

Abstract
Cytolethal distending toxins (CDTs) are proteins produced and secreted by facultative pathogenic strains of Gram-negative bacteria with potentially genotoxic effects. Mammalian cells exposed to CDTs undergo cell type-dependent cell-cycle arrest or apoptosis; however, the cell fate responses to such intoxication are mechanistically incompletely understood. Here we show that both normal and cancer cells (BJ, IMR-90 and WI-38 fibroblasts, HeLa and U2-OS cell lines) that survive the acute phase of intoxication by Haemophilus ducreyi CDT possess the hallmarks of cellular senescence. This characteristic phenotype included persistently activated DNA damage signalling (detected as 53BP1/γH2AX+ foci), enhanced senescence-associated β-galactosidase activity, expansion of promyelocytic leukaemia nuclear compartments and induced expression of several cytokines (especially interleukins IL-6, IL-8 and IL-24), overall features shared by cells undergoing replicative or premature cellular senescence. We conclude that analogous to oncogenic, oxidative and replicative stresses, bacterial intoxication represents another pathophysiological stimulus that induces premature senescence, an intrinsic cellular response that may mechanistically underlie the ‘distended’ morphology evoked by CDTs. Finally, the activation of the two anticancer barriers, apoptosis and cellular senescence, together with evidence of chromosomal aberrations (micronucleation) reported here, support the emerging genotoxic and potentially oncogenic effects of this group of bacterial toxins, and warrant further investigation of their role(s) in human disease.
Keywords: cellular senescence, DNA damage response, bacterial toxins, cytokines, genetic instability
Introduction
Haemophilus ducreyi, a Gram-negative coccobacillus causing the sexually transmitted disease chancroid, produces a potent toxin (HdCDT) from the family of cytolethal distending toxins (CDTs) [1]. CDTs are a subgroup of bacterial proteins secreted by facultative pathogenic strains of gram-negative bacteria, such as Escherichia coli, Campylobacter jejuni, Helicobacter hepaticus, Shigella dysenteriae and Actinobacillus actinomycetemcomitans[2–7]. The functional toxin is a tripartite complex in which CdtB is the active subunit possessing an intrinsic DNase I-like activity, while CdtA and CdtC subunits are required for the delivery of CdtB into the host cell [8–10]. The cells exposed to CDTs have been shown to arrest in the G1 and/or G2 phases of the cell cycle, or undergo apoptosis, depending on the cell type [11–14]. The CDT-induced cell-cycle arrest resembles the checkpoint response to ionizing radiation, characterized by activation of the ataxia telangiectasia mutated (ATM) kinase and ATM-dependent induction of the tumour suppressor p53, phoshorylation of histone H2AX and re-localization of the DNA repair proteins such as Mre11 and Rad50 to the sites of DNA double-strand breaks (DSBs) [10, 13, 15]. Although there is currently no direct proof that CDTs are involved in pathogenicity of their hosts (for a review see [16]), all the above mentioned data obtained with cell culture models should be considered as a warning sign that this type of toxins might be involved in serious pathologies including cancer development.
We and others have recently found that in early stages of human cancer development, the DNA damage response (DDR) machinery, including the ATM-Chk2-p53 pathway, is constitutively activated and serves as an inducible barrier against activated oncogenes and tumour progression [17–19]. The biological impact of the activated DDR barrier in human premalignant lesions and cellular models varies according to cell types and specific oncogenic insult, however the predominant cell fates induced by such oncogenic stress are either cell death (often apoptosis) or a long-term cell-cycle arrest state known as cellular senescence [20, 21]. Among the hallmarks of the senescence phenotype are permanent withdrawal from proliferative state yet with preserved metabolic activity, large flat cellular morphology, altered gene expression profile, activated DNA damage signalling, resistance to apoptosis and increase of ‘markers’ such as acidic β-galactosidase activity, and expansion of the promyelocytic leukaemia (PML) cellular bodies [22]. The persistent DNA damage checkpoint signalling, including the ATM-p53-p21 axis, appears to be critical for both the establishment and maintenance of the senescence phenotype, and it is shared by all types of cellular senescence reported to date [22]. Another apparently universal feature of senescence is the enhanced expression and secretion of pro-inflammatory cytokines, recently causally implicated in both replicative and oncogene-induced senescence [23–25].
From a broad biological perspective, cellular senescence plays important roles in fundamental processes such as stem cell and tissue homeostasis and ageing, and it has been implicated in pathogenesis of a range of devastating disorders including cancer [22]. Notably, whereas enhanced senescence may promote the organismal ageing and some ageing-related pathologies such as atherosclerosis or arthritis, it seems to play a dual role in tumorigenesis, by possibly creating a tumour-promoting microenvironment fuelled by the secreted cytokines [20, 21], while blocking the proliferation of nascent cancer cells themselves, the latter role consistent with the anti-oncogenic barrier provided by the activated DDR mentioned above [23–25]. The sources of elevated DNA damage in tumour cells encompass oncogene-induced replication stress, telomere attrition and oxidative stress that all evoke endogenous DNA lesions including DSBs, thereby also contributing to enhanced genetic instability [19]. Chronic bacterial infections have also been implicated in tumorigenesis, as exemplified by the established role of Helicobacter pylori in gastric cancer [26, 27]; however, whether bacterial intoxication is related to cellular senescence or genetic instability is unknown.
Based on the concept of DDR activation in response to oncogenic stress [26, 27], and intrigued by the emerging evidence of acute DNA damage evoked by the bacterial CDTs [10, 13, 15], we argued that such biological parallel between these two pathophysiological scenarios might extend beyond the early DNA damage signalling and induction of apoptosis. To test this working hypothesis, we designed the present study to examine the longer-term consequences of CDT exposure on multiple human cell types, both normal and transformed, with particular emphasis on the duration of the DDR signalling, potential evidence for features of genetic instability, production of pro-inflammatory cytokines and possible establishment of premature senescence as a cellular fate for cells that survive the acute phase of bacterial intoxication. As documented below by the results of these analyses, the data we obtained appear to support our hypothesis that bacterial intoxication may represent a genome-destabilizing and cellular senescence-inducing process.
Materials and methods
Toxin preparation and treatment
Preparation of recombinant H. ducreyi CdtA, CdtB and CdtC subunits and reconstitution of the active holotoxin (HdCDT) was previously described [28, 29]. The 100% activity of toxin preparation was estimated as the lowest cytopathic dose that caused complete irreversible G2/M block of ‘reference’ HeLa cell strain 24 hrs after intoxication. We used ‘balanced’ toxin dilutions to get optimal ratio of surviving cells with distended morphology to dead cells; i.e. 30% activity was used for HeLa and U2-OS cell lines, and 70% activity for normal WI-38, IMR-90 and BJ fibroblasts, which were less sensitive (see [30]). The medium was routinely changed 24 hrs after a single HdCDT-treatment.
Cell culture
Human IMR-90, BJ, WI-38, HeLa and U2-OS cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal calf serum (Gibco, Invitrogen, Carlsbad, CA, USA) and penicillin/streptomycin (Sigma, Saint Louis, MO, USA) in a humidified atmosphere of 5% CO2 at 37°C. The U2-OS-derived cell line with tetracycline-repressible expression of the dominant-negative p53 mutant (p53DD) [31] was grown in the same medium, further supplemented with puromycin, G418 and tetracycline (Sigma).
Immunofluorescence microscopy
For immunofluorescence microscopy, control or HdCDT-treated cells cultured on the cover slips were fixed in 4% paraformaldehyde at RT for 15 min., then permeabilized for 10 min. with 0.2% Triton X−100, washed and blocked for 30 min. in 10% foetal calf serum. Incubation with primary antibodies was for 60 min. at RT: rabbit anti−53BP1 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc-22760), mouse anti-γH2AX (1:500, Millipore, Billerica, MD, USA, #05–636), rabbit anti-P-Chk2 (Thr68) (1:300, Cell Signalling Technology, Danvers, MD, USA, #2661), mouse anti-PML (1:300, sc-966). Following a wash, the cover slips were incubated with goat anti-rabbit or anti-mouse Alexa Fluor 488 or Alexa Fluor 568 (1:1000, Molecular Probes, Invitrogen) secondary antibodies at RT for 60 min. Finally, nuclei were stained with DAPI (Sigma) and cover slips were mounted using anti−fading mounting reagent (Vectashield, Vector Laboratories, Burlingame, CA, USA). Images were captured by fluorescence microscope (Leica DMRXA, Wetzlar, Germany) equipped with digital camera or Olympus Soft Imaging Solutions GMBH using OSIS Scan® software (Münster, Germany).
BrdU proliferation assay
For BrdU incorporation, cells were incubated with 10 μM BrdU (Sigma) before fixation with 4% paraformaldehyde. After DNA denaturation in 2 M HCl for 30 min., cells were incubated with mouse anti-BrdU antibody (1:200, Amersham, GE Healthcare, UK, clone BU-1) and stained as described above.
Cell lysates and Western blotting
The cells were washed twice with PBS and lysed in 100 mM Tris-HCl buffer (pH 8.0) containing 140 mM NaCl, 1% Triton X-100, 1 mM ethylenediaminetetraacetic acid, 0.5 mM EGTA, 0.1% sodium deoxycholate, 0.1% SDS, protease and phosphatase inhibitors (cocktail from Roche Diagnostics, Meyland, France) followed by sonication and centrifugation at 10,000 ×g for 5 min. at 4°C. Supernatants were subjected to SDS-PAGE and proteins transferred onto nitrocellulose membranes. Membranes were blocked with 5% milk or bovine serum albumin in PBS containing 0.1% Tween-20 and probed with the following antibodies: rabbit anti-p53 (1:1000, a gift from B. Vojtesek, DEP, Brno, Czech Republic), rabbit anti-P-p53 (Ser15) (1:1000, Cell Signalling Technology, #9138), mouse anti-p21 (1:1000, DCS-60, [32]), rabbit anti-p16 (1:1000, sc-759), mouse anti-Rb (1:1000, Pharmingen, BD Biosciences, San Chose, CA, USA, #554136), mouse anti-GAPDH (1:1000, Gene Tex Inc., Irvine, CA, USA, #30666), GAR-HRP and GAM-HRP (1:10000, Bio-Rad Laboratories, Hercules, CA, USA). Antigens were then visualized by chemiluminescence using an X-ray film or a FUJIFILM LAS-3000 (Tokyo, Japan) and Aida Image Analyzer software (Straubenhardt, Germany).
Estimation of cellular senescence in vitro
Senescence associated-β-galactosidase assay (SA-β-gal) was performed according to [33] with modifications described in [34]. Images were captured by fluorescence microscope (Leica DMRXA) equipped with digital camera and contrast of images was adjusted in Adobe Photoshop software.
Quantitative PCR
Total DNA-free RNA was extracted using TRI Reagent (Sigma) and purified with TURBO DNA-free kit (Ambion, Applied Biosystems, Carlsbad, CA, USA). Reverse transcription was generated with High Capacity RNA-to-cDNA Kit (Applied Biosystems). PCR reactions were performed with Power Sybr Green Master Mix (Applied Biosystems) on Applied Biosystems 7300 Real Time PCR System with following primers: for interleukin (IL)-6 as forward 5′-GAT GAG TAC AAA AGT CCT GAT CCA-3′ and 5′-CTG CAG CCA CTG GTT CTG T-3′ as reverse; for IL-8 as forward 5′-GCC AGG ATC CAC AAG TCC T-3′ and 5′-TGG TGG CTA ATA CTT TTT CCA CT-3′ as reverse; for IL-24 as forward 5′-CGG AGA GCA TTC AAA CAG TTG-3′ and 5′-TGG TCT AGA CAT TCA GAG CTT GTA G-3′ as reverse; and for GAPDH as forward 5′-GTC GGA GTC AAC GGA TTT GG-3′ and 5′-AAA AGC AGC CCT GGT GAC C-3′ as reverse. GAPDH was used for normalization.
Human common cytokines – RT2 profiler PCR array
The Human Common Cytokines Arrays (SuperArray, SABiosciences, Frederick, MD, USA) were used following the manufacturer’s instructions. All PCR reactions were performed on Applied Biosystems 7300 Real Time PCR System. GAPDH was used for normalization.
Results
HdCDT intoxication causes persistent DNA damage and micronucleation
Given that CDTs can cause DNA breakage and ATM kinase mediated response in short-term experiments with mammalian cells monitored for up 48 hrs after intoxication [13, 35], we first decided to extend such previous analyses in three ways, by: (i) Examination of the kinetics of DNA damage signalling in time-course experiments over extended periods of time; (ii) Comparative analysis of the DDR parameters in G1 versus G2/M cells within the intoxicated cell population and (iii) Searching for potential features of CDT-induced genomic instability. To achieve these aims, we intoxicated various cultured human cell types with CDT prepared from H. ducreyi (HdCDT), and followed the presence of DNA DSBs detected as nuclear foci positive the phosphorylated histone variant H2AX (γH2AX) and 53BP1 [36] over time. In all cell types tested (HeLa, U2-OS, BJ, IMR-90 and WI-38), we found an increase in the number of 53BP1/γH2AX foci shortly after intoxication (4 hrs), consistent with previous studies [10, 29]. Notably, our prolonged experiments showed that the toxin-induced foci persisted for the entire duration of the 6–8-day experiments after HdCDT intoxication in 30–40% of the cells (Fig. 1A), although the number of foci gradually diminished, with concomitant increase of their size and intensity.
Fig 1.
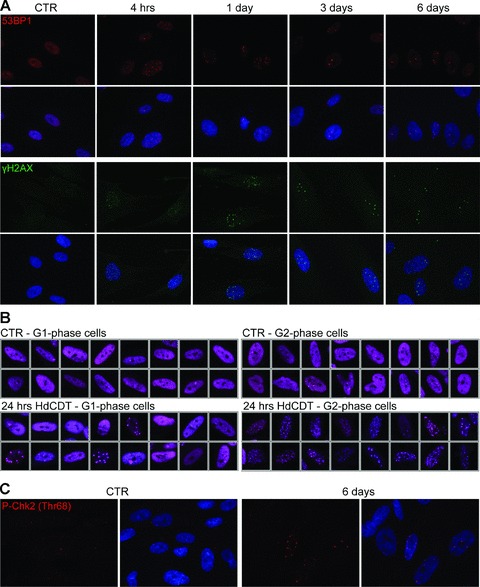
Persistent DNA damage foci in HdCDT-intoxicated cells. (A) BJ fibroblasts were fixed and stained with anti-53BP1 (red) or anti-γH2AX (green) antibody and DAPI (blue) at different times after HdCDT intoxication. (B) Control IMR-90 cells and IMR-90 cells exposed to HdCDT for 24 hrs were fixed and stained with anti-53BP1 (purple) and DAPI. Depending on an amount of incorporated DAPI dye into the DNA, the cells were separated into the G1- and G2-phase population. (C) Six days after the intoxication with HdCDT, IMR-90 fibroblasts were fixed and stained with anti-P-Chk2 (Thr68) (red) and DAPI (blue). Untreated cells were used as negative control (CTR).
Relative to cell cycle position, we found a marked difference in number and size of the 53BP1 foci detected in G1- versus G2-phase cells, particularly the first day after intoxication (Fig. 1B). The G1 cells contained a modest number of larger 53BP1 foci, in contrast to G2 cells with typically multiple small foci. Apart from somewhat elevated spontaneous foci formation in HeLa cells, analogous foci patterns with differences between G1- and G2-cells were seen in all intoxicated cell types examined, including IMR-90 fibroblasts pre-synchronized and intoxicated while in G0 or S phase (data not shown). These findings indicate that the toxin either caused DNA damage preferentially during the S and/or G2 phases, or the HdCDT-induced DNA damage remained unrecognized until the cells reached S/G2-phase of the cell cycle.
The long-term persistence of both markers of DNA damage foci was indicative of sustained DDR signalling in intoxicated cells, a conclusion that was further supported by persistent activatory phosphorylation of the DNA damage signalling kinase Chk2 (Fig. 1C), known to become acutely activated after toxin administration [13, 35].
Importantly, the clastogenic activity of HdCDT was indicated by increased numbers of fragmented nuclei with micronuclei formation in HdCDT-intoxicated fibroblasts. For example, compared to control mock-infected cell population, approximately 12-fold induction of micronuclei formation was observed at days 6–8 in intoxicated IMR-90 cells (Fig. 2A). The micronuclei scored commonly positive for markers of active DDR (Fig. 2B), indicating they originated following chromosomal breakage and missegregation of chromosomal fragments at mitosis, rather than from some defects of nuclear envelope formation.
Fig 2.
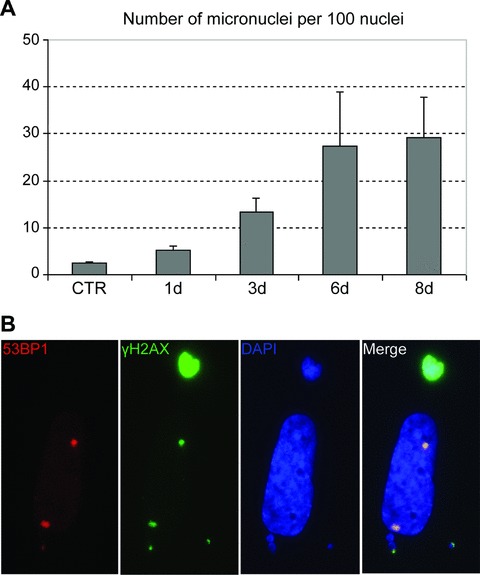
Increased fragmentation of nuclei in HdCDT-intoxicated fibroblasts. (A) Control or HdCDT-treated IMR-90 cells were fixed at different time-points upon intoxication and stained with DAPI. Numbers of micronuclei versus nuclei were calculated using the OSIS ScanR software. Approximately 1000 nuclei were calculated for each sample. Error bars represent the S.D. of three separate experiments. (B) Eight days after the HdCDT-intoxication IMR-90 fibroblasts were fixed and stained with anti-53BP1 (red) and anti-γH2AX (green) antibodies and DAPI (blue).
Apart from micronuclei formation in all cell types, we also observed formation of multinuclear cells after HdCDT intoxication, a phenomenon characteristic mainly for HeLa cell cultures, in contrast to only rare multinucleated cells in intoxicated populations of normal diploid fibroblasts. Multinucleation was also reported in HeLa cells intoxicated with Escherichia coli CDT [37].
Overall, we concluded that the rapidly induced activation of DNA damage signalling in both normal and cancerous human cells persisted for a number of days, and was often accompanied by micronuclei formation, indicating impaired reparability of the HdCTD-evoked DNA lesions and chromosomal instability.
Induction of CDK inhibitors and cell-cycle arrest after HdCTD intoxication
Consistent with a potential DDR-induced cell-cycle delay, HdCTD intoxication resulted in increased levels of p53 and its activated, Ser15-phosphorylated form P-p53(Ser15), accompanied by elevated p21waf/cip (p21), an inhibitor of cyclin-dependent kinases (CDKs) and p53 transcription target, in intoxicated U2-OS, IMR-90 and WI-38 cells. Indeed, the toxin-induced increase of p21 was dependent on p53, since inactivation of p53 by a tetracycline-regulatable dominant-negative form of p53 in U2-OS (U2-OS p53DD; [31]) abolished the p21 response (Fig. 3A).
Fig 3.
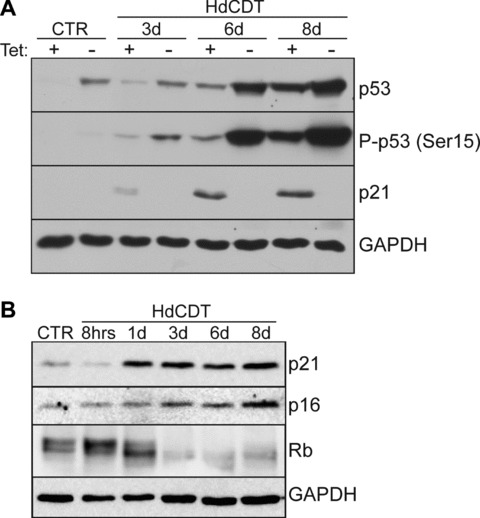
Activation of cell cycle inhibitors after HdCTD intoxication. (A) U2-OS p53DD cells were treated with HdCDT in the presence (p53DD OFF) or the absence (p53DD ON) of tetracycline (Tet off system) and lysed at different time-points after the intoxication. Cell extracts were separated by SDS-PAGE and analysed by Western blotting with anti-p53, anti-P-p53 (Ser15) and anti-p21 antibodies. GAPDH expression was used as a loading control. (B) Immunoblot analysis of cell lysates prepared from HdCDT-intoxicated IMR-90 fibroblasts after different time-points. Untreated cells were used as negative control (CTR). Blots were probed with anti-p21, anti-p16 and anti-Rb as indicated; GAPDH was used as a loading control.
Similarly to p21, the CDK inhibitor p16INK4a (p16) also increased with time after intoxication in IMR-90 and WI-38 fibroblasts (Fig. 3B, and data not shown). Simultaneously, the retinoblastoma protein (Rb) became gradually dephoshorylated by day 3 after intoxication, followed by Rb down-regulation (Fig. 3B).
To assess cell proliferation after HdCDT intoxication, we next examined BrdU incorporation by immunohistochemical staining, and DNA profiles by flow cytometry. In comparison to non-treated controls, HdCDT-treated cells examined during 8 days after intoxication showed rapid and dramatic decrease of BrdU incorporation, from days 2–3 onwards, consistent with a long-lasting cell cycle arrest. The decrease of BrdU incorporation was about 10-fold: from approximately 50% to 5%, and from 24% to 1% for U2-OS and IMR-90 cells, respectively. The flow cytometry profiles indicated that the arrested cells accumulated mainly in G2-phase (not shown), which is in agreement with previous reports [11–14]. Taken together, the surviving HdCDT-treated cells showed a prolonged activation of cell-cycle checkpoints resulting in durable cell cycle arrest.
HdCDT induced senescence-like phenotype in normal and transformed cells
The cellular response described above, together with the remarkable distension of cellular shape after bacterial intoxication were indicative of potential DNA damage-induced cellular senescence. To test this possibility, we investigated whether the other known markers attributable to cellular senescence can be found in HdCDT-treated cells. SA-β-gal, the commonly used marker of expanded lysosomal compartment in cells senescent in vitro[33, 38], was found enhanced in all cells exposed to HdCDT (Fig. 4A, and data not shown). In addition, the numerical expansion of specific sub-nuclear compartment, PML nuclear bodies, characteristic for replicative, oncogene- and drug-induced senescence [39–42] was also observed in all HdCDT-treated cell types (Fig. 4B, and data not shown).
Fig 4.
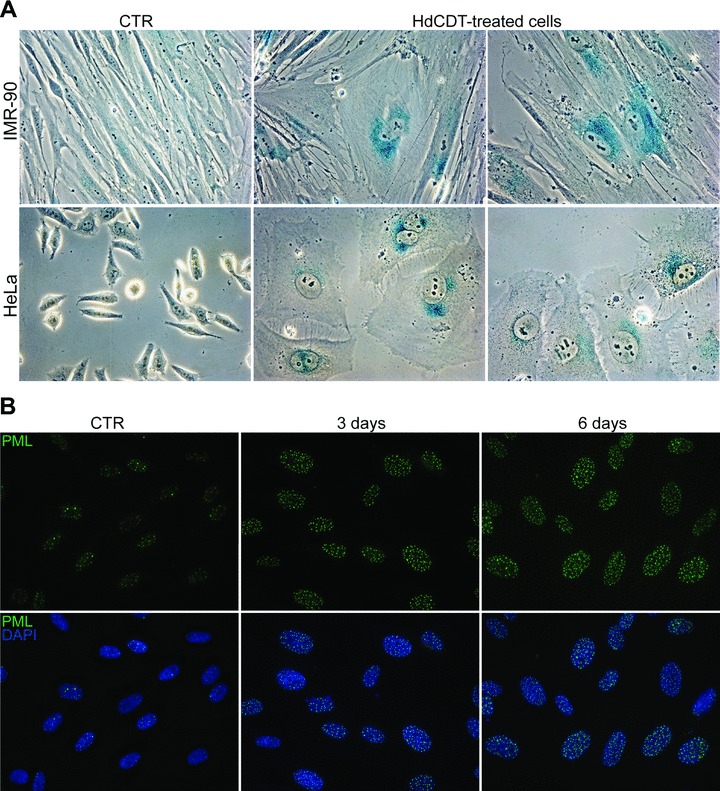
HdCDT induces SA-β-gal and increases number of PML bodies in normal and transformed cells. (A) Beta-galactosidase staining on IMR-90 and HeLa cells untreated or exposed to HdCDT for 6 to 8 days. (B) BJ fibroblasts, left untreated or exposed to HdCDT for the indicated periods of time, were fixed and stained with anti-PML (green) antibody and DAPI (blue).
Cells intoxicated with HdCDT express pro-inflammatory cytokines
The cellular senescence state in other scenarios is often accompanied by changes in secretory phenotype, referred to as senescence-associated secretome or senescence-messaging secretome [43], and including expression and secretion of several cytokine species.
Utilizing RT-PCR profiler assay ‘Common cytokines’ (see ‘Material and methods’), we compared the mRNA levels of different cytokine species in control and HdCDT intoxicated BJ fibroblasts at day 8 after exposition to HdCDT. Notably, the mRNA of several cytokine species (i.e. IL-24, TNFRSF11B, IL-1A, TNFSF4, IL-7, BMP4, INHBA, BMP2, including inflammatory IL-6 and IL-8) was significantly elevated in HdCDT-intoxicated, compared to untreated BJ cells (Table 1).
Table 1.
List of cytokines with transcripts up-regulated by HdCDT. Primary human BJ fibroblasts were exposed to HdCDT for 8 days and the expression levels of several cytokine transcripts were determined by RT2 Profiler PCR Array. The presented numbers indicate fold induction (measured in three separate arrays) over control cycling cells, arbitrarily defined as unity.
| Exp 1 | Exp 2 | Exp 3 | Average | |
|---|---|---|---|---|
| IL-24 | 685 | 19 | 10 | 238 |
| IL-6 | 267 | 100 | 290 | 219 |
| IL-8 | 320 | 47 | 118 | 162 |
| TNFRSF11B | 9 | - | 91 | 50 |
| IL-1A | 23 | 3 | 121 | 49 |
| TNFSF4 | - | 2 | 74 | 38 |
| IL-7 | 8 | - | 19 | 13 |
| BMP4 | 8 | - | 9 | 9 |
| INHBA | 11 | 2 | 5 | 6 |
| BMP2 | - | 2 | 5 | 4 |
The elevation of mRNA levels of IL-6, IL-8 and IL-24 was verified with separately designed set of primers during the time course of toxin-induced senescence in BJ and U2-OS cells (Fig. 5). The mRNA levels of all three cytokines gradually increased with time after intoxication and were elevated at day 8 ∼100 times for IL-6, ∼150 times for IL-8 and ∼170 times for IL-24 in BJ cells and ∼20, ∼400 and ∼10 times in U2-OS, respectively. The extent of IL-6 and IL-8 mRNA induction was comparable to that induced by BRAFE600 oncogene in primary human melanocytes [24].
Fig 5.
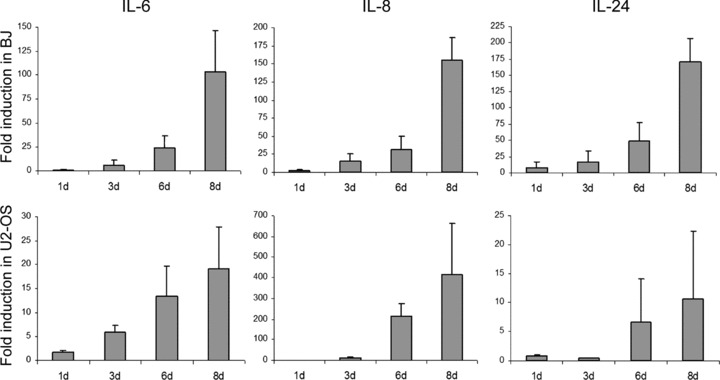
HdCDT evokes expression of pro-inflammatory cytokines IL-6, IL-8 and IL-24. BJ fibroblasts or U2-OS cells were intoxicated with HdCDT and after the indicated time-points the expression levels of IL-6, IL-8 and IL-24 transcripts were determined by RT-PCR. Levels are represented relative to those found in control cycling cells. Error bars represent the S.D. of three separate experiments.
Discussion
This study contributes to better understanding of cellular effects of toxins produced by pathogenic Gram-negative bacteria, an issue relevant to chronic inflammation, genetic stability and tumorigenesis. Our key novel finding is that human cells exposed to CDT that survive the acute intoxication phase undergo ‘premature cellular senescence’, a phenotype previously observed in response to oncogenes and diverse stresses including genotoxic chemicals [22], however so far not linked to bacterial intoxication. We interpret the HdCDT-induced cellular phenotype (observed here in multiple types of human cells – both normal and cancer derived) as senescence, based on the following criteria collectively regarded as hallmarks of senescence [20, 21, 23, 24, 33, 34, 39, 40, 42, 44]: (i) long-term cell cycle arrest; (ii) large flat cellular morphology; (iii) positivity for acidic β-galactosidase; (iv) activation of the two major tumour suppressor pathways – the p16/RB and p53/p21 cascades; (v) persistent DNA damage signalling; (vi) enhanced PML compartment and (vii) induced expression of pro-inflammatory cytokines.
Induction of cell death in a HdCTD-concentration dependent manner, and the morphological changes including distention of human cells surviving HdCDT intoxication, can be traced back to the report of Purven et al.[30]. Based on our data reported here, we would like to propose that the classical distended morphology of the intoxicated cells, that inspired the name of this class of bacterial toxins, reflects the underlying mechanism that leads to cellular senescence.
At the heart of this senescence phenotype is constitutive activation of DNA damage signalling, now commonly regarded as the mechanism underlying the long-term cell cycle checkpoint arrest shared by all known types of cellular senescence [20, 21]. DNA breakage and activation of the ATM-p53 signalling axis have already been reported for intoxicated cells resulting mostly in apoptosis [13], and attributed to the DNase-like activity of CDTs [8, 9], though genotoxic effects caused by reactive oxygen species production induced during intoxication cannot be fully excluded. The question whether these DNA lesions evoke persistent damage signalling because they are very difficult to repair or because the CDT enzyme can continuously create new lesions and hence outpace the DNA repair machinery of the host cell, remains to be elucidated.
Another major result we present is the broad spectrum of pro-inflammatory cytokines produced by the intoxicated cells, a feature recently reported for oncogene induced [24, 44, 45], and replicative senescence [23, 45], and further supported by our own results on drug-induced premature senescence (Novakova, unpublished data). Arguably the cytokines observed most commonly as part of the senescence phenotype include IL-6 and IL-8 [23, 24, 44, 45], consistent with our observation that both IL-6 and IL-8 were strongly up-regulated in several cell types after HdCDT intoxication. At least induction of IL-8 is shared by various CDTs, since IL-8 release was described in intestinal epithelial cells intoxicated with Campylobacter jejuni CDT [46, 47].
Notably, besides the up-regulation of already known senescence-associated cytokines, we identified several other cytokine species in HdCDT-treated cells (Table 1). Our finding of HdCDT-mediated up-regulation of IL-24 is particularly intriguing, since this cytokine possesses cancer cell selective pro-apoptotic, anti-angiogenic, growth and tumour suppressive properties [48, 49]. IL-24 enhances the tumour-suppressive activity of radiotherapy and chemotherapy, and it is currently under evaluation as an antitumour agent [50, 51]. Since we found IL-24 induction also in drug-induced senescent cells (Novakova, unpublished data), IL-24 transcriptional regulation may be part of the general cellular response to genotoxic stress, which mobilizes cellular ‘barrier’ mechanisms including the transmission of the alarm signal outside the injured cell. Taken together, this study further underscores the activation of cytokine network as the universal response to stress conditions associated with the development of senescent phenotype.
From a broader perspective, the defensive cellular responses to CDTs, including senescence and apoptosis, provide a biological advantage for the host organism, although the ensuing cellular loss may also undermine tissue homeostasis. However, it is unclear how can the genotoxic impact of CDT promote the bacterial life cycle. One possibility is that the senescence-associated cytokine secretion provides a niche that facilitates recruitment of infiltrating host cells including those in which bacteria might persist, thereby causing chronic inflammation. In contrast to bacteria, diverse pathogenic viruses evolved specific proteins that neutralize the ATM/ATR-mediated DDR of mammalian cells, thereby allowing viral DNA replication to occur without alarming the checkpoint and repair machinery of the host cell ([52] and references therein).
What is becoming evident is that CDTs are genotoxic and may trigger tumorigenesis. For example, a recent study of mouse liver carcinogenesis showed that the CDT produced by Helicobacter hepaticus was not required for induction of chronic hepatitis per se, but it was essential for the induction of dysplastic proliferative lesions that further develop into cancer [53]. The notion of genotoxic effects of CDTs is also supported by our present data on micronucleation (chromosomal aberrations) and activation of the senescence barrier known to be evoked by activated oncogenes and observed in a wide spectrum of early cancerous lesions [20, 21].
In conclusion, our study provides novel insights into the late cellular effects of HdCDT intoxication, which might contribute to the pathogenicity of CDT-producing bacteria. The toxin-induced activation of cellular barriers against tumorigenesis, i.e. cellular senescence and apoptosis, indicates potentially harmful effects of persistent infections with toxin-producing bacteria. Although these barriers need not be necessarily broken by the clastogenic effects of the toxin itself, chronic pro-inflammatory environment built up by toxin-provoked senescent cells can deregulate tissue homeostasis to pathologic conditions, including cancer. The association between bacterial infections and cancer is poorly understood. The only bacterium classified as human carcinogen is Helicobacter pylori, but a possible oncogenic impact has been suggested for other bacteria, such as the gram-negative CDT producing Salmonella typhi[54]. We believe that our study contributes to the emerging debate on such potential genotoxic and tumorigenic effects of chronic bacterial infections, an issue of broad medical and social concern.
Acknowledgments
The work was supported by Grant Agency of the Academy of Sciences of the Czech Republic (Project IAA500390501), Grant Agency of the Czech Republic (Projects 204/08/1418 and 301/08/0353), the Academy of Sciences of the Czech Republic (project AV0Z50520514), the European Community (projects TRIREME and INFLA-CARE), the Swedish Research Council and the Swedish Cancer Society.
References
- 1.Cope LD, Lumbley S, Latimer JL, et al. A diffusible cytotoxin of Haemophilus ducreyi. Proc Natl Acad Sci USA. 1997;94:4056–61. doi: 10.1073/pnas.94.8.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson WM, Lior H. A new heat-labile cytolethal distending toxin (CLDT) produced by Escherichia coli isolates from clinical material. Microb Pathog. 1988;4:103–13. doi: 10.1016/0882-4010(88)90052-6. [DOI] [PubMed] [Google Scholar]
- 3.Okuda J, Fukumoto M, Takeda Y, et al. Examination of diarrheagenicity of cytolethal distending toxin: suckling mouse response to the products of the cdtABC genes of Shigella dysenteriae. Infect Immun. 1997;65:428–33. doi: 10.1128/iai.65.2.428-433.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pickett CL, Pesci EC, Cottle DL, et al. Prevalence of cytolethal distending toxin production in Campylobacter jejuni and relatedness of Campylobacter sp cdtB gene. Infect Immun. 1996;64:2070–8. doi: 10.1128/iai.64.6.2070-2078.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott DA, Kaper JB. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect Immun. 1994;62:244–51. doi: 10.1128/iai.62.1.244-251.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young VB, Knox KA, Schauer DB. Cytolethal distending toxin sequence and activity in the enterohepatic pathogen Helicobacter hepaticus. Infect Immun. 2000;68:184–91. doi: 10.1128/iai.68.1.184-191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sugai M, Kawamoto T, Peres SY, et al. The cell cycle-specific growth-inhibitory factor produced by Actinobacillus actinomycetemcomitans is a cytolethal distending toxin. Infect Immun. 1998;66:5008–19. doi: 10.1128/iai.66.10.5008-5019.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lara-Tejero M, Galan JE. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 2000;290:354–7. doi: 10.1126/science.290.5490.354. [DOI] [PubMed] [Google Scholar]
- 9.Elwell CA, Dreyfus LA. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol Microbiol. 2000;37:952–63. doi: 10.1046/j.1365-2958.2000.02070.x. [DOI] [PubMed] [Google Scholar]
- 10.Li LQ, Sharipo A, Chaves-Olarte E, et al. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cellular Microbiology. 2002;4:87–99. doi: 10.1046/j.1462-5822.2002.00174.x. [DOI] [PubMed] [Google Scholar]
- 11.Comayras C, Tasca C, Peres SY, et al. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect Immun. 1997;65:5088–95. doi: 10.1128/iai.65.12.5088-5095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sert V, Cans C, Tasca C, et al. The bacterial cytolethal distending toxin (CDT) triggers a G2 cell cycle checkpoint in mammalian cells without preliminary induction of DNA strand breaks. Oncogene. 1999;18:6296–304. doi: 10.1038/sj.onc.1203007. [DOI] [PubMed] [Google Scholar]
- 13.Cortes-Bratti X, Karlsson C, Lagergard T, et al. The haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J Biol Chem. 2001;276:5296–302. doi: 10.1074/jbc.M008527200. [DOI] [PubMed] [Google Scholar]
- 14.Shenker BJ, McKay T, Datar S, et al. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J Immunol. 1999;162:4773–80. [PubMed] [Google Scholar]
- 15.Hassane DC, Lee RB, Pickett CL. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect Immun. 2003;71:541–5. doi: 10.1128/IAI.71.1.541-545.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cortes-Bratti X, Frisan T, Thelestam M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon. 2001;39:1729–36. doi: 10.1016/s0041-0101(01)00159-3. [DOI] [PubMed] [Google Scholar]
- 17.Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 18.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 19.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 20.Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 21.Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 22.Campisi J, D’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 23.Acosta JC, O’Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 24.Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 25.Bartek J, Hodny Z, Lukas J. Cytokine loops driving senescence. Nat Cell Biol. 2008;10:887–9. doi: 10.1038/ncb0808-887. [DOI] [PubMed] [Google Scholar]
- 26.Danesh J. Estimating the contribution of Helicobacter pylori to gastric cancer. Br J Cancer. 2000;83:970. doi: 10.1054/bjoc.2000.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giesecke J. International association between Helicobacter pylori and gastric cancer. Lancet. 1993;342:120–1. doi: 10.1016/0140-6736(93)91326-h. [DOI] [PubMed] [Google Scholar]
- 28.Frisan T, Cortes-Bratti X, Chaves-Olarte E, et al. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell Microbiol. 2003;5:695–707. doi: 10.1046/j.1462-5822.2003.00311.x. [DOI] [PubMed] [Google Scholar]
- 29.Guerra L, Teter K, Lilley BN, et al. Cellular internalization of cytolethal distending toxin: a new end to a known pathway. Cellular Microbiology. 2005;7:921–34. doi: 10.1111/j.1462-5822.2005.00520.x. [DOI] [PubMed] [Google Scholar]
- 30.Purven M, Lagergard T. Haemophilus ducreyi, a cytotoxin-producing bacterium. Infect Immun. 1992;60:1156–62. doi: 10.1128/iai.60.3.1156-1162.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mailand N, Falck J, Lukas C, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–9. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 32.Thullberg M, Welcker M, Bartkova J, et al. Monoclonal antibody probes for p21WAF1/CIP1 and the INK4 family of cyclin-dependent kinase inhibitors. Hybridoma. 2000;19:63–72. doi: 10.1089/027245700315806. [DOI] [PubMed] [Google Scholar]
- 33.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 35.Alby F, Mazars R, De Rycke J, et al. Study of the cytolethal distending toxin (CDT)-activated cell cycle checkpoint. Involvement of the CHK2 kinase. FEBS Lett. 2001;491:261–5. doi: 10.1016/s0014-5793(01)02205-0. [DOI] [PubMed] [Google Scholar]
- 36.DiTullio RA, Jr, Mochan TA, Venere M, et al. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol. 2002;4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 37.De Rycke J, Sert V, Comayras C, et al. Sequence of lethal events in HeLa cells exposed to the G2 blocking cytolethal distending toxin. Eur J Cell Biol. 2000;79:192–201. doi: 10.1078/S0171-9335(04)70022-9. [DOI] [PubMed] [Google Scholar]
- 38.Gary RK, Kindell SM. Quantitative assay of senescence-associated [beta]-galactosidase activity in mammalian cell extracts. Analytical Biochemistry. 2005;343:329–34. doi: 10.1016/j.ab.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Pearson M, Carbone R, Sebastiani C, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–10. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 40.Ferbeyre G, De Stanchina E, Querido E, et al. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000;14:2015–27. [PMC free article] [PubMed] [Google Scholar]
- 41.Langley E, Pearson M, Faretta M, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–96. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janderova-Rossmeislova L, Novakova Z, Vlasakova J, et al. PML protein association with specific nucleolar structures differs in normal, tumor and senescent human cells. J Struct Biol. 2007;159:56–70. doi: 10.1016/j.jsb.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 43.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 44.Minamino T, Yoshida T, Tateno K, et al. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation. 2003;108:2264–9. doi: 10.1161/01.CIR.0000093274.82929.22. [DOI] [PubMed] [Google Scholar]
- 45.Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hickey TE, McVeigh AL, Scott DA, et al. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect Immun. 2000;68:6535–41. doi: 10.1128/iai.68.12.6535-6541.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng J, Meng J, Zhao S, et al. Campylobacter-induced interleukin-8 secretion in polarized human intestinal epithelial cells requires Campylobacter-secreted cytolethal distending toxin- and Toll-like receptor-mediated activation of NF-kappaB. Infect Immun. 2008;76:4498–508. doi: 10.1128/IAI.01317-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang H, Su Z-Z, Lin JJ, et al. The melanoma differentiation associated gene mda-7 suppresses cancer cell growth. Proc Natl Acad Sci USA. 1996;93:9160–5. doi: 10.1073/pnas.93.17.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie Y, Sheng W, Xiang J, et al. Recombinant human IL-24 suppresses lung carcinoma cell growth via induction of cell apoptosis and inhibition of tumor angiogenesis. Cancer Biother Radiopharm. 2008;23:310–20. doi: 10.1089/cbr.2007.0453. [DOI] [PubMed] [Google Scholar]
- 50.Dent P, Yacoub A, Grant S, et al. MDA-7/IL-24 regulates proliferation, invasion and tumor cell radiosensitivity: a new cancer therapy? J Cell Biochem. 2005;95:712–9. doi: 10.1002/jcb.20502. [DOI] [PubMed] [Google Scholar]
- 51.Inoue S, Shanker M, Miyahara R, et al. MDA-7/IL-24-based cancer gene therapy: translation from the laboratory to the clinic. Curr Gene Ther. 2006;6:73–91. doi: 10.2174/156652306775515574. [DOI] [PubMed] [Google Scholar]
- 52.Carson CT, Orazio NI, Lee DV, et al. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J. 2009;28:652–62. doi: 10.1038/emboj.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ge Z, Rogers AB, Feng Y, et al. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 2007;9:2070–80. doi: 10.1111/j.1462-5822.2007.00939.x. [DOI] [PubMed] [Google Scholar]
- 54.Lax AJ. Opinion: Bacterial toxins and cancer–a case to answer? Nat Rev Microbiol. 2005;3:343–9. doi: 10.1038/nrmicro1130. [DOI] [PubMed] [Google Scholar]