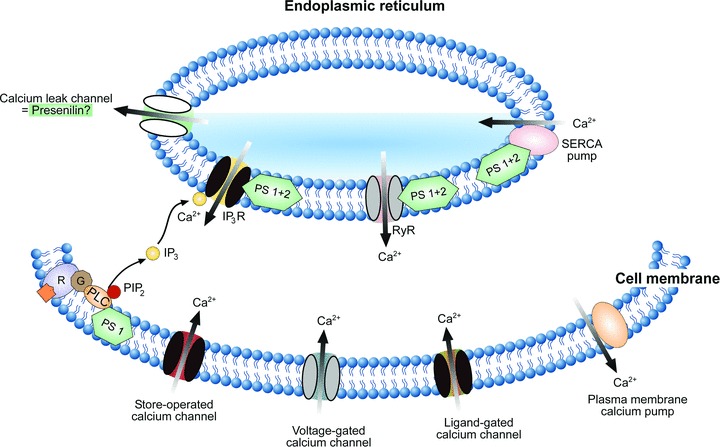
Presenilin-mediated regulation of the intracellular Ca2+ homeostasis. A scheme illustrating the mechanisms underlying intracellular regulation of [Ca2+]i. Ca2+ ions enter the cytosol through ligand-gated, voltage-gated or store-operated Ca2+ channels in the cell membrane. In addition, they are released from the intracellular Ca2+ stores of the ER via IP3 receptor channels (IP3R) or ryanodine receptor-channels (RyR). IP3 is produced from phosphatidylinositol-4,5-bisphosphate (PIP2) by PLC in response to the activation of metabotropic receptors (R). Intracellular Ca2+ levels (30–100 nM at rest, [130]) are controlled by plasma membrane Ca2+ pumps and by SERCAs. Presenilins are located both within the plasma membrane and the ER membrane and interact with many important elements of the Ca2+ signalling cascade (as indicated). Familiar AD mutations in presenilins (i) potentiate PLC activity, (ii) increase Ca2+ release through both IP3 and ryanodine receptors and (iii) modulate activity of SERCA pumps. In addition, presenilins may function as Ca2+ leak channels of the ER (indicated). AD-related mutations in presenilins have been shown to render these leak channels non-functional, thereby causing an overload of the ER Ca2+ stores.
As shown by studies in human SH-SY5Y neuroblastoma cells, presenilins may also regulate IP3 production by influencing basal activity of the IP3-producing enzyme phospholipase C (PLC) (Fig. 1, [77]). Familial PS1 mutations (PS1-ΔE9 and PS1 M146V) enhance PLC activity, thereby increasing IP3 levels within the cell which, in turn, mediate enhanced Ca2+ release from the intracellular Ca2+ stores [77, 78].
In addition to interaction with the IP3 receptor-mediated signalling, PS1 and PS2 were shown to interact with the ryanodine receptor at the cytoplasmic side of the ER membrane. For example, they increase Ca2+ flux through brain-type ryanodine receptor-channels incorporated into lipid bilayers (Fig. 1, [70, 71]). As documented in a recent study [79], this interaction seems to be important for the regulation of neurotransmitter release in the hippocampus. Using a genetic approach for selective inactivation of presenilins both in presynaptic and in postsynaptic neurons of the Schaeffer-collateral pathway, Zhang et al. demonstrated that presynaptic inactivation of presenilins impairs synaptic glutamate release and LTP. The underlying mechanism involves ryanodine receptor-mediated Ca2+-induced Ca2+ release from the ER. Presenilins seem to control this Ca2+ release, thereby modulating the release probability of neurotransmitters.
Along with Ca2+ release channels, Ca2+ pumps are the key component of the Ca2+ regulatory system. Since many presenilin mutations lead to enhanced filling of Ca2+ stores, presenilins might also regulate Ca2+ pumps. Indeed, using gain-of-function and loss-of-function approaches in both mammalian cell culture and Xenopus oocyte models, Green et al.[72] have demonstrated that presenilins physically associate with SERCA pumps and are necessary for their proper function (Fig. 1). Furthermore, modulating SERCA activity in CHO cells altered the amount of Aβ produced by these cells [72]. These data suggest that dysregulation of SERCA pumps (caused by AD-relevant presenilin mutations) may contribute to the pathogenesis of AD.
Presenilins not only modulate the function of the other Ca2+ regulating proteins, but also seem to form Ca2+-permeable ion channels themselves (Fig. 1). In a recent study, Tu et al.[80] report that in planar lipid bilayers, wild-type presenilins form low-conductance Ca2+-permeable channels, which account for ∼80% of passive Ca2+ leak from the ER. Notably, this ability of presenilins is disrupted by two AD-relevant presenilin mutations, PS1-M146V and PS2-N141I [80]. These results were confirmed by a subsequent study demonstrating that out of 6 familial AD mutations tested, five mutations abolished Ca2+ leak function of presenilins [81]. The consequence of an impaired Ca2+ leak from the ER would be again an overfilling of the intracellular Ca2+ stores [82] as well as deficits in capacitative calcium entry [83, 84].
It has to be mentioned that ER is not the only Ca2+ store within the cell. Intracellular Ca2+ is also buffered by mitochondria (for review see [85]). However, mitochondrial dysfunction in AD seems to be linked to Aβ rather than to presenilins and has recently been discussed in several review articles (see, e.g.[86, 87]). We would like to refer the reader to those papers for further details on this issue.
Taken together, in vitro data suggest that presenilins control the functional state of the intracellular Ca2+ stores through interaction with both SERCAs and multiple Ca2+ release mechanisms. These complex interactions might explain why presenilin mutations have such widespread effects on intracellular Ca2+ signalling. However, it remains unclear to which extent these processes also occur in vivo, in the intact brain tissue.
Dysregulation of Ca2+ homeostasis in vivo
Recently, three in vivo studies have investigated Ca2+ dynamics in the brains of AD mouse mutants. Using an adenoviral-based expression of the genetically encoded Ca2+ indicator Yellow Cameleon 3.6, Kuchibhotla et al.[88] observed increased resting intracellular Ca2+ levels in dendrites and dendritic spines of APPSwe/PS1-ΔE9 (APP/PS1) transgenic mice. This ‘Ca2+ overload’ was extremely evident in close proximity to amyloid plaques. Within 25 μm of a plaque, more than 30% of all neurites exhibited strongly elevated intracellular Ca2+ levels (yellow-red neurites in Fig. 2). Interestingly, the Ca2+ overload observed in APP/PS1 mice was associated with morphological neuritic alterations. These were mediated, at least in part, by activation of the Ca2+/ calmodulin-dependent phosphatase calcineurin [88]. These data support previous studies from the Bacskai/Hymans group [89] identifying plaque vicinity as a noxious factor in AD. However, the exact mechanism by which amyloid plaques affect [Ca2+]i in spines and neurites remains unclear.
Fig 2.
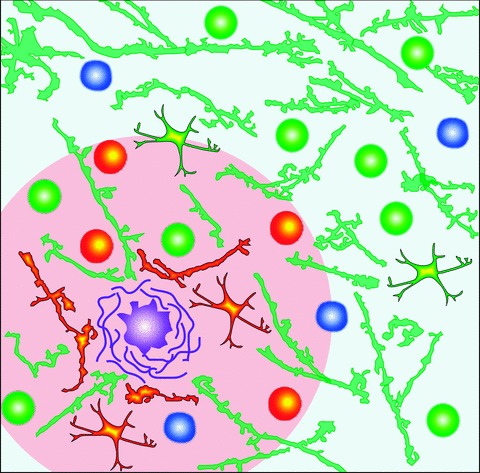
Pathological changes in brain parenchyma in the vicinity of amyloid plaques. Schematic drawing illustrating an amyloid plaque (purple) surrounded by neurons (circles), dendrites (branches) and astrocytes (branched cells). Three different types of neurons are illustrated: normally active neurons (white-green), hyperactive neurons (yellow-red) and silent neurons (white-blue; [90]). Dendritic branches with an increased intracellular Ca2+ concentration are coloured yellow-red while the ones with normal resting [Ca2+]i appear green [88]. The pacemaker astrocytes initiating propagating [Ca2+]i waves are shown in yellow-red and the rest of the astrocytes (having an increased resting [Ca2+]i) are shown in yellow-green [91]. Note that most of the changes in the intracellular Ca2+ homeostasis are restricted to the immediate plaque vicinity (an area situated less than 60 μm from the plaque border, coloured in pink).
Notably, in the absence of human APP mutations, mutant PS1 transgenic mice (PS1- ΔE9 or PS1M146V) did not exhibit any neuritic Ca2+ overload, suggesting that these mutations alone are not sufficient to induce Ca2+ overload in vivo[88]. This result is surprising in view of several in vitro studies pointing to a key role of presenilins in Ca2+ dyshomeostasis (see above).
Consistent with data from Kuchibhotla et al.[88], our Ca2+ imaging study on double-transgenic mice overexpressing APPSwe and mutant PS1 (G384A) revealed a profound functional impairment in 50% of layer 2/3 neurons [90]. Almost half of these neurons (21% of the total population) were ‘hyperactive’, i.e. displayed increased frequencies of spontaneous Ca2+ transients. The other 29% of cells were ‘silent’, showing no Ca2+ transients over a 6-min.-long recording period (Fig. 2). Interestingly, the hyperactive cells were found only in close proximity to plaques (<60 μm from the plaque border; yellow-red neurons in Fig. 2). In contrast to our initial expectation that Ca2+ transients in hyperactive cells would result from spontaneous Ca2+ release from overfilled intracellular Ca2+ stores (see above), these Ca2+ transients were tetrodotoxin-sensitive and thus caused by action potential firing. Further analyses have shown that these Ca2+ transients are of synaptic origin because they are completely and reversibly blocked by the glutamate receptor blockers CNQX (6-cyano-5-nitroquinoxaline-2,3-dione) and APV (D,L-2-amino-5-phosphonovaleric acid). It remains, however, to be established whether and to what extent the increased frequency of Ca2+ transients in hyperactive cells causes the Ca2+ overload observed by Kuchibhotla et al.[88].
In the latest in vivo Ca2+ imaging study, Bacskai and colleagues quantitatively determined resting [Ca2+]i in astrocytes of APP/PS1 mice and observed a global astrocytic response to plaque deposition [91]. Compared to wild-type mice, [Ca2+]i was globally increased in the astrocytic network of 6- to 8-month-old mutant mice (yellow-green astrocytes in Fig. 2). Furthermore, astrocytes in mutant mice exhibited a significant increase in spontaneous activity (see also [92]). Sometimes this activity was correlated over long distances (up to 200 μm), thus forming an intercellular Ca2+ wave. While the increase in [Ca2+]i and in the frequency of astrocytic Ca2+ transients was independent of plaque proximity, the pacemaker-astrocytes initiating the waves (yellow-red astrocytes in Fig. 2) were located 24.8 ± 7.8 μm from the three-dimensionally nearest amyloid plaque. These data suggest that plaques or plaque-associated bioactive species trigger these Ca2+ waves. Notably, increased astrocyte activity was not affected by blocking neuronal activity with tetrodotoxin. Therefore, increased astrocytic activity is not a simple reflection of neuronal hyperactivity in the plaque vicinity.
Taken together, these studies reveal a complex and widespread pattern of dysregulation of the intracellular Ca2+ homeostasis in vivo, involving several cellular/synaptic mechanisms in two different cell types. Some of these pathological changes are condensed in the plaque vicinity while others, like those in the astrocytic network, are diffusely distributed all over the brain.
AD-mediated hyperactivity and synaptic network dysfunction
The observation that amyloid plaques in vivo are surrounded by hyperactive cells [90] came as a surprise because of the wealth of data showing that AD is associated with synaptic dismantling and ‘synaptic failure’[93]. Already early histological studies of brain tissue from AD patients were able to reveal neuronal loss, shrinkage of dendritic trees and a decrease in the density of synapses [94–98]. Further studies in mutant mice have shown that elevated Aβ levels result in spine loss [17, 99–101], reduction in the number of excitatory synapses [102] and depressed glutamatergic synaptic transmission [103, 104] accompanied by glutamate receptor endocytosis [104, 105]. In cultured cortical neurons, application of Aβ induced internalization of NMDA receptors [105], whereas Aβ overexpression in organotypic hippocampal slices promoted AMPA receptor removal followed by the loss of dendritic spines and NMDA responses [104]. In double knock-in mice carrying human mutations in the genes for APP and PS1, only AMPA receptors were down-regulated, while NMDA receptors remained unaffected [106]. In agreement with these data, electrophysiological studies of triple transgenic mice harbouring PS1M146V, APPSwe and tauP301L[107] revealed deficits in LTP. The Aβ-mediated LTP blockade was also observed in vivo[16]. This elegant study demonstrated that inhibition of hippocampal LTP is attributable specifically to oligomers, not monomers or fibrils, of naturally secreted human Aβ. In view of the cumulative evidence described above, one might expect that accumulation of Aβ over the course of the disease decreases overall activity of neuronal networks.
Surprisingly, electrophysiological studies performed by Palop et al.[108] in freely moving hAPP transgenic mice revealed an increase in neuronal activity. By means of prolonged video-EEG monitoring, the authors observed generalized, sharp synchronous discharges in the cortex and in the hippocampus of mutant mice. Interestingly, some of the hippocampal discharges remained focal, with minimal spread to the neocortex [108]. Subsequently, similar epileptiform activity was observed in other AD mouse models, including Tg2576, hAPP/PS1 and APdE9 mice [109, 110].
Our data, obtained in the cortex of APPSwe/PS1 mice, additionally revealed a population of cells with augmented neuronal activity. Compared to control mice, the fraction of such hyperactive neurons in AD-mutants increased 16-fold [90]. Since application of the GABAA agonist diazepam reduced activity of hyperactive neurons to normal levels, and application of the GABAA antagonist gabazine was less effective in hyperactive cells compared to normal ones, our results support the hypothesis that hyperactivity is caused, at least in part, by impaired synaptic inhibition. The fact that some neurons become hyperactive and others silent [90] suggests that anatomical remodelling of both excitatory and inhibitory synapses underlies the changes in neuronal activity in AD. This conclusion is supported by the data of Palop et al.[108], showing that spontaneous epileptiform activity in cortical and hippocampal networks is accompanied by GABAergic sprouting in the dentate gyrus. Such abnormal sprouting, this time of excitatory entorhinal axons, was also observed by Phinney et al.[111] within the hippocampus and the thalamus of APP23 mice [112].
According to recent data, increased synaptic transmission enhances APP endocytosis in vivo via clathrin-mediated recycling of synaptic vesicles [113]. This, in turn, increases Aβ generation and its release into the brain interstitial fluid. Thus, neuronal hyperactivity is very likely to result in enhanced Aβ production and increased conversion of soluble Aβ into oligomers and/or plaques. This process may sustain a vicious cycle by further increasing neuronal hyperactivity.
Neuronal hyperactivity: implications for humans
Several studies have shown that AD patients are more susceptible to epileptic seizures than the control population. It is estimated that 7–21% of patients with sporadic AD will develop at least one clinically apparent seizure, which is about 5 to 10 times the risk for non-demented aged people [109, 114]. Patients with the early-onset (familiar) form of the disease, in particular those carrying a mutation in the presenilin genes, have a dramatically higher risk for developing seizures. Snider et al.[115], for example, have analysed 18 families with three or more family members exhibiting very early-onset AD (40 years or younger; 106 individuals in total). Out of these 18 families, 12 were analysed with respect to epileptic seizures. The latter were found in 75% of all cases, clearly indicating a close relation between the early-onset AD and seizure activity. Furthermore, the risk of developing seizures is substantially increased in patients with Down syndrome (who have three copies of the APP gene) [116]. Epileptiform activity has also been reported in non-demented carriers of the apolipoprotein E4, a known genetic risk factor of sporadic AD [117].
In many additional cases, neuronal hyperactivity may occur in a milder form (e.g. as interictal epileptiform discharges), thus remaining clinically undetected. Indeed, many clinical symptoms of AD, such as amnestic wandering, agitation, disorientation and, in particular, episodic fluctuations in functionality could be explained by sporadic hyperactivity of respective neuronal networks. Consistent with this hypothesis, a recent fMRI study in humans has reported the failure of task-induced inactivation of default network activity and hippocampal hyperactivation in asymptomatic and minimally impaired older individuals [118]. As revealed by simultaneous Pittsburg compound B imaging, this hyperactivity was associated with significant accumulation of fibrillar amyloid β-protein in the examined brain regions.
Taken together, data obtained in AD patients and mutant mice point towards a strong association between neuronal hyperactivity and AD. This suggests that neuronal (and glial) hyperactivity may represent a mechanism causally related to AD-mediated cognitive impairments.
Plaque vicinity
Although accumulation of cerebral amyloid plaques is the major hallmark of AD, the key question – how exactly do amyloid plaques impair brain function in AD patients – has not been answered. The principal argument against amyloid plaques as the primary toxic species stems from studies showing that the number of amyloid plaques does not correlate well with the severity of cognitive impairments. Indeed, several studies identified patients who had no overt symptoms of dementia antemortem, but postmortem were found to have many plaque deposits [119, 120]. Other studies indicate that the severity of the cognitive decline in AD patients is better correlated with the concentration of soluble Aβ oligomers than with the density of amyloid plaques [121, 122]. In vivo studies in mouse models of AD also confirmed that plaque formation is not necessary for learning and memory deficits [123, 124], thus suggesting that soluble Aβ oligomers (and not plaques) are the primary toxic species (reviewed in [2]).
Recent data, however, seem to challenge this hypothesis. Thus, in transgenic mouse models, loss of dendritic spines, shaft atrophy of dendrites and the development of large axonal varicosities were only observed inside and within 15–50 μm of amyloid plaques [99]. Furthermore, repeated two-photon imaging in vivo has shown that plaque deposition precedes neuritic deformation. Indeed, dystrophic neurites become visible only 3 to 4 days after the first appearance of a new plaque [89]. Further studies have shown that (i) increased resting Ca2+ concentration in neurites [88], (ii) synaptically driven neuronal hyperactivity [90], (iii) initiation of intercellular Ca2+ waves in astrocytes [91] and (iv) the already mentioned loss of dendritic spines [101] and excitatory synapses [102] are predominantly observed in close proximity to amyloid plaques (Fig. 2). Furthermore, the in vivo data in humans mentioned above [118] show a strong correlation between plaque-associated amyloid deposition and dysfunction of neural systems supporting the formation of new memories. Taken together, these recent in vivo data establish plaques as a critical mediator of cellular/network pathology in AD.
The mechanisms of this plaque-mediated toxicity remain unclear. On the one hand, the concentration of diffusible Aβ oligomers is increased within and in the immediate vicinity of amyloid plaques (about 6.5 μm from the edge of the plaque, [102]). The oligomer-rich volume is 180% larger than the volume of the dense core of the plaque and corresponds to the area with severe decrease in synapse density. Based on this observation, Koffie et al.[102] have suggested that senile plaques act as a potent local reservoir of oligomeric Aβ, which in turn acts as a toxic moiety to synapses in the cortex.
However, Aβ oligomers are not the only potentially toxic species in the plaque vicinity. As revealed by histological studies [125, 126], glial cells, namely activated astrocytes and microglia (the major immunocompetent cells in the brain), cluster around sites of amyloid deposition. These cells produce a wide variety of potentially neurotoxic substances, such as reactive oxygen and nitrogen species, inflammatory cytokines (e.g. interleukins, tumour necrosis factor-α and transforming growth factor-β), prostaglandins, complement system proteins and other inflammatory mediators [127]. Each of these factors may, alone or in concert, contribute to cellular/network dysfunction in the plaque vicinity.
Interestingly, a recent in vivo study has shown that acute application of lipopolysaccharides (the major toxins of gram-negative bacteria inducing inflammatory response in the central nervous system (for review see [128])) induces aberrant neuronal activity in the rat somatosensory cortex [129]. Notably, lipopolysaccharide-induced pathology manifested itself in a potentiation of somatosensory evoked potentials as well as epileptiform discharges and seizures. This aberrant cortical excitability was prevented by an interleukin-1 receptor antagonist, suggesting that interleukin-1, released by activated microglia, provokes neuronal hyperactivity.
Thus, an amyloid plaque, surrounded by synchronized hyperactive neurons and pacemaker astrocytes, represents a grain of hyperactivity within the brain parenchyma (Fig. 2). Such grains of hyperactivity may trigger a variety of pathological activities, including paradoxically increased fMRI signals [118], interictal epileptiform discharges and epileptic seizures.
Conclusions
Recent high-resolution analyses of cortical function in mouse models of AD revealed a marked dysregulation (mainly potentiation) of intracellular calcium homeostasis in vivo. This is reflected in (i) increased resting Ca2+ levels in neurons and astrocytes and (ii) increased frequency of spontaneous Ca2+ waves in neighbouring hyperactive neurons and in astrocytic networks. Notably, many of these pathological changes are either restricted to or governed from the immediate vicinity of amyloid plaques. Taken together, these new data identify hyperactive neurons and glia themselves as well as the ‘hyperactive’ plaque vicinity as important vicious species in AD.
Does the ‘hyperactive’ plaque vicinity play a major causal role in AD pathology? Cumulative evidence suggests that this may be the case, although more direct experiments are needed to definitively answer this question. The good news, however, is that this question is relatively easy to address because plaque vicinity can be targeted pharmacologically (e.g. by the drugs recognizing a fibrillar form of Aβ).
Acknowledgments
The authors thank A. Weible for the graphical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 596, GA 654/1–1).
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nature Rev Mol Cell Biol. 2007;8:101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. Medicine–The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters. Z ges Neurol Psychiatr. 1911;4:356–85. [Google Scholar]
- 5.Bachman DL, Wolf PA, Linn R, et al. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology. 1992;42:115–9. doi: 10.1212/wnl.42.1.115. [DOI] [PubMed] [Google Scholar]
- 6.Kukull WA, Bowen JD. Dementia epidemiology. Med Clin North Am. 2002;86:573–90. doi: 10.1016/s0025-7125(02)00010-x. [DOI] [PubMed] [Google Scholar]
- 7.Evans DA, Funkenstein HH, Albert MS, et al. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. JAMA. 1989;262:2551–6. [PubMed] [Google Scholar]
- 8.Cedazo-Minguez A. Apolipoprotein E and Alzheimer’s disease: molecular mechanisms and therapeutic opportunities. J Cell Mol Med. 2007;11:1227–38. doi: 10.1111/j.1582-4934.2007.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein-E type-4 allele and the risk of Alzheimers-disease in late-onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 10.Haass C. Take five–BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004;23:483–8. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steiner H, Haass C. Intramembrane proteolysis by presenilins. Nat Rev Mol Cell Biol. 2000;1:217–24. doi: 10.1038/35043065. [DOI] [PubMed] [Google Scholar]
- 12.Morrissette DA, Parachikova A, Green KN, et al. Relevance of transgenic mouse models to human alzheimer disease. J Biol Chem. 2009;284:6033–7. doi: 10.1074/jbc.R800030200. [DOI] [PubMed] [Google Scholar]
- 13.Walsh DM, Selkoe DJ. A beta oligomers – a decade of discovery. J Neurochem. 2007;101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 14.Chen YR, Glabe CG. Distinct early folding and aggregation properties of Alzheimer amyloid-beta peptides A beta 40 and A beta 42 – stable trimer or tetramer formation by A beta 42. J Biol Chem. 2006;281:24414–22. doi: 10.1074/jbc.M602363200. [DOI] [PubMed] [Google Scholar]
- 15.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–56. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 17.Shankar GM, Bloodgood BL, Townsend M, et al. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lacor PN, Buniel MC, Furlow PW, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lacor PN, Buniel MC, Chang L, et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24:10191–200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 22.Selkoe DJ. Clearing the brain’s amyloid cobwebs. Neuron. 2001;32:177–80. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 23.Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–8. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 24.Iwata N, Tsubuki S, Takaki Y, et al. Identification of the major A beta(1–42)-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–50. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 25.Leissring MA, Farris W, Chang AY, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–93. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 26.Choi DS, Wang D, Yu GQ, et al. PKC epsilon increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc Natl Acad Sci USA. 2006;103:8215–20. doi: 10.1073/pnas.0509725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tucker HM, Kihiko M, Caldwell JN, et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci. 2000;20:3937–46. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mueller-Steiner S, Zhou Y, Arai H, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron. 2006;51:703–14. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 29.Meilandt WJ, Cisse M, Ho K, et al. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic A beta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009;29:1977–86. doi: 10.1523/JNEUROSCI.2984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mouri A, Zou LB, Iwata N, et al. Inhibition of neprilysin by thiorphan (i.c.v.) causes an accumulation of amyloid beta and impairment of learning and memory. Behav Brain Res. 2006;168:83–91. doi: 10.1016/j.bbr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 31.Huang SM, Mouri A, Kokubo H, et al. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem. 2006;281:17941–51. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- 32.Iwata N, Takaki Y, Fukami S, et al. Region-specific reduction of A beta-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J Neurosci Res. 2002;70:493–500. doi: 10.1002/jnr.10390. [DOI] [PubMed] [Google Scholar]
- 33.Caccamo A, Oddo S, Sugarman MC, et al. Age- and region-dependent alterations in A beta-degrading enzymes: implications for A beta-induced disorders. Neurobiol Aging. 2005;26:645–54. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 34.Hellström-Lindahl E, Ravid R, Nordberg A. Age-dependent decline of neprilysin in Alzheimer’s disease and normal brain: inverse correlation with A beta levels. Neurobiol Aging. 2008;29:210–21. doi: 10.1016/j.neurobiolaging.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 35.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nature Rev Neurosci. 2002;3:862–72. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 36.Khachaturian ZS. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann N Y Acad Sci. 1989;568:1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x. [DOI] [PubMed] [Google Scholar]
- 37.Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann N Y Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- 38.Green KN, LaFerla FM. Linking calcium to A beta and Alzheimer’s disease. Neuron. 2008;59:190–4. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Arispe N, Rojas E, Pollard HB. Alzheimer-disease amyloid beta-protein forms calcium channels in bilayer-membranes–blockade by tromethamine and aluminum. Proc Natl Acad Sci USA. 1993;90:567–71. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–5. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 41.Lee G, Pollard HB, Arispe N. Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-beta-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides. 2002;23:1249–63. doi: 10.1016/s0196-9781(02)00060-8. [DOI] [PubMed] [Google Scholar]
- 42.Arispe N, Diaz JC, Simakova O. A beta ion channels. Prospects for treating Alzheimer’s disease with A beta channel blockers. Biochim Biophys Acta. 2007;1768:1952–65. doi: 10.1016/j.bbamem.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 43.Demuro A, Mina E, Kayed R, et al. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 44.De Felice FG, Velasco PT, Lambert MP, et al. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 45.Ueda K, Shinohara S, Yagami T, et al. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem. 1997;68:265–71. doi: 10.1046/j.1471-4159.1997.68010265.x. [DOI] [PubMed] [Google Scholar]
- 46.Nimmrich V, Grimm C, Draguhn A, et al. Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008;28:788–97. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu QS, Kawai H, Berg DK. beta-Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci USA. 2001;98:4734–9. doi: 10.1073/pnas.081553598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hensley K, Carney JM, Mattson MP, et al. A model for beta-amyloid aggregation and neurotoxicity based on free-radical generation by the peptide – relevance to alzheimer-disease. Proc Natl Acad Sci USA. 1994;91:3270–4. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid beta-peptide impairs ion-motive ATPase activities – evidence for a role in loss of neuronal Ca2+ homeostasis and cell-death. J Neurosci. 1995;15:6239–49. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mark RJ, Pang Z, Geddes JW, et al. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997;17:1046–54. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–63. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leissring MA, Murphy MP, Mead TR, et al. A physiologic signaling role for the gamma -secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA. 2002;99:4697–702. doi: 10.1073/pnas.072033799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao X, Südhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–20. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 54.Hebert SS, Serneels L, Tolia A, et al. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep. 2006;7:739–45. doi: 10.1038/sj.embor.7400704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Müller T, Meyer HE, Egensperger R, et al. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog Neurobiol. 2008;85:393–406. doi: 10.1016/j.pneurobio.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 56.Mattson MP, Cheng B, Culwell AR, et al. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–54. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 57.Furukawa K, Barger SW, Blalock EM, et al. Activation of K+ channels and suppression of neuronal activity by secreted beta-amyloid-precursor protein. Nature. 1996;379:74–8. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- 58.Querfurth HW, Selkoe DJ. Calcium ionophore increases amyloid-beta peptide production by cultured-cells. Biochemistry. 1994;33:4550–61. doi: 10.1021/bi00181a016. [DOI] [PubMed] [Google Scholar]
- 59.Querfurth HW, Jiang JW, Geiger JD, et al. Caffeine stimulates amyloid beta-peptide release from beta-amyloid precursor protein-transfected HEK293 cells. J Neurochem. 1997;69:1580–91. doi: 10.1046/j.1471-4159.1997.69041580.x. [DOI] [PubMed] [Google Scholar]
- 60.Buxbaum JD, Ruefli AA, Parker CA, et al. Calcium regulates processing of the alzheimer amyloid protein-precursor in a protein-kinase C-independent manner. Proc Natl Acad Sci USA. 1994;91:4489–93. doi: 10.1073/pnas.91.10.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pierrot N, Ghisdal P, Caumont AS, et al. Intraneuronal amyloid-beta 1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004;88:1140–50. doi: 10.1046/j.1471-4159.2003.02227.x. [DOI] [PubMed] [Google Scholar]
- 62.Dreses-Werringloer U, Lambert JC, Vingtdeux V, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, A beta levels, and Alzheimer’s disease risk. Cell. 2008;133:1149–61. doi: 10.1016/j.cell.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bertram L, Schjeide BMM, Hooli B, et al. No Association between CALHM1 and Alzheimer’s disease risk. Cell. 2008;135:993–4. doi: 10.1016/j.cell.2008.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sleegers K, Brouwers N, Bettens K, et al. No association between CALHM1 and risk for Alzheimer dementia in a Belgian population. Hum Mutat. 2009;30:E570–74. doi: 10.1002/humu.20990. [DOI] [PubMed] [Google Scholar]
- 65.Avila J, Perez M, Lim F, et al. Tau in neurodegenerative diseases: tau phosphorylation and assembly. Neurotox Res. 2004;6:477–82. doi: 10.1007/BF03033284. [DOI] [PubMed] [Google Scholar]
- 66.Stutzmann GE. The pathogenesis of Alzheimers disease – is it a lifelong “Calciumopathy”? Neuroscientist. 2007;13:546–59. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- 67.Stutzmann GE. Calcium dysregulation, IP3 signaling, and Alzheimer’s disease. Neuroscientist. 2005;11:110–5. doi: 10.1177/1073858404270899. [DOI] [PubMed] [Google Scholar]
- 68.Cheung KH, Shineman D, Muller M, et al. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–83. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stutzmann GE, Smith I, Caccamo A, et al. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–9. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rybalchenko V, Hwang SY, Rybalchenko N, et al. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 71.Hayrapetyan V, Rybalchenko V, Rybalchenko N, et al. The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction. Cell Calcium. 2008;44:507–18. doi: 10.1016/j.ceca.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 72.Green KN, Demuro A, Akbari Y, et al. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–16. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ito E, Oka K, Etcheberrigaray R, et al. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:534–8. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Etcheberrigaray R, Hirashima N, Nee L, et al. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol Dis. 1998;5:37–45. doi: 10.1006/nbdi.1998.0176. [DOI] [PubMed] [Google Scholar]
- 75.Guo Q, Fu W, Sopher BL, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 76.Leissring MA, Paul BA, Parker I, et al. Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999;72:1061–8. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- 77.Cedazo-Minguez A, Popescu BO, Ankarcrona M, et al. The presenilin 1 deltaE9 mutation gives enhanced basal phospholipase C activity and a resultant increase in intracellular calcium concentrations. J Biol Chem. 2002;277:36646–55. doi: 10.1074/jbc.M112117200. [DOI] [PubMed] [Google Scholar]
- 78.Popescu BO, Cedazo-Minguez A, Benedikz E, et al. Gamma-secretase activity of presenilin 1 regulates acetylcholine muscarinic receptor-mediated signal transduction. J Biol Chem. 2004;279:6455–64. doi: 10.1074/jbc.M306041200. [DOI] [PubMed] [Google Scholar]
- 79.Zhang C, Wu B, Beglopoulos V, et al. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–6. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tu H, Nelson O, Bezprozvanny A, et al. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–93. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nelson O, Tu H, Lei T, et al. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230–9. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garaschuk O, Yaari Y, Konnerth A. Release and sequestration of calcium by ryanodine-sensitive stores in rat hippocampal neurones. J Physiol. 1997;502:13–30. doi: 10.1111/j.1469-7793.1997.013bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 84.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–30. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 85.Parekh AB. Mitochondrial regulation of intracellular Ca2+ signaling: more than just simple Ca2+ buffers. News Physiol Sci. 2003;18:252–6. doi: 10.1152/nips.01458.2003. [DOI] [PubMed] [Google Scholar]
- 86.Celsi F, Pizzo P, Brini M, et al. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta. 2009;1787:335–44. doi: 10.1016/j.bbabio.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Starkov AA, Beal FM. Portal to Alzheimer’s disease. Nat Med. 2008;14:1020–1. doi: 10.1038/nm1008-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuchibhotla KV, Goldman ST, Lattarulo CR, et al. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–25. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Busche MA, Eichhoff G, Adelsberger H, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–9. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 91.Kuchibhotla KV, Lattarulo CR, Hyman BT, et al. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–5. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takano T, Han X, Deane R, et al. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann N Y Acad Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- 93.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 94.Whitehouse PJ, Price DL, Struble RG, et al. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–9. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 95.Buell SJ, Coleman PD. Dendritic growth in the aged human brain and failure of growth in senile dementia. Science. 1979;206:854–6. doi: 10.1126/science.493989. [DOI] [PubMed] [Google Scholar]
- 96.Hamos JE, DeGennaro LJ, Drachman DA. Synaptic loss in Alzheimer’s disease and other dementias. Neurology. 1989;39:355–61. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- 97.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 98.Thal DR, Griffin WS, Braak H. Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. J Cell Mol Med. 2008;12:1848–62. doi: 10.1111/j.1582-4934.2008.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tsai J, Grutzendler J, Duff K, et al. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–3. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- 100.Moolman DL, Vitolo OV, Vonsattel JP, et al. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–87. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- 101.Spires TL, Meyer-Luehmann M, Stern EA, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–87. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koffie RM, Meyer-Luehmann M, Hashimoto T, et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106:4012–7. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 104.Hsieh H, Boehm J, Sato C, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–43. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Snyder EM, Nong Y, Almeida CG, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 106.Chang EH, Savage MJ, Flood DG, et al. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci USA. 2006;103:3410–5. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 108.Palop JJ, Chin J, Roberson ED, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–40. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Minkeviciene R, Rheims S, Dobszay MB, et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–62. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Phinney AL, Deller T, Stalder M, et al. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–9. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sturchler-Pierrat C, Abramowski D, Duke M, et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–92. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cirrito JR, Kang JE, Lee J, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mendez M, Lim G. Seizures in elderly patients with dementia: epidemiology and management. Drug Aging. 2003;20:791–803. doi: 10.2165/00002512-200320110-00001. [DOI] [PubMed] [Google Scholar]
- 115.Snider BJ, Norton J, Coats MA, et al. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch Neurol. 2005;62:1821–30. doi: 10.1001/archneur.62.12.1821. [DOI] [PubMed] [Google Scholar]
- 116.Menendez M. Down syndrome, Alzheimer’s disease and seizures. Brain Dev. 2005;27:246–52. doi: 10.1016/j.braindev.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 117.Ponomareva NV, Korovaitseva GI, Rogaev EI. EEG alterations in non-demented individuals related to apolipoprotein E genotype and to risk of Alzheimer disease. Neurobiol Aging. 2008;29:819–27. doi: 10.1016/j.neurobiolaging.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 118.Sperling RA, Laviolette PS, O’Keefe K, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–88. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology. 1992;42:1681–8. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 120.Crystal HA, Dickson DW, Sliwinski MJ, et al. Pathological markers associated with normal aging and dementia in the elderly. Ann Neurol. 1993;34:566–73. doi: 10.1002/ana.410340410. [DOI] [PubMed] [Google Scholar]
- 121.Naslund J, Haroutunian V, Mohs R, et al. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–7. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 122.Lue LF, Kuo YM, Roher AE, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Koistinaho M, Ort M, Cimadevilla JM, et al. Specific spatial learning deficits become severe with age in beta -amyloid precursor protein transgenic mice that harbor diffuse beta -amyloid deposits but do not form plaques. Proc Natl Acad Sci USA. 2001;98:14675–80. doi: 10.1073/pnas.261562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Westerman MA, Cooper-Blacketer D, Mariash A, et al. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22:1858–67. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Itagaki S, McGeer PL, Akiyama H, et al. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–82. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 126.Wegiel J, Wisniewski HM. The complex of microglial cells and amyloid star in three-dimensional reconstruction. Acta Neuropathol. 1990;81:116–24. doi: 10.1007/BF00334499. [DOI] [PubMed] [Google Scholar]
- 127.Streit WJ. Microglia and Alzheimer’s disease pathogenesis. J Neurosci Res. 2004;77:1–8. doi: 10.1002/jnr.20093. [DOI] [PubMed] [Google Scholar]
- 128.Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 129.Rodgers KM, Hutchinson MR, Northcutt A, et al. The cortical innate immune response increases local neuronal excitability leading to seizures. Brain. 2009;132:2478–86. doi: 10.1093/brain/awp177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Airaksinen MS, Eilers J, Garaschuk O, et al. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin D28k gene. Proc Natl Acad Sci USA. 1997;94:1488–93. doi: 10.1073/pnas.94.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]