

Abstract
Atherosclerosis is the leading cause of illness and death. Therapeutic strategies aimed at reducing cholesterol plasma levels have shown efficacy in either reducing progression of atherosclerotic plaques and atherosclerosis-related mortality. The farnesoid-X-receptor (FXR) is a member of metabolic nuclear receptors (NRs) superfamily activated by bile acids. In entero-hepatic tissues, FXR functions as a bile acid sensor regulating bile acid synthesis, detoxification and excretion. In the liver FXR induces the expression of an atypical NR, the small heterodimer partner, which subsequently inhibits the activity of hepatocyte nuclear factor 4α repressing the transcription of cholesterol 7a-hydroxylase, the critical regulatory gene in bile acid synthesis. In the intestine FXR induces the release of fibroblast growth factor 15 (FGF15) (or FGF19 in human), which activates hepatic FGF receptor 4 (FGFR4) signalling to inhibit bile acid synthesis. In rodents, FXR activation decreases bile acid synthesis and lipogenesis and increases lipoprotein clearance, and regulates glucose homeostasis by reducing liver gluconeogenesis. FXR exerts counter-regulatory effects on macrophages, vascular smooth muscle cells and endothelial cells. FXR deficiency in mice results in a pro-atherogenetic lipoproteins profile and insulin resistance but FXR−/– mice fail to develop any detectable plaques on high-fat diet. Synthetic FXR agonists protect against development of aortic plaques formation in murine models characterized by pro-atherogenetic lipoprotein profile and accelerated atherosclerosis, but reduce HDL levels. Because human and mouse lipoprotein metabolism is modulated by different regulatory pathways the potential drawbacks of FXR ligands on HDL and bile acid synthesis need to addressed in relevant clinical settings.
Keywords: FXR, atherosclerosis, liver metabolism, vascular wall and macrophages
Introduction
Atherosclerosis is a pathogenic condition of the arterial vessel wall, characterized by lipid deposition, leucocyte infiltration and intimal thickening [1]. Inflammation plays a key role in the development of atherosclerosis. Immune cells dominate early atherosclerotic lesions, their effector molecules accelerate progression of the lesions and activation of inflammation can elicit acute coronary syndromes. Atherosclerotic lesions (atheromata) are asymmetric focal thickenings of the innermost layer of the artery, the intima. They consist of cells, connective-tissue elements, lipids and debris. Blood-borne inflammatory and immune cells constitute an important part of an atheroma, the remainder being vascular endothelial and smooth-muscle cells (SMC). In individuals with hyperlipidaemia and signs of systemic inflammation atherosclerosis is initiated by the activation and dysfunction of endothelial cells. Studies in animals and human beings have shown that hypercholesterolemia causes focal activation of endothelium in large- and medium-sized arteries [1]. The infiltration and retention of low-density lipoprotein (LDL) in the arterial intima initiate an inflammatory response in the artery wall. Modification of LDL, through oxidation or enzymatic degradation in the intima, leads to activation of endothelial cells preferentially at sites of hemodynamic strain. Patterns of hemodynamic flow typical for atherosclerosis-prone segments (low average shear but high oscillatory shear stress) cause increased expression of adhesion molecules and inflammatory genes by endothelial cells recruiting blood borne monocytes. Monocytes, which become loaded with cell-activating oxidized LDL (oxLDL) and other lipids, then accumulate in the evolving lesion and transform into foam cells to form early plaques (fatty streaks) in the intima. The relative expression of anti-inflammatory and pro-inflammatory factors will determine whether this early lesions progress into mature atherosclerotic plaques. Cytokines and chemokines-regulated adhesion molecules expressed on endothelial cells and macrophages are essential for recruiting blood-borne macrophages and lymphocytes to the aortic plaques. The vascular cell adhesion molecule (VCAM)-1 binds particularly classes of leucocytes found in the nascent atheroma: the monocyte and the T lymphocyte. The mechanism of VCAM-1 induction early after initiating an atherogenic diet depends on inflammation instigated by modified lipoprotein particles accumulating in the arterial intima in response to the hyperlipidaemia. Constituents of modified lipoprotein particles, among them certain oxidized phospholipids and short-chain aldehydes arising from lipoprotein oxidation, can induce transcriptional activation of the VCAM-1 gene mediated by nuclear factor-κB (NF-κB) (reviewed in [1]). Pro-inflammatory cytokines such as interleukin (IL)-1β or tumour-necrosis factor-α (TNF-α) induce VCAM-1 expression in endothelial cells by this pathway. Human atherosclerotic lesions contain inflammatory cytokines linking hypercholesterolaemia to VCAM-1 expression. A number of genes with potentially ‘atheroprotective’ properties contain shear-stress response elements in their promoter regions. Many of such atheroprotective genes modulate inflammation: superoxide dismutase, expressed at higher levels in regions of laminar flow, counteracts oxidative stress and hence limits VCAM-1 expression and other inflammatory pathways. Likewise, nitric oxide arising from endothelial nitric oxide synthase (eNOS), another shear stress-regulated gene, attenuates VCAM gene expression through a pathway involving inhibition of NF-κB, the central transcriptional checkpoint in vascular inflammation ([reviewed in [1]). Atherosclerosis is the leading cause of illness and death. Myocardial infarction and cerebrovascular accident are life-threatening complications of atherosclerosis. Prevention of atherosclerotic plaque formation and prevention or treatment of complication of atherosclerotic plaques rupture represents therefore a major therapeutic goal worldwide. In this context therapeutic strategies aimed at reducing cholesterol plasma levels have been shown to be efficient in reducing progression of atherosclerotic plaques as well as in reducing atherosclerosis- related mortality [1].
Bile acids synthesis from cholesterol is the predominant pathway for eliminating excess cholesterol in the body. Bile acids are synthesized in the liver and secreted into the intestine were they facilitate the absorption, transport and distribution of dietary lipids, lipid-soluble vitamins and steroids. Bile acids are reabsorbed in the ileum and transported via the enterohepatic circulation to the liver, where they inhibit their own synthesis [2]. In addition to their role in regulating dietary lipid absorption, bile acids have been demonstrated to act as signalling molecules by activating nuclear and cell surface receptor [3–8] in hepatocytes (Fig. 1). In 1999, bile acids were discovered to function as endogenous ligands for the farnesoid X receptor-α (FXR-α; NR1H4) [9–11]. Since then several other nuclear receptors (NRs), pregnane–X-receptors (PXR), constitutive androstane receptor (CAR) and the vitamin D receptor (VDR) have been shown to be activated by bile acids [7].
Fig 1.
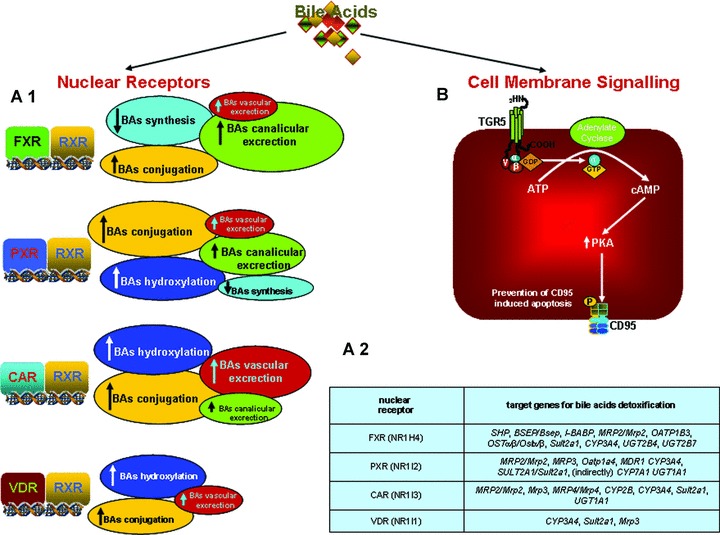
Bile acids and hepatocyte cell signalling. (A.1): In the liver bile acids activate NR involved bile acid synthesis, uptake and detoxification. In addition to FXR, a master gene that inhibits their uptake and synthesis, bile acids activate PXR, CAR and VDR. These NRs regulate the expression/activity of phase I and II detoxification enzymes and the induction of canalicular and alternative basolateral transporters (A.2). (B). In the muscle cells (human) and brown adipose tissue (mouse) bile acids activate a cell membrane receptor, TGR-5 (M-BAR), involved in regulation of thermogenesis and basal energy expenditure.
FXR is an adopted member of the metabolic NR superfamily whose expression is mainly restricted to liver, intestine, kidney and adrenals [12–15]. FXR functions as a bile acids sensor regulating the activity of genes encoding for genes/proteins involved in bile acids synthesis, transport, conjugation and excretion [8]. One of the key functions of FXR is the regulation of bile acid synthesis.
The bile acid feedback regulation is primarily achieved in hepatocytes through the transcriptional regulation of CYP7A1, the rate-limiting enzyme in the classic bile acid biosynthetic pathway. Bile acid-activated FXR induces the expression of the small heterodimer partner (SHP, NR0B2), an atypical NR that lacks the ligand binding domain, which interacts with the liver-related homologue-1 and hepatocyte nuclear factor 4α (HNF-4α) inhibiting the transcription of CYP7A1. However, because bile acid feeding of Shp knockout mice still reduces CYP7A1 mRNA levels to the same extent as that observed in wild-type mice, the requirement of SHP for this inhibitory effect appears to be dispensable [16, 17]. This finding indicates that other pathway(s), independent of SHP, are also involved in the repression of CYP7A1 by bile acids. In recent years it has been shown that activation of intestinal FXR increases the expression and secretion of fibroblast growth factor (FGF)-15 (FGF-19 in human beings) from enterocytes. Secreted FGF15 subsequently binds to the FGF receptor (FGF-R)-4, at the plasma membrane of hepatocytes, activating a JNK-mediated pathway that also represses CYP7A1 [18, 19].
A similar inhibitory loop has also been described in the liver where bile acid-activated FXR induces the expression of FGF-19 in human hepatocytes initiating an autocrine/paracrine loop activating the FGFR4. The hepatic FGF19/FGFR4/Erk1/2 pathway inhibit CYP7A1 independently of SHP [20]. In addition to these mechanisms it has been suggested that bile acids might activate additional pathways including a PKC/JNK pathway via transactivation of the epidermal growth factor receptor [21, 22] and the PXR (NR1I2) mediated pathway [23] that could also contribute to feed back inhibition of CYP7A1. In addition to this bile acid dependent regulation, CYP7A1 is regulated by a number of regulatory factors that connect the immune system with bile acids homeostasis. Indeed the same bile acids or lipopolysaccharide (LPS) by the activation of toll-like receptor 4 stimulate secretion of inflammatory cytokines, such as TNF-α and transforming grow factor β1 (TGF-β1) from Kupffer cells that activate the mitogen-activated protein kinase (MAPK)/JNK pathway to inhibit CYP7A1 expression. Together, these pathways establish a complex network of regulatory factors that modulate CYP7A1 activity and control of bile acid synthesis from cholesterol (Fig. 3).
Fig 3.
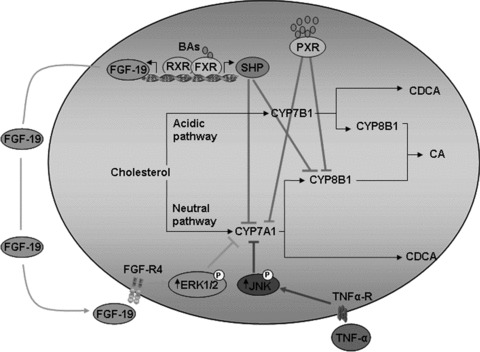
Bile acids regulate their own synthesis. In hepatocytes bile acids accumulation activates FXR leading to an SHP and FGF-19 (human) dependent repression of CYP7A1 and CYP8B1, whereas PXR activation decreases directly the transduction of CYP7A1 and CYP8B1.
Sterol 12α-hydroxylase (CYP8B1) catalyses the synthesis of cholic acid (CA) and controls the ratio of CA over chenodeoxycholic acid (CDCA) in the bile, an enzyme controlling the hydrophobicity of the bile acid pool. The transcription CYP8B1 is under the control of HNF-4α, a tissue-specific transcription factor, known to regulate a large number of genes in hepatocytes. Bile acids repress human CYP8B1 transcription by reducing the transactivation activity of HNF-4α through interaction of HNF-4α with SHP and by reducing the liver expression of HNF-4α[24].
In addition to the regulation of their synthesis, activation of FXR plays a role in inducing enzymes involved in bile acid detoxification. Bile acids are detergent molecules, are inherently cytotoxic for hepatocytes and their accumulation in liver is associated with hepatic disorders. FXR activation increases the expression of gene encoding proteins involved in bile acids detoxification (CYP3A4/Cyp3a11, UGT2Bs and Sult2a1), down-regulate basolateral bile acids uptake via repression of NTCP/Ntcp and OATP/Oatp 1 and 4 while stimulating the expression of both canalicular (MRP/Mrp2, BSEP/Bsep) and alternative basolateral efflux transporters (MDR3/Mdr3,MRP3/Mrp3 and OST/Ostα and β) (reviewed in (8) (see table in Fig. 1).
The others NRs activated by bile acids PXR, CAR and VDR, interact with FXR to modulate different pattern of genes involved in cell protection such as the phase I and II detoxification enzymes and canalicular and alternative basolateral bile acid transporters (reviewed in [8]) (see table in Fig. 1).
Molecular biology of FXR
FXR shares the common modular structure of all members of the metabolic-NR superfamily described in Fig. 2[25]. FXR functions as a heterodimer with the 9-cis-retinoic acid receptor (RXR) and after activation binds various FXR response elements (FXREs) [26, 27] regulating the expression of numerous genes (see Table 1) [26–28]. Upon ligand binding, FXR undergoes conformational changes to release corepressors such as NCor (nuclear co-repressor) and recruit coactivators such as SRC-1 (steroid receptor coactivator-1) [8, 25]Fig. 2). It should be emphasized that the bile acid pool is different from different animal species. In mice it consists mostly of hydrophilic bile acids, muricholic acids and CA, and is very different from the hydrophobic bile acid pool consisting predominantly CDCA, CA and deoxycholic acid (DCA) in human beings. Hydrophobic, but not hydrophilic bile acids are efficacious endogenous ligands of FXR, PXR and VDR (NR1I1) that play important roles in the regulation of bile acid synthesis and metabolism. CDCA is the most potent FXR ligand with an effective concentration (EC50) value of 10 μM [9]; however, secondary BAs such as DCA and lithocholic acid (LCA) could aloso activate the receptor [25]. In addition to these ‘classic’ ligands, in recent years several new synthetic FXR ligands have been described [29] and their increased potency compared with physiological ligands makes them useful tools for the study of FXR function.
Fig 2.
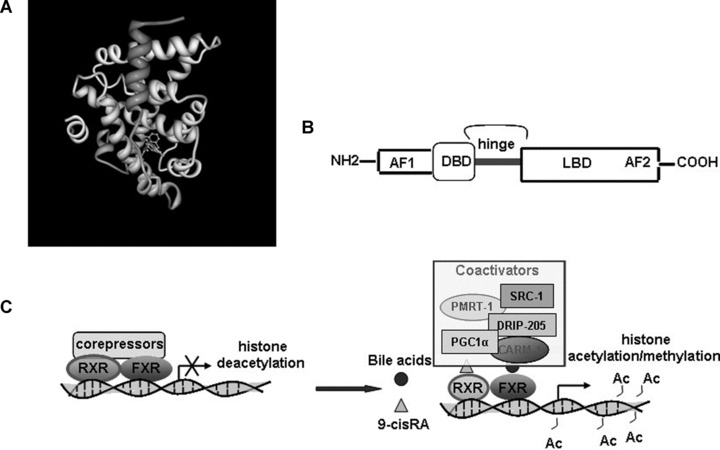
Molecular biology of FXR. (A) 3D structure of FXR. (http://www.rcsb.org/pdb/) in combination with an FXR synthetic agonist GW4064 (B) FXR molecular structure: includes a highly conserved DNA-binding domain in the N-terminal region and a moderately conserved Ligand Binding Domain (LBD) in the C-terminal region. The ligand-independent activation function-1 (AF-1) and ligand-dependent AF-2 are located in the N-terminal and C-terminal regions, respectively. (C) FXR molecular regulation: FXR bind DNA in correspondence to its FXREs. Coactivator and corepressor complexes are involved in FXR activation and repression, respectively. In the absence of a ligand, the FXR heterodimer associates with corepressor complexes, which recruit histone-deacetylase activities. Deacetylation of histone tails leads to chromatin compactation and transcriptional repression. Receptor activation causes the release of the corepressor complex and the AF-2-dependent recruitment of a coactivator complex that contains at least a p160 coactivator (such as SRC-1). These proteins possess histone-acetyltransferase activity that allows chromatin decompactation and gene activation. Multiple protein–protein interactions exist among the FXR and other coactivators such as PRMT-1 (protein arginine(R) methyl transferase-1) and CARM-1 (coactivator-associated arginine methyltransferase-1), inducing histone methylation and PGC-1α (ppar-γ coactivator-1α) and DRIP-205 (vitamin-D-receptor-interacting protein-205).
Table 1.
FXR regulated genes and FXRE
| Gene | Species | FXRE | Regulation |
|---|---|---|---|
| ALAS-1 | Human | IR-1 | + |
| A2-Crystallin | Human | IR-1 | + |
| ApoAI | Human | Negative FXRE | − |
| ApoAV | Human | IR-8 | + |
| ApoCII | Human | IR-1 | + |
| Human | IR-1 | + | |
| ApoCIII | Human | Negative FXRE | − |
| Mouse | DR-1 | − | |
| ASCT2 | Human | IR-1 | + |
| AT2R | Rat | IR-2 | + |
| BACS | Rat | IR-1 | + |
| BAT | Rat | IR-1 | + |
| BSEP | Human | IR-1 | + |
| Rat | + | ||
| Cyp3A4 | Human | IR-1, DR-3, ER-8 | + |
| Cyp8B1 | Human | IR-1 | − |
| CSE | Human | IR-1 | + |
| Decorin | Human | IR-8 | + |
| DDAH1 | Rat | IR-1 | + |
| DHEA Sulfotrasferase/SULT2A4 | Human | IR-0 | + |
| eNOS | Rat | IR-1 | + |
| FAS | Human | IR-1 | + |
| Fetuin-B | Human | IR-1 | + |
| FGF-15 | Mouse | IR-1 | + |
| FGF-19 | Human | IR-1 | + |
| GLUT-4 | Human | IR-1 half-site | + |
| Mouse | |||
| I-BABP | Mouse | IR-1 | + |
| Human | hND | + | |
| ICAM-1 | Mouse | IR-1 | + |
| INSIG-1 | Mouse | IR-1 | + |
| Kininogen | Human | IR-1 | + |
| MDR3, | Human | IR-1 | + |
| MRP2 | Rat | ER-8 | + |
| NaS-1/S1c13a1 | Mouse | IR-1 | + |
| OAT2 | Human | DR-1 | − |
| OATP8 | Human | IR-1 | + |
| Ost·/β | Human | IR-1 | + |
| Mouse | IR-1 | + | |
| PLTP | Human | IR-1 | + |
| Mouse | + | ||
| PPARα | Human | DR-5 Imperfect | + |
| PXR | Mouse | IR-1 | + |
| SHP | Human | IR-1 | + |
| STD | Rat | IR-0 | + |
| SYNDECAN | Human | DR-1 | − |
| UGT 2B4 | Human | B4-BARE | + |
| UGT2B7 | Human | Negative FXRE | − |
| VPAC1 | Human | IR-1 | + |
Is there a role for FXR in atherosclerosis?
Role of FXR in metabolism
Cholesterol metabolism
Cholesterol homeostasis is maintained by nutritional intake, intestinal absorption, de novo synthesis, catabolism, reverse transport and excretion into the bile. FXR plays a critical role in cholesterol homeostasis by regulating genes that are involved in each of these processes. FXR–/– mice fed high cholesterol diet (1% cholesterol) were distinguished from wild-type mice by elevated serum bile acid, cholesterol and triglycerides, increased hepatic cholesterol and triglycerides and a proatherogenic serum lipoprotein profile [30].
Bile synthesis
Bile acid pool size has a profound effect on lipid metabolism [31–33]. CDCA and CA are the main human bile acids. They are hydrophobic bile acids and strongly promote cholesterol absorption in the intestine [34]. The ratio of CDCA versus CA is determined by CYP8B1 activity. Tissue-selective deletion of FXR in the intestine results in the loss of CYP7A1 repression by a synthetic FXR agonist, while conversely FXR repression of CYP8B1 was more dependent upon hepatic FXR expression [35]. FXR also regulates biliary cholesterol secretion [36], in fact FXR–/– mice the liver ABCG5/8, that have been proposed to play a key role in the biliary excretion of sterols [37, 38], were down-regulated, moreover the expression of the cholesterol ester hydrolase (CEH), an enzyme responsible for the conversion of intra-cellular cholesterol ester (CE) into free cholesterol (FC) and the sterol carrier protein (SCP), a protein thought to be involved in the hepatocellular trafficking of FC to the canalicular membrane were also reduced in the liver of FXR–/– mice. Together, these data suggest that CEH-mediated mobilization, SCP-mediated intracellular transport and ABCG5/8-mediated biliary secretion of FC may be decreased in FXR–/– mice.
Cholesterol absorption
Intestinal epithelial cells absorb micelles of bile acids solubilized cholesterol by phagocytose. The phagocytosed cholesterol is esterified with fatty acids by acyl-CoA cholesterol acyl transferase (a microsomal enzyme). Cholesterol esters are used by microsomal triglyceride transfer protein to assemble chylomicrons which are secreted into the lymph. A significant part of cholesterol that is taken up by the intestinal epithelium is secreted back to the intestinal lumen via the ABC transporters. Both, ABC1 and heterodimeric ABCG5/ABCG8 transporters mediate re-secretion of absorbed cholesterol into the intestinal lumen. The expression of ABCG8 but not of ABCG5, is sharply decreased in the ileum of FXR–/– mice and this effect is associated with an increase of intestinal cholesterol absorption [36].
Liver cholesterol metabolism
Synthesis
Sterol regulatory element binding protein 2 (SREBP2) is involved in the de novo biosynthesis of cholesterol through a pathway that involves hydroxi-metylglutaryl CoA synthase (HMGCoAS) and hydroximetylglutaryl CoA reductase (HMGCoAR) [39]. Several studies [40–42] indicate that treatment with FXR agonist, decrease plasmatic cholesterol levels thanks to their ability to inhibit HMGCoA reductase liver levels.
LDL-cholesterol
In human hepatocytes CDCA induces the expression of LDL-(r)receptor by a MAPK mechanism [43] and it has recently been demonstrated [44] that FXR mediated a down-regulation of Pro-protein convertase subtilisin/kexin 9 (PCSK9). PCSK9 acts as a chaperone and promotes the intracellular degradation of the LDL-r by interfering with its recycling to the plasma membrane [45, 46]. PCSK9 has recently emerged as a central player in the regulation of cholesterol homeostasis [47]. Indeed PCSK9 mutations lead that to an increase of its function is associated a premature atherosclerosis lesion [48] and a ‘loss-of-function’ mutations are associated with low levels of LDL-c and confer protection against cardiovascular disease [49]. These studies indicated that FXR may reduce LDL plasmatic levels enhancing LDL-r activity.
HDL-cholesterol
FXR induces the expression of the hepatic scavenger receptor B1 (SR-B1), a protein that regulates high-density lipoprotein (HDL) uptake by liver and peripheral tissues [30, 36]. A reduced hepatic SR-BI expression in FXR-deficient mice [36] was associated with a decreased rate of HDL cholesterol clearance and high plasma levels of HDL [30, 36]. However, FXR strongly represses human apolipoprotein A-I (ApoA-I) gene expression in liver cells, both in vitro and in vivo[50]. ApoA-I is the primary protein constituent of HDL, defining its size and shape, solubilizing its lipid components, removing cholesterol from peripheral cells, activating the lecithin-cholesterol acyl transferase enzyme and delivering the resulting cholesterol esters to the liver. Consequently, feeding of hApoA-I transgenic mice with CA strongly reduces plasma HDL cholesterol and ApoA-I levels [50]. In contrast, in FXR–/– mice under normal feeding conditions, the hepatic expression of the murine ApoA-I gene appeared unchanged [36]. Pharmacological studies with synthetic FXR agonist (6-ECDCA and GW4064) [51–53], in three different models of hypercholesterolemic mice (ApoE–/–, ob/ob and db/db) have shown that FXR activation reduces HDL plasma levels.
Triglycerides metabolism
The first evidence that bile acids was able to reduce lipidaemia levels, date back to the early 1970s in individuals suffering from gallstones and in those with monogenic familial hypertriglyceridaemia treatment with CDCA reduced plasma triglyceride [33, 54]. The molecular mechanism underlying the hypotriglyceridaemic effect of CDCA has now been elucidated and found, at least in rodents, to be linked to FXR activation [55]. FXR-deficient mice are hypertriglyceridaemic, suggesting a role for FXR in the control of triglyceride metabolism [30]. FXR alters the transcription of several genes involved in fatty-acid and triglyceride synthesis and lipoprotein metabolism. Plasmatic triglyceride levels reflect the balance between production and clearance of triglyceride-rich lipoproteins such as very-LDL (VLDL) and chylomicrons. Lipoprotein lipase (LPL) is a key enzyme involved in the lipolysis of these particles; the activity of this enzyme is regulated by several lipoprotein such as ApoC-III that is an inhibitor whereas ApoC-II and ApoA-V are activators. Molecular biology experiments revealed that the FXR/RXR heterodimer represses [56] ApoC-III expression whereas induces ApoC-II expression and human ApoA-V promoter activity in liver cells [57–59]. Moreover, FXR induces the expression of the VLDL receptor [60], a protein that plays a major role in the metabolism of post-prandial lipoproteins by enhancing LPL mediated triglyceride hydrolysis. In addition, expression of syndecan-1, a transmembrane protein that binds remnant particles before their transfer to receptors, was found to be FXR sensitive [61]. All together these data indicate that FXR increases VLDL and chylomicrons clearance by modulating LPL activity. FXR is also involved in the control of hepatic lipogenesis, and its activation reduces the synthesis of FFA and decreases the secretion of VLDL from the liver [62]. The first mechanism identified in the regulation of triglyceride synthesis by FXR ligands (CA and GW4064) is represented by an SHP-mediated inhibition of SREBP-1c expression that is a transcription factor regulating lipogenic genes, including the fatty-acid synthase (FAS) [63]. However, the inhibitory action of FXR in triglycerides levels could be also mediated by a signalling cascade involving SHP-independent pathways because administering SHP–/– mice with an FXR agonist still decreases serum triglyceride levels [63, 64]. FXR also regulates the expression of peroxisome proliferator-activated receptor (PPAR)-α in human beings [65]. PPAR-α is an NR that is activated by fibrates, a class of synthetic hypolipidaemic drugs that decreases plasma triglyceride levels, probably by enhancing fat oxidation [66].
Glucose metabolism
Hepatic gluconeogenesis
Loss of FXR disrupts normal glucose homeostasis and leads to the development of insulin resistance and hyperglycaemia [67, 68]. In cultured hepatocytes, glucose was shown to induce the expression of FXR, whereas insulin counter-regulated this effect [67]. Activation of hepatic FXR results in repression of phosphoenoylpyruvate carboxykinase and glucose-6-phosphatase (G6Pase) and increased phosphorylation of GSK3b. Since phosphorylated GSK3b is inactive, the net effect of these activities is an increased level of the dephosphorylated/active glycogen synthase [68, 69]. FXR agonist attenuates the expression of multiple genes involved in neoglucogenesis in wild-type but not in FXR- and SHP-deficient mice – thus an FXR–SHP regulatory cascade appears involved in the regulation of gluconeogenesis [70]. Overall, these changes result in decreased hepatic gluconeogenesis, decreased plasma glucose levels and increased hepatic glycogen synthesis. Because type 2 diabetes is characterized by an increased hepatic glucose output, which contributes to fasting hyperglycaemia, FXR activation appears an attractive possibility for this disease.
Insulin sensitivity
FXR deficiency results in impaired insulin signalling and thereby leads to insulin resistance, such changes may result from the elevated free fatty acids noted in the plasma of FXR–/– mice [67–69]. In vitro experiments had demonstrated that FXR activation improves insulin signalling and insulin-stimulated glucose uptake in differentiated 3T3-L1 cells [71, 72]. The main insulin-responsive glucose transporter is GLUT-4 that plays a critical role in maintaining systemic glucose homeostasis and is subject to complicated metabolic regulation. GLUT-4 expression disorder might cause insulin resistance, and overexpression of GLUT-4 has been confirmed to ameliorate diabetes. Recently it has been demonstrated in adipocytes and hepatocytes cells line that FXR agonist induced GLUT-4 expression [26]. Based on these results, a prediction would be that FXR activation promotes insulin sensitivity, indeed an FXR agonism improves glucose tolerance and insulin sensitivity in db/db, KK-A(y) [70] and ob/ob [63] mice.
Role of FXR vessel wall
Recent works have indicated that FXR may play a role in the pathophysiology of atherosclerosis independent of its impact on plasma lipid levels. Expression of FXR has been detected in endothelial cells, muscle cells and macrophages involved in atherosclerotic plaque formation.
Endothelial cells
FXR is expressed in several endothelial cell lines such as human aortic endothelial cells, human umbilical vein endothelial cells [73], in rat pulmonary artery endothelial cells (RPAEC) and bovine aortic endothelial cells (BAEC) [74]. Bile acids exert either pro- or -anti-inflammatory activity on endothelial cells. Thus, bile acids increase the expression of intracellular adhesion molecule-1 (ICAM-1) and VCAM-1 via an FXR-independent NF-κB and p38 MAPK-dependent mechanism [75]. This pro-attivatory effects of bile acids on ICAM-1 and VCAM-1 was sufficient to promote the adhesion of monocytes to HUVEC, suggesting a contributory role for circulating bile acids to endothelium dysfunction. Because ICAM-1 is abundant in the endothelium, and the induction of ICAM-1 is known to contribute to the leucocyte-induced inflammation in vascular tissues, it is possible that under cholestatic conditions, elevated levels of bile acids in the circulation may cause endothelium dysfunction contributing to the initiation of early events associated with vascular lesions formation. The endothelium plays a crucial role in regulation of vascular tone and changes in the gene expression profile and an altered release of endothelium-derived factors occur in metabolic diseases characterized by an increased production of pro-inflammatory cytokines, decreased secretion of adiponectin from adipose tissue and high circulating levels of free fatty acids and hyperglycaemia [76, 77]. A dysfunctional endothelium displays uncoupling of eNOS accompanied with a decreased generation of nitric oxide and increased expression of endothelin 1 (ET-1). A growing body of evidence suggest that FXR is an important regulatory gene in endothelial cells. Thus, FXR is functional in RPAEC, as demonstrated by induction of its target genes such as SHP after treatment with FXR agonist. Interestingly, activation of FXR in these endothelial cells led to down-regulation of ET-1 expression via negatively interfering with AP-1 signalling [73]. Moreover treatment of BAEC with an FXR ligand up-regulates eNOS mRNA and protein and is associated with an increased production of nitrite/nitrate [74].
FXR might also interfere with endothelium-derived nitric oxide activity through modulation of serum asymmetric dimethylarginine (ADMA). ADMA is an endogenous NOS inhibitor [78] and elevated plasma ADMA levels are associated with reduced nitric oxide synthesis in patients with hypercholesterolemia, hypertriglyceridaemia, hypertension, type II diabetes, chronic renal failure and chronic heart failure; reduction of ADMA levels ameliorates nitric oxide synthesis [79–83]. In vitro studies have shown that FXR reduces ADMA formation by positively regulating the expression/activity of hepatic dimethylarginine dimethyl-aminohydrolase-1 (DDAH1), the major catabolic pathway of ADMA. The induction of liver DDAH1 gene expression correlates with a reduction of serum ADMA levels [78].
We have recently provided evidence that hydrogen sulphide (H2S), a gasotransmitter and vasodilator agent is also regulated by FXR [27]. In mammals, H2S production is catalysed by the activity of cystathionine β-synthase and/or cystathionine γ-lyase (CSE). CSE is expressed by vascular muscle cells. Deficiency in CSE expression increases blood pressure and significantly diminishes endothelium-dependent relaxation of resistance arteries, indicating that H2S is a physiologic vasorelaxant [84]. We have demonstrated [27] that liver expression of CSE is regulated by bile acids by an FXR-mediated mechanism. An FXR responsive element, IR-1, has been found in the CSE promoter and mutation of this responsive element abrogates induction of CSE expression caused by FXR ligands. FXR activation increases H2S production in the liver and reduces portal pressure attenuating the endothelial dysfunction of isolated and perfused cirrhotic rat livers. Interestingly, administration of H2S-donor to ApoE–/– mice inhibits the development of atherosclerotic lesions [85].
Vascular smooth muscle cells
Bishop-Bailey et al.[86] have reported that FXR, mRNA and protein, are expressed in atherosclerotic lesions and vascular SMC (VSMC) lines generated from rat aortic (RASMC) and human primary saphenous vein (hSVSMC). The functional roles of FXR in these cell lines are unclear. FXR activation in these cells regulates apoptosis, increases the reverse cholesterol transport from vessel wall to HDL by inducing the expression of PLTP and exerts an anti-inflammatory activity by targeting NF-κB activation in an SHP-depend manner [86, 87]. FXR ligands also reduce the migration of RASMC and human aortic VSMCs induced by platelet-derived growth factor (PDGF)-β[87] VSMCs are important in atherosclerosis because in addition to inflammation they are responsible for an inappropriate vascular remodelling ([reviewed in [1]). The fact that FXR activation down-regulates expression of pro-inflammatory mediators such as iNOS and COX-2 [87], as well as cell migration of VSMCs might have mechanistic relevance in explaining the ability of FXR activation in attenuating vascular inflammation, and remodelling of atherosclerotic plaques [86, 87]. Treatment of RASMC with an FXR ligand increases the expression of type 2 angiotensin receptor (AT2R) [88]. This receptor plays an antagonistic role in AT1R signalling [89–92] and its activation might explain the inhibitory effect exerted by FXR ligand on the VSMC proliferation induced by angiotensin II.
In sharp contrast, chronic administration of GW4064, an FXR ligand, impairs endothelium-dependent relaxation of SMCs. This effect associated with an impaired elevation of cGMP in response to stimulation with a nitric oxide donor, due to a reduction expression of α1- and β1-subunit of soluble guanylyl cyclase [93].
Finally in vitro experiments demonstrated that FXR might interfere in the remodelling of vessels by the up-regulation of the decorin expression in VSMCs [94]. Decorin, a class I small leucine-rich proteoglycans that also includes biglycan, is a component of the extracellular matrix (ECM) of a variety of tissues [95]. Decorin plays a number of important regulatory functions in cell adhesion, migration, proliferation and signalling [96]. Decorin binds to TGF-β and PDGF and inhibits their biological activity in a number of cell types [97–100]. Inhibition of TGF-β and PDGF by FXR might have a role in regulation of tissue remodelling, reducing intimal thickening caused by arterial injury altering the composition of the ECM and reducing ECM volume [97–100]. Indeed the arterial wall responds to damage through the induction of specific gene products, such as TGF-β and PDGF, that increases cellular proliferation and connective tissue formation and that might result in intimal hyperplasia, as observed in restenosis after balloon injury and the early phases of atherosclerosis [97–100].
Modulation of macrophages-inflammatory response
Activation of inflammatory genes in the vessel wall, with subsequent adhesion, chemoattraction, subendothelial migration, retention and activation of inflammatory and immune cells such as monocytes and T cells is an important step in the initiation, progression and destabilization of atherosclerotic plaques [1]. Macrophages are central to the development of the atherosclerotic lesion because of their ability to take up modified lipoproteins and to release inflammatory mediators [101]. Bile acids have been long known to exert direct regulatory function on cells of innate immunity. CDCA, the physiological ligand of FXR, attenuates IL-1β, IL-6 and TNF-α release from LPS primed monocytes [102]. Also results from three double knockout mice seem to indicate that FXR ablation might result in a pro-inflammatory phenotype. Thus, Guo et al.[103] have shown that peritoneal macrophages isolated from FXR-null mice and stimulated with LPS release higher levels of TNF-α and interferon-γ than wild-type macrophages. We have recently demonstrated [104] that FXR deficient mice develop a dysregulated immune response indicating that FXR could function as a braking signal for intestinal vascular immunity [51].
The modulatory effect exerted in vitro was maintained in ApoE–/– mice – indeed chronic administration of 6E-CDCA resulted in a significant down-regulation of IL-6 and IL-1β mRNAs expression in aortic plaques. Finally, in vivo treatment with 6E-CDCA caused a robust reduction of CD36 expression on circulating macrophages [51], and in isolated ApoE−/– macrophages exposure to an FXR agonist reduces ABCA1 mRNA expression, suggesting that FXR could interfere on foam cells formation by reducing cholesterol accumulation.
Lessons from FXR null mice and experimental models of atherosclerosis
The characterization of FXR deficient mice has highlighted the potential role of FXR as a regulatory gene in different metabolic pathways. FXR–/– mice demonstrate a specific lipid profile characterized by increased plasma triglyceride, free fatty acids, VLDL, HDL and LDL, glucose intolerance and insulin resistance [30, 53]. Moreover, FXR deficiency associates with impaired adipocytes differentiation [53] and hyperabsorption of cholesterol and triglycerides from the intestine [36]. Histopathological evaluation demonstrates that FXR deficiency results in a progressive liver injury characterized by hepatocytes vacuolization, lipid deposit, focal necrosis and infiltration of inflammatory cells [105]. Despite this pro-atherogenic lipids and lipoproteins profile, FXR−/– mice fail to develop any detectable plaques spontaneously and even when fed a Western (high fat, high cholesterol) diet [106]. However, FXR gene ablation represents a precipitating factor for development of atherosclerotic lesion in mice with altered lipid metabolism such as the ApoE and LDLr deficient mice. Thus, feeding male double FXR/ApoE knockouts mice with high-fat diet elevated plasma triglyceride and cholesterol levels and increased the extent of atherosclerotic lesions in comparison to ApoE−/– single knockout mice [106].
The animal gender, however, appears to be an important factor for development of atherosclerotic lesions in double FXR and ApoE knockout mice. Thus, in contrast to male mice, female FXR−/– ApoE−/– mice feed a high-fat diet are protected against development of atherosclerotic lesions in comparison to ApoE–/– mice, most likely as a result of reduced oxLDL-C uptake by macrophages [103].
A similar gender-related pro-atherogenetic effect was observed in the LDLr deficient mice. Male mice double knockout for FXR and LDLr fed with a high-fat diet, despite a 4-fold increase of plasma triglyceride levels, develop less aortic atherosclerotic lesions and display reduced macrophages lipid accumulation in comparison with LDLr–/–[107]. Female FXR–/–LDLr–/– mice fed high-fat diet had increased plasma triglyceride and cholesterol levels, and were not protected against atherosclerotic plaque formation [107]. In summary, it appears that FXR gene ablation results in an anti-atherogenetic phenotype in female ApoE−/– and male LDLr−/– mice, but favours the appearance of aortic plaques in male ApoE−/– mice. These mixed results suggest, at the maximum, a contributory role for FXR in the pathogenesis of plaque formation in rodent models of atherosclerosis. This role needs to be confirmed by appropriate analysis of FXR expression in appropriate subset of patients with atherosclerotic disorders.
While results from double knockout appear to cast doubts on the functional role of FXR in atherosclerotic plaques formation in these genetic models, pharmacological treatment with FXR agonists favours the hypothesis that FXR activation would protect against plaques formation. Indeed, recently two studies [51, 64] have demonstrated a beneficial effect of FXR activation in the treatment of rodent models of dyslipidaemia. In both studies treating mice with 6E-CDCA and WAY-362450 was found effective in protecting against development of aortic atherosclerotic lesions in male and female ApoE–/– and LDLr–/– mice. Interestingly, however, disruption of SHP in ApoE–/– and LDLr–/– mice, abrogates protection exerted by WAY-362450 in female mice. The observation that an FXR/SHP pathway is beneficial in female mice but not in male mice, has been linked to the finding that administration of WAY-362450 to male mice associated with a reduction of both plasma cholesterol levels and liver expression of CYP7A1/CYP8B1. The protective effect exerted by WAY-362450 was unrelated to its effects on triglycerides, because FXR activation exerted a similar triglyceride-lowering effect in both female and male LDLr−/– or ApoE−/– mice lacking SHP [64]. A sharp summary of these data indicate that FXR protects against formation of atherosclerotic plaques in male mice, but do the opposite in female ApoE−/– and LDLr−/– mice. The gender effect observed in these models is irrespective of the lipid profile, because FXR activation exerts a similar triglyceride-lowering effect in both sexes. These studies also show that FXR regulates plasma cholesterol levels by an SHP-dependent pathway, with gender difference, while reduces triglycerides levels by an SHP-independent mechanism that is gender indifferent.
Conclusion
The characterization of FXR as a ligand-induced transcription factor involved in the regulation of liver metabolism has led to a significant improvement of our understanding of the mechanisms controlling lipid homeostasis and has provided opportunities for the development of a novel class of therapeutic agents in the treatment of cardiovascular diseases. Several experimental findings indicated that FXR agonism would have beneficial effects in the treatment of atherosclerosis (summarized in Fig. 4). Thus FXR action decreases liver lipogenesis, increases liver GLUT-4 expression and decreases gluconeogenesis, attenuates the expression of inflammatory gene in macrophages and inhibits VSMC proliferation, migration and activation. Pre-clinical animal studies have demonstrated that synthetic FXR agonists protect against development of aortic plaques formation in murine models of atherogenesis [56, 64].
Fig 4.
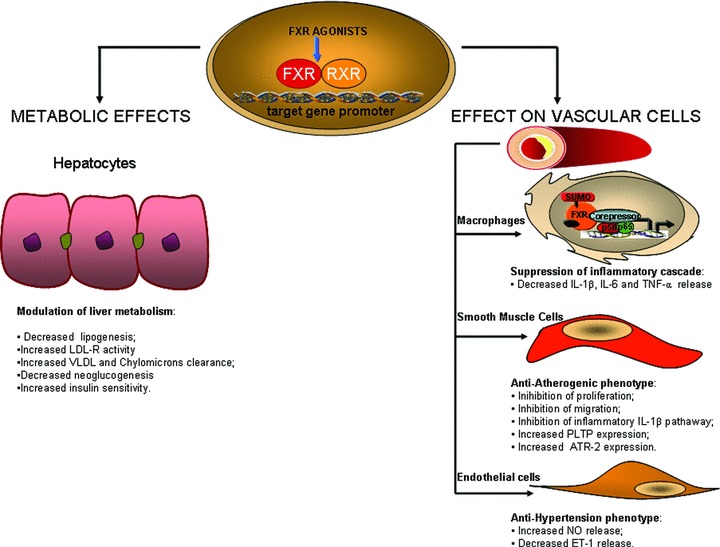
Metabolic and vascular effects of FXR. FXR heterodimerizes with RXR. The FXR/RXR heterodimer binds to FXR-responsive elements in the promoters of target genes. Following a ligand-induced activation, FXR induces the expression of genes encoding proteins involved in lipogenesis, lipoprotein clearance and glucose metabolism. In addition, FXR activation and sumoylation in macrophages stabilizes the nuclear co-repressor NCoR on NF-κB responsive element inhibiting the expression of inflammatory genes in macrophages, suppresses cell proliferation and migration and increases reverse cholesterol transport in VSMCs and endothelial cells. LDL-R, low-density lipoprotein receptor; VLDL, very low-density lipoprotein; IL-1β, interleukin 1β; IL-6, interleukin 6; TNF-α, tumour necrosis factor α; PLTP, phospholipid transfer protein; ATR-, angiotensin type 2 receptor; NO, nitric oxide and ET-1, endothelin 1.
However the role of FXR per se in the pathogenesis of atherosclerosis is likely secondary. Thus, the analysis of FXR null mice has demonstrated that despite a pro-atherosclerotic profile these mice did not spontaneously develop any detectable plaques even when feed a high-fat diet. Further, the gender effects observed in ApoE- and LDLr-deficient mice suggest that the role of FXR is not dichotomous with a simply positive or negative effect, but should be viewed as contributory, as frequently happens in the context of diseases implying the involvement of multiple genes. This also underlines a complex mechanistic role for FXR in the pathogenesis of atherosclerotic lesions that might arise from the combination of metabolic with vascular effects.
FXR activation might also have potential side effects. The major unwanted effect of FXR observed consistently in animal studies is the reduction of plasmatic HDL [51–53, 64]. Even if this effect could be linked to induction of SR-B1 in the liver, a reduction of HDL levels will impair the so-called reverse cholesterol transport. It has to be kept in mind, however, that in addition to its effects on mouse SB-R1, FXR exerts a direct negative effect on the synthesis of human ApoAI [50]. Because ApoAI is the primary protein constituent of HDL its inhibition will result in reduced plasma levels of HDL.
A potential drawback of the use of FXR agonists in clinical settings could also arise from the effect FXR exerts on CYP7A1. Human studies [108] have shown a correlation between CYP7A1 expression and plasma LDL cholesterol levels. Thus, CYP7A1-deficient individuals have elevated LDL cholesterol levels and, on the other hand, experimental studies have shown that increasing CYP7A1 expression lowers LDL cholesterol plasma levels in rodent species [109–111]. Further, pharmacological enhancement of cholesterol conversion into bile acids reduces LDL plasmatic levels [112]. Finally, inhibition of CYP7A1 could inhibit bile acid synthesis. In addition to these effects, unexpected effects of FXR in human beings should be taken into consideration keeping in mind that FXR is highly express in the human adrenal where it modulates steroids hormones synthesis by the induction of 3β-hydroxysteroid dehydrogenase [113, 114].
Several clinical trials (http://www.ClinicalTrials.gov) with synthetic FXR agonists are ongoing, which should increase our insight into potential utility of these new treatments for dyslipidaemia and eventually for cardiovascular disease.
References
- 1.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009 doi: 10.1194/jlr.R900010-JLR200. ; in press. PMID: 19346330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie W, Radominska-Pandya A, Shi Y, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA. 2001;98:3375–80. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makishima M, Lu TT, Xie W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–16. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 5.Kawamata Y, Fujii R, Hosoya M, et al. A G proteincoupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–44. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 6.Reinehr R, Häussinger D. Inhibition of bile salt-induced apoptosis by cyclic AMP involves serine/threonine phosphorylation of CD95. Gastroenterology. 2004;126:249–62. doi: 10.1053/j.gastro.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 7.Zollner G, Marschall HU, Wagner M, et al. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm. 2006;3:231–51. doi: 10.1021/mp060010s. [DOI] [PubMed] [Google Scholar]
- 8.Fiorucci S, Rizzo G, Donini A, et al. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol Med. 2007;13:298–309. doi: 10.1016/j.molmed.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–65. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 10.Parks K, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–68. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Chen J, Hollister K, et al. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–53. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 12.Goode E, Chen J, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–93. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 13.Huber RM, Murphy K, Miao B, et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene. 2002;290:35–43. doi: 10.1016/s0378-1119(02)00557-7. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Kast-Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem. 2003;278:104–10. doi: 10.1074/jbc.M209505200. [DOI] [PubMed] [Google Scholar]
- 15.Otte K, Kranz H, Kober I, et al. Identification of farnesoid X receptor β as a novel mammalian nuclear receptor sensing lanosterol. Mol Cell Biol. 2003;23:864–72. doi: 10.1128/MCB.23.3.864-872.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerr TA, Saeki S, Schneider M, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell. 2002;2:713–20. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Lee YK, Bundman D, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 18.Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–91. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–25. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 20.Song KH, Li T, Owsley E, et al. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49:297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stravitz RT, Rao YP, Vlahcevic ZR, et al. Hepatocellular protein kinase C activation by bile acids: implications for regulation of cholesterol 7 alpha-hydroxylase. Am J Physiol. 1996;271 doi: 10.1152/ajpgi.1996.271.2.G293. : G293–303 PMID: 8770045. [DOI] [PubMed] [Google Scholar]
- 22.De Fabiani E, Mitro N, Anzulovich AC, et al. The negative effects of bile acids and tumor necrosis factor-alpha on the transcription of cholesterol 7alpha-hydroxylase gene (CYP7A1) converge to hepatic nuclear factor-4: a novel mechanism of feedback regulation of bile acid synthesis mediated by nuclear receptors. J Biol Chem. 2001;276:30708–16. doi: 10.1074/jbc.M103270200. [DOI] [PubMed] [Google Scholar]
- 23.Li T, Chiang JY. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288:G74–84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 24.Zhang M, Chiang JY. Transcriptional regulation of the human sterol 12alpha-hydroxylase gene (CYP8B1): roles of heaptocyte nuclear factor 4alpha in mediating bile acid repression. Biol Chem. 2001;276:41690–9. doi: 10.1074/jbc.M105117200. [DOI] [PubMed] [Google Scholar]
- 25.Pellicciari R, Costantino G, Fiorucci S. Farnesoid X receptor: from structure to potential clinical applications. J Med Chem. 2005;48:5383–403. doi: 10.1021/jm0582221. [DOI] [PubMed] [Google Scholar]
- 26.Shen H, Zhang Y, Ding H, et al. Farnesoid X receptor induces GLUT4 expression through FXR response element in the GLUT4 promoter. Cell Physiol Biochem. 2008;22:1–14. doi: 10.1159/000149779. [DOI] [PubMed] [Google Scholar]
- 27.Renga B, Mencarelli A, Migliorati M, et al. Bile-acid-activated farnesoid X receptor regulates hydrogen sulfide production and hepatic microcirculation. World J Gastroenterol. 2009;15:2097–108. doi: 10.3748/wjg.15.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefebvre P, Cariou B, Lien F, et al. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–91. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 29.Fiorucci S, Mencarelli A, Distrutti E, et al. Targetting Farnesoid-X-receptor from medicinal chemistry to disease treatment. Curr Med Chem. 2009 doi: 10.2174/092986710790112666. ; in press. PMID: 19941473. [DOI] [PubMed] [Google Scholar]
- 30.Sinal CJ, Tohkin M, Miyata M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–44. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 31.Alberti S, Schuster G, Parini P, et al. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRbeta-deficient mice. J Clin Invest. 2001;107:565–73. doi: 10.1172/JCI9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crouse JR., 3rd Hypertriglyceridemia: a contraindication to the use of bile acid binding resins. Am J Med. 1987;83:243–48. doi: 10.1016/0002-9343(87)90692-9. [DOI] [PubMed] [Google Scholar]
- 33.Bateson MC, Maclean D, Evans JR, et al. Chenodeoxycholic acid therapy for hypertriglyceridaemia in men. Br J Clin Pharmacol. 1978;5:249–54. doi: 10.1111/j.1365-2125.1978.tb01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang DQ, Tazuma S, Cohen DE, et al. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: studies in the gallstone-susceptible mouse. Am J Physiol Gastrointest Liver Physiol. 2003;285 doi: 10.1152/ajpgi.00156.2003. : G 494–502 PMID: 12748061. [DOI] [PubMed] [Google Scholar]
- 35.Kim I, Ahn SH, Inagaki T, et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–72. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Lambert G, Amar MJ, Guo G, et al. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem. 2003;278:2563–70. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 37.Edwards PA, Kast HR, Anisfeld AM. BAREing it all: the adoption of LXR and FXR and their roles in lipid homeostasis. J Lipid Res. 2002;43 : 2–12 PMID: 11792716. [PubMed] [Google Scholar]
- 38.Repa JJ, Berge KE, Pomajzl C, et al. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. Biol Chem. 2002;277:18793–800. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- 39.Horton JD, Shimomura I, Brown MS, et al. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101:2331–9. doi: 10.1172/JCI2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niesor EJ, Flach J, Lopes-Antoni I, et al. The nuclear receptors FXR and LXRalpha: potential targets for the development of drugs affecting lipid metabolism and neoplastic diseases. Curr Pharm Des. 2001;7:231–59. doi: 10.2174/1381612013398185. [DOI] [PubMed] [Google Scholar]
- 41.Ong TP, Heidor R, De Conti A, et al. Farnesol and geraniol chemopreventive activities during the initial phases of hepatocarcinogenesis involve similar actions on cell proliferation and DNA damage, but distinct actions on apoptosis, plasma cholesterol and HMGCoA reductase. Carcinogenesis. 2006;27:1194–203. doi: 10.1093/carcin/bgi291. [DOI] [PubMed] [Google Scholar]
- 42.Hubbert ML, Zhang Y, Lee FY, et al. Regulation of hepatic Insig-2 by the farnesoid X receptor. Mol Endocrinol. 2007;21:1359–69. doi: 10.1210/me.2007-0089. [DOI] [PubMed] [Google Scholar]
- 43.Nakahara M, Fujii H, Maloney PR, et al. Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J Biol Chem. 2002;277:37229–34. doi: 10.1074/jbc.M206749200. [DOI] [PubMed] [Google Scholar]
- 44.Langhi C, Le May C, Kourimate S, et al. Activation of the farnesoid X receptor represses PCSK9 expression in human hepatocytes. FEBS. 2008;582:949–55. doi: 10.1016/j.febslet.2008.02.038. [DOI] [PubMed] [Google Scholar]
- 45.McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem. 2007;282:20799–803. doi: 10.1074/jbc.C700095200. [DOI] [PubMed] [Google Scholar]
- 46.Zhang DW, Lagace TA, Garuti R, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602–12. doi: 10.1074/jbc.M702027200. [DOI] [PubMed] [Google Scholar]
- 47.Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–7. doi: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 49.Cohen JC, Boerwinkle E, TH Mosley, Jr, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. New Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 50.Claudel T, Sturm E, Duez H, et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109:961–71. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mencarelli A, Renga B, Distrutti E, et al. Antiatherosclerotic effect of farnesoid X receptor. Am J Physiol Heart Circ Physiol. 2009;296:H272–81. doi: 10.1152/ajpheart.01075.2008. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Lee FY, Barrera G, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA. 2006;103:1006–11. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cariou B, Van Harmelen K, Duran-Sandoval D, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281:11039–49. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 54.Miller NE, Nestel PJ. Triglyceride-lowering effect of chenodeoxycholic acid in patients with endogenous hypertriglyceridaemia. Lancet. 1974;2:929–31. doi: 10.1016/s0140-6736(74)91134-9. [DOI] [PubMed] [Google Scholar]
- 55.Claudel T, Staels B, Kuipers F. The farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol. 2005;25:2020–30. doi: 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- 56.Claudel T, Inoue Y, Barbier O, et al. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125:544–55. doi: 10.1016/s0016-5085(03)00896-5. [DOI] [PubMed] [Google Scholar]
- 57.Fruchart-Najib J, Bauge E, Niculescu LS, et al. Mechanism of triglyceride lowering in mice expressing human apolipoprotein A5. Biochem Biophys Res Commun. 2004;319:397–404. doi: 10.1016/j.bbrc.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 58.Kast HR, Nguyen CM, Sinal CJ, et al. Farnesoid x-activated receptor induces apolipoprotein C-II transcription: a molecular mechanism linking plasma triglyceride levels to bile acids. Mol Endocrinol. 2001;15:1720–28. doi: 10.1210/mend.15.10.0712. [DOI] [PubMed] [Google Scholar]
- 59.Kardassis D, Roussou A, Papakosta P, et al. Synergism between nuclear receptors bound to specific hormone response elements of the hepatic control region-1 and the proximal apolipoprotein C-II promoter mediate apolipoprotein C-II gene regulation by bile acids and retinoids. Biochem J. 2003;372:291–304. doi: 10.1042/BJ20021532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sirvent A, Claudel T, Martin G, et al. The farnesoid X receptor induces very low density lipoprotein receptor gene expression. FEBS Lett. 2004;566:173–7. doi: 10.1016/j.febslet.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 61.Anisfeld AM, Kast-Woelbern HR, Meyer ME, et al. Syndecan-1 expression is regulated in an isoform-specific manner by the farnesoid-X receptor. J Biol Chem. 2003;278:20420–8. doi: 10.1074/jbc.M302505200. [DOI] [PubMed] [Google Scholar]
- 62.Hirokane H, Nakahara M, Tachibana S, et al. Bile acid reduces the secretion of very low density lipoprotein by repressing microsomal triglyceride transfer protein gene expression mediated by hepatocyte nuclear factor-4. J Biol Chem. 2004;279:45685–92. doi: 10.1074/jbc.M404255200. [DOI] [PubMed] [Google Scholar]
- 63.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–18. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hartman HB, Gardell SJ, Petucci CJ, et al. Activation of farnesoid X receptor prevents atherosclerotic lesion formation in LDLR-/- and apoE-/- mice. J Lipid Res. 2009;50:1090–100. doi: 10.1194/jlr.M800619-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pineda Torra I, Claudel T, Duval C, et al. Bile acids induce the expression of the human peroxisome proliferator- activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17:259–72. doi: 10.1210/me.2002-0120. [DOI] [PubMed] [Google Scholar]
- 66.Staels B, Dallongeville J, Auwerx J, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98 doi: 10.1161/01.cir.98.19.2088. : 2088–93 PMID: 9808609. [DOI] [PubMed] [Google Scholar]
- 67.Duran-Sandoval D, Mautino G, Martin G, et al. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53:890–8. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Lee FY, Barrera G, Lee H, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci USA. 2006;103:1006–11. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma K, Saha PK, Chan L, et al. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–9. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamagata K, Daitoku H, Shimamoto Y, et al. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem. 2004;279:23158–65. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 71.Cariou B, Van Harmelen K, Duran-Sandoval D, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem. 2006;281:11039–49. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 72.Rizzo G, Disante M, Mencarelli A, et al. The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo. Mol Pharmacol. 2006;70:1164–73. doi: 10.1124/mol.106.023820. [DOI] [PubMed] [Google Scholar]
- 73.He F, Li J, Mu Y, et al. Downregulation of endothelin-1 by farnesoid X receptor in vascular endothelial cells. Circ Res. 2006;98:192–9. doi: 10.1161/01.RES.0000200400.55539.85. [DOI] [PubMed] [Google Scholar]
- 74.Li J, Wilson A, Kuruba R, et al. FXR-mediated regulation of eNOS expression in vascular endothelial cells. Cardiovasc Res. 2008;77:169–77. doi: 10.1093/cvr/cvm016. [DOI] [PubMed] [Google Scholar]
- 75.Qin P, Tang X, Elloso MM, et al. Bile acids induce adhesion molecule expression in endothelial cells through activation of reactive oxygen species, NF-kappaB, and p38. Am J Physiol Heart Circ Physiol. 2006;291:H741–7. doi: 10.1152/ajpheart.01182.2005. [DOI] [PubMed] [Google Scholar]
- 76.Rask-Madsen C, King GL. Mechanisms of Disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- 77.Biddinger SB, Hernandez-Ono A, Rask-Madsen C, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–34. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu T, Chouinard M, Cox AL, et al. Farnesoid X receptor agonist reduces serum asymmetric dimethylarginine levels through hepatic dimethylarginine dimethylaminohydrolase-1 gene regulation. J Biol Chem. 2006;281:39831–8. doi: 10.1074/jbc.M606779200. [DOI] [PubMed] [Google Scholar]
- 79.Mallamaci F, Tripepi G, Maas R, et al. Analysis of the relationship between norepinephrine and asymmetric dimethyl arginine levels among patients with end-stage renal disease. J Am Soc Nephrol. 2004;15:435–41. doi: 10.1097/01.asn.0000106717.58091.f6. [DOI] [PubMed] [Google Scholar]
- 80.Ravani P, Tripepi G, Malberti F, et al. Asymmetrical dimethylarginine predicts progression to dialysis and death in patients with chronic kidney disease: a competing risks modeling approach. J Am Soc Nephrol. 2005;16:2449–55. doi: 10.1681/ASN.2005010076. [DOI] [PubMed] [Google Scholar]
- 81.Stählinger MC, Abbasi F, Chu JW, et al. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. J Am Med Assoc. 2002;287:1420–6. doi: 10.1001/jama.287.11.1420. [DOI] [PubMed] [Google Scholar]
- 82.Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler Suppl. 2003;4:33–40. doi: 10.1016/s1567-5688(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 83.Zoccali C, Kielstein JT. Asymmetric dimethylarginine: a new player in the pathogenesis of renal disease? Curr Opin Nephrol Hypertens. 2006;15:314–20. doi: 10.1097/01.mnh.0000222701.22583.e8. [DOI] [PubMed] [Google Scholar]
- 84.Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–90. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Y, Zhao X, Jin H, et al. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:173–9. doi: 10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 86.Bishop-Bailey D, Walsh DT, Warner TD. Expression and activation of the farnesoid X receptor in the vasculature. Proc Natl Acad Sci USA. 2004;101:3668–73. doi: 10.1073/pnas.0400046101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li YT, Swales KE, Thomas GJ, et al. Farnesoid x receptor ligands inhibit vascular smooth muscle cell inflammation and migration. Arterioscler Thromb Vasc Biol. 2007;27:2606–11. doi: 10.1161/ATVBAHA.107.152694. [DOI] [PubMed] [Google Scholar]
- 88.Zhang Q, He F, Kuruba R, et al. FXR-mediated regulation of angiotensin type 2 receptor expression in vascular smooth muscle cells. Cardiovasc Res. 2008;77:560–9. doi: 10.1093/cvr/cvm068. [DOI] [PubMed] [Google Scholar]
- 89.Nakajima M, Morishita R, Zhang L, et al. The angiotensin II type 2 receptor antagonizes the growth effects of the AT1 receptor: gain of function study using in vivo gene transfer. Proc Natl Acad Sci USA. 1995;92:10663–7. doi: 10.1073/pnas.92.23.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Levy BI. Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system. Circulation. 2004;109:8–13. doi: 10.1161/01.CIR.0000096609.73772.C5. [DOI] [PubMed] [Google Scholar]
- 91.Savoia C, Touyz RM, Volpe M, et al. Angiotensin type 2 receptor in resistance arteries of type 2 diabetic hypertensive patients. Hypertension. 2007;49:341–6. doi: 10.1161/01.HYP.0000253968.95136.b8. [DOI] [PubMed] [Google Scholar]
- 92.Molavi B, Chen J, Mehta JL. Cardioprotective effects of rosiglitazone are associated with selective overexpression of type 2 angiotensin receptors and inhibition of p42/44 MAPK. Am J Physiol Heart Circ Physiol. 2006;291:H687–93. doi: 10.1152/ajpheart.00926.2005. [DOI] [PubMed] [Google Scholar]
- 93.Kida T, Murata T, Hori M, et al. Chronic stimulation of farnesoid X receptor impairs nitric oxide sensitivity of vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2009;296:H195–201. doi: 10.1152/ajpheart.00679.2008. [DOI] [PubMed] [Google Scholar]
- 94.He F, Zhang Q, Kuruba R, et al. Upregulation of decorin by FXR in vascular smooth muscle cells. Biochem Biophys Res Commun. 2008;372:746–51. doi: 10.1016/j.bbrc.2008.05.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McEwan PA, Scott PG, Bishop PN, et al. Structural correlations in the family of small leucine-rich repeat proteins and proteoglycans. J Struct Biol. 2006;155:294–305. doi: 10.1016/j.jsb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 96.Xu G, Guimond MJ, Chakraborty C, et al. Control of proliferation, migration, and invasiveness of human extravillous trophoblast by decorin, a decidual product. Biol Reprod. 2002;67:681–9. doi: 10.1095/biolreprod67.2.681. [DOI] [PubMed] [Google Scholar]
- 97.Kolb M, Margetts PJ, Sime PJ, Gauldie J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1327–34. doi: 10.1152/ajplung.2001.280.6.L1327. [DOI] [PubMed] [Google Scholar]
- 98.Kolb M, Margetts PJ, Galt T, et al. Transient transgene expression of decorin in the lung reduces the fibrotic response to bleomycin. Am J Respir Crit Care Med. 2001;163:770–7. doi: 10.1164/ajrccm.163.3.2006084. [DOI] [PubMed] [Google Scholar]
- 99.Fischer JW, Kinsella MG, Clowes MM, et al. Local expression of bovine decorin by cell-mediated gene transfer reduces neointimal formation after balloon injury in rats. Circ Res. 2000;86:676–83. doi: 10.1161/01.res.86.6.676. [DOI] [PubMed] [Google Scholar]
- 100.Nili N, Cheema AN, Giordano FJ, et al. Decorin inhibition of PDGF-stimulated vascular smooth muscle cell function: potential mechanism for inhibition of intimal hyperplasia after balloon angioplasty. Am J Pathol. 2003;163:869–78. doi: 10.1016/S0002-9440(10)63447-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chodouri RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2005;2:309–31. doi: 10.1038/ncpcardio0195. [DOI] [PubMed] [Google Scholar]
- 102.Calmus Y, Guechot J, Podevin P, et al. Differential effects of chenodeoxycholic and ursodeoxycholic acids on interleukin 1, interleukin 6 and tumor necrosis factor-alpha production by monocytes. Hepatology. 1992;16:719–23. doi: 10.1002/hep.1840160317. [DOI] [PubMed] [Google Scholar]
- 103.Guo GL, Santamarina-Fojo S, Akiyama TE, et al. Effects of FXR in foam-cell formation and atherosclerosis development. Biochim Biophys Acta. 2006;1761:1401–9. doi: 10.1016/j.bbalip.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vavassori P, Mencarelli A, Renga B, et al. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. 2009;183:6251–61. doi: 10.4049/jimmunol.0803978. [DOI] [PubMed] [Google Scholar]
- 105.Yang F, Huang X, Yi T, et al. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–7. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- 106.Hanniman EA, Lambert G, McCarthy TC, et al. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. J Lipid Res. 2005;46:2595–604. doi: 10.1194/jlr.M500390-JLR200. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Y, Wang X, Vales C, et al. FXR deficiency causes reduced atherosclerosis in Ldlr-/- mice. Arterioscler Thromb Vasc Biol. 2006;26:2316–21. doi: 10.1161/01.ATV.0000235697.35431.05. [DOI] [PubMed] [Google Scholar]
- 108.Pullinger CR, Eng C, Salen G, et al. Human cholesterol 7α-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109–17. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spady D, Cuthbert JA, Willard MN, et al. Overexpression of cholesterol 7α-hydroxylase (CYP7A) in mice lacking the low density lipoprotein (LDL) receptor gene. J Biol Chem. 1998;273:126–32. doi: 10.1074/jbc.273.1.126. [DOI] [PubMed] [Google Scholar]
- 110.Miyake JH, Duong-Polk XT, Taylor JM, et al. Transgenic expression of cholesterol-7-α-hydroxylase prevents atherosclerosis in C57BL/6J mice. Arterioscler Thromb Vasc Biol. 2002;22:121–6. doi: 10.1161/hq0102.102588. [DOI] [PubMed] [Google Scholar]
- 111.Ratliff EP, Gutierrez A, Davis RA. Transgenic expression of CYP7A1 in LDL receptor-deficient mice blocks diet-induced hypercholesterolemia. J Lipid Res. 2006;47:1513–20. doi: 10.1194/jlr.M600120-JLR200. [DOI] [PubMed] [Google Scholar]
- 112.Sanyal S, Bavner A, Haroniti A, et al. Involvement of rerepressor complex subunit GPS2 in transcriptional pathways governing human bile acid biosynthesis. Proc Natl Acad Sci USA. 2007;40:15665–70. doi: 10.1073/pnas.0706736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang S, Lai K, Moy FJ, et al. The nuclear hormone receptor farnesoid X receptor (FXR) is activated by androsterone. Endocrinol. 2006;147:4025–33. doi: 10.1210/en.2005-1485. [DOI] [PubMed] [Google Scholar]
- 114.Xing Y, Saner-Amigh K, Nakamura Y, et al. The farnesoid X receptor regulates transcription of 3β-hydrosyxteroid dehydrogenase type 2 in human adrenal cells. Mol Cell Endocrinol. 2009;299:153–62. doi: 10.1016/j.mce.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]