

Abstract
Hepatic fibrosis is a common response to virtually all forms of chronic liver injury independent of the etiologic agent. Despite the relatively large population of patients suffering from hepatic fibrosis and cirrhosis, no efficient and well-tolerated drugs are available for the treatment of this disorder. The lack of efficient treatment options is at least partly because the underlying cellular mechanisms leading to hepatic fibrosis are only partly understood. It is thus of pivotal importance to better understand the cellular processes contributing to the progression of hepatic fibrosis. Interestingly in this perspective, a common feature of fibrotic disease of various organs is the activation of the coagulation cascade and hepatic fibrosis is also accompanied by a local hypercoagulable state. Activated blood coagulation factors directly target liver cells by activating protease-activated receptors (PAR) thereby inducing a plethora of cellular responses like (among others) proliferation, migration and extracellular matrix production. Coagulation factor driven PAR activation thus establishes a potential link between activation of the coagulation cascade and the progression of fibrosis. The current review focuses on blood coagulation factor Xa and summarizes the variety of cellular functions induced by factor Xa-driven PAR-2 activation and the subsequent consequences for tissue repair and hepatic fibrosis.
Keywords: protease-activated receptor-2, fibrosis, coagulation factor Xa
Introduction
Hepatic fibrosis results from a sustained wound healing response to chronic liver injury. Although even acute injury will activate mechanisms of fibrogenesis, the sustained signals associated with chronic liver disease caused by infection, drugs, metabolic disorders or auto-immune disease are required for the development of substantial fibrosis. Occasionally, fibrosis may be rapidly progressive over weeks to months, for example as a result of drug injury, hepatitis C infection after liver transplantation [1], or hepatitis C coinfection with HIV [2], but mostly fibrosis evolves over decades. Any chronic perturbation of hepatic homeostasis may elicit signals necessary to stimulate fibrogenesis [3]. The end-stage of liver fibrosis (cirrhosis), and its complications, i.e. portal hypertension, liver failure and hepatocellular carcinoma, represent a major healthcare issue worldwide. Despite the increasing burden of liver fibrosis and cirrhosis, the therapeutic repertoire for the treatment of these diseases remains limited, because no efficient and well-tolerated drugs are available [4]. The only treatment option currently available for severe end-stage liver disease is orthotopic liver transplantation. Therefore, there is a clear imperative to develop novel anti-fibrotic agents to combat these life threatening conditions.
At the cellular level, hepatic fibrosis is, similar to fibrosis in other organs such as lung, kidney, heart or skin, considered as an excessive wound healing response to chronic injury leading to excessive deposition of extracellular matrix (ECM). The distribution of fibrotic lesions during disease progression is primarily determined by the nature of the underlying hepatic injury. Thus, some fibrotic lesions are primarily periportal due to for instance chronic viral hepatitis, chronic cholestatic diseases and haemachromatosis, or they can be primarily centrilobular during steatohepatitis, and chronic venous outflow obstruction. Additionally, the fibrotic lesions can be divided into those that result in porto-portal (e.g. cholestatic liver injuries), porto-central (e.g. viral hepatitis) or centro-portal bridging (e.g. alcoholic liver disease). Irrespective of the actual location of the lesions, hepatic fibrosis is characterized by several key features: (1) the persistence of chronic damage to hepatocytes and/or cholangiocytes, (2) a complex inflammatory cell infiltrate and (3) activation of different types of ECM-producing cells with proliferative and synthetic features, finally leading to (4) marked changes in the quality and quantity of the hepatic ECM that distort the hepatic architecture and lead to an impairment of organ function [3, 5]. The essential difference between cirrhosis and (developing) fibrosis is that during cirrhosis more scar tissue is formed that distorts the liver parenchyma.
Activation of the coagulation cascade during liver fibrosis: a puzzling paradox
A common feature of fibrotic disease of various organs is the activation of the coagulation cascade [6, 7]. Accordingly, a local hypercoagulable state also accompanies liver fibrosis. Strikingly, patients diagnosed with liver fibrosis frequently also present with systemic coagulation deficiency [8]. These observations might at first appear as a puzzling paradox. One should realize however, that several procoagulant factors of the clotting cascade (factors (F)II (prothrombin), VII, IX and X), are synthesized in the liver by hepatocytes. This tissue specific expression is due to the positive regulation of their transcription by the liver-specific transcription factor HNF-4 [9–12]. In addition, these so-called vitamin K-dependent coagulation factors require post-ribosomal modification for biological activity. Indeed, they possess a specific number of glutamic acid residues that function as potential binding sites for calcium bridges in the NH2-terminal region. The glutamic acid residues are converted to γ-carboxyglutamic acids, and this modification is facilitated by a carboxylase that requires the fat-soluble vitamin K as cofactor (Fig. 1) [13]. In its naturally occurring state, vitamin K is in the quinone oxidation state and must be reduced to the hydroquinone form to become the active cofactor for the vitamin K-dependent carboxylase. The enzyme responsible for this reduction is present in the liver [14], and is known as vitamin K epoxide reductase, because it also reduces vitamin K epoxide formed during the carboxylation reaction. In severe liver disease, both HNF-4 and vitamin K epoxide reductase levels (and activity) are reduced as the synthetic capacity of the liver is lost. Consequently, severe liver disease is accompanied by acquired coagulation deficiencies. However, the main anticoagulant proteins (i.e. protein (P)C and PS) are also synthesized in the liver and also require γ-carboxylation for proper anticoagulant activity [13]. Thus, as liver disease progresses the levels of both pro- and anticoagulant proteins decrease and the balance between pro- and anticoagulation becomes precarious. The outcome of severe liver disease with respect to bleeding and/or thrombosis is thus dependent on this balance and difficult to predict. In addition to decreased PC and PS activity, hypercoagulability in liver disease may also be caused by decreased blood flow, and vasculopathy associated with chronic inflammation or endothelial dysfunction, which are all prominent features of liver fibrosis. Finally, increased expression of tissue factor, the initiator of blood coagulation [15], triggered by fibrosis within the hepatic parenchyma [16] is likely to play an important role in the hypercoagulable state.
Fig 1.
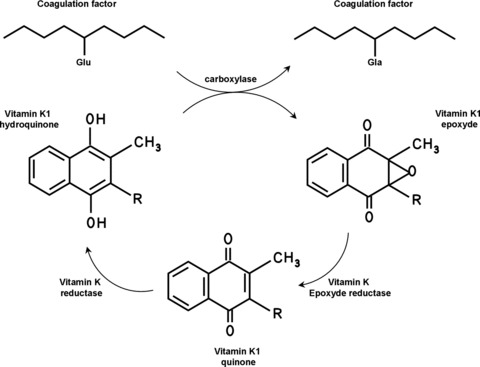
Vitamin K-dependent protein carboxylation. The gammacarboxylation of coagulation factors requires the conversion of reduced vitamin K1 hydroquinone into vitamin K1 epoxyde. The K1 epoxyde is then converted to vitamin K1 quinone by vitamin K1 epoxyde reductase and then recycled to vitamin K1 hydroquinone by the enzyme vitamin K reductase. R: phytyl group.
In keeping with the notion that activated coagulation accompanies liver fibrosis, morphological studies have shown that there is a close association between parenchymal remodelling and obliterative lesions of intrahepatic small portal and hepatic veins during the progression of chronic liver disease towards cirrhosis. These obliterative lesions are highly suggestive of intrahepatic thrombotic events. Indeed, microthrombi are present in the microvasculature of mice infected with hepatitis virus [17, 18] and similar findings were subsequently observed in patients with cirrhosis [19–21]. In a rat model of liver ischaemia reperfusion injury, which is an important pathological process that leads to hepatic damage after circulatory shock and major hepatic surgery [22–24], microthrombi were formed during ischaemia which inhibited the restoration of liver blood flow [25, 26]. Also, acute and chronic carbon tetrachloride-induced liver injury stimulates hepatic deposition of fibrin and fibrinogen within the microcirculation [27].
The potential relevance of hypercoagulablity in liver fibrosis has been demonstrated by observations that intimal fibrosis (which is highly suggestive of reorganized thrombosis) was observed in 70% of cirrhotic livers [19, 20]. Based on this observation, the authors suggested that microthrombi in the vasculature disrupt blood flow thereby leading to ischaemia and subsequent parenchymal extinction (as characterized by irreversible loss of contiguous hepatocytes due to hepatocyte apoptosis, collapse of the region between the central veins and their adjacent portal tract, and the replacement of hepatocytes by fibrotic lesions [19, 20]). Subsequently, small areas of parenchymal extinction accumulate and become confluent resulting in cirrhosis [19, 20, 28].
An alternative explanation by which microthrombi may contribute to cirrhosis comes from early studies reporting that, once associated with a thrombus, coagulation FXa retains its enzymatic activity and induces thrombin generation [29–31]. This is particularly relevant as it has been shown that both FXa and thrombin directly contribute to the progression of (among others) lung and kidney remodelling [6]. In keeping with this observation, patients with a thrombus at the site of vascular injury after percutaneous transluminal coronary angioplasty have a much higher risk of restenosis than patients without such a thrombus [32]. Accordingly, the presence of microthrombi is strongly related to disease severity in hepatic infection [33]. Moreover, thrombotic risk factors (deficiency of protein C, antithrombin III and plasminogen) are associated with the extent of fibrosis in non-alcoholic fatty liver disease and in chronic hepatitis B or C [34–36]. Finally, studies using thrombogenic FV Leiden mice show accelerated hepatic fibrogenesis [37]. Accordingly, the FV Leiden mutation is associated with an increased risk of fibrosis during chronic hepatitis C infection (odds ratio of 3.28) [38].
Protease-activated receptors: the link between coagulation cascade activation and liver fibrosis
The quandary as to how coagulation factors influence cellular responses was elucidated by the identification of a novel family of G-protein coupled receptors, the protease-activated receptors (PARs). These seven transmembrane domain receptors are activated in a unique manner by proteolytic cleavage of their N-terminal extremity, generating a new tethered ligand that binds to the second extracellular loop of the receptor [39, 40]. Activated PARs subsequently induce transmembrane signalling to G proteins triggering a cascade of downstream events involved in cellular functions implicated in (among others) cell survival, cell migration and inflammation [6]. To date, four PARs (PAR-1 to -4), with distinct N-terminal cleavage sites and tethered ligand pharmacology, have been identified. Strikingly, activated coagulation FVIIa, FXa and thrombin are able to activate these PARs. Indeed, thrombin signals through PAR-1, -3 and -4, FVIIa in association with tissue factor activates PAR-2, while FXa activates both PAR-1 and PAR-2 [41]. Coagulation factor driven PAR activation thus establishes a potential link between activation of the coagulation cascade and the progression of fibrosis. This link might be most relevant for FXa as this coagulation factor occupies a unique position at the crossroads between coagulation and signalling. On the one hand, FXa leads to thrombin generation via its procoagulant properties after which thrombin induces profibrotic responses via PAR-1 activation. On the other hand, FXa induces fibrotic responses by PAR-2 dependent signalling in a way which is not reminiscent of thrombin signalling.
The importance of thrombin-induced PAR-1 activation in liver fibrosis has been established recently (for review, see [42]); however, the role of PAR-2 and the cellular consequences of its activation by coagulation FXa has remained remarkably underexplored in the context of liver fibrosis. This is mainly because it has long been thought that FXa signalling (via PAR-1) was only reminiscent of thrombin signalling. The expression of the thrombin receptors PAR-1, -3 and -4 has consequently been investigated in different settings of liver fibrosis [16, 43–45], whereas the expression of PAR-2 in the setting of liver fibrosis remains underexplored. This omission seems important as several lines of evidence point to PAR-2 as a key player in the progression of a wide pattern of pathologies in which coagulation cascade activation plays a crucial role (like among others, tissue remodelling, fibrosis and cancer [6]). The variety of cellular functions regulated by FXa and its role in tissue repair and fibrosis, together with the differential PAR-2 expression in healthy and fibrotic liver, forms the basis for the current review on FXa/PAR-2 in liver fibrosis.
Expression and distribution of human PAR-2 in normal and pathological liver tissue
The liver lobule is formed by parenchymal cells, i.e. hepatocytes, and non-parenchymal cells. In contrast to hepatocytes that occupy most of the total liver volume and perform the majority of liver functions, non-parenchymal liver cells contribute only to a minor portion of the liver volume, but they make up almost 40% to the total number of liver cells. These non-parenchymal cells include cells of the supporting stroma, components of the biliary system and the sinusoidal compartment. The walls of the hepatic sinusoid are lined by three different cell types: sinusoidal endothelial cells (SECs), Kupffer cells (KCs) and hepatic stellate cells (HSCs) which are located in the space of Disse between the hepatocytes and the hepatic sinusoids [46].
Ongoing research in our laboratory shows that under normal conditions PAR-2 is expressed by hepatocytes, KCs, the endothelium of large vessels and by bile duct epithelial cells (personal observation; Fig. 2). Because of strong PAR-2 staining on hepatocytes in healthy liver, we cannot yet discriminate between PAR-2 expression on hepatocytes and/or HSCs. However, in rat studies, PAR-2 expression was also observed on HSCs physiologically [44, 45] suggesting that these cells may express PAR-2 in human beings as well. Interestingly, PAR-2 expression changes during the progression of liver fibrosis (Fig. 2D–F). Expression in hepatocytes remains relatively unchanged, but PAR-2 expression is strongly increased in proliferating biliary duct epithelium to an extent that correlates with the severity and/or the duration of liver disease. PAR-2 is also expressed by the endothelium and vascular smooth muscle cells of newly formed blood vessels in fibrotic septa, and by (myo)fibroblasts in fibrotic areas (Fig. 2F), whereas several cells within the inflammatory infiltrate also show PAR-2 expression.
Fig 2.

Immunostaining for PAR-2 in normal and cirrhotic human liver. (A–C) In normal liver, a very intense PAR-2 staining is present in hepatocytes (A, 100×), and more weakly in the endothelium of larger vessels (B, **, 400×), and in bile duct epithelial cells (B, *). Staining was strong in KCs (C, arrows, 400×). (D–F). In fibrotic liver, PAR-2 staining remained strong in hepatocytes in regenerative nodules (D, 40×). Staining was strongly increased in the epithelium of newly formed biliary structures (E, arrows, 100×) and in the endothelium and smooth muscle cells of small vascular structures (F, arrows, 400×), compared with weaker staining in pre-existing bile ducts (E+F,*).
It is noteworthy that in a rodent model of acute liver injury induced by LPS administration, PAR-2 expression was also induced. However, in this inflammatory condition, PAR-2 expression was predominantly induced on KCs and (although to a lesser extent) hepatocytes, with discrete expression on endothelium and bile duct epithelium [47]. This observation emphasizes the differential cellular responses involved in acute or chronic liver injury.
The differential expression of PAR-2 in healthy and diseased liver tissue, with up-regulation during the pathogenesis of fibrosis, is in line with studies showing that PAR-2 contributes to fibrotic disease. For instance, high expression of pulmonary PAR-2 is observed in acute and chronic lung injury suggesting that PAR-2 participates in inflammation and fibroproliferation during lung fibrosis [48]. In renal interstitial fibrosis, the expression of α-SM actin and PAR-2 is correlated [49], whereas PAR-2 expression is increased in biopsies of patients with IgA nephropathy [50].
FXa signalling on PAR-2 expressing cells, and the relevance for liver fibrosis
Following PAR-2 activation, FXa typically induces phosphoinositides hydrolysis leading to calcium oscillation. FXa also triggers the phosphorylation of extracellular-signal related kinase and JNK pathways, leading to initiation of different transcriptional programs promoting proliferation, differentiation, migration and ECM deposition [6], all of obvious relevance in the context of hepatic fibrosis. It should be kept in mind that FXa signal transduction shows a degree of cell type specificity, but importantly it seems that FXa-induced signalling pathways are rather similar in cells derived from the same lineage (e.g. mesenchymal; for a review, see [6]). Consequently, one could obtain significant insight on the role of FXa signalling in liver fibrosis by addressing the role of FXa on cells with similar characteristics as hepatic fibroblasts, epithelial cells, KCs and SECs. The next paragraph provides an overview of the known signalling events provoked by FXa in fibroblasts, epithelial cells, KCs and SECs, and this overview supports the hypothesis that FXa is an essential player in the pathogenesis of liver fibrosis.
FXa triggers fibroproliferative and pro-inflammatory responses in mesenchymal cells via PAR-2 activation
Activated fibroblasts or myofibroblasts are key mediators of organ fibrosis. Wound healing in general, and pathological fibrosis in particular, primarily results from myofibrolast-mediated ECM synthesis and deposition. The phenotype of the myofibroblast population in a diseased tissue therefore influences the extent of fibrosis. In fibrotic livers, myofibroblasts are considered to derive via activation and proliferation of resident stellate cells and periportal fibroblasts [3]. Recent evidence however suggests that fibroblasts are more heterogeneous than traditionally thought and additional mechanisms such as recruitment of bone marrow derived cells and differentiation of resident epithelial cells are described to contribute to the accumulation of myofibroblasts in fibrotic tissues [51]. Further understanding of the underlying pathogenic mechanisms involved in the accumulation of scar-forming myofibroblasts might help to stop progressive liver fibrosis, which is currently not yet possible.
HSCs are without question the most studied fibrogenic cells in the liver. Located in the space of Disse between the hepatocytes and the hepatic sinusoids, these cells are indeed major mediators of the fibrotic process during the wound healing response [52]. The discovery of HSC differentiation into myofibroblasts remains among the most informative discoveries to date in unlocking the basis for hepatic fibrogenesis. In normal healthy liver, HSCs show minimal proliferation and collagen synthesis. In injured liver, HSCs transform to myofibroblasts and subsequently migrate and accumulate at the site of tissue repair where they synthesize various ECM components [53–58].
A first insight into the role of PAR-2 in HSC activation came from the observation that specific PAR-2 agonist peptides (synthetic hexapeptides corresponding to the N-terminus of the tethered ligand) are able to activate HSCs leading to cell proliferation and increased collagen synthesis in rodents [44, 45]. In keeping with this observation, FXa promotes similar fibroproliferative responses in a number of other mesenchymal cells. Indeed, FXa is mitogenic on embryonic, dermal and lung fibroblasts of different species, on rat pancreatic stellate cells, on murine myoblasts and smooth muscle cells of both rodent and human origin, and on human mesangial cells [41, 59–65]. Moreover, FXa enhances the production of the cytokines/chemoattractants interleukin (IL)-6, IL-8 and MCP-1 in dermal and embryonic fibroblasts and in murine myoblasts [65, 66]. FXa-induced PAR-2 activation also leads to the release of PAI-1 and transforming growth factor (TGF)-β (a powerful profibrotic cytokine) in both human mesangial cells and in embryonic fibroblasts [50, 65]. Finally, FXa acts as a powerful chemoattractant for murine embryonic fibroblasts and contributes to differentiation of these fibroblasts into myofibroblasts [65].
Direct proof for the importance of FXa-dependent PAR-2 signalling in portal fibroblasts and/or other resident fibroblasts during hepatic fibrosis has not yet been obtained. However, as already indicated, PAR-2 is expressed on these cell types in fibrotic liver tissue (Fig. 2). It is therefore tempting to speculate that FXa signalling indeed activates hepatic fibroblasts and consequently aggravates fibrosis.
FXa triggers pro-inflammatory responses and modulates the survival of epithelial cells via PAR-2 activation
The liver contains two relevant epithelial cell types: hepatocytes and cholangiocytes (epithelial cells of the bile duct). The contribution of hepatocytes to liver fibrosis is considered to be mainly mediated by paracrine effects of the released inflammatory cytokines leading to the activation of HSCs and KCs. In addition, however, apoptotic hepatocytes are ingested by KCs leading to the release of TGF-β and subsequent HSC activation [67, 68]. These activated HSCs subsequently induce profibrotic responses [3].
Activated cholangiocytes also activate HSCs by the secretion of pro-inflammatory and chemotactic proteins. In addition, proliferation of the cholangiocytes also aggravates fibrosis [5]. Interestingly, the fact that PAR-2 mRNA expression is increased in cholangiocytes of bile duct ligated rats might suggest that PAR-2 activation in these cells would contribute to liver fibrosis [45]. PAR-2 expression in cholangiocytes does however not provide proof that its activation would contribute to fibrosis, and future experiments should pursue this hypothesis.
Most likely because the concept of FXa signalling in pathology emerged only recently, the functional consequences of FXa signalling in liver epithelium and especially its potential pro-inflammatory role has never been investigated. However, the effects of FXa-induced PAR-2 activation, especially with respect to pro-inflammatory responses, on different epithelial cells have been well described. For instance, FXa induces the activation of the pro-inflammatory transcription factor NF-κB in HeLa cells (derived from cervix adenocarcinoma) [69], and the inflammatory cytokines such as IL-6, IL-8, MCP-1 or cyclooxygenase 2, respectively, in endothelial, mesangial and in epithelial and fibroblastic cell lines [70–72]. In addition, FXa-induced PAR-2 activation enhances PAI-1 gene induction and TGF-β production in epithelial kidney cells [50]. Next to inducing inflammation, FXa also modulates cell survival. While FXa induces PAR-1 dependent apoptosis in a broad range of epithelial cells [73–75] (which is consistent with thrombin-induced apoptosis in epithelial cells [76]), FXa-dependent PAR-2 activation in cancer cells with an epithelial origin is generally associated with cell proliferation (this has for instance been shown using colon, cervix, breast, pancreas and skin cancer cells [41, 77–82]. Interestingly, PAR-2 expression is strongly up-regulated on cholangiocytes in fibrotic liver tissue (Fig. 2) and it is thus tempting to speculate that PAR-2 activation will lead to cholangiocyte proliferation thereby aggravating fibrosis.
FXa and PAR-2 signalling in inflammatory cells
KCs are the resident macrophages in the liver. They release a variety of inflammatory mediators, free radicals, fibrogenic cytokines, as well as ECM proteinases that alter the normal ECM structure during early stages of liver injury. As already indicated, KCs play a critical role in promoting HSC activation [83] and they express PAR-2 in fibrotic conditions. The relevance of PAR-2 expression on KCs in the progression of fibrosis might be supported by a recent study showing that sepsis-induced PAR-2 expression on KCs is correlated with an inflammatory response. Moreover, PAR-2 inhibition in this rat model of acute liver injury limits tumour necrosis factor-α production and improves outcome [47].
An overwhelming amount of data indicate that FXa and PAR-2 signalling trigger pro-inflammatory responses in inflammatory cells. Agonists of PAR-2 modulate neutrophil cytokine secretion, expression of cell adhesion molecules, rolling and migration [84, 85]. PAR-2 activation also enhances the adhesion of peripheral blood mononuclear cells to endothelial cells [70]. Furthermore, PAR-2 activation in the lung induces airway constriction, lung inflammation and protein-rich pulmonary oedema [86]. Accordingly, PAR-2 deficiency reduces inflammation and mortality in different mouse models of inflammation [84, 87, 88]. Also, FXa signalling in leucocytes induces tissue factor expression [89], as well as IL-1, IL-6, IL-8, MCP-1 and CCR-2 release [66, 90–92]. Interestingly, FXa enhances neutrophil chemoattractant production after ischaemia/reperfusion in the rat liver [93]. Taken together, these observations suggest that the prominent expression of PAR-2 on KCs is potentially related to, or underlies, the acute inflammatory response induced by FXa.
FX-induced PAR-2 activation modulates endothelial barrier permeability
SECs line the sinusoids and are in constant contact with sinusoidal blood flow and closely associated with hepatocytes, HSCs and KCs. Changes in SECs can already be detected before fibrosis using electron microscopy [94–96]. Whether SECs might drive or initiate fibrosis, as has been suggested [94, 97], is still a matter of debate. However, SECs are known to play a crucial role in capillarization of the sinusoids which is a hallmark of liver fibrosis. Indeed, in the fibrotic milieu, the fenestrae in normal SECs are markedly decreased in number and size, leading to decreased porosity of the endothelial barrier [98]. Additionally, a discontinuous basement membrane on the basal side of SECs is replaced by a continuous basement membrane, accompanied by abundant collagen fibres that accumulate in the space of Disse.
In vitro experimental evidence indicates that FXa signalling modifies the endothelial barrier function. Indeed, FXa enhances endothelial barrier integrity via both PAR-1 and PAR-2 [99] and FXa might therefore contribute to the decreased porosity of the endothelium observed during liver fibrosis. Moreover, FXa induces a proangiogenic phenotype in fibroblasts by stimulating VEGF expression which subsequent induces tube formation [100], a feature which is reminiscent of the altered topography of the vascularized fibrotic septa, leading to the establishment of intrahepatic shunts between afferent (portal vein and hepatic artery) and efferent (hepatic vein) vessels of the liver during fibrosis [101–104]. In addition, in a variety of endothelial in vitro systems, FXa induces a wide array of pro-inflammatory responses, thereby contributing to the formation of a vicious cycle which sustains fibrosis progression. Indeed, via PAR-2 activation mainly, FXa induces the deposition of connective tissue growth factor [105], it leads to the activation of NF-κB, and the release of IL-6, IL-8, and MCP-1 [41, 106, 107], which might contribute to leucocyte recruitment. Furthermore, FXa induces the expression of E-selectin and the adhesion molecules ICAM-1 and VCAM-1 resulting in leucocyte adhesion [108]. Finally, very recent data show that agonist peptide-driven PAR-2 activation triggers ROS generation by endothelial cells [109]. Indeed, oxidative stress plays an important role in producing liver damage and initiating hepatic fibrogenesis. Increased ROS is commonly detected in the liver of patients with chronic liver disease and in most types of experimental liver fibrosis [110, 111]. Therefore, it is tempting to speculate that the release of ROS following FXa-induced PAR-2 activation in the endothelium might further enhance the fibrotic response, for instance, by inducing apoptosis of hepatocytes, amplification of the inflammatory response as discussed in [110], the production of profibrotic mediators from KCs or by fibroproliferative responses of (KC-) activated HSCs [112].
Targeting FXa in animal models of liver fibrosis
Proof of concept for a role of FXa-PAR-2 signalling in liver fibrosis seems to be derived from rodent models of liver fibrosis in which different FXa inhibitors were used. Indeed, administration of the low molecular weight heparins enoxaparin, nadroparin and tinzaparin all significantly reduced liver fibrosis in a rat bile duct ligation model [113]. Moreover, dalteparin administration markedly ameliorated fibrosis in a rat model of carbon tetrachloride induced hepatic fibrogenesis. Importantly, however, it should be realized that targeting FXa also reduces downstream thrombin formation. Consequently, the reduced fibrotic response might not be solely FXa-PAR-2 dependent but also due to diminished thrombin-induced PAR-1 signalling as this latter signalling pathway is known to be profibrogenic as well [42].
Summary and conclusions
Several lines of evidence strongly suggest that coagulation FXa driven PAR-2 activation might play an important role in the progression of hepatic fibrosis. First, a local hypercoagulable state frequently accompanies liver fibrosis. Second, PAR-2 expression is increased in fibrotic liver tissue compared to healthy tissue. Third, FXa-induced signalling promotes profibrotic responses, like proliferation, differentiation, migration and ECM deposition, by fibroblasts, epithelial cells, inflammatory cells and endothelial cells of different origin. Most of these in vitro studies have however not been performed with specific liver cells and future studies need to establish the exact fibrotic program induced by FXa in stellate cells, hepatocytes, cholangiocytes, KCs and SECs. Fourth, FXa targeting limits hepatic fibrosis in different rodent models.
Based on these data, we propose that FXa-PAR-2 signalling contributes to the progression of fibrosis via multiple pathways (summarized in Fig. 3). (i) FXa induces pro-inflammatory responses in hepatocyes, cholangiocytes and KCs; (ii) It decreases endothelial barrier porosity; (iii) FXa leads to proliferation, migration, ECM production and differentiation into myofibroblasts of portal fibroblasts and HSCs; (iv) In a paracrine manner it activates portal fibroblasts HSCs thereby inducing proliferation, migration, ECM production and differentiation into myofibroblasts of these cells.
Fig 3.
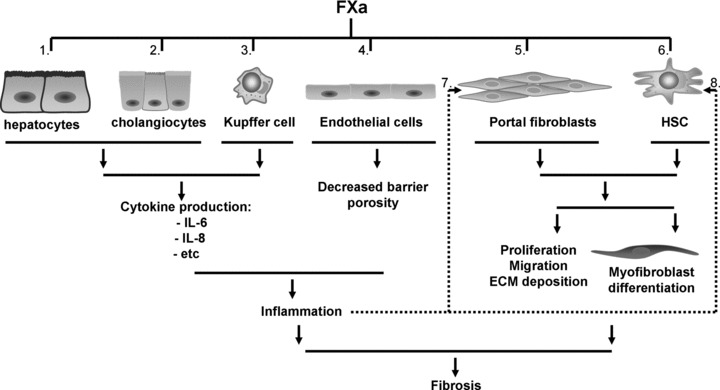
Model for the contribution of FXa-PAR-2 signalling in liver fibrosis. Depicted are the different PAR-2 expressing liver cells and their potential contribution to the progression of fibrosis as a functional consequence of FXa-signalling. FXa induces cytokine release in hepatocytes (1), cholangiocytes (2) and KCs (3), it decreases barrier porosity in SECs (4), and it leads to proliferation, migration, ECM production and differentiation into myofibroblasts of portal fibroblasts (5) and HSCs (6). Moreover, FXa-induced paracrine activation of portal fibroblast (7) and HSCs (8) also leads to proliferation, migration, ECM production and differentiation into myofibroblasts.
The pathophysiological significance of these novel findings needs to be established in the upcoming years however. It is of great importance to fully understand the contribution of FXa-driven PAR-2 activation to hepatic fibrosis and to prove or refute an essential role of this agonist–receptor pair in hepatic fibrosis. Such studies would pave the way for novel anticoagulant treatment strategies to reduce the progression of fibrosis. In this perspective, targeting FXa would be of greatest interest as FXa plays a dual role in hepatic fibrosis. Through its procoagulant effect it generates thrombin, the profibrotic effects of which are well known, and via PAR-2-dependent signalling it induces profibrotic responses by itself. Although anticoagulant treatment thus seems to be a very exciting possibility to modify the progression of hepatic fibrosis, careful monitoring of adverse events (i.e. bleeding complications) will be particularly important in view of the complex dysregulation of the coagulation system that is known to accompany advanced liver fibrosis. The advent of novel oral anticoagulants with a better safety profile is already a first important step to bringing the clinical applicability of anticoagulants for hepatic fibrosis closer to the clinic.
References
- 1.Schluger LK, Sheiner PA, Thung SN, et al. Severe recurrent cholestatic hepatitis C following orthotopic liver transplantation. Hepatology. 1996;23:971–6. doi: 10.1002/hep.510230505. [DOI] [PubMed] [Google Scholar]
- 2.Bonnard P, Lescure FX, Amiel C, et al. Documented rapid course of hepatic fibrosis between two biopsies in patients coinfected by HIV and HCV despite high CD4 cell count. J Viral Hepat. 2007;14:806–11. doi: 10.1111/j.1365-2893.2007.00874.x. [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lotersztajn S, Julien B, Teixeira-Clerc F, et al. Hepatic fibrosis: molecular mechanisms and drug targets. Annu Rev Pharmacol Toxicol. 2005;45:605–28. doi: 10.1146/annurev.pharmtox.45.120403.095906. [DOI] [PubMed] [Google Scholar]
- 5.Svegliati-Baroni G, De Minicis S, Marzioni M. Hepatic fibrogenesis in response to chronic liver injury: novel insights on the role of cell-to-cell interaction and transition. Liver Int. 2008;28:1052–64. doi: 10.1111/j.1478-3231.2008.01825.x. [DOI] [PubMed] [Google Scholar]
- 6.Borensztajn K, Peppelenbosch MP, Spek CA. Factor Xa: at the crossroads between coagulation and signaling in physiology and disease. Trends Mol Med. 2008;14:429–40. doi: 10.1016/j.molmed.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Borensztajn K, Peppelenbosch MP, Spek CA. Coagulation factor Xa signaling: the link between coagulation and inflammatory bowel disease? Trends Pharmacol Sci. 2009;30:8–16. doi: 10.1016/j.tips.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Northup PG, Sundaram V, Fallon MB, et al. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost. 2008;6:2–9. doi: 10.1111/j.1538-7836.2007.02772.x. [DOI] [PubMed] [Google Scholar]
- 9.Bancroft JD, McDowell SA, Degen SJ. The human prothrombin gene: transcriptional regulation in HepG2 cells. Biochemistry. 1992;31:12469–76. doi: 10.1021/bi00164a025. [DOI] [PubMed] [Google Scholar]
- 10.Huang MN, Hung HL, Stanfield-Oakley SA, et al. Characterization of the human blood coagulation factor X promoter. J Biol Chem. 1992;267:15440–6. [PubMed] [Google Scholar]
- 11.Reijnen MJ, Peerlinck K, Maasdam D, et al. Hemophilia B Leyden: substitution of thymine for guanine at position -21 results in a disruption of a hepatocyte nuclear factor 4 binding site in the factor IX promoter. Blood. 1993;82:151–8. [PubMed] [Google Scholar]
- 12.Erdmann D, Heim J. Orphan nuclear receptor HNF-4 binds to the human coagulation factor VII promoter. J Biol Chem. 1995;270:22988–96. doi: 10.1074/jbc.270.39.22988. [DOI] [PubMed] [Google Scholar]
- 13.Furie B, Bouchard BA, Furie BC. Vitamin K-dependent biosynthesis of gamma-carboxyglutamic acid. Blood. 1999;93:1798–808. [PubMed] [Google Scholar]
- 14.Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–41. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 15.Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53:505–18. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 16.Rullier A, Senant N, Kisiel W, et al. Expression of protease-activated receptors and tissue factor in human liver. Virchows Arch. 2006;448:46–51. doi: 10.1007/s00428-005-0078-0. [DOI] [PubMed] [Google Scholar]
- 17.Levy GA, MacPhee PJ, Fung LS, et al. The effect of mouse hepatitis virus infection on the microcirculation of the liver. Hepatology. 1983;3:964–73. doi: 10.1002/hep.1840030614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacPhee PJ, Schmidt EE, Keown PA, et al. Microcirculatory changes in livers of mice infected with murine hepatitis virus. Evidence from microcorrosion casts and measurements of red cell velocity. Microvasc Res. 1988;36:140–9. doi: 10.1016/0026-2862(88)90014-3. [DOI] [PubMed] [Google Scholar]
- 19.Wanless IR, Liu JJ, Butany J. Role of thrombosis in the pathogenesis of congestive hepatic fibrosis (cardiac cirrhosis) Hepatology. 1995;21:1232–7. [PubMed] [Google Scholar]
- 20.Wanless IR, Wong F, Blendis LM, et al. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology. 1995;21:1238–47. [PubMed] [Google Scholar]
- 21.Ben-Ari Z, Panagou M, Patch D, et al. Hypercoagulability in patients with primary biliary cirrhosis and primary sclerosing cholangitis evaluated by thrombelastography. J Hepatol. 1997;26:554–9. doi: 10.1016/s0168-8278(97)80420-5. [DOI] [PubMed] [Google Scholar]
- 22.Atalla SL, Toledo-Pereyra LH, MacKenzie GH, et al. Influence of oxygen-derived free radical scavengers on ischemic livers. Transplantation. 1985;40:584–90. doi: 10.1097/00007890-198512000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Hasselgren PO. Prevention and treatment of ischemia of the liver. Surg Gynecol Obstet. 1987;164:187–96. [PubMed] [Google Scholar]
- 24.Keller GA, West MA, Cerra FB, et al. Macrophage-mediated modulation of hepatic function in multiple-system failure. J Surg Res. 1985;39:555–63. doi: 10.1016/0022-4804(85)90124-6. [DOI] [PubMed] [Google Scholar]
- 25.Kuroe K, Kurokawa T, Nishikimi M, et al. Effects of thromboxane A2 synthetase inhibitor on postischemic liver injury in rats. Eur Surg Res. 1991;23:20–6. doi: 10.1159/000129132. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi Y, Yoshimura N, Nakamura K, et al. Expression of tissue factor in hepatic ischemic-reperfusion injury of the rat. Transplantation. 1998;66:708–16. doi: 10.1097/00007890-199809270-00004. [DOI] [PubMed] [Google Scholar]
- 27.Neubauer K, Knittel T, Armbrust T, et al. Accumulation and cellular localization of fibrinogen/fibrin during short-term and long-term rat liver injury. Gastroenterology. 1995;108:1124–35. doi: 10.1016/0016-5085(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 28.Anstee QM, Wright M, Goldin R, et al. Parenchymal extinction: coagulation and hepatic fibrogenesis. Clin Liver Dis. 2009;13:117–26. doi: 10.1016/j.cld.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Eisenberg PR, Siegel JE, Abendschein DR, et al. Importance of factor Xa in determining the procoagulant activity of whole-blood clots. J Clin Invest. 1993;91:1877–83. doi: 10.1172/JCI116404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herault JP, Bernat A, Pflieger AM, et al. Comparative effects of two direct and indirect factor Xa inhibitors on free and clot-bound prothrombinase. J Pharmacol Exp Ther. 1997;283:16–22. [PubMed] [Google Scholar]
- 31.Meddahi S, Bara L, Fessi H, et al. Determination of prothombinase activation after adding human purified prothrombin to human clot: comparison of hirudin, an activated factor II inhibitor, with DX9065a, an activated factor X inhibitor, on clot-associated thrombin and on prothrombin activation. Blood Coagul Fibrinolysis. 2005;16:125–33. doi: 10.1097/01.mbc.0000161566.82011.94. [DOI] [PubMed] [Google Scholar]
- 32.Bauters C, Lablanche JM, McFadden EP, et al. Relation of coronary angioscopic findings at coronary angioplasty to angiographic restenosis. Circulation. 1995;92:2473–9. doi: 10.1161/01.cir.92.9.2473. [DOI] [PubMed] [Google Scholar]
- 33.Marsden PA, Ning Q, Fung LS, et al. The Fgl2/fibroleukin prothrombinase contributes to immunologically mediated thrombosis in experimental and human viral hepatitis. J Clin Invest. 2003;112:58–66. doi: 10.1172/JCI18114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Assy N, Bekirov I, Mejritsky Y, et al. Association between thrombotic risk factors and extent of fibrosis in patients with non-alcoholic fatty liver diseases. World J Gastroenterol. 2005;11:5834–9. doi: 10.3748/wjg.v11.i37.5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papatheodoridis GV, Papakonstantinou E, Andrioti E, et al. Thrombotic risk factors and extent of liver fibrosis in chronic viral hepatitis. Gut. 2003;52:404–9. doi: 10.1136/gut.52.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poujol-Robert A, Rosmorduc O, Serfaty L, et al. Genetic and acquired thrombotic factors in chronic hepatitis C. Am J Gastroenterol. 2004;99:527–31. doi: 10.1111/j.1572-0241.2004.04092.x. [DOI] [PubMed] [Google Scholar]
- 37.Anstee QM, Goldin RD, Wright M, et al. Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. J Thromb Haemost. 2008;6:1336–43. doi: 10.1111/j.1538-7836.2008.03015.x. [DOI] [PubMed] [Google Scholar]
- 38.Wright M, Goldin R, Hellier S, et al. Factor V Leiden polymorphism and the rate of fibrosis development in chronic hepatitis C virus infection. Gut. 2003;52:1206–10. doi: 10.1136/gut.52.8.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macfarlane SR, Seatter MJ, Kanke T, et al. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–82. [PubMed] [Google Scholar]
- 40.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 41.Ruf W, Dorfleutner A, Riewald M. Specificity of coagulation factor signaling. J Thromb Haemost. 2003;1:1495–503. doi: 10.1046/j.1538-7836.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 42.Calvaruso V, Maimone S, Gatt A, et al. Coagulation and fibrosis in chronic liver disease. Gut. 2008;57:1722–7. doi: 10.1136/gut.2008.150748. [DOI] [PubMed] [Google Scholar]
- 43.Marra F, DeFranco R, Grappone C, et al. Expression of the thrombin receptor in human liver: up-regulation during acute and chronic injury. Hepatology. 1998;27:462–71. doi: 10.1002/hep.510270221. [DOI] [PubMed] [Google Scholar]
- 44.Gaca MD, Zhou X, Benyon RC. Regulation of hepatic stellate cell proliferation and collagen synthesis by proteinase-activated receptors. J Hepatol. 2002;36:362–9. doi: 10.1016/s0168-8278(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 45.Fiorucci S, Antonelli E, Distrutti E, et al. PAR1 antagonism protects against experimental liver fibrosis. Role of proteinase receptors in stellate cell activation. Hepatology. 2004;39:365–75. doi: 10.1002/hep.20054. [DOI] [PubMed] [Google Scholar]
- 46.Kmiec Z. Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol. 2001;161:III–XIII. doi: 10.1007/978-3-642-56553-3. , 1–151. [DOI] [PubMed] [Google Scholar]
- 47.Jesmin S, Gando S, Zaedi S, et al. Chronological expression of PAR isoforms in acute liver injury and its amelioration by PAR2 blockade in a rat model of sepsis. Thromb Haemost. 2006;96:830–8. [PubMed] [Google Scholar]
- 48.Cederqvist K, Haglund C, Heikkila P, et al. High expression of pulmonary proteinase-activated receptor 2 in acute and chronic lung injury in preterm infants. Pediatr Res. 2005;57:831–6. doi: 10.1203/01.PDR.0000161416.63314.70. [DOI] [PubMed] [Google Scholar]
- 49.Xiong J, Zhu Z, Liu J, et al. Role of protease activated receptor-2 expression in renal interstitial fibrosis model in mice. J Huazhong Univ Sci Technolog Med Sci. 2005;25:523–6. doi: 10.1007/BF02896006. [DOI] [PubMed] [Google Scholar]
- 50.Grandaliano G, Pontrelli P, Cerullo G, et al. Protease-activated receptor-2 expression in IgA nephropathy: a potential role in the pathogenesis of interstitial fibrosis. J Am Soc Nephrol. 2003;14:2072–83. doi: 10.1097/01.asn.0000080315.37254.a1. [DOI] [PubMed] [Google Scholar]
- 51.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–3. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 52.Friedman SL, Roll FJ, Boyles J, et al. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci USA. 1985;82:8681–5. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milani S, Herbst H, Schuppan D, et al. Cellular localization of type I III and IV procollagen gene transcripts in normal and fibrotic human liver. Am J Pathol. 1990;137:59–70. [PMC free article] [PubMed] [Google Scholar]
- 54.Milani S, Herbst H, Schuppan D, et al. Procollagen expression by nonparenchymal rat liver cells in experimental biliary fibrosis. Gastroenterology. 1990;98:175–84. doi: 10.1016/0016-5085(90)91307-r. [DOI] [PubMed] [Google Scholar]
- 55.Ramadori G, Veit T, Schwogler S, et al. Expression of the gene of the alpha-smooth muscle-actin isoform in rat liver and in rat fat-storing (ITO) cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1990;59:349–57. doi: 10.1007/BF02899424. [DOI] [PubMed] [Google Scholar]
- 56.Rockey DC, Boyles JK, Gabbiani G, et al. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J Submicrosc Cytol Pathol. 1992;24:193–203. [PubMed] [Google Scholar]
- 57.Marra F, Arrighi MC, Fazi M, et al. Extracellular signal-regulated kinase activation differentially regulates platelet-derived growth factor’s actions in hepatic stellate cells, and is induced by in vivo liver injury in the rat. Hepatology. 1999;30:951–8. doi: 10.1002/hep.510300406. [DOI] [PubMed] [Google Scholar]
- 58.Svegliati-Baroni G, Saccomanno S, Van Goor H, et al. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver. 2001;21:1–12. doi: 10.1034/j.1600-0676.2001.210101.x. [DOI] [PubMed] [Google Scholar]
- 59.Monno R, Grandaliano G, Faccio R, et al. Activated coagulation factor X: a novel mitogenic stimulus for human mesangial cells. J Am Soc Nephrol. 2001;12:891–9. doi: 10.1681/ASN.V125891. [DOI] [PubMed] [Google Scholar]
- 60.Bretschneider E, Braun M, Fischer A, et al. Factor Xa acts as a PDGF-independent mitogen in human vascular smooth muscle cells. Thromb Haemost. 2000;84:499–505. [PubMed] [Google Scholar]
- 61.Bretschneider E, Schror K. Cellular effects of factor Xa on vascular smooth muscle cells–inhibition by heparins? Semin Thromb Hemost. 2001;27:489–93. doi: 10.1055/s-2001-17956. [DOI] [PubMed] [Google Scholar]
- 62.Kaiser B. DX-9065a, a direct inhibitor of factor Xa. Cardiovasc Drug Rev. 2003;21:91–104. doi: 10.1111/j.1527-3466.2003.tb00108.x. [DOI] [PubMed] [Google Scholar]
- 63.Tanaka M, Arai H, Liu N, et al. Role of coagulation factor Xa and protease-activated receptor 2 in human mesangial cell proliferation. Kidney Int. 2005;67:2123–33. doi: 10.1111/j.1523-1755.2005.00317.x. [DOI] [PubMed] [Google Scholar]
- 64.Masamune A, Kikuta K, Satoh M, et al. Protease-activated receptor-2-mediated proliferation and collagen production of rat pancreatic stellate cells. J Pharmacol Exp Ther. 2005;312:651–8. doi: 10.1124/jpet.104.076232. [DOI] [PubMed] [Google Scholar]
- 65.Borensztajn K, Stiekema J, Nijmeijer S, et al. Factor Xa stimulates proinflammatory and profibrotic responses in fibroblasts via protease-activated receptor-2 activation. Am J Pathol. 2008;172:309–20. doi: 10.2353/ajpath.2008.070347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bachli EB, Pech CM, Johnson KM, et al. Factor Xa and thrombin, but not factor VIIa, elicit specific cellular responses in dermal fibroblasts. J Thromb Haemost. 2003;1:1935–44. doi: 10.1046/j.1538-7836.2003.00363.x. [DOI] [PubMed] [Google Scholar]
- 67.Fadok VA, Bratton DL, Konowal A, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Canbay A, Taimr P, Torok N, et al. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655–63. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- 69.Riewald M, Kravchenko VV, Petrovan RJ, et al. Gene induction by coagulation factor Xa is mediated by activation of protease-activated receptor 1. Blood. 2001;97:3109–16. doi: 10.1182/blood.v97.10.3109. [DOI] [PubMed] [Google Scholar]
- 70.Pan SL, Tao KY, Guh JH, et al. The p38 mitogen-activated protein kinase pathway plays a critical role in PAR2-induced endothelial IL-8 production and leukocyte adhesion. Shock. 2008;30:496–502. doi: 10.1097/SHK.0b013e3181673233. [DOI] [PubMed] [Google Scholar]
- 71.Tanaka Y, Sekiguchi F, Hong H, et al. PAR2 triggers IL-8 release via MEK/ERK and PI3-kinase/Akt pathways in GI epithelial cells. Biochem Biophys Res Commun. 2008;377:622–6. doi: 10.1016/j.bbrc.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 72.Wilson BJ, Harada R, LeDuy L, et al. CUX1 transcription factor is a downstream effector of the proteinase-activated receptor 2 (PAR2) J Biol Chem. 2009;284:36–45. doi: 10.1074/jbc.M803808200. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki T, Moraes TJ, Vachon E, et al. Proteinase-activated receptor-1 mediates elastase-induced apoptosis of human lung epithelial cells. Am J Respir Cell Mol Biol. 2005;33:231–47. doi: 10.1165/rcmb.2005-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borensztajn KS, Bijlsma MF, Groot AP, et al. Coagulation factor Xa drives tumor cells into apoptosis through BH3-only protein Bim up-regulation. Exp Cell Res. 2007;313:2622–33. doi: 10.1016/j.yexcr.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 75.Borensztajn KS, Spek CA. Protease-activated receptors, apoptosis and tumor growth. Pathophysiol Haemost Thromb. 2008;36:137–47. doi: 10.1159/000175152. [DOI] [PubMed] [Google Scholar]
- 76.Flynn AN, Buret AG. Proteinase-activated receptor 1 (PAR-1) and cell apoptosis. Apoptosis. 2004;9:729–37. doi: 10.1023/B:APPT.0000045784.49886.96. [DOI] [PubMed] [Google Scholar]
- 77.Jiang X, Bailly MA, Panetti TS, et al. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J Thromb Haemost. 2004;2:93–101. doi: 10.1111/j.1538-7836.2004.00545.x. [DOI] [PubMed] [Google Scholar]
- 78.Shimamoto R, Sawada T, Uchima Y, et al. A role for protease-activated receptor-2 in pancreatic cancer cell proliferation. Int J Oncol. 2004;24:1401–6. [PubMed] [Google Scholar]
- 79.Darmoul D, Gratio V, Devaud H, et al. Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem. 2004;279:20927–34. doi: 10.1074/jbc.M401430200. [DOI] [PubMed] [Google Scholar]
- 80.Jiang X, Guo YL, Bromberg ME. Formation of tissue factor-factor VIIa-factor Xa complex prevents apoptosis in human breast cancer cells. Thromb Haemost. 2006;96:196–201. [PubMed] [Google Scholar]
- 81.Uchima Y, Sawada T, Hirakawa K. Action of antiproteases on pancreatic cancer cells. Jop. 2007;8:479–87. [PubMed] [Google Scholar]
- 82.Sanchez-Hernandez PE, Ramirez-Duenas MG, Albarran-Somoza B, et al. Protease-activated receptor-2 (PAR-2) in cervical cancer proliferation. Gynecol Oncol. 2008;108:19–26. doi: 10.1016/j.ygyno.2007.08.083. [DOI] [PubMed] [Google Scholar]
- 83.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–86. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 84.Lindner JR, Kahn ML, Coughlin SR, et al. Delayed onset of inflammation in protease-activated receptor-2-deficient mice. J Immunol. 2000;165:6504–10. doi: 10.4049/jimmunol.165.11.6504. [DOI] [PubMed] [Google Scholar]
- 85.Shpacovitch VM, Varga G, Strey A, et al. Agonists of proteinase-activated receptor-2 modulate human neutrophil cytokine secretion, expression of cell adhesion molecules, and migration within 3-D collagen lattices. J Leukoc Biol. 2004;76:388–98. doi: 10.1189/jlb.0503221. [DOI] [PubMed] [Google Scholar]
- 86.Su X, Camerer E, Hamilton JR, et al. Protease-activated receptor-2 activation induces acute lung inflammation by neuropeptide-dependent mechanisms. J Immunol. 2005;175:2598–605. doi: 10.4049/jimmunol.175.4.2598. [DOI] [PubMed] [Google Scholar]
- 87.Blackhart BD, Emilsson K, Nguyen D, et al. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem. 1996;271:16466–71. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- 88.Takizawa T, Tamiya M, Hara T, et al. Abrogation of bronchial eosinophilic inflammation and attenuated eotaxin content in protease-activated receptor 2-deficient mice. J Pharmacol Sci. 2005;98:99–102. doi: 10.1254/jphs.scz050138. [DOI] [PubMed] [Google Scholar]
- 89.Akahane K, Okamoto K, Kikuchi M, et al. Inhibition of factor Xa suppresses the expression of tissue factor in human monocytes and lipopolysaccharide-induced endotoxemia in rats. Surgery. 2001;130:809–18. doi: 10.1067/msy.2001.116452. [DOI] [PubMed] [Google Scholar]
- 90.Busch G, Seitz I, Steppich B, et al. Coagulation factor Xa stimulates interleukin-8 release in endothelial cells and mononuclear leukocytes: implications in acute myocardial infarction. Arterioscler Thromb Vasc Biol. 2005;25:461–6. doi: 10.1161/01.ATV.0000151279.35780.2d. [DOI] [PubMed] [Google Scholar]
- 91.Jones A, Geczy CL. Thrombin and factor Xa enhance the production of interleukin-1. Immunology. 1990;71:236–41. [PMC free article] [PubMed] [Google Scholar]
- 92.Pertosa G, Simone S, Soccio M, et al. Coagulation cascade activation causes CC chemokine receptor-2 gene expression and mononuclear cell activation in hemodialysis patients. J Am Soc Nephrol. 2005;16:2477–86. doi: 10.1681/ASN.2004070621. [DOI] [PubMed] [Google Scholar]
- 93.Yamaguchi Y, Okabe K, Liang J, et al. Thrombin and factor Xa enhance neutrophil chemoattractant production after ischemia/reperfusion in the rat liver. J Surg Res. 2000;92:96–102. doi: 10.1006/jsre.2000.5884. [DOI] [PubMed] [Google Scholar]
- 94.Bardadin KA, Desmet VJ. Ultrastructural observations on sinusoidal endothelial cells in chronic active hepatitis. Histopathology. 1985;9:171–81. doi: 10.1111/j.1365-2559.1985.tb02433.x. [DOI] [PubMed] [Google Scholar]
- 95.Horn T, Junge J, Christoffersen P. Early alcoholic liver injury: changes of the Disse space in acinar zone 3. Liver. 1985;5:301–10. doi: 10.1111/j.1600-0676.1985.tb00253.x. [DOI] [PubMed] [Google Scholar]
- 96.Diaz R, Kim JW, Hui JJ, et al. Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum Pathol. 2008;39:102–15. doi: 10.1016/j.humpath.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 97.Rescan PY, Loreal O, Hassell JR, et al. Distribution and origin of the basement membrane component perlecan in rat liver and primary hepatocyte culture. Am J Pathol. 1993;142:199–208. [PMC free article] [PubMed] [Google Scholar]
- 98.McGuire RF, Bissell DM, Boyles J, et al. Role of extracellular matrix in regulating fenestrations of sinusoidal endothelial cells isolated from normal rat liver. Hepatology. 1992;15:989–97. doi: 10.1002/hep.1840150603. [DOI] [PubMed] [Google Scholar]
- 99.Feistritzer C, Lenta R, Riewald M. Protease-activated receptors-1 and -2 can mediate endothelial barrier protection: role in factor Xa signaling. J Thromb Haemost. 2005;3:2798–805. doi: 10.1111/j.1538-7836.2005.01610.x. [DOI] [PubMed] [Google Scholar]
- 100.Borensztajn K, Aberson H, Peppelenbosch MP, et al. FXa-induced intracellular signaling links coagulation to neoangiogenesis: potential implications for fibrosis. Biochim Biophys Acta. 2009;1793:798–805. doi: 10.1016/j.bbamcr.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 101.Ankoma-Sey V, Wang Y, Dai Z. Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells. Hepatology. 2000;31:141–8. doi: 10.1002/hep.510310122. [DOI] [PubMed] [Google Scholar]
- 102.Corpechot C, Barbu V, Wendum D, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology. 2002;35:1010–21. doi: 10.1053/jhep.2002.32524. [DOI] [PubMed] [Google Scholar]
- 103.Medina J, Arroyo AG, Sanchez-Madrid F, et al. Angiogenesis in chronic inflammatory liver disease. Hepatology. 2004;39:1185–95. doi: 10.1002/hep.20193. [DOI] [PubMed] [Google Scholar]
- 104.Wang YQ, Luk JM, Ikeda K, et al. Regulatory role of vHL/HIF-1alpha in hypoxia-induced VEGF production in hepatic stellate cells. Biochem Biophys Res Commun. 2004;317:358–62. doi: 10.1016/j.bbrc.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 105.Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci USA. 2001;98:7742–7. doi: 10.1073/pnas.141126698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hezi-Yamit A, Wong PW, Bien-Ly N, et al. Synergistic induction of tissue factor by coagulation factor Xa and TNF: evidence for involvement of negative regulatory signaling cascades. Proc Natl Acad Sci USA. 2005;102:12077–82. doi: 10.1073/pnas.0504526102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Daubie V, Cauwenberghs S, Senden NH, et al. Factor Xa and thrombin evoke additive calcium and proinflammatory responses in endothelial cells subjected to coagulation. Biochim Biophys Acta. 2006;1763:860–9. doi: 10.1016/j.bbamcr.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 108.Senden NH, Jeunhomme TM, Heemskerk JW, et al. Factor Xa induces cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells. J Immunol. 1998;161:4318–24. [PubMed] [Google Scholar]
- 109.Banfi C, Brioschi M, Barbieri SS, et al. Mitochondrial reactive oxygen species: a common pathway for PAR1- and PAR2-mediated tissue factor induction in human endothelial cells. J Thromb Haemost. 2009;7:206–16. doi: 10.1111/j.1538-7836.2008.03204.x. [DOI] [PubMed] [Google Scholar]
- 110.Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. doi: 10.1016/s0168-8278(01)00142-8. [DOI] [PubMed] [Google Scholar]
- 111.Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124:778–90. doi: 10.1053/gast.2003.50087. [DOI] [PubMed] [Google Scholar]
- 112.Galli A, Svegliati-Baroni G, Ceni E, et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology. 2005;41:1074–84. doi: 10.1002/hep.20683. [DOI] [PubMed] [Google Scholar]
- 113.Salam OM, Baiuomy AR, Ameen A, et al. A study of unfractionated and low molecular weight heparins in a model of cholestatic liver injury in the rat. Pharmacol Res. 2005;51:59–67. doi: 10.1016/j.phrs.2004.04.009. [DOI] [PubMed] [Google Scholar]