

Abstract
Regulated intramembrane proteolysis (RIP) is a signaling mechanism through which transmembrane precursor proteins are cleaved to liberate their cytoplasmic and/or luminal /extracellular fragments from membranes so that these fragments are able to function at a new location. Recent studies have indicated that this proteolytic reaction plays an important role in host-virus interaction. On one hand, RIP transfers the signal from the endoplasmic reticulum (ER) to nucleus to activate antiviral genes in response to alteration of the ER caused by viral infection. On the other hand, RIP can be hijacked by virus to process transmembrane viral protein precursors and to destroy transmembrane antiviral proteins. Understanding this Yin and Yang side of RIP may lead to new strategies to combat viral infection.
Keywords: Regulated intramembrane proteolysis, Site-2 protease, Signal peptide-peptidase, CREB3L1, CREB3, virus
1 Introduction
There is growing evidence indicating that membrane proteins can be cleaved within the plane of the membrane. Such cleavage liberates cytoplasmic and/or lumenal /extracellular fragments of transmembrane precursor proteins and allows these fragments to function at a new location. This mechanism, termed regulated intramembrane proteolysis (RIP), influences processes as diverse as cellular differentiation, lipid metabolism, and the response to unfolded proteins [1]. Three classes of protease, namely Site-2 protease (S2P), rhomboid, and aspartyl intramembrane proteases, are known to catalyze the intramembrane cleavage [2]. Aspartyl intramembrane proteases are a large family of proteins that include γ-secretase, signal peptide peptidase (SPP), and SPP-like proteins (SPPLs) [3]. Emerging evidence has now demonstrated the importance of RIP in pathogen-host interaction during viral infection.
Infection of mammalian cells by almost all classes of virus induces endoplasmic reticulum (ER) stress by massive synthesis of ER-associated viral proteins [4]. Viral infection also frequently leads to alteration in the ER structure to generate membrane vesicles that protect the viral replication machinery [5]. Thus, the signaling pathway transmitting the signal generated through changes in the ER caused by viral infection to nucleus where genes combating viral infection are activated should be important for antiviral immunity. Indeed, recent studies have pointed out the importance of S2P-catalyzed RIP, which plays a critical role to transfer signal from the ER to nucleus [2], in antiviral immunity [6-8].
In addition to S2P, SPP and SPPLs-mediated RIP has also been reported to mediate virus-host interactions. RIP catalyzed by these proteases regulates functions of immune cells [9-11]. Nevertheless, SPP has also been shown to be hijacked by virus to facilitate the progression of its life cycle. The protease is used by several viruses to process the viral protein precursors and to degrade transmembrane proteins involved in antiviral immune responses [12-14].
This article will discuss the RIP process that plays an important role in antiviral immunity as well as that facilitates viral infection.
2 Host-virus interactions mediated by S2P-catalyzed RIP
2.1 Overview of S2P-catalyzed RIP
S2P is a polytopic membrane protein with the characteristics of a membrane-embedded zinc metalloprotease [15]. The protease cleaves transcription factors synthesized as membrane-bound precursors [2]. Cleavage mediated by S2P is tightly coupled to proteolysis catalyzed by Site-1 protease (S1P): The S2P-catalyzed cleavage does not take place until the bulk of the luminal portion of the RIP substrates has been removed by S1P-mediated proteolysis, as a short luminal segment is a prerequisite for S2P-catalyed intramembrane cleavage [16-18]. Notably, S1P also cleaves protein precursors of Lassa virus and Crimean-Congo hemorrhagic fever virus to produce mature viral surface glycoproteins that are required for viral infection [19,20]. However, since these cleavages only require S1P but not S2P, they should not be categorized as RIP.
The most extensively studied proteins cleaved by S2P are sterol regulatory element binding proteins (SREBPs), a family of transcription factors critical for regulation of cholesterol metabolism [21,22]. SREBPs are inserted in membranes of the endoplasmic reticulum (ER) through two transmembrane helices in a hairpin fashion with both the NH2- and COOH-terminal of the protein facing the cytosol. In cells deprived of cholesterol, SREBPs are transported from the ER to Golgi complex where the proteins are cleaved by S1P followed by S2P, both of which are Golgi-resident proteins [23,24]. This cleavage releases the NH2-terminal domain of SREBPs from membranes, allowing them to activate all genes required for cholesterol synthesis and uptake in nucleus [22]. Accumulation of cholesterol in the ER causes SREBPs to be trapped in the ER so that they are separated from S1P and S2P [25]. Consequently, SREBPs are not proteolytically activated, resulting in declination of transcription of genes required for cholesterol synthesis and uptake.
Another well-characterized transcription factor proteolytically activated by S2P is activation transcription factor 6 (ATF6). ATF6 is a type II membrane protein with a single transmembrane domain [26]. The NH2-terminal domain of the protein faces cytosol [26]. Under resting condition, ATF6 is not cleaved as it is retained in the ER [27]. Upon accumulation of unfolded proteins in the ER, ATF6 is translocated from the ER to Golgi, where it is cleaved sequentially by S1P and S2P [17,27,28]. The intramembrane proteolysis carried out by S2P liberates the NH2-terminal domain of ATF6 from membranes, allowing it to enter nucleus where it activates transcription of chaperons that facilitate protein folding in the ER [26]. This regulatory pathway helps cells to survive conditions of ER stress.
RIP of SREBPs and AFT6 plays important roles for viral infection, but the effect is indirectly related to cholesterol metabolism and ER stress. Thus, it will not be further discussed in this article. Instead, two RIP reactions catalyzed by S1P and S2P that directly counter viral infections will be discussed in detail below.
2.2 Antiviral responses mediated by RIP of cAMP response element binding protein 3-like 1 (CREB3L1)
Similar to ATF6, CREB3L1 (also known as OASIS) contains a single transmembrane helix with the NH2-terminal cytosolic domain resembling a transcription factor of the basic leucine zipper family [29]. CREB3L1 has been reported to stimulate synthesis of collagen required for bone formation [30]. The first clue that CREB3L1 may be involved in antiviral responses came from the observation that infection of West Nile virus reduced the amount of this protein in neurons [31]. The importance of CREB3L1 in antiviral immunity has been subsequently recognized through observations that expression of the protein has to be silenced in cells permissive for hepatitis C virus (HCV) replication [7,32]
HCV is a positive stranded RNA virus accounting for most cases of chronic liver disease worldwide and is the major risk factor for development of hepatocarcinoma [33,34]. The replication of HCV in cultured cells was first studied through HCV subgenomic replicon, which consists of HCV RNA engineered to express a selectable marker gene, neo, in place of a portion of the viral RNA that is not required for viral replication [35]. When Huh7 cells, a line of human hepatoma cells, were transfected with the HCV subgenomic replicon followed by selection with G418, subclones of Huh7 cells were established in which HCV RNA was constantly replicating [35]. However, only a few cells survived the selection, an observation suggesting that Huh7 cells are not very permissive for HCV replication [35,36]. When HCV RNA was eliminated from the cells harboring the HCV replicon through interferon treatment, the cured cells showed dramatically enhanced permissiveness for HCV RNA replication as demonstrated by the large number of cells that survived G418 selection following re-transfection with the HCV replicon RNA [36]. Thus, transfection of HCV replicon followed by G418 treatment selected a small population of Huh7 cells permissive for HCV replication [32]. Compared to their parental Huh7 cells, expression of CREB3L1 protein is dramatically reduced in various independent subclones of Huh7 cells permissive for HCV replication [7,32].
Further studies revealed that HCV infection triggered sequential cleavage of CREB3L1 catalyzed by S1P and S2P [7] (Figures 1 and 2). The S2P-catalyzed final cleavage liberates the NH2-terminal domain of CREB3L1 from membranes, allowing it to activate genes encoding cell cycle inhibitors to block proliferation of the virus-infected cells [7] (Figures 1 and 2). Inasmuch as HCV replication in Huh7-derived cells requires active division of the host cells [37,38], RIP of CREB3L1 not only blocks proliferation of HCV-infected cells but also inhibits viral replication [7]. These results explain why expression of CREB3L1 has to be silenced in subclones of Huh7 cells permissive for HCV replication, as these cells have to divide in the presence of efficient replication of the viral RNA.
Figure 1.
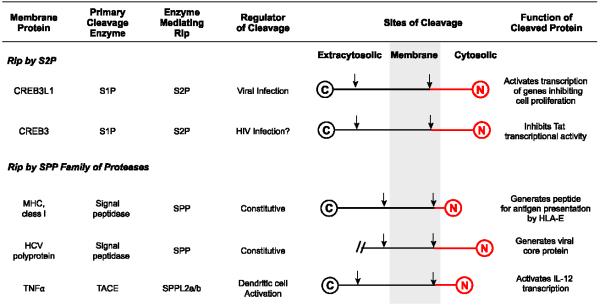
RIP reactions important for virus-host interactions
The cleaved protein fragments are highlighted in red. For simplicity, proteins are not drawn to scale. For HCV polyprotein, we show only the sequence linked by the first transmembrane helix.
Figure 2.

Viral infection-induced RIP of CREB3L1
Viral infection-induced production of ceramide and/or ER stress caused by massive synthesis of virus-encoded membrane proteins may stimulate translocation of CREB3L1 precursor from the ER to Golgi, where the protein is sequentially cleaved by S1P and S2P. This cleavage releases the NH2-terminal domain of CREB3L1 from membranes, allowing it to enter the nucleus where it activates genes encoding cell cycle inhibitors to block proliferation of the virus-infected cells.
The effect of CREB3L1 on cell proliferation is not restricted to HCV-infected cells. Infection with other RNA virus such as West Nile and Sendai virus and DNA virus such as murine γ-herpesvirus 68 also triggered RIP of CREB3L1, resulting in blockage in proliferation of the cells infected by the virus [7]. Thus, CREB3L1 may play an important role to prevent viral spreading by inhibiting proliferation of virus-infected cells, and the protein may be particularly effective in combating infection of virus whose replication depends on active division of the host cells. While replication of HCV in cultured cells requires proliferation of the host cells [37,38], such requirement in virus-infected patients remains to be determined. The best studied virus whose infection in vivo depends on mitosis of the host cells is human papillomavirus (HPV) [39], a DNA virus that is a predominant risk factor for development of cervical and oropharyngeal cancers [40]. Therefore, CREB3L1 could be a major obstacle for HPV infection. To overcome this obstacle, HPV is likely to develop strategies to inactivate CREB3L1. HPV is known to encode viral proteins that inhibit cellular proteins controlling progression of cell cycle [39]. One of the viral proteins E5, which stimulates cell cycle progression [39], is a transmembrane protein located in the ER and Golgi complex [41]. Considering that ER to Golgi translocation is required for transmembrane precursor proteins to be proteolytically activated by S2P-catalyzed RIP, E5 may be an inhibitor for RIP of CREB3L1. If so, relieving this inhibition could be a novel strategy to treat HPV infection.
Inasmuch as CREB3L1 activates multiple genes encoding inhibitors of cell cycle, the protein may be considered as a tumor suppressor. Further evidence supporting this notion came from the observation that RIP of CREB3L1 is required for doxorubicin, an extensively used chemotherapeutic reagent, to block proliferation of cancer cells [42], and the observation that inactivation of CREB3L1 through chromosome fusion is associated with development of low-grade fibromyxoid sarcoma [43]. Considering that CREB3L1 is proteolytically activated in virus-infected cells, the protein may play an important role in preventing virus-induced tumorigenesis. If this is indeed the case, tumors induced by viral infection may have impaired RIP of CREB3L1 and consequently become resistant to doxorubicin treatment. Thus, understanding the role of CREB3L1 in virus-induced tumor formation may provide more insight into selection of chemotherapeutic reagents to treat these tumors.
It remains unclear how viral infection triggers RIP of CREB3L1. It was reported previously that ER stress triggers cleavage of CREB3L1 [44]. Since viral infection is known to induce ER stress through massive synthesis of ER-associated viral protein [4], it is possible that viral infection triggers cleavage of CREB3L1 through ER stress (Figure 2). Viral infection was also known to trigger production of ceramide [45,46]. Inasmuch as ceramide is a potent stimulator for RIP of CREB3L1 [42], viral infection-induced accumulation of ceramide may also be involved in stimulating RIP of CREB3L1 (Figure 2).
2.3 Antiviral responses mediated by RIP of CREB3
CREB3 (also known as Lzip and Luman), which adopts a membrane topology similar to that of CREB3L1, is another transcription factor activated through RIP catalyzed by S1P and S2P [47]. Similar to CREB3L1, CREB3 may also be involved in inhibition of cell cycle, as loss of CREB3 function triggers transformation of NIH-3T3 cells [48]. Expression of HCV core protein stimulates transformation of the cells by excluding CREB3 from nucleus [48]. While the mechanism of this reaction has not been identified, it is likely that core protein inhibits RIP of CREB3. However, it should be noted that cellular transformation caused by core-mediated inhibition of CREB3 was observed in cells overexpressing the viral core protein alone but not in those infected by HCV [48]. Thus, the contribution of core-mediated inactivation of CREB3 in HCV-induced hepatocarcinoma remains unclear.
The CREB3-mediated antiviral response has been best illustrated by its activity in inhibiting infection of human immunodeficiency virus (HIV), a retrovirus [6]. HIV envelope glycoprotein (Env) is synthesized as a 160 kDa precursor and is processed during its passage through the secretory pathway by a host cell protease, furin, to produce the extracellular subunit (gp120) and the transmembrane subunit (gp41) [49]. CREB3 was identified as a protein that binds to gp41, and this interaction leads to degradation of CREB3 [6].
To determine the functional significance of gp41-mediated degradation of CREB3, the nuclear form of CREB3 corresponding to its NH2-terminal domain was transfected to cells infected by HIV. The result revealed that the cleaved nuclear form of CREB3 bound to another HIV protein Tat that activates transcription of viral proteins required for assembly of viral particles [6]. This interaction inhibited the function of Tat, leading to reduced production of infectious viral particles [6]. These observations suggest that RIP of CREB3 is likely to be stimulated in response to retroviral infection in order to inhibit assembly of viral particles by blocking the Tat activity (Figure 1). To counter this antiviral response, HIV uses gp41 subunit of its envelop protein to stimulate degradation of CREB3 precursor. According to this scenario, blocking the interaction between gp41 and CREB3 could be a novel strategy to treat HIV infection. Nevertheless, it should be emphasized that RIP of CREB3 has never been observed in HIV-infected cells owing to the rapid degradation of CREB3 precursor in these cells.
CREB3 may also be involved in inhibition of lytic infection of herpes simplex virus (HSV), a class of DNA virus [8]. Infection of epithelial cells by HSV results in massive synthesis of viral genome and infectious viral particles, eventually leading to death of the host cells. This phase of the viral life cycle, which is referred as lytic infection, requires viral protein VP-16-activated transcription of viral genes [50,51]. In order to activate its target gene, VP-16 has to bind several host proteins including human factor C1 (HCF) [52]. The membrane-bound CREB3 precursor was identified as a cellular protein interacting with HCF [53,54]. This interaction retained HCF in the ER, preventing HCF from entering nucleus to facilitate transcription activated by VP-16 [8]. As a result, cells overexpressing CREB3 were resistant to lytic infection of HSV [8].
In addition to lytic infection, HSV can establish latent infection in neurons within the trigeminal ganglia, during which only small portions of the viral genome are transcribed [55]. The virus is quiescent during latent infection but can be reactivated periodically to replicate and move back along the axons to resume lytic infection in epithelial cells [55]. It is proposed that high level of CREB3 expression in neurons may suppress lytic infection by sequestering HCF in the ER so that latent infection can be established in these cells [8]. According to this hypothesis, RIP of CREB3 may trigger reactivation of HSV by releasing HCF into nucleus. Thus, identifying the conditions activating RIP of CREB3 may lead to novel strategies to prevent reactivation of HSV from latency.
RIP of CREB3 has been observed during maturation of dendritic cells, the major antigen-presenting cells that play an important role in antiviral adapt immunity [56,57]. However, the mechanism through which CREB3 is proteolytic activated during dendritic cell maturation and the physiological significance of such activation remains unclear.
3 Host-virus interaction mediated by SPP and SPPLs
3.1 Overview of SPP and SPPLs
SPP and SPPLs are a family of membrane-embedded aspartic protease [3,58]. In mammalian cells, the family contains five members: SPP, SPPL2a, SPPL2b, SPPL2c and SPPL3 [3]. Similar to S2P, SPP and SPPLs cleave type II transmembrane protein, and in most cases the proteolysis also requires a primary cleavage that removes bulk of the protease substrates on the extracytoplasmic (lumenal or extracellular) side of the membranes [2]. While SPP locates in the ER, SPPL2a and SPPL2b reside in endosomes, lysosomes and plasma membranes [3]. The difference in their localization is consistent with the findings that these proteases do not share the same substrates [3]. SPP and SPPLs are involved in regulation of innate and adaptive immune responses, but they can also be hijacked by viruses to facilitate the progression of their life cycle (See section 3.2-3.5).
3.2 SPP-catalyzed production of HLA-E ligand
The best-studied example of RIP mediated by SPP is the proteolytic processing of signal peptides from major histocompatibility complex (MHC) class I molecules such as HLA-A [9]. HLA-A is targeted to the secretory pathway through its signal sequence. During its translocation through the ER membrane, the signal sequence is cleaved off from the pre-protein by signal peptidase. The cleaved signal peptide, which remains membrane-bound with a type II orientation, is then cleaved by SPP in the middle of the membrane to liberate the NH2-terminal half of the signal peptide into the cytosol [9] (Figure 1). This cytosolic fragment is then transported into the ER lumen where it binds to HLA-E, a nonclassical MHC class I molecule [59]. The HLA-E/peptide complexes travel to the cell surface where they bind to CD94/NKG2A receptors on natural killer (NK) cells and inhibit NK cell-mediated lysis [60]. This pathway protects cells expressing MHC class I molecules from killing by NK cells.
Remarkably, this SPP-mediated pathway is hijacked by human cytomegalovirus (HCMV) to protect virus-infected cells from NK cell-induced cytotoxicity. HCMV, a DNA virus, encodes UL40 that contains within its signal sequence a stretch of peptide identical to the ligand of HLA-E derived from signal sequence of the MHC class 1 molecules [61,62]. Following cleavage by signal peptidase, the signal sequence of UL40 is further cleaved by SPP to produce the ligand for HLA-E [63]. Enhanced presentation of HLA-E/peptide complex at the cell surface protects the virus-infected cells from NK cell –mediated lysis [62].
3.3 SPP-mediated degradation of MHC class I molecules in HCMV-infected cells
HCMV is known to encode multiple proteins to evade immune system. In addition to UL40 discussed in section 3.2, HCMV also encodes a viral protein US2 to stimulate degradation of MHC class I molecules in order to hide viral infection from detection by the immune system [64]. US2 triggers retrotranslocation of MHC class I molecules from ER membranes to cytosol so that the proteins can be degraded by proteasomes [64]. To identify the mechanism of the US2-mediated evasion of the immune system, proteins associated with wild type US2 were compared to those bound to a mutant inactive form of the viral protein. This analysis identified SPP as a protein interacting with wild type but not the mutant US2 [14]. Knockdown of SPP blocked membrane dislocation and subsequent proteasomal degradation of MHC class I molecules in cells expressing US2, indicating SPP is involved in ER-associated degradation of MHC class I molecules induced by the viral protein [14]. Importantly, SPP did not stimulate degradation of MHC class I molecules in cells that did not express US2 [14]. Thus, this process is completely different from SPP-mediated RIP of MHC class I molecules (see section 3.2) that takes place in cells free of viral infection. Indeed, it remains unclear whether the protease activity of SPP is required for this process. SPP was found to be in two distinct complexes: One contained signal peptides capable to be cleaved by SPP, while the other one contained unfolded ER membrane proteins that are not substrates for SPP but are destined for proteasomal degradation [65,66]. Thus, SPP may not only cleave signal peptide but also participate in ER-associated degradation. Future studies are required to determine whether US2 recruits MHC class I molecules into the SPP complex involved in proteasomal degradation of membrane proteins in the ER.
3.4 SPP-catalyzed maturation of HCV core protein
HCV encodes a polyprotein that is processed by viral and cellular proteases to produce at least 10 viral proteins required for replication of viral RNA and assembly of viral particles [34]. The core protein corresponds to the first 169 amino acids of the viral polyprotein (Figure 3). It is localized at cytosolic side of the ER membrane and is linked to the remainder of the viral protein via a signal peptide-like transmembrane sequence at its COOH terminus (Figures 1 and 3). After cleavage by signal peptidase in the ER lumen, SPP cleaves the signal peptide-like transmembrane domain [12,67,68] (Figures 1 and 3). This cleavage liberates the core protein from the ER, allowing it to associate with lipid droplets where it assembles viral particles into a virus-lipoprotein complex (Figure 3) [12,69-72]. Consistent with these observations, inhibition of SPP-catalyzed maturation of the core protein blocked production of infectious HCV particles [68,73].
Figure 3.
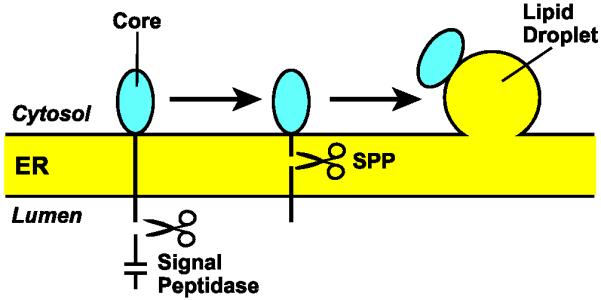
SPP-catalyzed maturation of HCV core protein
Signal peptidase cleaves HCV-encoded polyprotein in the ER lumen immediately after the signal peptide-like transmembrane sequence located at the COOH-terminal end of the core protein. This cleavage allows SPP to cleave in the middle of the transmembrane domain so that the mature core protein can be transported to lipid droplets where it is assembled into virus-lipoprotein particles.
In addition to HCV, SPP is also required to process viral polyprotein to produce the mature core protein in virus related to HCV, such as classical swine fever virus and GB virus-B [13,74].
3.5 SPPL2a/b-catalyzed RIP of tumor necrosis factor α (TNFα) and foamy virus envelope protein
SPPL2a and SPPL2b play an important role for activated dendritic cells to induce T-helper 1 (Th1) cell-mediated antiviral adaptive immunity. Upon activation by viral infection, dendritic cells produce TNFα, a type 2 membrane protein [10] (Figure 1). Following cleavage by TNFα converting enzyme (TACE) that sheds the majority of the extracellular domain, SPPL2a/b cleaves inside the transmembrane domain to release the intracellular domain of TNFα from membranes, allowing it to enter the nucleus to activate transcription of IL-12 [10,75] (Figure 1). Since IL-12 secreted from activated dendritic cells promotes differentiation of Th1 cells [76], RIP of TNFα may be required for the antiviral immunity mediated by Th1 cells. However, this hypothesis has not been tested in vivo.
SPPL2a/b and SPPL3 were also reported to cleave the 18-kDa leader peptide of the foamy virus envelope protein (LP18) [77]. While SPPL2a/b catalyze the intramembrane proteolysis, SPPL3 in this case may catalyze the primary cleavage that removes bulk of the LP18 at the luminal side [77]. The functional significance of this RIP reaction has not been identified.
4 Conclusion
RIP has been identified as one of the major signaling pathways that mediate virus-host interactions. Viruses can hijack the pathway to facilitate progression of their life cycle. They can use the intramembrane protease such as SPP to process the viral polyprotein, and to evade immune system by cleaving cellular transmembrane proteins critical for antiviral immunity. On the other hand, RIP is crucial for host to orchestrate antiviral responses, either directly by combating viral infection in virus-infected cells, or indirectly by regulating the functions of lymphocytes and leukocytes. In order to establish successful infection, viruses encode viral proteins to inhibit these antiviral responses. Understanding this Yin and Yang side of RIP may lead to new strategies to combat viral infection.
Highlights.
Regulated intramembrane proteolysis (RIP) is a signaling mechanism for host cells to orchestrate antiviral responses.
Intramembrane proteases involved in RIP can be hijacked by virus to facilitate progression of the viral life cycle.
Understanding the roles of RIP in host-virus interactions may reveal novel strategies to combat viral infection.
Acknowledgement
JY is supported by research grants from the NIH (HL-20948, AI 090119).
Abbreviation
- ATF6
activation transcription factor 6
- CREB3
cAMP response element binding protein 3
- CREB3L1
CREB3-like 1
- ER
endoplasmic reticulum
- HCMV
human cytomegalovirus
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HPV
human papillomavirus
- HSV
herpes simplex virus
- RIP
regulated intramembrane proteolysis
- S1P
Site-1 protease
- S2P
Site-2 protease
- SPP
signal peptide peptidase
- SPPLs
SPP-like proteins
- SREBPs
sterol response element binding proteins
- TNFα
tumor necrosis factor α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell. 2000;100:391. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 2.Ye J, Brown MS, Goldstein JL. Regulated Intramembrane Proteolysis (Rip) Encyclopedia of Biological Chemistry. 2004;3:665. [Google Scholar]
- 3.Golde TE, Wolfe MS, Greenbaum DC. Signal peptide peptidases: A family of intramembrane-cleaving proteases that cleave type 2 transmembrane proteins. Semin. Cell Dev. Biol. 2009;20:225. doi: 10.1016/j.semcdb.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 5.Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Micro. 2008;6:363. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blot G, Lopez-Vergès S, Treand C, Kubat NJ, croix-Genéte D, Emiliani S.p., Benarous R, Berlioz-Torrent C. Luman, a new partner of HIV-1 TMgp41, interferes with Tat-mediated transcription of the HIV-1 LTR. J. Mol. Biol. 2006;364:1034. doi: 10.1016/j.jmb.2006.09.080. [DOI] [PubMed] [Google Scholar]
- 7.Denard B, Seemann J, Chen Q, Gay A, Huang H, Chen Y, Ye J. The membrane-bound transcription factor CREB3L1 is activated in response to virus infection to inhibit proliferation of virus-infected cells. Cell Host & Microbe. 2011;10:65. doi: 10.1016/j.chom.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu R, Misra V. Potential role for Luman, the cellular homologue of Herpes Simplex Virus VP16 (α Gene trans-inducing factor), in herpesvirus latency. J. Virol. 2000;74:934. doi: 10.1128/jvi.74.2.934-943.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemberg MK, Bland FA, Weihofen A, Braud VM, Martoglio B. Intramembrane proteolysis of signal peptides: An essential step in the generation of HLA-E epitopes. J Immunol. 2001;167:6441. doi: 10.4049/jimmunol.167.11.6441. [DOI] [PubMed] [Google Scholar]
- 10.Friedmann E, Hauben E, Maylandt K, Schleeger S, Vreugde S, Lichtenthaler SF, Kuhn PH, Stauffer D, Rovelli G, Martoglio B. SPPL2a and SPPL2b promote intramembrane proteolysis of TNFα in activated dendritic cells to trigger IL-12 production. Nat. Cell Biol. 2006;8:843. doi: 10.1038/ncb1440. [DOI] [PubMed] [Google Scholar]
- 11.Schneppenheim J, Dressel R, Hüttl S, Lüllmann-Rauch R, Engelke M, Dittmann K, Wienands J, Eskelinen EL, Hermans-Borgmeyer I, Fluhrer R, Saftig P, Schröder B. The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J. Exp. Med. 2013;210:41. doi: 10.1084/jem.20121069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLauchlan J, Lemberg MK, Hope G, Martoglio B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002;21:3980. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heimann M, Sosa GR, Martoglio B, Thiel HJ, Rümenapf T. Core protein of pestiviruses Is processed at the C terminus by signal peptide peptidase. J. Virol. 2006;80:1915. doi: 10.1128/JVI.80.4.1915-1921.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loureiro J, Lilley BN, Spooner E, Noriega V, Tortorella D, Ploegh HL. Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature. 2006;441:894. doi: 10.1038/nature04830. [DOI] [PubMed] [Google Scholar]
- 15.Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell. 1997;1:47. doi: 10.1016/s1097-2765(00)80006-4. [DOI] [PubMed] [Google Scholar]
- 16.Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, Goldstein JL. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell. 1996;85:1037. doi: 10.1016/s0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- 17.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 2000;6:1355. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 18.Shen J, Prywes R. Dependence of Site-2 protease cleavage of ATF6 on prior Site-1 protease digestion is determined by the size of the luminal domain of ATF6. J. Biol. Chem. 2004;279:43046. doi: 10.1074/jbc.M408466200. [DOI] [PubMed] [Google Scholar]
- 19.Lenz O, ter Meulen J, Klenk HD, Seidah NG, Garten W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA. 2001;98:12701. doi: 10.1073/pnas.221447598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vincent MJ, Sanchez AJ, Erickson BR, Basak A, Chretien M, Seidah NG, Nichol ST. Crimean-Congo Hemorrhagic Fever Virus glycoprotein proteolytic processing by subtilase SKI-1. J. Virol. 2003;77:8640. doi: 10.1128/JVI.77.16.8640-8649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA. 1999;96:11041. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. USA. 2003;100:12027. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeBose-Boyd RA, Brown MS, Li WP, Nohturfft A, Goldstein JL, Espenshade PJ. Transport-dependent proteolysis of SREBP: Relocation of Site-1 Protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell. 1999;99:703. doi: 10.1016/s0092-8674(00)81668-2. [DOI] [PubMed] [Google Scholar]
- 24.Nohturfft A, Yabe D, Goldstein JL, Brown MS, Espenshade PJ. Regulated step in cholesterol feedback localized to budding of SCAP from ER membranes. Cell. 2000;102:315. doi: 10.1016/s0092-8674(00)00037-4. [DOI] [PubMed] [Google Scholar]
- 25.Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008;8:512. doi: 10.1016/j.cmet.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 1999;10:3787. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 2002;3:99. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 28.Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002;277:13045. doi: 10.1074/jbc.M110636200. [DOI] [PubMed] [Google Scholar]
- 29.Omori Y, Imai J.i., Suzuki Y, Watanabe S, Tanigami A, Sugano S. OASIS is a transcriptional activator of CREB/ATF family with a transmembrane domain. Biochem. Biophys. Res. Commun. 2002;293:470. doi: 10.1016/S0006-291X(02)00253-X. [DOI] [PubMed] [Google Scholar]
- 30.Murakami T, Saito A, Hino S.i., Kondo S, Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga K, Saitoh M, Nishimura R, Yoneda T, Kou I, Furuichi T, Ikegawa S, Ikawa M, Okabe M, Wanaka A, Imaizumi K. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 2009;11:1205. doi: 10.1038/ncb1963. [DOI] [PubMed] [Google Scholar]
- 31.van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, Power C. West Nile Virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J. Virol. 2007;81:10933. doi: 10.1128/JVI.02422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Q, Denard B, Huang H, Ye J. Epigenetic silencing of antiviral genes renders clones of Huh-7 cells permissive for hepatitis C virus replication. J. Virol. 2013;87:659. doi: 10.1128/JVI.01984-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Appel N, Schaller T, Penin F, Bartenschlager R. From structure to function: new insights into hepatitis C virus RNA replication. J. Biol. Chem. 2006;281:9833. doi: 10.1074/jbc.R500026200. [DOI] [PubMed] [Google Scholar]
- 34.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat. Rev. Micro. 2007;5:453. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 35.Lohmann V, Korner F, Koch JO, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 36.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for hepatitis C virus genomic and subgenomic RNA replication. J. Virol. 2002;76:13001. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pietschmann T, Lohmann V, Rutter G, Kurpanek K, Bartenschlager R. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 2001;75:1252. doi: 10.1128/JVI.75.3.1252-1264.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scholle F, Li K, Bodola F, Ikeda M, Luxon BA, Lemon SM. Virus-host cell interactions during hepatitis C virus RNA replication: Impact of polyprotein expression on the cellular transcriptome and cell cycle association with viral RNA synthesis. J. Virol. 2004;78:1513. doi: 10.1128/JVI.78.3.1513-1524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer. 2010;10:550. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 40.Lowy DR, Schiller JT. Reducing HPV-associated cancer globally. Cancer Prev. Res. 2012;5:18. doi: 10.1158/1940-6207.CAPR-11-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conrad M, Bubb VJ, Schlegel R. The human papillomavirus type 6 and 16 E5 proteins are membrane-associated proteins which associate with the 16-kilodalton pore-forming protein. J. Virol. 1993;67:6170. doi: 10.1128/jvi.67.10.6170-6178.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denard B, Lee C, Ye J. Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1. ELife Sciences. 2012;1:e00090. doi: 10.7554/eLife.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mertens F, Fletcher CDM, Antonescu CR, Coindre JM, Colecchia M, Domanski HA, Downs-Kelly E, Fisher C, Goldblum JR, Guillou L, Reid R, Rosai J, Sciot R, Mandahl N, Panagopoulos I. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS//CREB3L1 fusion gene. Lab Invest. 2005;85:408. doi: 10.1038/labinvest.3700230. [DOI] [PubMed] [Google Scholar]
- 44.Murakami T, Kondo S, Ogata M, Kanemoto S, Saito A, Wanaka A, Imaizumi K. Cleavage of the membrane-bound transcription factor OASIS in response to endoplasmic reticulum stress. J. Neurochem. 2006;96:1090. doi: 10.1111/j.1471-4159.2005.03596.x. [DOI] [PubMed] [Google Scholar]
- 45.Jan JT, Chatterjee S, Griffin DE. Sindbis virus entry into cells triggers apoptosis by activating sphingomyelinase, leading to the release of ceramide. J. Virol. 2000;74:6425. doi: 10.1128/jvi.74.14.6425-6432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grassmé H, Riehle A, Wilker B, Gulbins E. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J. Biol. Chem. 2005;280:26256. doi: 10.1074/jbc.M500835200. [DOI] [PubMed] [Google Scholar]
- 47.Raggo C, Rapin N, Stirling J, Gobeil P, Smith-Windsor E, O’Hare P, Misra V. Luman, the cellular counterpart of Herpes Simplex Virus VP16, is processed by regulated intramembrane proteolysis. Mol. Cell. Biol. 2002;22:5639. doi: 10.1128/MCB.22.16.5639-5649.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin DY, Wang HL, Zhou Y, Chun ACS, Kibler KV, Hou YD, Kung HF, Jeang KT. Hepatitis C virus core protein-induced loss of LZIP function correlates with cellular transformation. EMBO J. 2000;19:729. doi: 10.1093/emboj/19.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hunter E, Swanstrom R. Retrovirus envelope glycoproteins. Curr. Top. Microbiol. Immunol. 1990;157:187. doi: 10.1007/978-3-642-75218-6_7. [DOI] [PubMed] [Google Scholar]
- 50.O’Hare P, Goding CR. Herpes simplex virus regulatory elements and the immunoglobulin octamer domain bind a common factor and are both targets for virion transactivation. Cell. 1988;52:435. doi: 10.1016/s0092-8674(88)80036-9. [DOI] [PubMed] [Google Scholar]
- 51.Triezenberg SJ, LaMarco KL, McKnight SL. Evidence of DNA: protein interactions that mediate HSV-1 immediate early gene activation by VP16. Genes Dev. 1988;2:730. doi: 10.1101/gad.2.6.730. [DOI] [PubMed] [Google Scholar]
- 52.Wilson AC, LaMarco K, Peterson MG, Herr W. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell. 1993;74:115. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 53.Freiman RN, Herr W. Viral mimicry: common mode of association with HCF by VP16 and the cellular protein LZIP. Genes Dev. 1997;11:3122. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu R, Yang P, Padmakumar S, Misra V. The Herpesvirus transactivator VP16 mimics a human basic domain leucine zipper protein, Luman, in its interaction with HCF. J. Virol. 1998;72:6291. doi: 10.1128/jvi.72.8.6291-6297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones C. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv. Virus Res. 1998;51:81. doi: 10.1016/s0065-3527(08)60784-8. [DOI] [PubMed] [Google Scholar]
- 56.Eleveld-Trancikova D, Sanecka A, van Hout-Kuijer MA, Looman MWG, Hendriks IAM, Jansen BJH, Adema GJ. DC-STAMP interacts with ER-resident transcription factor LUMAN which becomes activated during DC maturation. Mol. Immunol. 2010;47:1963. doi: 10.1016/j.molimm.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 57.Sanecka A, Ansems M, van Hout-Kuijer MA, Looman MWG, Prosser AC, Welten S, Gilissen C, Sama IE, Huynen MA, Veltman JA, Jansen BJH, Eleveld-Trancikova D, Adema GJ. Analysis of genes regulated by the transcription factor LUMAN identifies ApoA4 as a target gene in dendritic cells. Mol. Immunol. 2012;50:66. doi: 10.1016/j.molimm.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B. Identification of signal peptide peptidase, a Presenilin-type aspartic protease. Science. 2002;296:2215. doi: 10.1126/science.1070925. [DOI] [PubMed] [Google Scholar]
- 59.Lee N, Goodlett DR, Ishitani A, Marquardt H, Geraghty DE. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J Immunol. 1998;160:4951. [PubMed] [Google Scholar]
- 60.Braud VM, Allan DSJ, O’Callaghan CA, Soderstrom K, D’Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI, Phillips JH, Lanier LL, McMichael AJ. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 61.Tomasec P, Braud VM, Rickards C, Powell MB, McSharry BP, Gadola S, Cerundolo V, Borysiewicz LK, McMichael AJ, Wilkinson GWG. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 2000;287:1031. doi: 10.1126/science.287.5455.1031. [DOI] [PubMed] [Google Scholar]
- 62.Ulbrecht M, Martinozzi S, Grzeschik M, Hengel H, Ellwart JW, Pla M, Weiss EH. The human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J. Immunol. 2000;164:5019. doi: 10.4049/jimmunol.164.10.5019. [DOI] [PubMed] [Google Scholar]
- 63.Prod’homme V, Tomasec P, Cunningham C, Lemberg MK, Stanton RJ, McSharry BP, Wang ECY, Cuff S, Martoglio B, Davison AJ, Braud VM, Wilkinson GWG. Human cytomegalovirus UL40 signal peptide regulates cell surface expression of the NK cell ligands HLA-E and gpUL18. J. Immunol. 2012;188:2794. doi: 10.4049/jimmunol.1102068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiertz EJHJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec6l-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 65.Schrul B, Kapp K, Sinning I, Dobberstein B. Signal peptide peptidase (SPP) assembles with substrates and misfolded membrane proteins into distinct oligomeric complexes. Biochem. J. 2010;427:523. doi: 10.1042/BJ20091005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crawshaw SG, Martoglio B, Meacock SL, High S. A misassembled transmembrane domain of a polytopic protein associates with signal peptide peptidase. Biochem. J. 2004;384:9. doi: 10.1042/BJ20041216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okamoto K, Moriishi K, Miyamura T, Matsuura Y. Intramembrane proteolysis and endoplasmic reticulum retention of hepatitis C virus core protein. J. Virol. 2004;78:6370. doi: 10.1128/JVI.78.12.6370-6380.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oehler V, Filipe A, Montserret R, da Costa D, Brown G, Penin F, McLauchlan J. Structural analysis of hepatitis C virus Core-E1 signal peptide and requirements for cleavage of the genotype 3a signal sequence by signal peptide peptidase. J. Virol. 2012;86:7818. doi: 10.1128/JVI.00457-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 2007;282:37158. doi: 10.1074/jbc.M707329200. [DOI] [PubMed] [Google Scholar]
- 70.Boulant S, Targett-Adams P, McLauchlan J. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J. Gen. Virol. 2007;88:2204. doi: 10.1099/vir.0.82898-0. [DOI] [PubMed] [Google Scholar]
- 71.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007;9:1089. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 72.Ye J. Hepatitis C virus: a new class of virus associated with particles derived from very low-density lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2012;32:1099. doi: 10.1161/ATVBAHA.111.241448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Targett-Adams P, Hope G, Boulant S, McLauchlan J. Maturation of hepatitis C virus core protein by signal peptide peptidase is required for virus production. J. Biol. Chem. 2008;283:16850. doi: 10.1074/jbc.M802273200. [DOI] [PubMed] [Google Scholar]
- 74.Targett-Adams P, Schaller T, Hope G, Lanford RE, Lemon SM, Martin A, McLauchlan J. Signal peptide peptidase cleavage of GB virus B core protein is required for productive infection in vivo, J. Biol. Chem. 2006;281:29221. doi: 10.1074/jbc.M605373200. [DOI] [PubMed] [Google Scholar]
- 75.Fluhrer R, Grammer G, Israel L, Condron MM, Haffner C, Friedmann E, Bohland C, Imhof A, Martoglio B, Teplow DB, Haass C. A γ-secretase-like intramembrane cleavage of TNFα by the GxGD aspartyl protease SPPL2b. Nat. Cell Biol. 2006;8:894. doi: 10.1038/ncb1450. [DOI] [PubMed] [Google Scholar]
- 76.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity. 2003;19:641. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 77.Voss M, Fukumori A, Kuhn PH, Künzel U, Klier B, Grammer G, Haug-Kröper M, Kremmer E, Lichtenthaler SF, Steiner H, Schröder B, Haass C, Fluhrer R. Foamy virus envelope protein is a substrate for signal peptide peptidase-like 3 (SPPL3) J. Biol. Chem. 2012;287:43401. doi: 10.1074/jbc.M112.371369. [DOI] [PMC free article] [PubMed] [Google Scholar]