

Abstract
E10A, a replication-defective adenovirus carrying human endostatin gene, has finished Phase I clinical trials for solid cancers. We assessed whether the combination of E10A with docetaxel would enhance antiangiogenic activities and inhibit prostate cancer growth and metastases. Combination use of conditioned medium from prostate cancer cells infected by E10A and docetaxel exerted synergistic inhibition of HUVECs proliferation, migration and tube formation, compared with either agent alone. In prostate cancer s.c. xenograft models, combined therapy resulted in significant tumour growth inhibition and survival improvement. The antitumoural effect was tightly correlated with a remarkable decrease in tumour cell proliferation, microvessel, especially immature vasculature and significant increase in apoptosis induction. Systemic administration of E10A and docetaxel also effectively inhibited orthotopic growth and metastases of prostate cancer and achieved better in vivo antiangiogenic effects than either agent alone. Our data indicate that E10A in combination with docetaxel exert enhanced antiangiogenic activities and inhibit prostate cancer growth and metastases. Therefore, this approach may be an effective treatment for advanced prostate cancer and deserves more extensive investigation.
Keywords: prostate cancer, antiangiogensis, endostatin, adenovirus, docetaxel
Introduction
Prostate cancer is the most common lethal malignancy diagnosed and the second leading cause of male cancer death in the United States [1]. Most advanced prostate cancer ultimately progresses to an androgen-independent state resulting in death due to widespread metastases [2]. The conventional treatment, including androgen ablation, prostatectomy, radiotherapy and chemotherapy, has improved survival rate. But the therapeutic efficacy in patients with advanced prostate cancer was limited. Accordingly, novel therapeutic strategies must be developed to improve therapeutic effectiveness against advanced prostate cancer that is resistant to conventional treatment.
The growth and metastasis of prostate cancer are angiogenesis-dependent. Therefore, microvascular endothelial cells recruited by a tumour have become an important target in cancer therapy [3]. Endostatin, a 20-kD C-terminal fragment of collagen XVIII, inhibits endothelial cell proliferation and tumour angiogenesis [4] and confers survival advantage on cancer patients [5]. But long-term systemic delivery of recombinant protein is expensive, is a painful experience for patients, and may be insufficient to obtain high concentrations of the therapeutic molecule in the tumour tissue [6]. Antiangiogenic gene therapy can overcome the defects and become an effective and promising approach in the treatment of cancer [7]. Recently, we developed a recombinant replication-defective adenovirus, E10A, that carries the human endostatin gene. E10A has been shown to inhibit tumour growth and angiogenesis in a wide variety of animal tumour models [8, 9]. This virus was registered by the U.S. Food and Drug Administration (FDA, Rockville, MD, USA) in 2005 (ClinicalTrials.gov identifier, NCT00262327) and has finished Phase I clinical trials for the treatment of advanced solid tumours [10]. Docetaxel is a member of the taxane family, a class of cytotoxic chemotherapeutic agents that bind to β tubulin, thereby stabilizing microtubules and causing cell-cycle arrest and apoptosis. It has become the standard first-line therapy for hormone refractory prostate cancer [2, 11].
In this study, we evaluated the effect E10A in combination with docetaxel on HUVECs in vitro and human prostate cancer s.c. xenograft, orthotopic tumours and angiogenic models in vivo. Combination treatment with E10A and docetaxel enhanced the inhibition effects on endothelial cell function and tumour growth and metastasis of prostate cancer. The results of tumour microvascular analysis, alginate capsule and Matrigel plug models revealed that the combined therapy significantly enhanced the antiangiogenic efficacy, compared with E10A or docetaxel alone.
Materials and methods
Cell culture
HUVECs were isolated from umbilical cords as described previously [12] and cultured to a passage of 3–7. Cells were grown in M199 medium, supplemented with 15% foetal bovine serum (Hyclone Laboratories Inc, Logan, UT, USA), 4 ng/ml bFGF (Promega, Madison, WI, USA), 0.05 mg/ml ECGS (BD Biosciences, Bedford, MA, USA) and 0.1 mg/ml heparin. Human prostate cancer PC-3 and DU-145 cells (American Type Culture Collection, Rockville, MD, USA) were cultured in Ham’F12 medium and α-MEM medium contained 10% foetal bovine serum, respectively.
Recombinant adenoviruses and docetaxel
Replication-defective recombinant adenoviral vectors carrying the human endostatin gene (E10A) and β-galactosidase (Ad-LacZ) were generated as described previously [13]. The cDNA for human endostatin was inserted following an IL-2 signal sequence. The transgene expression was driven by the cytomegalovirus (CMV) promoter. The viruses were titred using Adeno-XTM Rapid titer Kit (BD Biosciences Clontech, USA) and stored at −70°C. Docetaxel (Aventis Pharmaceuticals, Bridgewater, NJ, USA) was purchased from the Cancer Centre Pharmacy of Sun Yat-sen University. The supernatants of PC-3 cells after 72 hrs of infection with E10A or Ad-LacZ at MOI of 100 were harvested as conditioned medium (CM) and evaluated endostatin protein using a human endostatin ELISA kit (R&D systems, Minneapolis, MN, USA) according to the recommendation of the manufacturer. CM from Ad-LacZ served as negative control, which did not affect HUVECs biological function in vitro compared with untreated group.
In vitro determination of proliferation
Effect of endostatin and docetaxel either alone or in combination on endothelial cell proliferation was determined by isobolographic analysis as described previously [14]. Recombinant endostatin expressed in 293 cells was purified from the culture supernatants by chromatography. HUVECs (3000 per well) were dispensed in each well of a 96 culture plate (BD Falcon, CA, USA). After allowing for attachment overnight, HUVECs were treated with endostatin for 30 min., and docetaxel was added in an array to quintuplicate sets of cultures. After 72 hrs, cell proliferation was measured with Cell Counting Kit-8 (Dojindo Molecular Technologies, INC., Gaithersburg, MD, USA) according to the instruction of the manufacturer. For in vitro experiments, a docetaxel dose of 0.1 nM (IC90) was chosen. In proliferation assay, after cells were placed into plate overnight, the medium was replaced with 50 μl of CM. Thirty minutes later, HUVECs complete medium (150 μl) in the presence or absence of 0.1 nM docetaxel was added.
Tube formation assay
HUVECs (10,000 per well) were suspended in 25 μl of CM and gently added to Matrigel-coated wells. Thirty minutes later, HUVECs complete medium (75 μl) containing 20 ng/ml vascular endothelial growth factor (VEGF) (Peprotech, London, UK) with or without 0.1 nM docetaxel was added. After 16 hrs, images were captured under ×50 magnification, and tube formation was scored as follows: a three-branch-point event was scored as one tube [15].
Boyden chamber assay of endothelial cell migration
Transwell inserts with 8-μm-pore size (Costar, MD, USA) were coated with 0.78 mg/ml Matrigel (Becton Dickinson, Bedford, MA, USA). Harvested HUVECs (20,000 per well) were preincubated with CM for 30 min. before 150 μl of serum-free M199 with or without 0.1 nM docetaxel was added to the upper chamber of the inserts and HUVECs complete medium (500 μl) containing 50 ng/ml VEGF was added to the lower wells. After 6 hrs of incubation, the upper surface of the insert was swabbed to remove nonmigrated cells. HUVECs were fixed by cold 70% ethanol and fluorescently stained with DAPI (Roche Molecular Biochemicals, Germany). Cell migration was quantitated by counting the number of migrated cells under ×100 magnification with a DMIRB Leica fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Prostate cancer s.c. xenografts model
Male athymic nude mice (4–6 weeks old) were obtained from Beijing vitalriver Experimental Animals Co. Ltd. After 1 week of adaptation, human prostate cancer cells (PC-3, DU-145) were injected s.c. into the flanks of mice (5 × 106 cells in 100 μl of PBS). When 150-250 mm3 tumours had formed, mice were randomly assigned to groups and treatment was initiated. For antitumour study, six groups of animals with PC-3 xenograft were used, each group had 10 animals. The first group of animals was left untreated. The second group and fifth group of animals were injected intratumourally (i.t.) with Ad-LacZ (1 × 109 pfu) once a week. The third and sixth groups of animals were injected i.t. with E10A (1 × 109 pfu) once a week. The fourth group of animals was treated i.p. with docetaxel (10 mg/kg) weekly 24 hrs after each vector injection. In addition to the virus injection, the fifth and sixth groups of animals were also treated with docetaxel at the same dose and schedule as described previously. Four groups of animals with DU-145 cells xenograft (n= 10) were treated with Ad-LacZ, E10A, docetaxel plus Ad-LacZ, E10A plus docetaxel, respectively, with the same protocol as animals with PC-3 xenografts. All animals were treated for 4 weeks. Tumour volume (V) was determined twice a week by direct measurement with callipers and was calculated by the formula: V = L×W2/2 (L, length; W, width). Upon termination of the experiment, the tumours were removed. One part of the tumour tissue was fixed in 10% neutral buffered formalin. Another part of the tumour tissue was embedded in OCT compound for storage at –70°C. For survival studies, there were four groups mice with PC-3 xenograft (n= 10). After 4 weeks of treatment, animals either found dead or killed when tumours were observed by palpation to approach 10% body weight or individual animals seemed to be stressed by weight loss, ruffled skins and/or lethargy [16].
Histology analysis
For immunohistochemical studies, tumours were fixed in buffered formalin and embedded in paraffin. Paraffin-embedded tissue sections were incubated with rabbit anti-human Ki-67, rabbit anti-mouse CD31 (Neomarkers, Lab Vision, CA, USA) and rat anti-mouse LYVE-1 (Santa Cruz Biotechnology Inc., Berkeley, CA, USA). The sections were visualized with DAB using DAKO Envision system (Dako, Carpinteria, CA, USA). Apoptosis analysis was performed with in situ Cell Death Detection kit (TUNEL) according to the manufacturer’s instruction (Roche Molecular Biochemicals, Mannheim, Germany). To quantify tumour cell proliferation, apoptosis and MVD, 10 random fields at ×200 were captured for imaging within tumour tissue. The characteristics of blood vessels from the tumours were analysed by dual immunofluorescence staining for endothelial cells and pericytes [17]. Pericyte-coated mature vasculatures were differentially indentified by staining with a mouse monoclonal anti-α-smooth muscle actin (Dako) and rabbit anti-mouse CD31. Frozen tissue sections were immunostained with a mixture of primary antibodies, and then with a combination of TRITC-conjugated goat anti-rabbit IgG and FITC-conjugated goat anti-mouse IgG (Santa Cruz). Endothelial cells were identified by red fluorescence, and pericytes were detected by green fluorescence. Ten independent fields at ×400 were used in morphometric analysis to determine vessel length positive for pericyte coating.
Orthotopic prostate cancer model
Orthotopic prostate cancer model was established in male athymic nude mice by surgical implantation [18]. Mice were anaesthetized by chloral hydrate and placed in the supine position. A low midline abdominal incision was made and 1 × 106 PC-3 cells in 20 μl of phosphate buffer solution (PBS) were injected into the dorsal lobe of the prostate using a 30-gauge needle with a dissecting microscope. The surgical wound was closed in two layers with interrupted sutures. Two weeks after surgical orthotopic implantation, the mice were randomized into six groups. Group and treatment were performed with the same protocol as PC-3 s.c. xenografts model, except that the method of adenovirus administration was intravenous injection instead of intratumoural injection. After 4 weeks of treatment, at autopsy, the weight of the primary tumours was recorded, and the metastasis to regional lymph nodes was determined. The endostatin levels and VEGF in the serum were determined using ELISA kits (R&D systems, Minneapolis, MN, USA) according to the recommendation of the manufacturer.
Alginate-encapsulated tumour cells assay
Alginate-encapsulated tumour cells assay was performed in vivo as previously described [19, 20]. Four alginate beads (1 × 105 PC-3 cells per bead) were implanted s.c. into an incision made on the dorsal side of athymic mice. Mice were randomized into four groups (n= 4). Twelve hours later, the mice were intravenously injected with Ad-LacZ or E10A (1 × 109 pfu/week) and 10 mg/kg docetaxel was given i.p. 24 h after each virus injection for a total of 20 mg/kg. On 15 day after implanting, the mice were injected with 0.1 ml of 100 mg/kg FITC-dextran (Sigma Chemical Co., St. Louis, MO, USA) solution via tail vein. Alginate beads were rapidly removed and photographed 20 min. after FITC-dextran injection. The uptake of FITC-dextran was measured against a standard curve of FITC-dextran.
Statistical analysis
Results were evaluated using t-test and one-way ANOVA with SPSS 11.0 software (SPSS, Inc., Chicago, IL, USA), unless otherwise specified. The incidence of lymph node metastasis between two groups was compared using Fisher’s exact test. Results of survival were evaluated using Kaplan–Meier. P < 0.05 was considered statistically significant.
Results
Inhibition of HUVECs proliferation
In vitro, endostatin did not affect PC-3 cells proliferation. Isobolographic analysis demonstrated combination of endostatin and docetaxel could synergistically inhibit HUVECs proliferation (data not shown). To evaluate the effect of CM on HUVECs biological function in vitro, HUVECs were incubated with CM at a final concentration of 25%. The expression levels of endostatin in CM-E10A were 392 ± 21 ng/ml. We found that incubation of HUVECs with CM-E10A inhibited proliferation by 20%, in comparison with CM-Ad-LacZ control (Fig. 1A). However, combination of E10A and 0.1 nM docetaxel (IC90) appeared to have higher antiproliferative rate of 59% on HUVECs, compared with CM-E10A or docetaxel plus CM-Ad-LacZ (P < 0.001).
Fig 1.
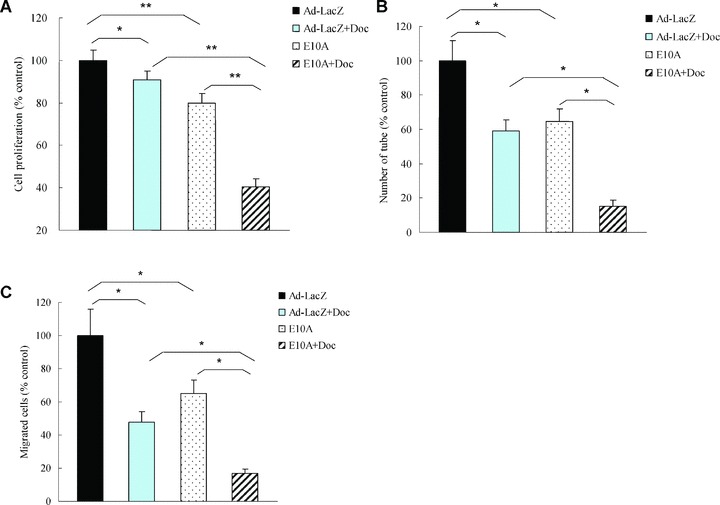
The effects of E10A and docetaxel on biological function of HUVECs. (A) Proliferation assay. HUVECs were treated with CM from infected PC-3 cells or/and 0.1 nM docetaxel, and cell proliferation was evaluated after 72 hrs. Columns, cell proliferation normalized against Ad-LacZ control (mean ± S.D.). (B) Tube formation assay. HUVECs were treated with CM or/and 0.1 nM docetaxel. Cells were plated on 96-well plates with Matrigel. After 16 hrs of incubation, images of tube formation were captured, and tube formation was scored in one ×50 microscopic field. The number of tube formation was quantitated, and the data are presented as the mean ± S.D. per field (×50) and were normalized to Ad-LacZ-treated control (n= 5). *P < 0.001; (C) Migration assay. HUVECs were treated CM or/and 0.1 nM docetaxel and pipetted into inserts of Matrigel-coated transwells. Addition of chemoattractant, HUVECs complete medium containing 50 ng/ml VEGF, to the lower well of Boyden chamber stimulated endothelial migration to the underside of the transwell membrane. After 6 hrs of incubation, migrated cells were stained by DAPI. The number of cells that migrated was counted by microscopy, and the data are presented as the mean ± S.D. per field (×100) and were normalized to Ad-LacZ-treated control (n= 6), *P < 0.001.
Inhibition of HUVECs migration and tube formation
We investigated the effect of E10A and docetaxel on HUVECs tube formation. Control HUVECs, grew on Matrigel, aligned to form tube-like structures over 16 hrs and connected tubes with multicentric junctions. Either CM-E10A or docetaxel inhibited tube formation by 35% and 41%, respectively (Fig. 1B), compared with the control. When combined, however, E10A and docetaxel inhibited tube formation to a greater extent (85%) than either agent alone (P < 0.001).
Boyden chamber assay was used to assess the effect of combination of E10A and docetaxel on HUVECs migration. Stimulation of HUVECs along a directional gradient of VEGF resulted in migration to the underside of the insert, the migration of HUVECs treated with CM-E10A or docetaxel plus CM-Ad-LacZ was significantly reduced by 35%, 52%, respectively, in comparison with the control (Fig. 1C). Interestingly, combination of CM-E10A and docetaxel showed stronger inhibitory effect on HUVECs migration (P < 0.001), compared with CM-E10A or docetaxel plus CM-Ad-LacZ.
E10A and docetaxel inhibited growth of prostate cancer s.c. xenografts
We investigated the antitumour efficacy of E10A in combination with docetaxel in the prostate cancer s.c. xenograft models. We found that tumour growth was significantly delayed by E10A alone or docetaxel plus Ad-LacZ, compared with Ad-LacZ control (Fig. 2A). For the PC-3, tumour growth delay was significant from day 14 after the beginning of therapy onward (P < 0.05). For the DU-145, the growth delay was significant from day 10 onward (P < 0.05). However, the combination of E10A and docetaxel resulted in a significantly (P < 0.05) greater tumour growth delay than was produced by other groups, starting on day 7 for DU-145 and day 14 for PC-3, until the end of experiments. The inhibition rate of the treated group was determined according to tumour weight (Fig. 2B). In the PC-3 xenograft models, the inhibition rates of E10A, docetaxel plus Ad-LacZ and E10A plus docetaxel groups were 46%, 31%, and 74%, respectively. In the DU-145 models, the corresponding inhibition rates were 43%, 32% and 70%, respectively. In the case of PC-3, there was no significant effect on growth kinetics and tumour weight in tumours treated with Ad-LacZ compared with untreated group, treated with docetaxel compared with docetaxel plus Ad-LacZ (P > 0.05).
Fig 2.
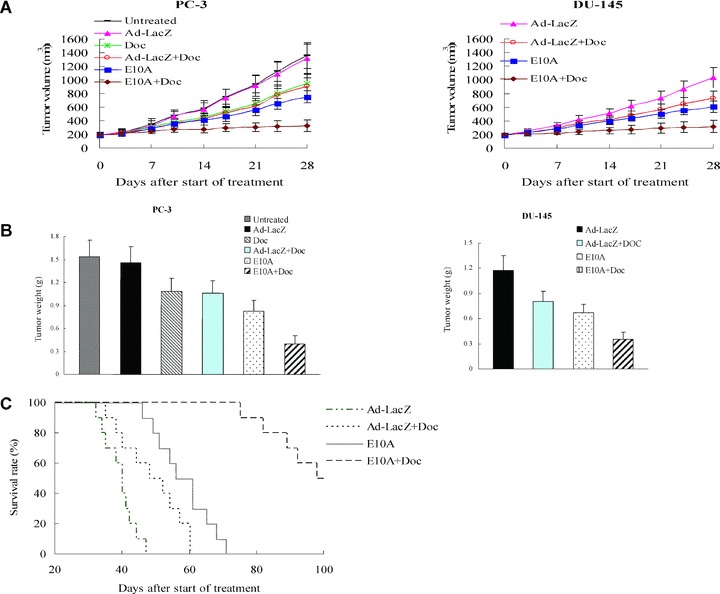
The effect of combined with E10A and docetaxel on the growth of s.c. xenografts of human prostate cancer in nude mice. When 150-250 mm3 s.c. xenografts had formed, mice were randomized groups and then subjected to 4 weeks treatment. (A) Time-dependent evolution of tumour volume in athymic mice with PC-3 and DU-145 cells xenografts (n= 10). (B) Tumour weight estimate of antitumour effect (n= 10). Mice were killed after 4 weeks of treatment and tumours were resected and weighted. (C) Kaplan–Meier estimate of survival time (n= 10). Survival of mice injected with E10A in combination with docetaxel had a better survival survival rate compared with additional groups of mice included Ad-LacZ control, E10A alone, Ad-LacZ plus docetaxel (P < 0.001 by log-rank method).
The long-term outcome of treatment was evaluated by survival rate of tumour-burden mice. There was no additional treatment after 4 weeks of treatment. Figure 2C shows the survival curve of treated mice. Although E10A alone prolonged median survival over Ad-LacZ control, and docetaxel plus Ad-LacZ animals (56 days versus 40, 48 days, respectively), there were no animals alive past day 71. However, after combination treatment with E10A and docetaxel, median survival was significantly increased to 98 days (P < 0.001 by log rank compared with E10A alone or docetaxel plus Ad-LacZ).
Evaluation of tumour cell proliferation and apoptosis
Representative tumours harvested from each group were processed for immunohistochemical analyses (Fig. 3A). Cell proliferation and apoptosis were evaluated using Ki-67 stain and TUNEL method, respectively. Results are shown in Table 1. The median number of Ki-67-positive tumour cells counted per 200× field was decreased from 596 in control tumours to 370 after therapy with E10A alone (P < 0.001). Treatment with docetaxel did not result in a significant reduction in cell proliferation when compared with the control (P= 0.62). The combination of E10A and docetaxel significantly inhibited proliferation compared with the other treatment group (P < 0.05). The mean number of apoptotic cells counted per 200× field was significantly increased from 6 in Ad-LacZ control tumours to 28, 25 after therapy with E10A or docetaxel plus Ad-LacZ, respectively (P < 0.001). The tumours from combination exhibited the greatest apoptotic cells in all groups (P < 0.001).
Fig 3.
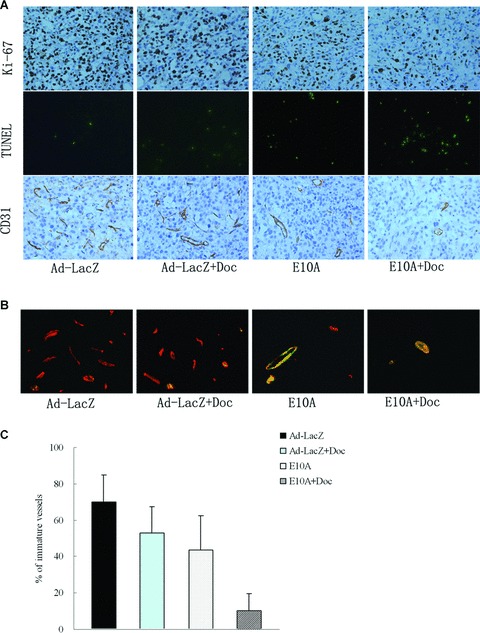
Immunohistochemical analyses PC-3 s.c. tumours in different treatment groups. (A) The sections were immunostained for expression of Ki-67 (cell proliferation), TUNEL (cell apoptosis) and CD31 (endothelial cells). Representative images were shown (×200). (B) Vessels were stained with rabbit anti-CD31 (red) and anti-mouse α-smooth muscle actin (green) to identify whether vessels are mature. Representative images of vessels positive for CD31 (endothelium cells) and α-smooth muscle actin (pericytes) were shown (×400). (C) Determination of immature vessels. Ten independent fields at ×400 were used in morphometric analysis to determine whether vessel is mature. Columns, mean ± S.D.
Table 1.
Immuohistochemical analysis of human prostate cancer PC-3 s.c. xenogrft tumours
| Treatment group | Ki-67a | TUNELa | CD31a |
|---|---|---|---|
| Ad-LacZ | 596 ± 144 | 6 ± 2 | 52 ± 15 |
| Ad-LacZ +Doc | 512 ± 113 | 25 ± 6 b | 42 ± 11 |
| E10A | 370 ± 100 b | 28 ± 9 b | 21 ± 4 b |
| E10A+Doc | 228 ± 78 c | 67 ± 18 d | 10 ± 3 d |
Median number proliferating cells (Ki-67 stain), apoptotic cells (TUNEL stain) and vessels (CD31 stain), respectively, per high-power field (×200).
P < 0.05, compared with Ad-LacZ control.
P < 0.05, compared with E10A and docetaxel plus Ad-LacZ.
Inhibition of tumour neovascularization
To investigate whether the antitumour effect is associated with a reduction in the tumour angiogenesis, MVD was determined by CD31-positive cells per 200× field. MVD was not significantly reduced in tumours of mice treated with docetaxel plus Ad-LacZ, but was in tumours of mice treated with E10A alone or in combination with docetaxel (21 and 10, respectively; P < 0.001). The effect of antiangiogenesis was enhanced in mice treated with E10A in combination with docetaxel.
We sought out to further characterize tumour vessels by dual immunofluorescence staining. Immature vessels were defined as vessel structures composed of endothelial cells, whereas mature vessels were defined as vessel structures containing endothelial cells and pericytes. Figure 3B shows representative images from the tumour in treated mice. The results are summarized in Fig. 3C. Control mice treated with Ad-LacZ showed that majority of the vessels were devoid of pericytes (70% of the total vessels), indicating that the vast majority of the vessels are immature. Treated mice showed lesser degree of angiogenesis in the tumours. E10A or docetaxel plus Ad-LacZ treatment reduced the relative amount of immature vessels to 44%, 53% of the total vessels. However, a combination of E10A and docetaxel treatment nearly eliminated the immature vessels in the tumour, reducing to 10% of the total vasculature.
Effect of orthotopic prostate cancer growth and metastasis by E10A and docetaxel
To evaluate whether systemic administration of E10A plus docetaxel could inhibit prostate cancer metastasis, orthotopic prostate cancer models were established. Four weeks after the start of treatment, all mice were killed. The data summarized in Table 2 show that weekly therapy with either docetaxel, docetaxel plus Ad-LacZ or the combination significantly decreased tumour weight when compared with the Ad-LacZ control. In addition, the tumours in combination treatment group were a greater reduction of tumour burden than use of either agent alone (P < 0.05) and it was possible to separate the seminal vesicles and bladder indicating that the grade of tumour progression was lower. Tumour aggressions to intestines were observed in 3 of 6 untreated mice, 3 of 8 mice treated with Ad-LacZ and 1 of 8 mice treated with E10A, whereas no tumour aggressions were detected in the intestines of mice given with docetaxel plus Ad-LacZ and combination treatment. It is interesting to note that histologically positive regional lymph node metastases were found in 7 of 8 mice treated with Ad-LacZ, 5 of 8 mice treated with docetaxel plus Ad-LacZ, 4 of 8 mice treated with E10A and 1 of 8 mice given combination therapy (Table 2). Hence, the metastatic spread of primary tumour appeared to be significantly inhibited by combination therapy (P≤ 0.01). Neither gross nor microscopic metastases were detected in the liver, lung, kidney, heart or brain. All of the treatments were tolerated well, and no difference in animal behaviour or weight was found between groups in s.c. xenograft and orthotopic tumour models.
Table 2.
The effect of combination therapy with E10A plus docetaxel on inhibition of growth and metastasis of orthotopic prostate cancer in nude mice
| Treatment group | n | Tumour weight (mg) | LN metastasis |
|---|---|---|---|
| Untreated | 6 | 446 ± 107 | 6/6 |
| Ad-LacZ | 8 | 427 ± 138 | 7/8 |
| Doc | 6 | 213 ± 54a | 3/6 |
| Ad-LacZ+Doc | 8 | 194 ± 60 a | 5/8 |
| E10A | 8 | 234 ± 59 | 4/8 |
| E10A+Doc | 8 | 104 ± 33 b,c | 1/8b |
P < 0.05, compared with untreated and Ad-LacZ control.
P≤ 0.01 compared with untreated and Ad-LacZ control.
P < 0.05, compared with single agent and docetaxel plus Ad-LacZ.
Lymphatic vessels were identified using LYVE-1 immunostatining, and lymphatic vessel density (LVD) was evaluated positive cells per 200× field (Fig. 4A). The number of LVD in control tumours was 36 ± 9. LVD was not significantly reduced in tumours of mice treated with docetaxel plus Ad-LacZ (33 ± 12, P > 0.05), but was in tumours of mice treated with E10A alone or in combination with docetaxel (18 ± 7 and 7 ± 3, respectively; P < 0.01). Moreover, the tumours that had received combination of E10A and docetaxel showed a greatest marked reduction of LVD (P < 0.01).
Fig 4.
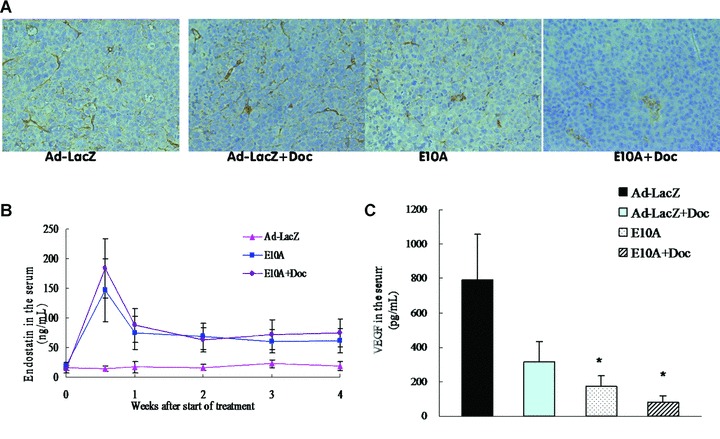
(A) LYVE-1-positive lymphatic vessels in orthotopic tumours received the different treatment. Representative images were shown (×200). (B) Serum endostatin levels in the mice with orthotopic tumours. After systemic delivery of E10A, serum endostatin levels were determined by ELISA. Serum endostatin levels in mice treated with Ad-LacZ were used as control. (C) Serum VEGF levels in different groups of mice. Mice were killed after 4 weeks of treatment and serum were collected for determination of VEGF levels by ELISA. Columns, mean ± S.D. *P < 0.05, compared with Ad-LacZ control.
To evaluate gene expression after combination treatment, we measured serum levels of endostatin in orthotopic tumour-bearing mice treated with E10A and/or docetaxel. Four days after the first injection, serum endostatin levels reached the observed peak. One week after each injection, serum endostatin levels stabilized within a range of 60.7–87.9 ng/ml, which exhibited approximately three- five-fold higher levels, compared with Ad-LacZ group (Fig. 4B). To determine whether the combination treatment affects proangiogenic environment, VEGF secreted by the tumour cells was measured in the serum. Control group of mice showed 793.8 ± 260.8 pg/ml human VEGF (Fig. 4C). E10A-treated mice had 4.6-fold less VEGF in the serum compared with the control group (P= 0.028). About 10-fold reduction in serum VEGF was seen in the combination treatment group compared with the serum levels of VEGF in the control group (P= 0.019).
In vivo testing of antiangiogenic effects
The inhibition of angiogenesis stimulated by tumour cells was evaluated in the alginate-encapsulated tumour cell assay. Growth factors produced by the encapsulated PC-3 cells induce vascularization of beads over 14 days (Fig. 5A), which can then be measured by uptake of FITC-dextran. FITC-dextran uptake was significantly decreased of 51%, 23% in E10A alone or docetaxel plus Ad-LacZ group, compared with Ad-LacZ control group. However, the most striking difference was observed in mice treated with combination of E10A and docetaxel (P < 0.001, Fig. 5B). This group of mice had a significant decrease by 77% compared with control group.
Fig 5.
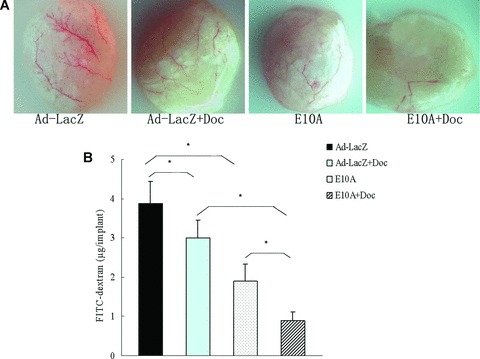
Alginate-encapsulated tumour cells assay. Alginate beads containing 1 × 105 tumour cells were implanted s.c. into the backs of athymic mice (four beads per mice). Mice treated with E10A or Ad-LacZ i.v. at 1 × 109 pfu, or/and docetaxel i.p. at 10 mg/kg once a week for 2 weeks. On day 15 after inoculation, beads were surgically removed. FITC-dextran was quantified. (A) Representative figures of alginate bead are shown. (B) FITC-dextran uptake of beads, Columns, mean ± S.D., *P < 0.001.
Discussion
E10A, a replication-defective adenovirus carrying human endostatin gene, had shown to be effective against a wide variety of tumour models, but the results of Phase I clinical trial demonstrated E10A exhibited mild antitumour effect [21]. In addition, antiangiogenic therapy could develop the appearance of ‘acquired drug resistance’[22] due to the activation of hypoxia-inducible factor [23]. To improve the efficacy of this form of treatment, combined strategies with other therapies are desirable. In previous studies, antiangiogenic drugs combined with each other [24–26], as well as with chemotherapy [17, 27, 28] and radiation [29, 30] against tumour. To date, most clinical trials indicate that they are the most effective when piggybacked onto traditional therapies, especially chemotherapy [31, 32]. In addition to proliferating cancer cells, chemotherapeutic drugs affect the endothelium of the growing tumour vasculature. The antiangiogenic efficacy of chemotherapy seems to be optimized by administering low doses of drug on a frequent schedule, known as metronomic chemotherapy [33]. Docetaxel is one of the most important cytotoxic chemotherapeutic agents used today in the treatment of prostate cancer. However, the clinical efficacy and utility of docetaxel at conventional treatment doses are often compromised due to adverse effects such as haematologic toxicity. Consistent with recent reports [34, 35], our studies showed that docetaxel also exhibit antiangiogenic properties when used at very low doses by inhibiting endothelial cell proliferation, migration and tube formation. In order to enhance therapeutic effect of E10A, we investigated combined therapy using docetaxel on endothelial cell in vitro, on human prostate cancer and angiogenic models in vivo.
In vitro, when given together, E10A and docetaxel synergistically inhibited endothelial cell function including proliferation, migration and tube formation. In vivo, combination treatment resulted in enhanced inhibition of tumour growth and survival improvement in androgen independent human prostate cancer s.c. xenograft and orthotropic tumour models. The enhanced antitumour efficacy in the present study may result from the increased induction of the apoptosis and enhanced reduction in tumour cellular proliferation and microvasculature.
Compared to the normal prostate, VEGF expression is increased in prostate carcinomas [36]. VEGF plays a critical role in tumour angiogenesis and is a potent inducer of dilated, tortuous, disorganized and leaky vasculature in tumours [5]. The abnormal vessels impair the delivery of oxygen and chemotherapeutic agents, which may be one of the causes of tumour resistant to radiotherapy and chemotherapy. Moreover, VEGF induces lymphanaiogenesis to promote cancer metastasis [37], and has autocrine function acting as a survival factor for tumour cells protecting them from chemotherapy [38]. Therefore, neutralization of VEGF and inhibition of VEGF pathway can be useful methods of treatment for prostate cancer. Endostatin interferes with VEGF signalling by down-regulating VEGF [39] and direct interaction with KDR/Flk-1 [40]. Antiangiogenic drugs would prune immature growing blood vessels – generally considered to be the vessels most sensitive to such drugs – leaving behind an increased proportion of mature, functional vessels [41]. Residue immature vessels might mature rapidly as a result of the recruitment of pericytes. Vasculature normalization provides an opportunity to transiently improve blood flow in tumours and better deliver chemotherapeutic drugs to cancer cells, which result in further elimination of vessels. Consistent with other reports [17], combination therapy significantly reduced MVD, especially immature vessels in tumour tissues. Interestingly, endostatin inhibited lymphangiogensis and lymph node metastasis by suppressing the expression of VEGF-C in tumour cells [42]. Our results provided evidence that combined with endostatin and docetaxel could produce enhanced decrease in the incidence of prostate cancer lymphatic meatstasis.
The therapeutic dose of docetaxel (10 mg/kg/week) was determined by efficacy and minimal body weight loss, according to our initial experiments and the previous studies of Huang et al.[28].
To achieve maximum therapeutic concentration of endostatin within the tumour microenvironment, intratumoural administration of E10A was performed in s.c. pre-established tumours. Orthotopic prostate cancer models, which are essential to evaluation of metastasis, were injected intravenously with adenovirus. Sauter et al. [43] applied systemic administration of adenovirus carrying endostatin that produced potent antimetastatic effect in lung cancer models. Our studies demonstrated serum endostatin levels dropped significantly a week after E10A injection, but weekly systemic administration of E10A resulted in sustained therapeutic concentration [9, 44]. E10A treatment when combined with docetaxel significantly reduced tumour burden and inhibited cancer cell activity, which may contribute to the marked decrease in serum VEGF. Systemic administration of E10A and docetaxel resulted in remarkable antiangiogenic and antitumour effect, which provide an effective strategy for metastatic prostate cancer.
Clinical studies of endostatin showed little toxicity at the doses studied with evidence of antitumour effects [45]. Similar results were observed in preclinical toxicity studies [13] and Phase I clinical trails of E10A [21, 46]. In the prostate cancer model, no significant adverse effects were observed in combination treatment group of mice, similar to other treatment groups, which suggests that combined therapy with E10A and docetaxel can be safely administered.
There are at least three different mechanisms that may contribute to the better therapeutic efficacy mediated by combination therapy with E10A and docetaxel. First, Chemotherapeutic and antiangiogenic agents target tumour and endothelial cells, respectively. Independent targeting can enhance the total effect of either of the treatment alone. Endostatin inhibits tumour angiogenesis to prevent rapid tumour cell repopulation after cytotoxic chemotherapy and lessen the possibility that resistant tumour cells emerge. Second, endostatin normalizes the tumour vasculature so that chemotherapeutic drugs could reach the tumour tissue more efficiently to eliminate tumour cells. Third, endostatin and docetaxel synergistically inhibit angiogenesis and lymphangiogenesis, and exert enhanced anti-metastatic effect. In summary, we have developed a promising strategy for advanced prostate cancer and metastatic disease with a replication-defective adenovirus carrying human endostatin gene, E10A, in combination with conventional chemotherapeutic agents. This combination therapy resulted in enhanced antitumour and antiangiogenic effect, which warrants more extensive investigation.
Acknowledgments
This work was supported by the National Basic Research Program of China (973 Program) grant 2004CB518801, the Guangzhou Science Foundation grant 2004Z3-E4011, the High Tech Research Program of China (863 Program) grant 2007AA021202, 2007AA021203. We thank Miss Jingjing Wu, Hongyun Jia and Yufang Zuo (Sun Yat-sen University, Guangzhou, China) for helpful discussion. We also thank Miss Jiemin Chen, Miaola Ke (Sun Yat-sen University, Guangzhou, China) for their excellent technical assistance.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Pienta KJ, Smith DC. Advances in prostate cancer chemotherapy: a new era begins. CA Cancer J Clin. 2005;55:300–18. doi: 10.3322/canjclin.55.5.300. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain PK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 4.O’Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–85. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 5.Cao Y, Liu Q. Tumor-produced angiogenic factors and their roles in promoting tumor angiogenesis. Avd Cancer Res. 2007;97:203–24. doi: 10.1016/S0065-230X(06)97009-2. [DOI] [PubMed] [Google Scholar]
- 6.Torimura T, Ueno T, Kin M, et al. Gene transfer of kringle 1–5 suppresses tumor development and improves prognosis of mice with hepatocellular carcinoma. Gastroenterology. 2006;130:1301–10. doi: 10.1053/j.gastro.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Cao Y. Antiangiogenesis gene therapy. Gene Ther Regul. 2000;1:123–40. [Google Scholar]
- 8.Li L, Huang JL, Liu QC. Endostatin gene therapy for liver cancer by a recombinant adenovirus delivery. World J Gastroenterol. 2004;10:1867–71. doi: 10.3748/wjg.v10.i13.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Liu RY, Huang JL, et al. Adenovirus-mediated intra-tumoral delivery of the human endostatin gene inhibits tumor growth in nasopharyngeal carcinoma. Int J Cancer. 2006;118:2064–71. doi: 10.1002/ijc.21585. [DOI] [PubMed] [Google Scholar]
- 10.Passey S. Endostatin gene therapy inhibits tumour growth. Lancet Oncol. 2006;7:199. doi: 10.1016/s1470-2045(06)70599-x. [DOI] [PubMed] [Google Scholar]
- 11.Montero A, Fossella F, Hortobagyi G, et al. Docetaxel for treatment of solid tumours: a systematic review of clinical data. Lancet Oncol. 2005;6:229–39. doi: 10.1016/S1470-2045(05)70094-2. [DOI] [PubMed] [Google Scholar]
- 12.Hotchkiss KA, Ashton AW, Mahmood R, et al. Inhibition of endothelial cell function in vitro and angiogenesis in vivo by docetaxel (Taxotere): association with impaired repositioning of the microtubule organizing center. Mol Cancer Ther. 2002;1:1191–200. [PubMed] [Google Scholar]
- 13.Huang BJ, Liu RY, Huang JL, et al. Long-term toxicity studies in canine of E10A, an adenoviral vector for human endostatin gene. Hum Gene Ther. 2007;18:207–21. doi: 10.1089/hum.2006.149. [DOI] [PubMed] [Google Scholar]
- 14.Berenbaum MC. What is synergy. Pharmacol Rev. 1989;41:93–141. [PubMed] [Google Scholar]
- 15.Reimer CL, Agata N, Tammam JG, et al. Antineoplastic effects of chemotherapeutic agents are potentiated by NM-3, an inhibitor of angiogenesis. Cancer Res. 2002;62:789–95. [PubMed] [Google Scholar]
- 16.Wu J, Zhao P, Xue G, et al. Minicircle-IFNgamma induces antiproliferative and antitumoral effects in human nasopharyngeal carcinoma. Clin Cancer Res. 2006;12:4702–13. doi: 10.1158/1078-0432.CCR-06-0520. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian IV, Bui Nguyen TM, Truskinovsky AM, et al. Adeno-associated virus-mediated delivery of a mutant endostatin in combination with carboplatin treatment inhibits orthotopic growth of ovarian cancer and improves long-term survival. Cancer Res. 2006;66:4319–28. doi: 10.1158/0008-5472.CAN-05-3297. [DOI] [PubMed] [Google Scholar]
- 18.Stephenson RA, Dinney CP, Gohji K. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84:951–7. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmann J, Schirner M, Menrad A, et al. A highly sensitive model for quantification of in vivo tumor angiogenesis induced by alginate-encapsulated tumor cells. Cancer Res. 1997;57:3847–51. [PubMed] [Google Scholar]
- 20.Li G, Tian L, Hou JM, et al. Improved therapeutic effectiveness by combining recombinant CXC chemokine ligand 10 with Cisplatin in solid tumors. Clin Cancer Res. 2005;11:4217–24. doi: 10.1158/1078-0432.CCR-04-2117. [DOI] [PubMed] [Google Scholar]
- 21.Lin X, Huang H, Li S, et al. A Phase I clinical trial of an adenovirus-mediated endostatin gene (E10A) in patients with solid tumors. Cancer Biol Ther. 2007;6:648–53. doi: 10.4161/cbt.6.5.4004. [DOI] [PubMed] [Google Scholar]
- 22.Folkman J. Endogenous angiogenesis inhibitors. APMIS. 2004;112:496–507. doi: 10.1111/j.1600-0463.2004.apm11207-0809.x. [DOI] [PubMed] [Google Scholar]
- 23.Blagosklonny MV. Antiangiogenic therapy and tumor progression. Cancer Cell. 2004;5:13–7. doi: 10.1016/s1535-6108(03)00336-2. [DOI] [PubMed] [Google Scholar]
- 24.Abdollahi A, Lipson KE, Sckell A, et al. Combined therapy with direct and indirect angiogenesis inhibition results in enhanced antiangiogenic and antitumor effects. Cancer Res. 2003;63:8890–8. [PubMed] [Google Scholar]
- 25.Raikwar SP, Temm CJ, Raikwar NS, et al. Adenoviral vectors expressing human endostatin-angiostatin and soluble Tie2: enhanced suppression of tumor growth and antiangiogenic effects in a prostate tumor model. Mol Ther. 2005;12:1091–100. doi: 10.1016/j.ymthe.2005.07.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graepler F, Verbeek B, Graeter T, et al. Combined endostatin/sFlt-1 antiangiogenic gene therapy is highly effective in a rat model of HCC. Hepatology. 2005;41:879–86. doi: 10.1002/hep.20613. [DOI] [PubMed] [Google Scholar]
- 27.Plum SM, Hanson AD, Volker KM, et al. Synergistic activity of recombinant human endostatin in combination with adriamycin: analysis of in vitro activity on endothelial cells and in vivo tumor progression in an orthotopic murine mammary carcinoma model. Clin Cancer Res. 2003;9:4619–26. [PubMed] [Google Scholar]
- 28.Huang SF, Kim SJ, Lee AT, et al. Inhibition of growth and metastasis of orthotopic human prostate cancer in athymic mice by combination therapy with pegylated interferon-alpha-2b and docetaxel. Cancer Res. 2002;62:5720–6. [PubMed] [Google Scholar]
- 29.Mauceri HJ, Hanna NN, Beckett MA, et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature. 1998;394:287–91. doi: 10.1038/28412. [DOI] [PubMed] [Google Scholar]
- 30.Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6:553–63. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 31.Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer. Science. 2006;312:1171–5. doi: 10.1126/science.1125950. [DOI] [PubMed] [Google Scholar]
- 32.Medina MA, Muñoz-Chápuli R, Quesada AR. Challenges of antiangiogenic cancer therapy: trials and errors, and renewed hope. J Cell Mol Med. 2007;11:374–82. doi: 10.1111/j.1582-4934.2007.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4:423–36. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- 34.Belotti D, Vergani V, Drudis T, et al. The microtubule-affecting drug paclitaxel has antiangiogenic activity. Clin Cancer Res. 1996;2:1843–9. [PubMed] [Google Scholar]
- 35.Grant DS, Williams TL, Zahaczewsky M, et al. Comparison of antiangiogenic activities using paclitaxel (taxol) and docetaxel (taxotere) Int J Cancer. 2003;104:121–9. doi: 10.1002/ijc.10907. [DOI] [PubMed] [Google Scholar]
- 36.Mazzucchelli R, Montironi R, Santinelli A, et al. Vascular endothelial growth factor expression and capillary architecture in high-grade PIN and prostate cancer in untreated and androgen-ablated patients. Prostate. 2000;45:72–9. doi: 10.1002/1097-0045(20000915)45:1<72::aid-pros9>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 37.Cao Y. Emerging mechanisms of tumour lymphangiogenesis and lymphatic metastasis. Nat Rev Cancer. 2005;5:735–43. doi: 10.1038/nrc1693. [DOI] [PubMed] [Google Scholar]
- 38.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9:777–94. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hajitou A, Grignet C, Devy L, et al. The antitumoral effect of endostatin and angiostatin is associated with a down-regulation of vascular endothelial growth factor expression in tumor cells. FASEB J. 2002;16:1802–4. doi: 10.1096/fj.02-0109fje. [DOI] [PubMed] [Google Scholar]
- 40.Kim YM, Hwang S, Kim YM, et al. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem. 2002;277:27872–9. doi: 10.1074/jbc.M202771200. [DOI] [PubMed] [Google Scholar]
- 41.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–9. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 42.Fukumoto S, Morifuji M, Katakura Y, et al. Endostatin inhibits lymph node metastasis by a down-regulation of the vascular endothelial growth factor C expression in tumor cells. Clin Exp Metastasis. 2005;22:31–8. doi: 10.1007/s10585-005-3973-5. [DOI] [PubMed] [Google Scholar]
- 43.Sauter BV, Martinet O, Zhang WJ, et al. Adenovirus-mediated gene transfer of endostatin in vivo results in high level of transgene expression and inhibition of tumor growth and metastases. Proc Natl Acad Sci USA. 2000;97:4802–7. doi: 10.1073/pnas.090065597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Celik I, Surucu O, Dietz C, et al. Therapeutic efficacy of endostatin exhibits a biphasic dose-response curve. Cancer Res. 2005;65:11044–50. doi: 10.1158/0008-5472.CAN-05-2617. [DOI] [PubMed] [Google Scholar]
- 45.Herbst RS, Hess KR, Tran HT, et al. Phase I study of recombinant human endostatin in patients with advanced solid tumors. J Clin Oncol. 2002;20:3792–803. doi: 10.1200/JCO.2002.11.061. [DOI] [PubMed] [Google Scholar]
- 46.Li HL, Li S, Shao JY, et al. Pharmacokinetic and pharmacodynamic study of intratumoral injection of an adenovirus encoding endostatin in patients with advanced tumors. Gene Ther. 2008;15:247–56. doi: 10.1038/sj.gt.3303038. [DOI] [PubMed] [Google Scholar]