

Abstract
Non-small-cell lung cancer (NSCLC) with mutations in the epidermal growth factor receptor (EGFR) is a distinct subgroup of NSCLCs that is particularly responsive to EGFR tyrosine-kinase inhibitors (TKIs). A weighted pooled analysis of available studies was performed to evaluate clinical outcome in patients with EGFR-mutated NSCLC who were treated with chemotherapy or EGFR TKIs. Median progression-free survival (PFS) times were pooled from prospective or retrospective studies that evaluated chemotherapy or single-agent EGFR TKIs (erlotinib or gefitinib) in patients with NSCLC and EGFR mutations. Among the studies identified for inclusion in the analysis, 12 evaluated erlotinib (365 patients), 39 evaluated gefitinib (1069 patients) and 9 evaluated chemotherapy (375 patients). Across all studies, the most common EGFR mutations were deletions in exon 19 and the L858R substitution in exon 21. In the weighted pooled analysis, the overall median PFS was 13.2 months with erlotinib, 9.8 months with gefitinib and 5.9 months with chemotherapy. Using a two-sided permutation, erlotinib and gefitinib produced a longer median PFS versus chemotherapy, both individually (P= 0.000 and P= 0.002, respectively) and as a combined group (EGFR TKI versus chemotherapy, P= 0.000). EGFR TKIs appear to be the most effective treatment for patients with advanced EGFR-mutant NSCLC. Ongoing prospective trials comparing the efficacy of first-line chemotherapy and EGFR TKIs in EGFR-mutant disease should provide further insight into the most appropriate way to treat this specific group of patients.
Keywords: adenocarcinoma, chemotherapy, epidermal growth factor receptor, erlotinib, gefitinib, lung cancer, mutation, tyrosine kinase inhibitor
Introduction
Molecular techniques have allowed us to redefine the ways in which we describe cancer. In the lung cancer setting, it is becoming increasingly apparent that non-small-cell lung cancer (NSCLC) may not only be subgrouped according to tumour histology but also by the aberrant coding, expression or activation of various proteins. The best studied example of this is the subgroup of tumours with mutations in the gene encoding the epidermal growth factor receptor (EGFR). These mutations may generate a distinct molecular pathway in the pathogenesis of NSCLC, responsible for the aetiology seen in this group of patients, and appear to confer sensitivity to the EGFR tyrosine-kinase inhibitors (TKIs) erlotinib [1] and gefitinib [1–3].
Erlotinib and gefitinib have been extensively investigated in unselected patients with NSCLC. In the phase III BR.21 study, which was performed in advanced NSCLC patients who had failed previous chemotherapy, erlotinib produced a significant overall survival (OS) benefit relative to placebo in the whole study population (P < 0.001), as well as significant improvements in progression-free survival (PFS), overall response rate, disease control rate, tumour-related symptoms and patient quality of life [4, 5]. In contrast, the pivotal phase III placebo-controlled study of gefitinib in previously treated advanced NSCLC patients, Iressa Survival Evaluation in Lung Cancer (ISEL), failed to demonstrate a statistically significant improvement in OS with gefitinib in the overall study population [6]. Second-line gefitinib was shown to be non-inferior to docetaxel in a multi-national study Iressa Non-Small-Cell Lung Cancer Trial Evaluating Response and Survival Against Taxotere (INTEREST) [7], but not in a similarly designed trial performed in Japanese patients (V15–32) [8]. A recent meta-analysis including data from INTEREST, V15–32 and two other (open-label) studies of gefitinib versus docetaxel (second-line indication of gefitinib in NSCLC [SIGN][9] and Iressa as Second-Line Therapy in Advanced NSCLC-Asia [ISTANA][10]) concluded that the efficacy of gefitinib was similar to that of docetaxel in previously treated advanced NSCLC (hazard ratio [HR] for OS 1.03, 95% confidence interval [C.I.] 0.93–1.13) [11]. As two of the studies included in this meta-analysis were conducted exclusively in Asia, 43% of the 2257 patients were of Asian ethnicity.
Biomarker analyses in the BR.21, ISEL, INTEREST and sequential Tarceva in UN resectable NSCLC (SATURN) studies [12–15] have indicated that patients with EGFR mutations have a pronounced response to EGFR TKIs; however, the relatively low rate of mutations and the limited availability of tumour samples from randomized trials make it difficult to conduct large-scale investigations of possible associations between EGFR mutations and therapeutic outcome. For this reason, we have performed an in-depth literature review to examine the evidence surrounding EGFR-mutated NSCLC and have then conducted a pooled analysis of available studies to evaluate clinical outcome in patients with EGFR-mutated NSCLC, who have been treated with chemotherapy or EGFR TKIs.
Literature review
An in-depth review of the published literature was undertaken. The PubMed database was searched on June 11, 2009 using the search string: ((lung[title] OR NSCLC[title] OR adenocarcinoma[title]) AND (‘epidermal growth factor receptor’[title] OR EGFR[title]) AND (mutation*)) AND English[language]. A total of 499 papers were identified. Titles were scanned for relevant papers that add to the body of evidence describing EGFR-mutated NSCLC for inclusion in the literature review. Reports from the 2009 Annual Meeting of the American Society of Clinical Oncology (ASCO) were also included in the review in order to accommodate the latest information in the field. Reference lists in key reviews were also scanned to identify any other papers of interest. Because of space restrictions, only key or summary papers have been cited in this literature review.
Molecular biology of EGFR mutations
EGFR is a 170-kDa member of the ErbB or human epidermal receptor (HER) family of tyrosine kinase (TK) growth factor receptors. Following binding of specific ligands, the transmembrane receptor homodimerizes with another EGFR protein or heterodimerizes with related proteins (primarily HER2/ErbB2). Once the dimer is formed, the intracellular enzymatic subunit of one EGFR phosphorylates several tyrosines of the other protein. This leads to the recruitment of additional intracellular signalling molecules, which begin a cascade that activates specific cellular growth and differentiation pathways [16]. EGFR has also been identified as a cellular proto-oncogene with close sequence homology to the viral oncogene v-erb-B[17] and is expressed in a variety of solid tumours, including lung cancer [18]. Although the mechanisms by which EGFR contributes to a malignant phenotype have not been fully elucidated, it is clear that tumourigenic pathways such as the ras-raf-MEK-ERK pathway and the PI3K/AKT pathway are mediated by EGFR activation, leading to tumour progression, proliferation, evasion from apoptosis, angiogenesis, invasion and metastatic spread [19–21].
Mutations in the region of the EGFR gene that encodes the TK domain of the receptor were first reported in patients with NSCLC in 2004 [2, 3]. Paez and colleagues amplified and sequenced the EGFR gene from 119 unselected patients with NSCLC. Somatic deletion and missense mutations were identified in 16 of the samples. When EGFR was sequenced in five patients who had responded to gefitinib and four patients who had progressed on gefitinib, mutations were found in all of the responders but none of those with disease progression. Lynch’s group published a report on the same day that revealed somatic heterozygous mutations clustered around the adenosine triphosphate (ATP)-binding pocket of the TK domain in eight of nine patients with primary NSCLC who had responded to gefitinib, while no mutations were found in tumour cells from patients who had not responded to gefitinib. Furthermore, in vitro studies showed that mutant receptors were functional, more responsive to endogenous ligands, and more sensitive to gefitinib than wild-type receptors. In a similar fashion, Pao et al. sequenced exons 18−24 of the EGFR gene from seven patients with bronchioloalveolar carcinoma (BAC) who had responded to erlotinib in a phase II trial, five of whom were shown to possess mutations [1]. In contrast, analysis of tumour tissue from 10 patients in the trial who did not respond to erlotinib did not reveal any EGFR mutations.
The discovery of somatic EGFR mutations in some patients with NSCLC was a very significant breakthrough in the understanding of this disease. Further molecular studies performed by a number of research groups have shown that EGFR mutations occur almost exclusively within exons 18−21 of the gene, which encodes the amino lobe and part of the carboxy lobe of the receptor (Fig. 1). Murray and colleagues have compiled an extensive database of published literature, containing data from over 12,000 patients, that has identified 254 independent somatic mutations in this region of the gene [22]. The most common mutations involve point mutations in exon 18, deletions and/or insertions in exon 19, insertions/duplications and point mutations in exon 20 and point mutations in exon 21. One mutation in exon 20 (T790M) appears to confer resistance to EGFR TKIs, although this is very rare among EGFR TKI-naïve patients [22]. Other mutations, occurring in the region of the EGFR gene encoding the TK domain, appear to confer sensitivity to EGFR TKIs. The best characterized of these are deletions in exon 19 around the ATP-binding cleft of the receptor (particularly E746-A750del) and a missense mutation in exon 21 (L858R) (Fig. 1); in this review we focus on these mutations.
Fig 1.
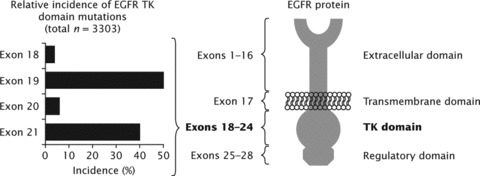
Sites of common activating mutations in exons 18−21 of the epidermal growth factor receptor gene [22, 23].
Mutations in the EGFR gene may lead to the stimulation of oncogenic pathways. For example, NIH-3T3 cells expressing EGFR with L858R and G719S mutations underwent oncogenic transformation, even when no ligand was present [24]. Furthermore, injection of the clonal, transformed NIH-3T3 fibroblasts into immunocompromised mice led to the formation of tumours. In contrast, no transformation or tumour growth occurred when similar experiments were performed with wild-type EGFR. Ji and colleagues have provided further evidence to support the oncogenic potential of mutated EGFR: they created bitransgenic mice with inducible expression of two common EGFR mutants seen in human lung cancer. Both transgenic lines developed lung adenocarcinoma after sustained mutant EGFR expression, with tumour maintenance dependent on the continued expression of the mutated proteins [25]. Similar observations in mutant EGFR transgenic mice were reported by Politi’s group [26].
The mechanism by which mutated EGFR induces an oncogenic state has not been fully elucidated. The mutated receptors may be constitutively activated, take longer to de-phosphorylate following ligand activation, or lead to the aberrant phosphorylation of other proteins [2, 27–34]. Experiments with the kinase domain of EGFR have shown that the L858R mutated form is a faster enzyme than the wild-type form. Specifically, the turnover number kcat is increased by an order of magnitude and the mutation seems to disrupt autoinhibitory interactions so that the enzyme is ‘locked’ in the constitutively active conformation [35]. Mutated EGFR may also preferentially form heterodimers with different proteins to wild-type EGFR; for example, mutated EGFR is associated with increased expression of HER3 (ErbB3) [36] and, unlike wild-type EGFR, may preferentially bind with this protein [37]. These aberrant activities may generate sustained activation of intracellular pathways and promotion of a cancer phenotype. It has been shown that mutated EGFR is a potent activator of particular downstream pathways, including ras-raf-MEK-ERK, PI3K/AKT, STAT3 and STAT5 [36, 38–41]. The ras-raf-MEK-ERK pathway is particularly involved in cell proliferation and the PI3K/AKT pathway is particularly involved in cell survival. Indeed, EGFR-mutant tumours appear to be strongly dependent on activation of the PI3K/AKT pathway for survival, leading to the suggestion that mutated EGFR is an ‘addicting oncogene’– in other words, tumours with EGFR mutations may actually require hyperactivated EGFR for survival [25, 26, 42]. This has implications for therapy, as blockade of EGFR may be necessary in such patients to re-establish apoptotic signalling and undermine tumour maintenance or growth.
Clinical characteristics of NSCLC patients with EGFR mutations
NSCLC patients with specific clinical features appear to have a greater likelihood of having a mutated form of EGFR. Analysis of 14 studies involving 2880 patients showed EGFR mutations occur more frequently in women than men (38%versus 10%), Asian than non-Asian patients (32%versus 7%), never-smokers than current or former smokers (47%versus 7%) and patients with adenocarcinoma histology than non-adenocarcinoma histology (30%versus 2%) [43]. Furthermore, EGFR mutations have been largely associated with specific adenocarcinoma histologies: BAC, invasive adenocarcinomas with prominent lepidic growth and papillary adenocarcinomas [44–52]. In particular, EGFR mutations may be more common in the macropapillary subset of lung adenocarcinomas, a particularly aggressive lung cancer type [44, 53, 54].
While EGFR mutations appear more frequently in patients with certain clinical characteristics, no characteristics have been identified that are sufficient or necessary for such mutations to occur. This can be demonstrated by analysis of the clinical characteristics of patients enrolled in a recent trial performed by the Spanish Lung Cancer Group: while all patients had advanced NSCLC with EGFR mutations, a notable portion were male (30%), former smokers (26%) or had non-adenocarcinoma/BAC histology (9%) [55]. This suggests that it is not appropriate to rule the possibility of an EGFR mutation in or out on the grounds of gender, ethnicity, histology or smoking status. Therefore, in the future, diagnoses will need to determine not only the type of disease and histology of tumours, but also their molecular characteristics, including EGFR mutation status. As a consequence, the role of pathologists is likely to evolve to incorporate relevant molecular techniques.
Genetic characteristics of EGFR-mutated tumours
While it is not yet known why certain patients are susceptible to the development of EGFR mutations, EGFR-mutated tumours may have distinct genetic characteristics from tumours expressing wild-type EGFR. Several studies have indicated that there is a strong correlation between EGFR mutations and increased EGFR gene copy number, as determined by fluorescence in situ hybridization (FISH) or chromogenic in situ hybridization [56–58]. Further to this, a Japanese group used laser-capture microdissection and array comparative genomic hybridization to show that adenocarcinomas with EGFR mutations have significantly more genetic copy number alterations than wild-type tumours (P= 0.01) [59]. This study focused on 800 chromosomal loci containing cancer-related genes and discovered 58 loci that showed significant differences in the frequency of copy number alterations between EGFR wild-type and mutated tumours, including amplification of the mutated EGFR gene itself. When a supervised hierarchical clustering technique was used to classify tumours according to 46 selected loci, two distinct groups were formed: one containing wild-type tumours only and one containing primarily EGFR-mutated tumours. Another recent study showed a distinct pattern of alteration in tumour samples from patients with EGFR mutations compared with patients with wild-type EGFR and KRAS mutations [60]. In this analysis, genome-wide single nucleotide polymorphism assay screening for allelic imbalance in patients with adenocarcinoma revealed one region on chromosome 14 (14q21.3) and three regions on chromosome 7 (7p21.3-p21.2, 7p21.3 and 7p21.2–7p15.3) that were significantly amplified in EGFR-mutated tumours compared with wild-type tumours. EGFR-mutated tumours also showed homozygous deletions at CDKN2A, which encodes the tumour suppressor cyclin-dependent kinase inhibitor 2A, and loss of heterozygosity at RB1, the gene encoding the tumour-suppressing retinoblastoma protein. Patients with EGFR mutations have also been shown to have a distinct pattern of methylation of several tumour suppressor genes compared with patients with KRAS mutations, which may affect the function of the tumour suppressor genes [61]. Further adding to the hypothesis that EGFR-mutated NSCLC may be a distinct subtype of disease is the results of a recent integrated genomic analysis of lung adenocarcinoma [62]. In this study, EGFR-mutant tumours were clustered according to genomic copy number alterations into two particular classifications, different from the clusters that KRAS-mutant tumours fell into. This non-random pattern of copy number alterations associated with different mutations suggests that distinct oncogenic pathways may be differentiated by co-ordinated genetic alterations. Furthermore, this analysis showed that EGFR-mutant adenocarcinomas are associated with underexpression of DUSP4, a protein involved in negative feedback of EGFR signalling. This underexpression may further perpetuate EGFR hyperactivity in EGFR-mutant disease. In contrast, KRAS-mutant tumours are associated with overexpression of DUSP4.
Interestingly, there appears little association between the presence of EGFR mutations and mutations seen in other NSCLCs [63] including BRCA mutations [62, 64], KRAS mutations [48, 50, 62, 65], BRAF mutations [62], HER2 mutations [62], LKB1 mutations [62], p16 homozygous deletions [66] and p53 mutations [65]. EGFR mutations may therefore be clonal events with high oncogenic potential that reduces the likelihood of further mutations and leads to oncogenic addiction of cancer cells. This suggests that EGFR-mutated NSCLC has a different genetic basis to other forms of NSCLC and this may have implications for treatment.
Prognostic significance of NSCLC with EGFR mutations
The prognostic significance of EGFR mutations in NSCLC, independent of other features, is not clear. However, the available data suggest that any prognostic impact of EGFR mutations is likely to be small. In a large cohort of Japanese patients with lung adenocarcinoma who had undergone potentially curative pulmonary resection, the presence of an EGFR mutation was associated with longer survival than the absence of a mutation in a univariate analysis; however, the significance of this association was lost in multivariate analysis [65]. Likewise, in a series of unselected patients with NSCLC from Japan, Taiwan, USA and Australia, OS was not significantly longer in patients with EGFR mutations compared with those without mutations, although there was a trend for decreased survival in patients with exon 19 deletions and increased survival in patients with L858R mutations [46]. Analysis of the recent Iressa Pan-ASia Study (IPASS), which compared first-line gefitinib with carboplatin/paclitaxel in East Asian patients with adenocarcinoma who were never-smokers or light ex-smokers, found that there were no major differences in OS among patients with or without EGFR mutations in the chemotherapy group [67].
These observations are further validated by a biomarker analysis of the recently completed phase III SATURN study that evaluated erlotinib as maintenance therapy in patients with NSCLC treated with first-line platinum-containing chemotherapy. In the group of patients who received four cycles of chemotherapy followed by placebo, the presence of an EGFR mutation did not influence PFS (HR 0.78; 95% C.I. 0.52−1.22) [15].
How do structural changes in EGFR lead to greater sensitivity to EGFR TKIs?
Seminal studies by Paez and Lynch’s groups showed that NSCLC cell lines expressing EGFR with a L858R missense mutation are more sensitive to gefitinib than wild-type receptors [2, 3], a finding that has also been demonstrated by other groups [24, 33, 35]. Analyses of the kinetic parameters of wild-type EGFR and the L858R and del747–753 mutants have revealed that the mutant forms bind ATP less tightly than the wild-type enzyme [35, 68]. Since erlotinib and gefitinib bind to the same pocket as ATP, it may be expected that they are affected in a similar manner to ATP by mutations in this domain. Surprisingly, however, the opposite is the case: depending on the assay, erlotinib and gefitinib bind similarly [69] or even more tightly [35, 68] to the mutant forms of EGFR. It is this unique property that allows the TKIs to target the mutated EGFR so effectively. While both the L858R mutation and del747−753 are highly sensitive to both erlotinib and gefitinib [70], some in vitro studies have found that EGFR with exon 19 deletions or L858R, G719S, V742A or R766C substitutions are more sensitive to erlotinib [70, 71]. As previously discussed, EGFR mutations may be associated with EGFR amplification [56–58, 72]. Such gains in gene copy number may also be related to EGFR TKI sensitivity [73], and this highlights the need to consider the contribution of patients with EGFR mutation-positive disease when analysing other potential biomarkers.
In the clinical setting, NSCLC patients with EGFR mutations have shown dramatic responses to EGFR TKIs. In IPASS, gefitinib produced a response rate of 71% among 132 Asian patients with EGFR mutations [67]. In a Spanish study involving 197 evaluable patients with EGFR mutations, erlotinib produced a response rate of 71% (including 12% complete response), with a further 20% achieving stable disease [55]. The recent phase III SATURN study has given particular insight into the benefits of erlotinib in EGFR-mutant disease [15]. In this placebo-controlled study where erlotinib was used as maintenance therapy following first-line platinum-based chemotherapy, patients with both wild-type EGFR and mutated EGFR derived a significant PFS benefit from erlotinib; however, the impact of erlotinib on PFS was particularly profound in EGFR-mutated patients (HR 0.10; 95% C.I. 0.04−0.25). Some studies have found that patients with exon 19 deletions may have a better response to EGFR TKIs than those with L858R mutations [55, 74, 75], although this has not been shown in all reports [76, 77].
Taken together, the evidence suggests that NSCLC with mutations in the TK domain of the EGFR is a distinct subgroup of NSCLCs. This appears to be a disease characterized by ‘oncogene addiction’, with tumour cells dependent on hyperactivated EGFR for survival, and is more common in, but not restricted to, Asians, non-smokers, women and patients with adenocarcinoma histology. Patients with EGFR-mutated disease appear to have distinct genetic characteristics, different from patients with wild-type EGFR.
In the pre-biological era, survival outcome in patients with EGFR mutations appears to be similar to those with wild-type disease. However, the advent of EGFR TKIs has potentially changed the prognosis of patients with EGFR-mutant NSCLC. A pooled analysis of available studies has been performed to evaluate clinical outcome in patients with EGFR-mutated NSCLC who have been treated with chemotherapy or EGFR TKIs.
Methods
Selection criteria
All prospective or retrospective studies were eligible for the study pool if they evaluated chemotherapy or single-agent EGFR TKIs (erlotinib or gefitinib) in patients with NSCLC and EGFR mutations. Among the studies identified for inclusion, a variety of techniques were used to determine the EGFR mutation status of tumours; the methods used in individual reports were not critically assessed as part of this analysis.
Search strategy
The medical literature was reviewed to identify appropriate clinical studies for pooled analysis inclusion. The Datastar Web search engine was used to search Medline, Biosis previews and Embase (excluding reviews, and non-English language) on June 11, 2009 using the search string (‘epidermal growth factor’ OR EGFR) AND (lung OR NSCLC) AND (mutation OR mutations). Searches were limited to studies published in 2004 or later (given that EGFR TK domain mutations were first identified in NSCLC in 2004). Non-English language papers and reviews were excluded. Search results were initially filtered by scanning titles. The abstracts of papers of potential relevance were reviewed and papers that were clearly relevant were selected for further analysis. Studies presented at the 2008 and 2009 ASCO meetings were also searched to ensure that the most up-to-date data were included in the analysis.
Data extraction and synthesis
PFS (or time to progression [TTP]) was chosen as the most appropriate end-point to compare between studies. Data from all eligible publications were extracted by three individuals and tabulated in an Excel spreadsheet. Entries from one individual were validated by at least one of the other two individuals. Predefined exclusion codes were assigned for studies that did not include the minimal necessary information: PFS or TTP values for the group with EGFR mutations and the associated sample size. Furthermore, studies that were performed in the maintenance or adjuvant treatment settings or involved sequential administration of multiple EGFR TKIs were excluded. To avoid inclusion of duplicate data, papers were checked for overlap in authorship, institutions and reported recruitment dates. For trials that were published in more than one paper, data from the most recent publication was used, with prior publications used to verify data.
Statistical analysis
The majority of published papers included in this analysis did not report individual patient data, and so only high-level information could be extracted for analysis. The statistical approach therefore required a number of simplifying assumptions to be made, as detailed below.
The main focus of the analysis was to obtain an estimate of the pooled median PFS by a weighted average of the single study medians. Median PFS estimates, 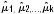 obtained in each eligible study, with group sizes N1, N2,…, Nk, were summed to yield Nall. The pooled median PFS was then estimated as the group-size weighted average, as follows:
obtained in each eligible study, with group sizes N1, N2,…, Nk, were summed to yield Nall. The pooled median PFS was then estimated as the group-size weighted average, as follows:
In one study, where PFS information was given only as the percentage of progression-free survivors at a specified time-point [78], an estimate of median PFS was calculated through the simplifying assumption that PFS times were exponentially distributed, as described by the survival function 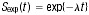 , where λ is the constant hazard rate. A further study reported only mean PFS [79]: in this case, the pertaining median
, where λ is the constant hazard rate. A further study reported only mean PFS [79]: in this case, the pertaining median 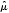 was also estimated through the exponential assumption leading to
was also estimated through the exponential assumption leading to 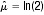 . mean. The influence of these assumptions on the overall pooled median PFS was checked in a sensitivity analysis.
. mean. The influence of these assumptions on the overall pooled median PFS was checked in a sensitivity analysis.
C.I.s could not be calculated based only on high level median PFS data. Therefore the exponential distribution was used to estimate surrogate ‘accuracy intervals’. The observed median PFS was considered as an approximate (maximum likelihood) estimate of the median of an exponential distribution. Based on this, a 90% accuracy interval for single studies and 95% accuracy interval for pooled median estimates were calculated. This approximative method does not provide a true C.I. and censoring could not be accounted for, although this may not be a strong deficiency for the PFS end-point. Therefore, the interval reported from this analysis is referred to as an ‘accuracy interval’ as its size reflects the accuracy of the relevant estimate.
Permutation testing was performed to allow the comparison of median pooled PFS for each specific therapy [80]. Random permutations across studies were generated for 1000 iterations to test the null hypothesis that the difference in median PFS between treatments was zero (i.e. that the treatment effects were identical). This statistical test is not biased but is based solely on the selected study pool, and cannot be extrapolated to any study pool.
The potential for publication bias in reported median PFS values was assessed using funnel plots, with the appropriate accuracy intervals.
Sensitivity analysis
Sensitivity analyses were performed to test the robustness of the calculated accuracy intervals. This was achieved using a re-sampling technique known as ‘bootstrap’[80]. As with permutation testing, the results of this analysis can only be applied to the selected study and cannot be extrapolated to a wider pool.
Results
Figure 2 summarizes the process of identifying studies eligible for inclusion in our analysis. We reviewed the full text from 175 published studies and meeting abstracts identified. A total of 54 studies met the criteria for inclusion (Table 1). Of these, 12 evaluated erlotinib (365 patients), 39 evaluated gefitinib (1069 patients) and 9 evaluated chemotherapy (375 patients) in EGFR-mutant NSCLC (some studies reported data for more than one regimen).
Fig 2.

Flow diagram showing citations retrieved from literature searches and number of trials included in analysis.
Table 1.
Characteristics of included studies for the pooled studies that evaluated the effects of chemotherapy or EGFR TKIs in patients with EGFR-mutant NSCLC
| Study | Design | Patients | Treatment | PFS/TTP/TTF |
|---|---|---|---|---|
| Erlotinib | ||||
| Ahn et al.[81] | Prospective | n= 24: Korean; ≥1 prior treatment; exon 19 deletion (n= 17); L858R (n= 5); exon 20 mutation (n= 1); exon 18 mutation and exon 19 deletion (n= 1) | Erlotinib 150 mg/day | TTP: 8.6 months |
| Amann et al.[82] | Ph II, single-arm | n= 3: chemo-naïve | Erlotinib 150 mg/day | PFS: 13.1 months |
| Hirsch et al.[83] | Ph II; randomized comparison with erlotinib intercalated with carboplatin/ paclitaxel | n= 13: chemo-naïve; EGFR mutation | Erlotinib 150 mg/day | PFS: 11.04 months |
| Jackman et al.[84] | Ph II, single-arm | n= 9: primarily white; chemo-naïve; ≥70 years; exon 19 deletion (n= 3); L858R (n= 5); L861Q and exon 19 deletion (n= 1) | Erlotinib 150 mg/day | TTP: 13 months |
| Jackman et al.[85] | Ph II, single-arm | n= 33: female; chemo-naïve; adenocarcinoma; EGFR mutations | Erlotinib 150 mg/day | TTP: 12.6 months |
| Massuti et al.[55] | Prospective | n= 217: Spanish; 0−2 prior treatments; exon 19 deletion (n= 134); L858R (n= 83) | Erlotinib 150 mg/day | PFS: 14 months |
| Miller et al.[86] | Ph II, single-arm | n= 18: BAC and adenocarcinoma, BAC subtype; 0−1 prior treatments; exon 19 or 21 EGFR mutation | Erlotinib 150 mg/day | PFS: 13 months |
| Pirker et al.[87] | Prospective (TRUST study) | n= 12: primarily white; chemo-naïve or previously treated; exon 19 deletion (n= 7); L858R (n= 5) | Erlotinib 150 mg/day | PFS: 405 days |
| Riely et al.[75] | Retrospective | n= 12: primarily white; chemo-naïve or previously treated; exon 19 deletion (n= 8); L858R (n= 4) | Erlotinib 150 mg/day | PFS: 12 months |
| Rosell et al.[88] | Prospective, ph II | n= 12: Spanish; non-squamous cell carcinoma; exon 19 or 21 EGFR mutation | Erlotinib 150 mg/day | PFS: 13 months |
| Schneider et al.[89] | Prospective (TRUST study) | n= 6: German; chemo-naïve or previously treated; exon 19 deletion (n= 2); L858R (n= 4) | Erlotinib 150 mg/day | PFS: 12.4 months |
| Zhou et al.[90] | Retrospective | n= 6: Chinese; ≥1 previous treatment; EGFR mutations | Erlotinib 150 mg/day | TTP: 15.8 months |
| Gefitinib | ||||
| Asahina et al.[76] | Ph II, single-arm | n= 16: Japanese; chemo-naïve; exon 19 deletion (n= 13); L858R; (n= 3) | Gefitinib 250 mg/day | PFS: 8.9 months |
| Bell et al.[91] | Retrospective (ph II IDEAL studies) | n= 14: ≥1 previous treatment; exon 19 deletion (n= 11); L858R (n= 2); InsG771 (n= 1) | Gefitinib 250 or 500 mg/day | TTP: 3.8 months |
| Buckingham et al.[92] | Retrospective | n= 17: ≥1 previous treatment; EGFR mutation | Gefitinib 250 mg/day | PFS: 13.6 months |
| Cappuzzo et al.[93] | Prospective, Ph II, single-arm | n= 21: Italian; chemo-naïve or previously treated; never smokers and EGFR FISH+ or p-AKT+, or any smoking history and both EGFR FISH- and p-AKT−; exon 19 deletion (n= 13); exon 21 mutation (n= 4); exon 19 and 21 mutation (n= 3); exon 19 and 20 mutation (n= 1) | Gefitinib 250 mg/day | TTP: 7.1 months |
| Chou et al.[94] | Retrospective | n= 33: Taiwanese; prior platinum therapy; exon 18 substitution (n= 4); exon 19 deletion (n= 11); exon 20 substitution or deletion (n= 4); exon 21 substitution (n= 12); ≥1 mutation (n= 2) | Gefitinib 250 mg/day | PFS: 7.6 months |
| Cortes-Funes et al.[95] | Retrospective | n= 10: Spanish; ≥1 previous treatment; exon 19 deletion (n= 8); L858R (n= 2) | Gefitinib 250 mg/day | TTP: 12.3 months |
| D’Addario et al.[96] | Ph II, single-arm | n= 4: Swiss; chemo-naïve; exon 19 deletion (n= 2); L858R (n= 2) | Gefitinib 250 mg/day | TTP: 7.5 months |
| Dongiovanni et al.[97] | Retrospective | n= 9: Italian; chemo-naïve or previously treated; exon 19 deletion (n= 8); L858R (n= 1) | Gefitinib 250 mg/day | TTP: 14.9 months |
| Douillard et al.[14] | Ph III INTEREST study; randomized, comparison with docetaxel | n= 19: primarily white; prior platinum chemotherapy; EGFR mutation | Gefitinib 250 mg/day | PFS: 7.0 months |
| Fukuoka et al.[98] | Ph III IPASS; randomized comparison with carboplatin/ paclitaxel | n= 132: East-Asian; adenocarcinoma; never-smokers; chemo-naïve; EGFR mutation | Gefitinib 250 mg/day | PFS: 9.5 months |
| Han et al.[99] | Retrospective | n= 21: Korean; previously treated; exon 19 deletion (n= 12); L858R (n= 6); G719A (n= 3) | Gefitinib 250 mg/day | TTP: 13.8 months |
| Hirsch et al.[100] | Pooled analysis of [101] and [102] | n= 43: Italian or US; chemo-naïve or previously treated; exon 21 mutations (n= 31); exon 19 deletions (n= 11); mutations in exons 19 and 21 (n= 1) | Gefitinib 250 or 500 mg/day | PFS: 3 months |
| Hong et al.[103] | Prospective, Ph II, single-arm | n= 3: Korean; exon 19 deletion (n= 2); L858R (n= 1) | Gefitinib 250 mg/day | PFS: 5.8 months |
| Ichihara et al.[104] | Retrospective | n= 30: Japanese; chemo-naïve and previously treated; exon 19 deletion (n= 16); L858R (n= 14) | Gefitinib 250 mg/day | PFS: 11.3 months |
| Inoue et al.[77] | Ph II, non-randomized comparison with standard chemotherapy | n= 16: Japanese; chemo-naïve; exon 19 deletion (n= 9); L858R (n= 7) | Gefitinib 250 mg/day | PFS: 9.7 months |
| Inoue et al.[105] | Ph II, single-arm | n= 29: Japanese; chemo-naïve; poor performance status; exon 19 deletion (n= 18); L858R (n= 10), L861Q (n= 1) | Gefitinib 250 mg/day | PFS: 6.5 months |
| Kim et al.[106] | Retrospective | n= 6: Korean; ≥1 previous treatment; exon 19 deletion (n= 5); L858R (n= 1) | Gefitinib 250 mg/day | TTP: 12.6 months |
| Kim et al.[78] | Prospective, ph II, single-arm | n= 45: Korean; chemo-naïve; adenocarcinoma; exon 19 deletion (n= 29); L858R (n= 15); L861Q (n= 1) | Gefitinib 250 mg/day | PFS at 6 months: 75% |
| Kimura et al.[107] | Prospective, single-arm | n= 9: Japanese; chemo-naïve and previously treated; exon 19 deletion (n= 4); L858R (n= 4); V689L (n= 1) | Gefitinib 250 mg/day | PFS: 6.4 months |
| Kobayashi et al.[108] | Ph III; randomized comparison with carboplatin/paclitaxel | n= 98: chemo-naïve; EGFR mutation | Gefitinib 250 mg/day | PFS: 10.4 months |
| Koyama et al.[79] | Retrospective | n= 18: Japanese; chemo-naïve or previous treatment; G719C (n= 2); G719C and W731R (n= 1); P733S (n= 1); exon 19 deletion (n= 6); V738–I744 ins (n= 2); S768C (n= 1); T790M (n= 1); Q812R (n= 1); V843I (n= 1); L858R (n= 2) | Gefitinib 250 mg/day | Mean TTP: 13.7 months |
| Massarelli et al.[109] | Retrospective | n= 7: Asian or Caucasian; chemo-naïve or previous treatment; exon 19 deletion (n= 6); G719A (n= 1) | Gefitinib 250 mg/day | TTP: 9.3 months |
| Oshita et al.[110] | Retrospective | n= 11: Japanese; ≥1 previous treatment; EGFR mutation | Gefitinib 250 mg/day | PFS: 16 months |
| Pallis et al.[111] | Retrospective | n= 11: Greek; ≥1 previous treatment; exon 19 deletion (n= 6); L858R (n= 3); G719D (n= 1); E746V (n= 1) | Gefitinib 250 mg/day | TTP: 14.7 months |
| Riely et al.[75] | Retrospective | n= 22: primarily white; chemo-naïve or previously treated; exon 19 deletion (n= 15); L858R (n= 7) | Gefitinib 250 mg/day | PFS: 12 months |
| Sequist et al.[112] | Ph II, single-arm | n= 31: primarily non-Asian; chemo-naïve; exon 19 deletion (n= 17); L858R (n= 8); atypical mutation (n= 6) | Gefitinib 250 mg/day | PFS: 9.2 months |
| Shao et al.[113] | Ph II, single-arm | n= 51: Taiwanese; chemo-naïve; EGFR mutation | Gefitinib 250 mg/day | PFS: 8.8 months |
| Shoji et al.[114] | Retrospective | n= 20; Japanese; chemo-naïve and previously treated; exon 19 deletion (n= 10); L858R (n= 8); E709A and G719S (n= 1); L858R and Y725Y (n= 1) | Gefitinib 250 mg/day | PFS: 14 months |
| Sugio et al.[115] | Ph II, single-arm | n= 19: Japanese; exon 19 deletion (n= 7); L858R (n= 10); exon 19 deletion and L858R (n= 1); exon 19 deletion and G796A (n= 1) | Gefitinib 250 mg/day | PFS: 7.1 months |
| Sunaga et al.[116] | Ph II, single-arm | n= 21: Japanese; chemo-naïve or previously treated; exon 19 deletion (n= 17); L858R (n= 4) | Gefitinib 250 mg/day | PFS: 12.9 months |
| Sutani et al.[117] | Ph II, single-arm | n= 27: Japanese; 0−1 previous treatments; exon 19 deletion; L858R, L861Q | Gefitinib 250 mg/day | TTP: 9.4 months |
| Takano et al.[118] | Retrospective | n= 85: Japanese; chemo-naïve or previously treated; exon 19 deletion (n= 49); L858R (n= 36) | Gefitinib 250 mg/day | PFS: 9.2 months |
| Tamura et al.[119] | Ph II, single-arm | n= 28: Japanese; 0−2 previous treatments; exon 19 deletion (n= 14); L858R (n= 14) | Gefitinib 250 mg/day | PFS: 11.5 months |
| Varella-Garcia et al.[120] | Retrospective | n= 27: Japanese; chemo-naïve or previously treated; EGFR mutations | Gefitinib 250 mg/day | TTP: 10.2 months |
| Wu et al.[121] | Retrospective | n= 16: Taiwanese; chemo-naïve or previously treated; EGFR mutation | Gefitinib 250 mg/day | PFS: 8.1 months |
| Wu et al.[122] | Retrospective | n= 32: Chinese; ≥1 previous treatment; EGFR mutation | Gefitinib 250 mg/day | PFS: 8 months |
| Xu et al.[123] | Retrospective | n= 32: Chinese; chemo-naïve and previous treatment; exon 19 deletion (n= 11); exon 19–not deletion (n= 6); L858R (n= 6); exon 18 mutation (n= 6); exon 20 mutation (n= 2); exon 23 mutation (n= 1) | Gefitinib 250 mg/day | TTP: 15 months |
| Yoshida et al.[124] | Prospective | n= 21: Japanese; chemo-naïve and previously treated; exon 19 deletion (n= 8); L858 (n= 13) | Gefitinib 250 mg/day | PFS: 7.7 months |
| Zhang et al.[125] | Retrospective | n= 12: Chinese; ≥1 previous treatment; exon 19 deletion (n= 4); L858 (n= 8) | Gefitinib 250 mg/day | PFS: 10 months |
| Chemotherapy | ||||
| Bell et al.[91] | Retrospective (ph III INTACT studies; randomized comparison with gefitinib) | n= 9: primarily white; chemo-naïve; exon 19 deletion; L858R; other mutations | Paclitaxel/carboplatin or gemcitabine/cisplatin | PFS: 6.7 months |
| Douillard et al.[14] | Ph III INTEREST study; randomized, comparison with gefitinib | n= 19; primarily white; prior platinum chemotherapy; EGFR mutation | Docetaxel | PFS: 4.1 months |
| Eberhard et al.[126] | Retrospective (ph III TRIBUTE study; randomized comparison with erlotinib plus carboplatin/ paclitaxel) | n= 14: primarily white; chemo-naïve; exon 19 deletion; L858R; other mutations | Carboplatin/paclitaxel | TTP: 6.6 months |
| Fukuoka et al.[98] | Prospective, ph III IPASS; randomized comparison with gefitinib | n= 129: East-Asian; adenocarcinoma; never-smokers; chemo-naïve; EGFR mutation | Carboplatin/paclitaxel | PFS: 6.3 months |
| Inoue et al.[77] | Ph II, non-randomized comparison with gefitinib | n= 9: Japanese; chemo-naïve; exon 19 deletions (n= 8); L858R (n= 1) | Standard chemotherapy | PFS: 7.6 months |
| Kobayashi et al.[108] | Prospective, ph III; randomized comparison with gefitinib | n= 100: chemo-naïve; EGFR mutation | Carboplatin/paclitaxel | PFS: 5.5 months |
| Lee et al.[127] | Retrospective | n= 14: Korean; chemo-naïve; patients receiving platinum-based chemotherapy; EGFR mutation | Platinum-based chemotherapy | TTP: 8 months paclitaxel, 9.7 months; gemcitabine, 7.4 months |
| Tambo et al.[128] | Retrospective | n= 26: Japanese; chemo-naïve; EGFR mutations | Chemotherapy | PFS: 8.4 months |
| Wu et al.[122] | Retrospective | n= 55: Chinese; chemo-naïve; exon 19 deletion (n= 32); L858R (n= 21); exon 19 deletion and L858R (n= 2) | Chemotherapy | PFS: 4 months |
Ph = phase.
Study characteristics
As expected, the majority of studies evaluating chemotherapy in patients with EGFR mutations were conducted in the first-line treatment setting (Table 1). Many studies involving EGFR TKIs did not report the specific line of treatment for patients with EGFR mutations, although this information was generally presented for the overall population. It is, therefore, only possible to estimate the proportion of patients included in this analysis who received an EGFR TKI as first-line treatment. The estimated proportion of patients in this analysis who received first-line treatment with erlotinib, gefitinib and chemotherapy was 57%, 57%, and 95%, respectively. For chemotherapy, three studies were prospective analyses performed as part of phase III trials, while the remaining studies were retrospective or phase II trials; these studies included a cross-section of East Asian and Caucasian patients. For EGFR TKIs, the majority of studies were retrospectively analysed cohorts, often involving patients from a single ethnic group (Table 1). Across all studies, the most common EGFR mutations were deletions in exon 19 and the L858R substitution in exon 21.
PFS analysis
For patients treated in any line of therapy, median PFS ranged from 8.6−15.8 months in patients treated with erlotinib, 3−16 months in patients treated with gefitinib and 4−8.4 months in patients treated with chemotherapy (Fig. 3). In the weighted, pooled analysis, shown in Fig. 4, the overall median PFS was 13.2 months with erlotinib, 9.8 months with gefitinib, and 5.9 months with chemotherapy. As discussed previously, it was not possible to assess outcome according to line of therapy, as many reports did not provide data on this aspect specifically for patients with EGFR mutations. In order to estimate the effect of treatments in the first-line setting, an analysis was performed that included only studies where 90% or more of the included patients (regardless of EGFR mutation status) received the treatment in question as first-line therapy. This analysis showed similar pooled median PFS in the first-line setting to that in the overall analysis for all lines of therapy (Table 2).
Fig 3.
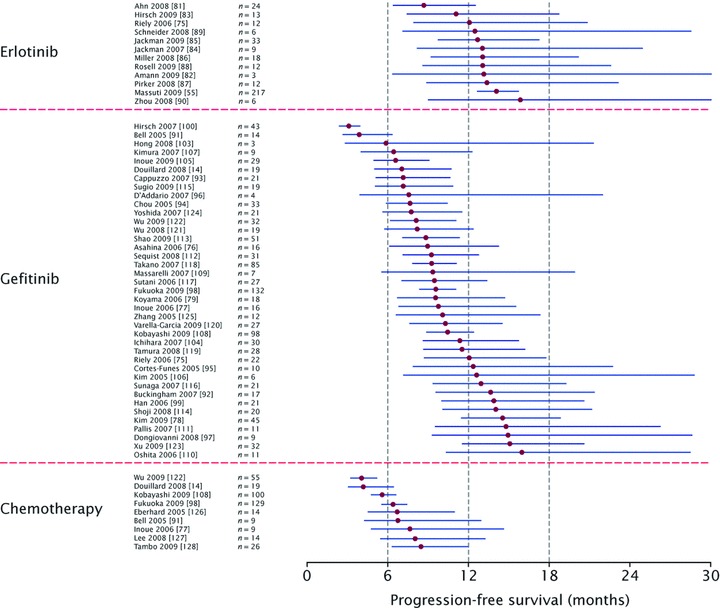
Forest plot showing analysis of median pooled PFS or TTP and 90% accuracy intervals during treatment with single-agent erlotinib, single-agent gefitinib or chemotherapy, in patients with EGFR-mutant NSCLC.
Fig 4.
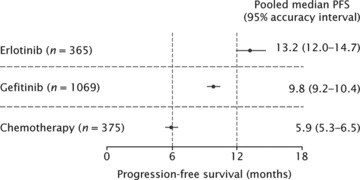
Forest plot showing pooled analysis of median PFS or TTP and 95% accuracy intervals during treatment with single-agent erlotinib, single-agent gefitinib or chemotherapy, in patients with EGFR-mutant NSCLC.
Table 2.
Pooled median PFS and 95% accuracy intervals for single-agent erlotinib, single-agent gefitinib or chemotherapy, in patients with EGFR-mutant NSCLC with chemotherapy: all lines of therapy, and studies in which ≥90% of patients received treatment in the first-line setting
| Single-agent erlotinib | |
|---|---|
| All lines of therapy (n= 365) | 13.2 (12.0−14.7) |
| Predominantly first-line (n= 70) | 12.5 (10.0−16.0) |
| Single-agent gefitinib | |
| All patients (n= 1069) | 9.8 (9.2−10.4) |
| Predominantly first-line (n= 520) | 9.9 (9.0−10.9) |
| Chemotherapy | |
| All patients (n= 375) | 5.9 (5.3−6.5) |
| Predominantly first-line (n= 359) | 6.0 (5.4−6.7) |
Permutation testing was performed to determine whether there was any difference between outcome with each treatment strategy in this study pool. Using a two-sided permutation, erlotinib and gefitinib produced a longer median PFS compared with chemotherapy, both individually (P= 0.000 and P= 0.002, respectively) and as a combined group (EGFR TKI versus chemotherapy, P= 0.000). The permutation P-value for the comparison of erlotinib versus gefitinib was 0.005 in this study pool.
Sensitivity analysis
A bootstrap analysis using 1000 runs for the estimated pooled median PFS found similar pooled median values and 95% accuracy intervals to the estimated pooled median values and 95% accuracy intervals in the original analysis; bootstrap median PFS and 95% accuracy intervals: erlotinib 13.2 (11.1–13.8) months; gefitinib 9.8 (8.9–10.8) months; chemotherapy 5.9 (5.0–6.8) months.
Publication bias
Potential publication bias was assessed using funnel plots with PFS or TTP as the outcome. The funnel plots were symmetrical for each of the treatment groups (Fig. 5A–C), indicating a lack of publication bias.
Fig 5.
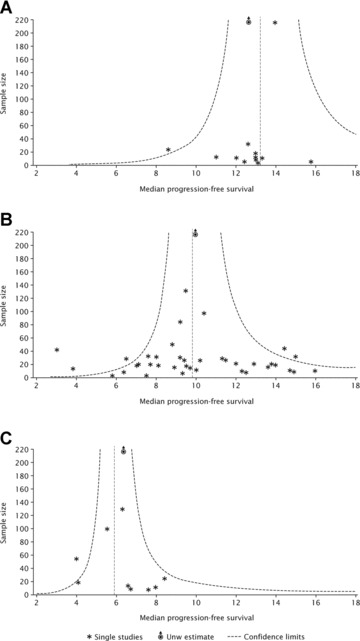
Funnel plots using PFS or TTP as an outcome for (A) erlotinib; (B) gefitinib and (C) chemotherapy.
Discussion
The principal finding of this pooled analysis is that patients with EGFR-mutated NSCLC appear to have a longer PFS when treated with erlotinib (13.2 months) or gefitinib (9.8 months) than with cytotoxic chemotherapy (5.9 months).
Permutation testing of the studies included in our analysis found that both erlotinib and gefitinib were associated with a significantly longer PFS than chemotherapy. This analysis should be interpreted in the light of permutation testing, which is based solely on the selected study pool and cannot be extrapolated to any study pool.
This analysis adds to our understanding of the place of EGFR TKIs in the treatment of patients with NSCLC. At present, only four studies have directly compared EGFR TKI monotherapy with cytotoxic chemotherapy [7, 8, 67, 108] and only one of these [108] has been adequately powered to detect any difference in outcome according to EGFR mutation status. In this phase III study, chemonaïve patients with EGFR mutations were randomized to gefitinib or carboplatin plus paclitaxel. The full results of the study are not yet available; however, a preliminary analysis performed in 198 patients found that gefitinib was associated with significantly longer PFS than chemotherapy. This supports the findings of other analyses of phase III comparative studies. In the INTEREST study [7], which compared second-line gefitinib with second-line docetaxel, retrospective analysis found that PFS was significantly longer in the EGFR-mutated patients treated with gefitinib than docetaxel [14]. However, in the V15–32 study, which performed a similar comparison between gefitinib and docetaxel in Japanese patients, there were no significant differences in PFS between treatment groups in EGFR-mutation-positive patients, although actual median PFS values were not published [8]. The low number of patients with EGFR mutations in this study should also be noted. Analysis of IPASS, which compared first-line gefitinib with carboplatin/paclitaxel in East Asian patients with adenocarcinoma who were never-smokers or light ex-smokers, found that EGFR mutations were a strong driver of PFS in the gefitinib group: median PFS in EGFR mutation-positive patients was longer with gefitinib than chemotherapy (HR 0.48: 95% C.I. 0.36–0.64; P < 0.0001); however, in EGFR mutation-negative patients, PFS was longer with chemotherapy than gefitinib (HR 2.85: 95% C.I. 2.05–3.98; P < 0.0001) [67]. This is consistent with the preclinical findings of Gandhi and colleagues that EGFR wild-type cell lines are resistant to gefitinib [73].
The results of this study are consistent with a smaller analysis of gefitinib studies recently undertaken [129]. This combined analysis of individual patient data from seven trials conducted in Japan, involving 148 patients, found that median PFS was 9.7 months with gefitinib in EGFR-mutated patients treated in the first- or subsequent-line setting. This is consistent with the 9.8 months seen with gefitinib in our review.
Our analysis is broad in scope, including patients of a number of different ethnicities in different clinical settings. While clinical characteristics, such as female gender, non-smoking history, adenocarcinoma histology and Asian ethnicity, have previously been used to guide which patients should be selected for EGFR TKI therapy, it is evident that the benefits of treatment are not restricted to these patient subgroups. Indeed, this supports the previous findings of the erlotinib BR.21 study [4, 130] and SATURN study [15], which showed that erlotinib is effective in a proportion of patients in all clinical and biomarker subgroups. Furthermore, given the impressive response to EGFR TKIs in patients with EGFR mutations, it is becoming more apparent that it is inappropriate to use clinical characteristics alone as a surrogate for mutation testing. This has been further emphasized by IPASS, which included the best possible group for mutation prediction (Asian patients with adenocarcinoma who were predominantly female and had never been smokers); however, only 60% of these patients were mutation-positive. As previously stated, those patients who were mutation-positive had a better outcome with gefitinib, while those who were mutation-negative achieved better results on cytotoxic chemotherapy. This highlights the critical need to determine EGFR mutation status before making clinical decisions regarding the use of first-line EGFR TKIs.
In our study, PFS was chosen as the most appropriate end-point to pool. OS was not evaluated, as data are often immature at the time of trial publication and median duration of OS may be influenced by subsequent therapies. While most studies report response data, this end-point does not share the reputation and weight of PFS, particularly given that treatment with EGFR TKIs has been shown to prolong PFS but does not necessarily lead to an objective response measured according to standard criteria. Furthermore, concentrating on the single end-point of PFS allows avoidance of the well-known multiplicity trap.
To evaluate the sensitivity of our analysis, a bootstrap test was performed. This showed very similar findings to the weighted analysis, providing confidence in the validity of the main analysis.
As with any analysis, there are several limitations with our study. As many of the included studies were retrospective in nature with the inherent potential for bias, it is possible that this bias would have also affected our pooled analysis. No quality analysis of the included studies was undertaken; therefore, it is not possible to determine the quality of the data that were actually included. Furthermore, while PFS was considered the most appropriate end-point to pool to provide the best estimate of efficacy, it must be noted that PFS is not assessed in the same way in all studies and is likely to be influenced by the frequency and timings of tumour measurement. Consequently, PFS values from prospective trials are likely to be more accurate than those from retrospective trials because of the stipulation for regular assessment using pre-specified criteria. Similarly, different methods, with different sensitivities, were used between studies to identify patients with EGFR mutations.
Nevertheless, the findings of this study are relevant as we continue to learn how best to tailor treatment for patients with NSCLC. The median PFS achieved with EGFR TKIs in this setting is dramatic, reaching a median of 13.2 months with erlotinib and 9.8 months with gefitinib. This is particularly notable, given that standard first-line platinum-based chemotherapy offers PFS in the range of 3 to 5 months in the general NSCLC population [131]. Indeed, the response of some EGFR-mutant patients with poor performance status to EGFR TKIs has been described as a ‘Lazarus response’– the returning to life after resuscitation has been given up [132]. By identifying patients with EGFR mutations and treating them with EGFR TKIs rather than chemotherapy, it is not only likely that they will have a superior outcome but also that they will not be subject to the debilitating toxicities associated with cytotoxic agents. Ideally, all patients who present with NSCLC should be tested for EGFR mutations, given that these are not restricted to a group of patients with certain clinical characteristics. The feasibility of this will be greatly improved when non-invasive tests (such as serum-based assays) become available.
Phase III trials are currently underway that will prospectively evaluate EGFR TKIs as first-line therapy in patients with EGFR-mutant disease. For example, the European Trial of Tarceva versus Chemotherapy (EURTAC; NCT00446225) is evaluating erlotinib versus platinum doublet chemotherapy, with a targeted accrual of 173 patients and a primary end-point of PFS. The phase III West Japan Thoracic Oncology Group study 3405 is assessing gefitinib versus cisplatin/docetaxel, also with PFS as the primary end-point. The outcome of these studies will be essential in further helping to determine the impact of EGFR TKIs in the treatment of patients with EGFR-mutant NSCLC.
Conclusions
This extensive review of the literature has shown that NSCLC associated with EGFR mutations presents as a distinct disease that is dependent on hyperactivated EGFR for survival. Because of this, it is not surprising that blockade of EGFR TK activity appears to be the most effective treatment for this subgroup of NSCLC. Ongoing trials that are prospectively comparing the efficacy of chemotherapy and EGFR TKIs as first-line therapy in EGFR-mutant disease should provide further insight into the most appropriate way to treat NSCLC in patients with EGFR mutations.
Acknowledgments
We thank Wolfgang von der Saal Ph.D. for helpful discussions and comments on this manuscript, Zuzana Hermann M.D. for assistance with data collection and input, Professor Theo Gasser for important input on the statistical analysis and Walter Köhler for valuable support with statistical programming. We also thank Mark Smith Ph.D. of Gardiner-Caldwell Communications for editorial assistance funded by F. Hoffmann-La Roche.
L.P.-A. has acted as a consultant for, and received honoraria from, AstraZeneca, Roche and Eli Lilly. Several authors (I.M., L.K., B.K.) are employees of F. Hoffmann-La Roche or related companies. J.M. is a former employee of F. Hoffmann-La Roche, and is now a consultant statistician for Roche. D.S. has participated in advisory boards and speaker bureau for, and received research funding from Roche. T.M. has acted as a consultant for AstraZeneca, Roche, Pfizer, Eli Lilly and Novartis, and has received honoraria from AstraZeneca, Roche, Pfizer, Eli Lilly and Eisai.
References
- 1.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 5.Bezjak A, Tu D, Seymour L, et al. Symptom improvement in lung cancer patients treated with erlotinib: quality of life analysis of the National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2006;24:3831–7. doi: 10.1200/JCO.2006.05.8073. [DOI] [PubMed] [Google Scholar]
- 6.Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–37. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 7.Kim ES, Hirsh V, Mok T, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet. 2008;372:1809–18. doi: 10.1016/S0140-6736(08)61758-4. [DOI] [PubMed] [Google Scholar]
- 8.Maruyama R, Nishiwaki Y, Tamura T, et al. Phase III study, V-15–32, of gefitinib versus docetaxel in previously treated Japanese patients with non-small-cell lung cancer. J Clin Oncol. 2008;26:4244–52. doi: 10.1200/JCO.2007.15.0185. [DOI] [PubMed] [Google Scholar]
- 9.Cufer T, Vrdoljak E, Gaafar R, et al. Phase II, open-label, randomized study (SIGN) of single-agent gefitinib (IRESSA) or docetaxel as second-line therapy in patients with advanced (stage IIIb or IV) non-small-cell lung cancer. Anticancer Drugs. 2006;17:401–9. doi: 10.1097/01.cad.0000203381.99490.ab. [DOI] [PubMed] [Google Scholar]
- 10.Lee D, Kim S, Park K, et al. A randomized open-label study of gefitinib versus docetaxel in patients with advanced/metastatic non-small cell lung cancer (NSCLC) who have previously received platinum-based chemotherapy. J Clin Oncol. 2008;26:430s. : Abstract 8025. [Google Scholar]
- 11.Shepherd FA, Douillard J, Fukuoka M, et al. Comparison of gefitinib and docetaxel in patients with pretreated advanced non-small cell lung cancer (NSCLC): meta-analysis from four clinical trials. J Clin Oncol. 2009;27:409s. [Google Scholar]
- 12.Zhu CQ, Da Cunha Santos G, Ding K, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26:4268–75. doi: 10.1200/JCO.2007.14.8924. [DOI] [PubMed] [Google Scholar]
- 13.Hirsch FR, Varella-Garcia M, Bunn PA, Jr, et al. Molecular predictors of outcome with gefitinib in a phase III placebo-controlled study in advanced non-small-cell lung cancer. J Clin Oncol. 2006;24:5034–42. doi: 10.1200/JCO.2006.06.3958. [DOI] [PubMed] [Google Scholar]
- 14.Douillard J, Hirsh V, Mok TS, et al. Molecular and clinical subgroup analyses from a phase III trial comparing gefitinib with docetaxel in previously treated non-small cell lung cancer (INTEREST) J Clin Oncol. 2008;26:424s. [Google Scholar]
- 15.Brugger W, Triller N, Blasinska-Morawiec M, et al. Biomarker analyses from the phase III placebo-controlled SATURN study of maintenance erlotinib following first-line chemotherapy for advanced NSCLC. J Clin Oncol. 2009;27:411s. [Google Scholar]
- 16.Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–43. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- 17.Downward J, Yarden Y, Mayes E, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307:521–7. doi: 10.1038/307521a0. [DOI] [PubMed] [Google Scholar]
- 18.Pavelic K, Banjac Z, Pavelic J, et al. Evidence for a role of EGF receptor in the progression of human lung carcinoma. Anticancer Res. 1993;13:1133–7. [PubMed] [Google Scholar]
- 19.Arteaga CL. Overview of epidermal growth factor receptor biology and its role as a therapeutic target in human neoplasia. Semin Oncol. 2002;29:3–9. doi: 10.1053/sonc.2002.35642. [DOI] [PubMed] [Google Scholar]
- 20.Salomon DS, Brandt R, Ciardiello F, et al. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 21.Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37:S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 22.Murray S, Dahabreh IJ, Linardou H, et al. Somatic mutations of the tyrosine kinase domain of epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database. J Thorac Oncol. 2008;3:832–9. doi: 10.1097/JTO.0b013e31818071f3. [DOI] [PubMed] [Google Scholar]
- 23.Ciesielski MJ, Fenstermaker RA. Structure of the epidermal growth factor receptor gene and intron recombination in human gliomas. Curr Genomics. 2003;4:1–12. doi: 10.2174/138920207780833838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji H, Li D, Chen L, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–95. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 26.Politi K, Zakowski MF, Fan PD, et al. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guha U, Chaerkady R, Marimuthu A, et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proc Natl Acad Sci USA. 2008;105:14112–7. doi: 10.1073/pnas.0806158105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Gureasko J, Shen K, et al. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–49. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Zimmer S, Kahl P, Buhl TM, et al. Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J Cancer Res Clin Oncol. 2009;135:723–30. doi: 10.1007/s00432-008-0509-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Meter AJ, Rodriguez AS, Bowman ED, et al. Laser capture microdissection and protein microarray analysis of human non-small cell lung cancer: differential epidermal growth factor receptor (EGPR) phosphorylation events associated with mutated EGFR compared with wild type. Mol Cell Proteomics. 2008;7:1902–24. doi: 10.1074/mcp.M800204-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ono M, Hirata A, Kometani T, et al. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004;3:465–72. [PubMed] [Google Scholar]
- 32.Han SW, Hwang PG, Chung DH, et al. Epidermal growth factor receptor (EGFR) downstream molecules as response predictive markers for gefitinib (Iressa, ZD1839) in chemotherapy-resistant non-small cell lung cancer. Int J Cancer. 2005;113:109–15. doi: 10.1002/ijc.20550. [DOI] [PubMed] [Google Scholar]
- 33.Mukohara T, Engelman JA, Hanna NH, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97:1185–94. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 34.Mulloy R, Ferrand A, Kim Y, et al. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007;67:2325–30. doi: 10.1158/0008-5472.CAN-06-4293. [DOI] [PubMed] [Google Scholar]
- 35.Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–27. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akca H, Tani M, Hishida T, et al. Activation of the AKT and STAT3 pathways and prolonged survival by a mutant EGFR in human lung cancer cells. Lung Cancer. 2006;54:25–33. doi: 10.1016/j.lungcan.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Engelman JA, Janne PA, Mermel C, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci USA. 2005;102:3788–93. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sordella R, Bell DW, Haber DA, et al. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki S, Igarashi S, Hanawa M, et al. Diversity of epidermal growth factor receptor-mediated activation of downstream molecules in human lung carcinomas. Mod Pathol. 2006;19:986–98. doi: 10.1038/modpathol.3800619. [DOI] [PubMed] [Google Scholar]
- 40.Alvarez JV, Greulich H, Sellers WR, et al. Signal transducer and activator of transcription 3 is required for the oncogenic effects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res. 2006;66:3162–8. doi: 10.1158/0008-5472.CAN-05-3757. [DOI] [PubMed] [Google Scholar]
- 41.Haura EB, Zheng Z, Song L, et al. Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin Cancer Res. 2005;11:8288–94. doi: 10.1158/1078-0432.CCR-05-0827. [DOI] [PubMed] [Google Scholar]
- 42.Sharma SV, Gajowniczek P, Way IP, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425–35. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitsudomi T, Kosaka T, Yatabe Y. Biological and clinical implications of EGFR mutations in lung cancer. Int J Clin Oncol. 2006;11:190–8. doi: 10.1007/s10147-006-0583-4. [DOI] [PubMed] [Google Scholar]
- 44.Ninomiya H, Hiramatsu M, Inamura K, et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas Significance of the micropapillary pattern and the hobnail cell type. Lung Cancer. 2009;63:235–40. doi: 10.1016/j.lungcan.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 45.Yokose T, Suzuki K, Nagai K, et al. Favorable and unfavorable morphological prognostic factors in peripheral adenocarcinoma of the lung 3 cm or less in diameter. Lung Cancer. 2000;29:179–88. doi: 10.1016/s0169-5002(00)00103-3. [DOI] [PubMed] [Google Scholar]
- 46.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–46. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Xu ML, Zhong HH, et al. EGFR mutations are more frequent in well-differentiated than in poor-differentiated lung adenocarcinomas. Pathol Oncol Res. 2008;14:373–9. doi: 10.1007/s12253-008-9113-1. [DOI] [PubMed] [Google Scholar]
- 48.Sonobe M, Manabe T, Wada H, et al. Mutations in the epidermal growth factor receptor gene are linked to smoking-independent, lung adenocarcinoma. Br J Cancer. 2005;93:355–63. doi: 10.1038/sj.bjc.6602707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomizawa Y, Iijima H, Sunaga N, et al. Clinicopathologic significance of the mutations of the epidermal growth factor receptor gene in patients with non-small cell lung cancer. Clin Cancer Res. 2005;11:6816–22. doi: 10.1158/1078-0432.CCR-05-0441. [DOI] [PubMed] [Google Scholar]
- 50.Tam IY, Chung LP, Suen WS, et al. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res. 2006;12:1647–53. doi: 10.1158/1078-0432.CCR-05-1981. [DOI] [PubMed] [Google Scholar]
- 51.Sasaki H, Endo K, Takada M, et al. L858R EGFR mutation status correlated with clinico-pathological features of Japanese lung cancer. Lung Cancer. 2006;54:103–8. doi: 10.1016/j.lungcan.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 52.Blons H, Cote JF, Le Corre D, et al. Epidermal growth factor receptor mutation in lung cancer are linked to bronchioloalveolar differentiation. Am J Surg Pathol. 2006;30:1309–15. doi: 10.1097/01.pas.0000213285.65907.31. [DOI] [PubMed] [Google Scholar]
- 53.De Oliveira Duarte Achcar R, Nikiforova MN, Yousem SA. Micropapillary lung adenocarcinoma: EGFR, K-ras, and BRAF mutational profile. Am J Clin Pathol. 2009;131:694–700. doi: 10.1309/AJCPBS85VJEOBPDO. [DOI] [PubMed] [Google Scholar]
- 54.Motoi N, Szoke J, Riely GJ, et al. Lung adenocarcinoma: modification of the 2004 WHO mixed subtype to include the major histologic subtype suggests correlations between papillary and micropapillary adenocarcinoma subtypes, EGFR mutations and gene expression analysis. Am J Surg Pathol. 2008;32:810–27. doi: 10.1097/PAS.0b013e31815cb162. [DOI] [PubMed] [Google Scholar]
- 55.Massuti B, Morán T, Porta R, et al. Multicenter prospective trial of customized erlotinib for advanced non-small cell lung cancer (NSCLC) patients (p) with epidermal growth factor receptor (EGFR) mutations: final results of the Spanish Lung Cancer Group (SLCG) trial. J Clin Oncol. 2009;27:412s. [Google Scholar]
- 56.Morinaga R, Okamoto I, Fujita Y, et al. Association of epidermal growth factor receptor (EGFR) gene mutations with EGFR amplification in advanced non-small cell lung cancer. Cancer Sci. 2008;99:2455–60. doi: 10.1111/j.1349-7006.2008.00962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang JW, Liu HP, Hsieh MH, et al. Increased epidermal growth factor receptor (EGFR) gene copy number is strongly associated with EGFR mutations and adenocarcinoma in non-small cell lung cancers: a chromogenic in situ hybridization study of 182 patients. Lung Cancer. 2008;61:328–39. doi: 10.1016/j.lungcan.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 58.Li AR, Chitale D, Riely GJ, et al. EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J Mol Diagn. 2008;10:242–8. doi: 10.2353/jmoldx.2008.070178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shibata T, Uryu S, Kokubu A, et al. Genetic classification of lung adenocarcinoma based on array-based comparative genomic hybridization analysis: its association with clinicopathologic features. Clin Cancer Res. 2005;11:6177–85. doi: 10.1158/1078-0432.CCR-05-0293. [DOI] [PubMed] [Google Scholar]
- 60.Blons H, Pallier K, Le Corre D, et al. Genome wide SNP comparative analysis between EGFR and KRAS mutated NSCLC and characterization of two models of oncogenic cooperation in non-small cell lung carcinoma. BMC Med Genomics. 2008;1:25. doi: 10.1186/1755-8794-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toyooka S, Tokumo M, Shigematsu H, et al. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res. 2006;66:1371–5. doi: 10.1158/0008-5472.CAN-05-2625. [DOI] [PubMed] [Google Scholar]
- 62.Chitale D, Gong Y, Taylor BS, et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene. 2009;28:2773–83. doi: 10.1038/onc.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marks JL, Golas B, Kirchoff T, et al. EGFR mutant lung adenocarcinomas in patients with germline BRCA mutations. J Thorac Oncol. 2008;3:805. doi: 10.1097/JTO.0b013e31817e4664. [DOI] [PubMed] [Google Scholar]
- 65.Kosaka T, Yatabe Y, Endoh H, et al. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–23. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 66.Iwakawa R, Kohno T, Anami Y, et al. Association of p16 homozygous deletions with clinicopathologic characteristics and EGFR/KRAS/p53 mutations in lung adenocarcinoma. Clin Cancer Res. 2008;14:3746–53. doi: 10.1158/1078-0432.CCR-07-4552. [DOI] [PubMed] [Google Scholar]
- 67.Mok T, Wu Y-L, Thongprasert S, et al. Phase III, randomised, open-label, first-line study of gefitinib vs carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer (IPASS) Ann Oncol. 2008;19:viii1. [Google Scholar]
- 68.Carey KD, Garton AJ, Romero MS, et al. Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res. 2006;66:8163–71. doi: 10.1158/0008-5472.CAN-06-0453. [DOI] [PubMed] [Google Scholar]
- 69.Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 70.Kancha RK, Von Bubnoff N, Peschel C, et al. Functional analysis of epidermal growth factor receptor (EGFR) mutations and potential implications for EGFR targeted therapy. Clin Cancer Res. 2009;15:460–7. doi: 10.1158/1078-0432.CCR-08-1757. [DOI] [PubMed] [Google Scholar]
- 71.Costa DB, Schumer ST, Tenen DG, et al. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol. 2008;26:1182–6. doi: 10.1200/JCO.2007.14.9039. [DOI] [PubMed] [Google Scholar]
- 72.Tang X, Varella-Garcia M, Xavier AC, et al. Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas. Cancer Prev Res. 2008;1:192–200. doi: 10.1158/1940-6207.CAPR-08-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gandhi J, Zhang J, Xie Y, et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4:e4576. doi: 10.1371/journal.pone.0004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jackman DM, Yeap BY, Sequist LV, et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:3908–14. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 75.Riely GJ, Pao W, Pham D, et al. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–44. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 76.Asahina H, Yamazaki K, Kinoshita I, et al. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor mutations. Br J Cancer. 2006;95:998–1004. doi: 10.1038/sj.bjc.6603393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Inoue A, Suzuki T, Fukuhara T, et al. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol. 2006;24:3340–6. doi: 10.1200/JCO.2005.05.4692. [DOI] [PubMed] [Google Scholar]
- 78.Kim D, Lee S, Lee J, et al. A multicenter phase II study to evaluate efficacy and safety of gefitinib as the first-line treatment for Korean patients (pts) with advanced pulmonary adenocarcinoma harboring EGFR mutations. J Clin Oncol. 2009;27:423s. doi: 10.1016/j.lungcan.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 79.Koyama N, Jinn Y, Takabe K, et al. The characterization of gefitinib sensitivity and adverse events in patients with non-small cell lung cancer. Anticancer Res. 2006;26:4519–25. [PubMed] [Google Scholar]
- 80.Hesterberg T, Moore DS, Monaghan S. Bootstrap methods and permutation tests. In: Moore DS, McCabe GP, et al., editors. Introduction to the practice of statistics. 5th ed. New York: WHFreeman; 2006. pp. 14–170. [Google Scholar]
- 81.Ahn MJ, Park BB, Ahn JS, et al. Are there any ethnic differences in molecular predictors of erlotinib efficacy in advanced non-small cell lung cancer? Clin Cancer Res. 2008;14:3860–6. doi: 10.1158/1078-0432.CCR-07-4608. [DOI] [PubMed] [Google Scholar]
- 82.Amann JM, Lee J, Roder H, et al. Genetic and proteomic features associated with survival after treatment with erlotinib in first-line therapy of non-small cell lung cancer. J Clin Oncol. 2009;27:429s. doi: 10.1097/JTO.0b013e3181c8cbd9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hirsch FR, Dziadziuszko R, Varella-Garcia L, et al. Randomized phase II study of erlotinib (E) or intercalated E with carboplatin/paclitaxel (CP) in chemotherapy-naive advanced NSCLC: correlation of biomarker status and clinical benefit. J Clin Oncol. 2009;27:413s. [Google Scholar]
- 84.Jackman DM, Yeap BY, Lindeman NI, et al. Phase II clinical trial of chemotherapy-naive patients > or = 70 years of age treated with erlotinib for advanced non-small-cell lung cancer. J Clin Oncol. 2007;25:760–6. doi: 10.1200/JCO.2006.07.5754. [DOI] [PubMed] [Google Scholar]
- 85.Jackman DM, Cioffredi L, Lindeman NI, et al. Phase II trial of erlotinib in chemotherapy-naive women with advanced pulmonary adenocarcinoma. J Clin Oncol. 2009;27:423s. [Google Scholar]
- 86.Miller VA, Riely GJ, Zakowski MF, et al. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol. 2008;26:1472–8. doi: 10.1200/JCO.2007.13.0062. [DOI] [PubMed] [Google Scholar]
- 87.Pirker R, Su W, Rooneem R, et al. Clinical outcomes with erlotinib in relation to biomarker status: analyses from the open -label TRUST study in advanced non-small-cell lung cancer (NSCLC) Ann Oncol. 2008:19. [Google Scholar]
- 88.Rosell R, Perez-Roca L, Sanchez JJ, et al. Customized treatment in non-small-cell lung cancer based on EGFR mutations and BRCA1 mRNA expression. PLoS One. 2009;4:e5133. doi: 10.1371/journal.pone.0005133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schneider CP, Heigener D, Schott-von-Romer K, et al. Epidermal growth factor receptor-related tumor markers and clinical outcomes with erlotinib in non-small cell lung cancer: an analysis of patients from german centers in the TRUST study. J Thorac Oncol. 2008;3:1446–53. doi: 10.1097/JTO.0b013e31818ddcaa. [DOI] [PubMed] [Google Scholar]
- 90.Zhou S, Zhou C, Ren S, et al. Predictive effects of serum VEGF, TGF-a and EGFR mutations in advanced non-small cell lung cancer treated by erlotinib. J Clin Oncol. 2008;26:716s. [Google Scholar]
- 91.Bell DW, Lynch TJ, Haserlat SM, et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol. 2005;23:8081–92. doi: 10.1200/JCO.2005.02.7078. [DOI] [PubMed] [Google Scholar]
- 92.Buckingham LE, Coon JS, Morrison LE, et al. The prognostic value of chromosome 7 polysomy in non-small cell lung cancer patients treated with gefitinib. J Thorac Oncol. 2007;2:414–22. doi: 10.1097/01.JTO.0000268675.02744.b0. [DOI] [PubMed] [Google Scholar]
- 93.Cappuzzo F, Ligorio C, Janne PA, et al. Prospective study of gefitinib in epidermal growth factor receptor fluorescence in situ hybridization-positive/phospho-Akt-positive or never smoker patients with advanced non-small-cell lung cancer: the ONCOBELL trial. J Clin Oncol. 2007;25:2248–55. doi: 10.1200/JCO.2006.09.4300. [DOI] [PubMed] [Google Scholar]
- 94.Chou TY, Chiu CH, Li LH, et al. Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non-small cell lung cancer. Clin Cancer Res. 2005;11:3750–7. doi: 10.1158/1078-0432.CCR-04-1981. [DOI] [PubMed] [Google Scholar]
- 95.Cortes-Funes H, Gomez C, Rosell R, et al. Epidermal growth factor receptor activating mutations in Spanish gefitinib-treated non-small-cell lung cancer patients. Ann Oncol. 2005;16:1081–6. doi: 10.1093/annonc/mdi221. [DOI] [PubMed] [Google Scholar]
- 96.D’Addario G, Rauch D, Stupp R, et al. Multicenter phase II trial of gefitinib first-line therapy followed by chemotherapy in advanced non-small-cell lung cancer (NSCLC): SAKK protocol 19/03. Ann Oncol. 2008;19:739–45. doi: 10.1093/annonc/mdm564. [DOI] [PubMed] [Google Scholar]
- 97.Dongiovanni D, Daniele L, Barone C, et al. Gefitinib (ZD1839): therapy in selected patients with non-small cell lung cancer (NSCLC)? Lung Cancer. 2008;61:73–81. doi: 10.1016/j.lungcan.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 98.Fukuoka M, Wu Y, Thongprasert S, et al. Biomarker analyses from a phase III, randomized, open-label, first-line study of gefitinib (G) versus carboplatin/paclitaxel (C/P) in clinically selected patients (pts) with advanced non-small cell lung cancer (NSCLC) in Asia (IPASS) J Clin Oncol. 2009;27:408s. doi: 10.1200/JCO.2010.33.4235. [DOI] [PubMed] [Google Scholar]
- 99.Han SW, Kim TY, Lee KH, et al. Clinical predictors versus epidermal growth factor receptor mutation in gefitinib-treated non-small-cell lung cancer patients. Lung Cancer. 2006;54:201–7. doi: 10.1016/j.lungcan.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 100.Hirsch FR, Varella-Garcia M, Cappuzzo F, et al. Combination of EGFR gene copy number and protein expression predicts outcome for advanced non-small-cell lung cancer patients treated with gefitinib. Ann Oncol. 2007;18:752–60. doi: 10.1093/annonc/mdm003. [DOI] [PubMed] [Google Scholar]
- 101.Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–55. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 102.Hirsch FR, Varella-Garcia M, McCoy J, et al. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group Study. J Clin Oncol. 2005;23:6838–45. doi: 10.1200/JCO.2005.01.2823. [DOI] [PubMed] [Google Scholar]
- 103.Hong J, Kim Y, Park S, et al. Epidermal growth factor-receptor mutations and the clinical outcome in male smokers with squamous cell carcinoma of lung. J Clin Oncol. 2008;26:759s. doi: 10.3346/jkms.2009.24.3.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ichihara S, Toyooka S, Fujiwara Y, et al. The impact of epidermal growth factor receptor gene status on gefitinib-treated Japanese patients with non-small-cell lung cancer. Int J Cancer. 2007;120:1239–47. doi: 10.1002/ijc.22513. [DOI] [PubMed] [Google Scholar]
- 105.Inoue A, Kobayashi K, Usui K, et al. First-line gefitinib for patients with advanced non-small-cell lung cancer harboring epidermal growth factor receptor mutations without indication for chemotherapy. J Clin Oncol. 2009;27:1394–400. doi: 10.1200/JCO.2008.18.7658. [DOI] [PubMed] [Google Scholar]
- 106.Kim KS, Jeong JY, Kim YC, et al. Predictors of the response to gefitinib in refractory non-small cell lung cancer. Clin Cancer Res. 2005;11:2244–51. doi: 10.1158/1078-0432.CCR-04-2081. [DOI] [PubMed] [Google Scholar]
- 107.Kimura H, Suminoe M, Kasahara K, et al. Evaluation of epidermal growth factor receptor mutation status in serum DNA as a predictor of response to gefitinib (IRESSA) Br J Cancer. 2007;97:778–84. doi: 10.1038/sj.bjc.6603949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kobayashi K, Inoue A, Maemondo M, et al. First-line gefitinib versus first-line chemotherapy by carboplatin (CBDCA) plus paclitaxel (TXL) in non-small cell lung cancer (NSCLC) patients (pts) with EGFR mutations: a phase III study (002) by North East Japan Gefitinib Study Group. J Clin Oncol. 2009;27:411s. [Google Scholar]
- 109.Massarelli E, Varella-Garcia M, Tang X, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13:2890–6. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 110.Oshita F, Matsukuma S, Yoshihara M, et al. Novel heteroduplex method using small cytology specimens with a remarkably high success rate for analysing EGFR gene mutations with a significant correlation to gefitinib efficacy in non-small-cell lung cancer. Br J Cancer. 2006;95:1070–5. doi: 10.1038/sj.bjc.6603396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pallis AG, Voutsina A, Kalikaki A, et al. ‘Classical’ but not ‘other’ mutations of EGFR kinase domain are associated with clinical outcome in gefitinib-treated patients with non-small cell lung cancer. Br J Cancer. 2007;97:1560–6. doi: 10.1038/sj.bjc.6604068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–9. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 113.Shao Y, Lin Z, Hu F, et al. Quality of life in advanced non-small cell lung cancer patients receiving first-line gefitinib monotherapy. J Clin Oncol. 2009;27:511s. [Google Scholar]
- 114.Shoji F, Yano T, Yoshino I, et al. The characteristics and failure pattern of gefitinib responders with postoperative recurrence of pulmonary adenocarcinoma. Eur J Surg Oncol. 2008;34:89–93. doi: 10.1016/j.ejso.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 115.Sugio K, Uramoto H, Onitsuka T, et al. Prospective phase II study of gefitinib in non-small cell lung cancer with epidermal growth factor receptor gene mutations. Lung Cancer. 2009;64:314–8. doi: 10.1016/j.lungcan.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 116.Sunaga N, Tomizawa Y, Yanagitani N, et al. Phase II prospective study of the efficacy of gefitinib for the treatment of stage III/IV non-small cell lung cancer with EGFR mutations, irrespective of previous chemotherapy. Lung Cancer. 2007;56:383–9. doi: 10.1016/j.lungcan.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 117.Sutani A, Nagai Y, Udagawa K, et al. Gefitinib for non-small-cell lung cancer patients with epidermal growth factor receptor gene mutations screened by peptide nucleic acid-locked nucleic acid PCR clamp. Br J Cancer. 2006;95:1483–9. doi: 10.1038/sj.bjc.6603466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Takano T, Ohe Y, Tsuta K, et al. Epidermal growth factor receptor mutation detection using high-resolution melting analysis predicts outcomes in patients with advanced non small cell lung cancer treated with gefitinib. Clin Cancer Res. 2007;13:5385–90. doi: 10.1158/1078-0432.CCR-07-0627. [DOI] [PubMed] [Google Scholar]
- 119.Tamura K, Okamoto I, Kashii T, et al. Multicentre prospective phase II trial of gefitinib for advanced non-small cell lung cancer with epidermal growth factor receptor mutations: results of the West Japan Thoracic Oncology Group trial (WJTOG0403) Br J Cancer. 2008;98:907–14. doi: 10.1038/sj.bjc.6604249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Varella-Garcia M, Mitsudomi T, Yatabe Y, et al. EGFR and HER2 genomic gain in recurrent non-small cell lung cancer after surgery: impact on outcome to treatment with gefitinib and association with EGFR and KRAS mutations in a Japanese cohort. J Thorac Oncol. 2009;4:318–25. doi: 10.1097/JTO.0b013e31819667a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu SG, Chang YL, Hsu YC, et al. Good response to gefitinib in lung adenocarcinoma of complex epidermal growth factor receptor (EGFR) mutations with the classical mutation pattern. Oncologist. 2008;13:1276–84. doi: 10.1634/theoncologist.2008-0093. [DOI] [PubMed] [Google Scholar]
- 122.Wu M, Zhao J, Song SW, et al. EGFR mutations are associated with prognosis but not with the response to front-line chemotherapy in the Chinese patients with advanced non-small cell lung cancer. Lung Cancer. 2009 doi: 10.1016/j.lungcan.2009.04.011. DOI: 10.1016/j.lungcan.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 123.Xu JM, Han Y, Duan HQ, et al. EGFR mutations and HER2/3 protein expression and clinical outcome in Chinese advanced non-small cell lung cancer patients treated with gefitinib. J Cancer Res Clin Oncol. 2009;135:771–82. doi: 10.1007/s00432-008-0512-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yoshida K, Yatabe Y, Park JY, et al. Prospective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer. J Thorac Oncol. 2007;2:22–8. [PubMed] [Google Scholar]
- 125.Zhang XT, Li LY, Mu XL, et al. The EGFR mutation and its correlation with response of gefitinib in previously treated Chinese patients with advanced non-small-cell lung cancer. Ann Oncol. 2005;16:1334–42. doi: 10.1093/annonc/mdi340. [DOI] [PubMed] [Google Scholar]
- 126.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–9. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 127.Lee KH, Han SW, Hwang PG, et al. Epidermal growth factor receptor mutations and response to chemotherapy in patients with non-small-cell lung cancer. Jpn J Clin Oncol. 2006;36:344–50. doi: 10.1093/jjco/hyl039. [DOI] [PubMed] [Google Scholar]
- 128.Tambo Y, Kasahara K, Sone T, et al. Prognostic and predictive impact of EGFR and K-ras mutation, and EGFR gene copy number in patients with advanced non-small cell lung cancer (NSCLC) who received first-line cytotoxic chemotherapy. J Clin Oncol. 2009;27:430s. [Google Scholar]
- 129.Morita S, Okamoto I, Kobayashi K, et al. Combined survival analysis of prospective clinical trials of gefitinib for non-small cell lung cancer with EGFR mutations. Clin Cancer Res. 2009;15:4493–8. doi: 10.1158/1078-0432.CCR-09-0391. [DOI] [PubMed] [Google Scholar]
- 130.Tsao AS, Tang XM, Sabloff B, et al. Clinicopathologic characteristics of the EGFR gene mutation in non-small cell lung cancer. J Thorac Oncol. 2006;1:231–9. doi: 10.1016/s1556-0864(15)31573-2. [DOI] [PubMed] [Google Scholar]
- 131.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–8. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 132.Langer CJ. The “Lazarus response” in treatment-naive, poor performance status patients with non-small-cell lung cancer and epidermal growth factor receptor mutation. J Clin Oncol. 2009;27:1350–4. doi: 10.1200/JCO.2008.20.4859. [DOI] [PubMed] [Google Scholar]