

Abstract
Gluconacetobacter europaeus, one of the microorganisms most commonly used for vinegar production, produces the unfavorable flavor compound acetoin. Since acetoin reduction is important for rice vinegar production, a genetic approach was attempted to reduce acetoin produced by G. europaeus KGMA0119 using specific gene knockout without introducing exogenous antibiotic resistance genes. A uracil-auxotrophic mutant with deletion of the orotate phosphoribosyltransferase gene (pyrE) was first isolated by positive selection using 5-fluoroorotic acid. The pyrE disruptant designated KGMA0704 (ΔpyrE) showed 5-fluoroorotic acid resistance. KGMA0704 and the pyrE gene were used for further gene disruption experiments as a host cell and a selectable marker, respectively. Targeted disruption of aldC or als, which encodes α-acetolactate decarboxylase or α-acetolactate synthase, was attempted in KGMA0704. The disruption of these genes was expected to result in a decrease in acetoin levels. A disruption vector harboring the pyrE marker within the targeted gene was constructed for double-crossover recombination. The cells of KGMA0704 were transformed with the exogenous DNA using electroporation, and genotypic analyses of the transformants revealed the unique occurrence of targeted aldC or als gene disruption. The aldC disruptant KGMA4004 and the als disruptant KGMA5315 were cultivated, and the amount of acetoin was monitored. The acetoin level in KGMA4004 culture was significantly reduced to 0.009% (wt/vol) compared with KGMA0119 (0.042% [wt/vol]), whereas that of KGMA5315 was not affected (0.037% [wt/vol]). This indicates that aldC disruption is critical for acetoin reduction. G. europaeus KGMA4004 has clear application potential in the production of rice vinegar with less unfavorable flavor.
INTRODUCTION
Acetic acid bacteria (AAB) are Gram-negative and obligately aerobic bacteria that generally have high ethanol-oxidizing ability and high acetic acid resistance (1). Several unique AAB are involved in cellulose production (2, 3), symbiosis with insects (4), nitrogen fixing in the rhizosphere (5), and chronic granulomatous disease (6). Gluconacetobacter europaeus is representative of vinegar producers and is used for the production of high-acidity vinegar (acid concentration, >10%) in submerged bioreactors because it has extremely strong ethanol-oxidizing ability and ethanol/acetic acid resistance compared with other species (7). Therefore, G. europaeus is an invaluable bacterium for industrial vinegar production.
Despite its importance in vinegar production, molecular-genetic and biochemical analyses of G. europaeus are limited. Although a draft genome sequence of the G. europaeus type strain, LMG 18890T, was recently determined (8), targeted gene disruption systems in the bacterium remain to be developed. It would be beneficial to establish gene knockout techniques for the analysis of gene function and metabolic modification in G. europaeus. Three methods of transformation with plasmids have been reported for other AAB: calcium chloride (9), electroporation (10), and conjugation with Escherichia coli (11). However, exogenous antibiotic resistance genes were the main selectable markers used to screen transformants in these studies, and these markers often remain in the chromosome or plasmid. Since these transformants are genetically chimeric and are defined as transgenic organisms, there are many challenges to overcome before they can be approved for use in food production. Specific gene knockout is also applicable using a bactericidal orotic acid analogue, 5-fluoroorotic acid (5-FOA), as a source of selective pressure, along with pyrimidine-biosynthetic pathway genes mutated to generate a selectable marker system. This approach does not require any exogenous drug resistance genes. Orotate phosphoribosyltransferase (OPRTase) (encoded by pyrE) and orotidine-5′-monophosphate decarboxylase (OMPdecase) (encoded by pyrF) catalyze the last two steps of pyrimidine biosynthesis. 5-FOA is converted to 5-fluorouridine monophosphate (5-FUMP) by both enzymes, and 5-FUMP can be incorporated into RNA, which results in disruption of transcription. Mutants with defects in either of these enzymes are resistant to 5-FOA but are uracil auxotrophs. A selection system utilizing these properties has been successfully established in several microbes (12–14), including hyperthermophilic archaea (15–17).
In general, the process of rice vinegar production is divided into three steps, which are each accomplished by a different microorganism. The first step is saccharification of carbohydrates by Aspergillus, the second is alcohol fermentation by yeast, and the third is acetic acid fermentation by AAB. Therefore, the flavor of rice vinegar is characterized not only by the source, but also by a large number of flavor compounds, including various organic acids and volatile compounds, that are produced by these microorganisms during the fermentative processes. Among these flavor compounds, acetoin and diacetyl, which have a strong butter/cream/cheese-like odor, are undesirable volatile compounds (perceived as “off flavor”) in Japanese sake and rice vinegar. Thus, it is important to manage the amounts of acetoin and diacetyl for the quality control of rice vinegar.
Many microorganisms, including several lactic acid bacteria (LAB) and Bacillus subtilis, produce acetoin as a product of fermentative or overflow metabolism (18–20). In these microorganisms, acetoin is produced from pyruvate via α-acetolactate, and the enzymes responsible for acetoin formation are α-acetolactate synthase (Als) and α-acetolactate decarboxylase (AldC), as shown in Fig. 1C. Als catalyzes the condensation of two molecules of pyruvate to form one molecule of α-acetolactate, and AldC decarboxylates α-acetolactate to give acetoin. On the other hand, diacetyl is generated by the chemical decarboxylation of α-acetolactate under aerobic conditions because of its chemical instability. Acetoin is also produced enzymatically from diacetyl by diacetyl reductase (Fig. 1C) (18). The acetoin pathway shares intermediary metabolites (pyruvate and α-acetolactate) with the branched-chain amino acid (BCAA)-biosynthetic pathway, and Als catalyzes the same reaction as acetohydroxyacid synthase (AHAS), which is the rate-limiting enzyme of BCAA biosynthesis (21). The roles of the acetoin pathway are to repress the intracellular and environmental acidification caused by excess carbohydrates and unbalanced carbon fluxes in the central pathway (20, 22, 23) and to store carbon in the form of acetoin, which can be used in case of nutrient starvation (19). In AAB, it has long been known that the addition of lactic acid to the growth medium results in the formation of acetoin via pyruvate and α-acetolactate (24). This suggests that the acetoin pathway of AAB has a function similar to those of other bacteria.
Fig 1.

G. europaeus pyrE locus, aldC gene cluster, and putative acetoin pathway predicted from draft genome sequences of the type strain, LMG18890T. (A) Gene arrangement of pyrE encoding orotate phosphoribosyltransferase on contig 26 (CADP01000026.1). The arrows above and below the scheme indicate primers used for the construction of a pyrE disruption vector and a pyrE marker cassette, respectively (Fig. 3). cs, citrate synthase; gr, glutamate racemase. (B) The aldC gene cluster located on contig 6 (CADP01000006.1). The arrows above and below the scheme indicate primers used for the construction of als and aldC disruption vectors, respectively (Fig. 3). aldC, α-acetolactate decarboxylase; als, α-acetolactate synthase; h, hypothetical protein; ldh, lactate dehydrogenase; pdc, pyruvate decarboxylase. (C) Putative acetoin pathway in G. europaeus. AHAS, acetohydroxyacid synthase; AldC, α-acetolactate decarboxylase; Als, α-acetolactate synthase; DR, diacetyl reductase; Ldh, lactate dehydrogenase; Pdc, pyruvate decarboxylase.
This study aimed to construct an acetoin-nonproducing strain by aldC or als disruption in G. europaeus. For this purpose, we first constructed a strain with pyrE deleted displaying 5-FOA resistance and uracil auxotrophy. Using this strain as a host cell, the mutant with aldC or als disrupted, in which each locus was replaced by a pyrE marker cassette through double-crossover homologous recombination, was isolated and the acetoin productivities were investigated.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains of G. europaeus and the plasmids used in this study are listed in Table 1. G. europaeus KGMA0119 (wild type), previously designated M0119 (25), was isolated from rice vinegar. The 16S rRNA sequence of KGMA0119 is 100% identical to that of the type strain, LMG 18890T (8), and other properties also indicate that KGMA0119 belongs to the species G. europaeus (data not shown). KGMA0119 and its derivatives were cultivated in yeast-peptone-dextrose broth (YPD) (rich medium) or minimal medium (described below) at 30°C with reciprocal shaking at 150 rpm. YPD was composed of 30 g/liter glucose, 5 g/liter yeast extracts (Nihon Seiyaku, Tokyo, Japan), and 2 g/liter polypeptone (Nihon Seiyaku). The minimal medium consisted of 30 g/liter glucose, 10 g/liter l-(+)-monosodium glutamate monohydrate, 0.1 g/liter K2HPO4, 0.5 g/liter KH2PO4, 0.1 g/liter KCl, 0.1 g/liter CaCl2 · 2H2O, 0.25 g/liter MgSO4 · 7H2O, 2 mg/liter calcium (+)-pantothenate, and 1.0 ml/liter trace mineral solution (5 g FeCl3, 50 g ZnSO4 · 7H2O, 0.5 g Na2MoO4 · 2H2O, 2.5 g CuSO4 · 5H2O, 0.5 g H3BO3, 10 g MnSO4 · 5H2O, and 0.5 g CoCl2 per liter). Unless otherwise indicated, 0.4% (wt/vol) ethanol and 0.5% (wt/vol) acetic acid were added to both YPD and the minimal medium. For plate culture, 0.9% (wt/vol) agar was added. When necessary, 5-FOA (Wako Pure Chemicals, Osaka, Japan) and uracil were added at final concentrations of 0.2% (wt/vol) and 60 μg/ml, respectively.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics |
|---|---|
| E. coli | |
| DH5α | λ− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 |
| G. europaeus | |
| KGMA0119 | Wild type isolated from rice vinegar; previously designated M0119 (25) |
| KGMA0704 | ΔpyrE derived from KGMA0119 |
| KGMA4004 | ΔpyrE ΔaldC::pyrE derived from KGMA0704 |
| KGMA5315 | ΔpyrE Δals::pyrE derived from KGMA0704 |
| Plasmids | |
| pUC19 | Ampr |
| pBR322 | Ampr |
| pUC19-ΔpyrE | pUC19 derivative; ΔpyrE cassette composed of 5′ upstream and 3′ downstream regions of pyrE (1,943 bp) |
| pBR322-ΔaldC::pyrE | pBR322 derivative; ΔaldC::pyrE cassette composed of 5′ upstream and 3′ downstream regions of aldC and a pyrE cassette (3,538 bp) |
| pBR322-Δals::pyrE | pBR322 derivative; Δals::pyrE cassette composed of 5′ upstream and 3′ downstream regions of als and a pyrE cassette (3,411 bp) |
E. coli strain DH5α (TaKaRa Bio, Ohtsu, Shiga, Japan), which was used to generate disruption vectors, was routinely cultivated at 37°C in Luria-Bertani medium containing 50 μg/ml ampicillin.
DNA manipulation and DNA sequencing.
General DNA manipulations were performed as described by Green and Sambrook (26). The DNA sequences of G. europaeus pyrE, pyrF, aldC, and als were predicted from draft genome sequences of G. europaeus LMG 18890T (8) by BLAST searches (TBLASTN) using the corresponding amino acid sequences of Gluconacetobacter xylinus NBRC 3288 (27) as queries (pyrE, YP_004867468.1; pyrF, YP_004868701.1; aldC, YP_004868152.1; als, YP_004868151.1). PCR was carried out using KOD plus (Toyobo, Osaka, Japan) or Pfu-X (Greiner Bio-One, Frickenhausen, Germany) as the DNA polymerase, and primers were designed based on the draft genome sequences of G. europaeus LMG18890T. Restriction enzymes and modifying enzymes were purchased from TaKaRa Bio or Nippon Gene (Tokyo, Japan). After gel electrophoresis, DNA fragments were extracted using the NucleoSpin Gel and PCR Cleanup Kit (TaKaRa Bio). For the construction of targeted gene disruption vectors, pUC19 or pBR322 (TaKaRa Bio) was used. Plasmid DNA was isolated using the Plasmid Midi Kit (Qiagen, Hilden, Germany). DNA sequencing was performed using a BigDye Terminator Cycle Sequencing kit, ver. 3.1, and a model 3130 capillary sequencer (Applied Biosystems, Foster City, CA).
Construction of disruption vectors.
The primers used in this study are listed in Table 2 and are shown in Fig. 1 (also see Fig. 3). Vectors for targeted gene disruption through double-crossover homologous recombination were constructed as follows. A 2.7-kb fragment containing the pyrE open reading frame (ORF) together with its 5′ (1.0-kb) and 3′ (1.0-kb) flanking regions was amplified from KGMA0119 genomic DNA using primers EU-F and ED-R. The amplified fragment was then subcloned into pUC19 between the EcoRI and BamHI sites. The 5′ and 3′ flanking regions of pyrE and the plasmid backbone, excluding 672 bp of the pyrE ORF, were then amplified from the intermediary plasmid using primers E5′-R and E3′-F, which were phosphorylated before use. The PCR fragment obtained was self-ligated, and the resulting plasmid was designated pUC19-ΔpyrE (4,614 bp) (see Fig. 3A).
Table 2.
Primers used in this study
| Primer | Sequence (5′–3′)a | Use |
|---|---|---|
| EU-F | GGAATTCGATCGCCATCCACGACGAAT | pyrE disruption |
| ED-R | CGGGATCCAGCCCGGAAAACATTCAGCA | pyrE disruption |
| E5′-R | GGAGCCTGTTGAAAGTCCAG | pyrE disruption |
| E3′-F | GAAGAAGCACTGGCGCTGAA | pyrE disruption |
| EP-F2 | CTGCCATATCCCGTGTTCGT | pyrE marker cassette |
| E-R4 | TCGCCATAGGGAAAGACTGC | pyrE marker cassette, genotyping |
| EP-F3 | ATCCCCACCAGCATGTTCAC | Genotyping |
| aldC-F | AGAATTCGATCACGCTCGAAACCCTGT | aldC disruption |
| aldC-R | AAATCGATCGATATCCCCCACCAGTTCA | aldC disruption |
| aldC-i1 | ATTCACGAAGCCATTCGCGTGGCTG | aldC disruption |
| aldC-i2 | GATTGCCCGAGAATGGTGAAGCAGG | aldC disruption |
| aldC-F2 | CCTGAACCTTCATTTCAATGGTGCG | Genotyping |
| aldC-R2 | GTCCATGCTCTGGTGCGTAAGCTTC | Genotyping |
| als-F2 | AAATCGATAGGAGACTCAGGTCTTGAAGCTGAA | als disruption, genotyping |
| als-R2 | AAGGATCCGTTCCAGTTGATGCTGGATTCG | als disruption |
| als-i1 | AATCACCTGCTGATGAAGCCGCTG | als disruption |
| als-i2 | GTCAAACAGGCGGTCCACCTTC | als disruption |
| ldh-i2 | AGTACACGCCACTGTTCCACCAG | Genotyping |
| pyrE-probe-F1 | CCTTCATCTATCCGCGCAAC | Probe for Southern blot analysis |
| pyrE-probe-R1 | ACAGGTCGCCTGCATGATCT | Probe for Southern blot analysis |
| aldC-probe-F1 | AATGTGCCACAGGACGAGGT | Probe for Southern blot analysis |
| aldC-probe-R1 | TGAAGGTTCAGGACGACACG | Probe for Southern blot analysis |
Restriction enzyme sites are underlined.
Fig 3.

Schematic diagrams of the isolation of specific gene disruptants. (A) Disruption of pyrE. The arrowheads indicate PvuII recognition sites. (B) Disruption of aldC. The arrowheads indicate SmaI recognition sites. (C) Disruption of als. The black and gray arrowheads indicate SmaI and EcoRI recognition sites, respectively. The construction of each disruption vector and the procedure for transformation are described in Materials and Methods. The genotypes of all strains were investigated by PCR and Southern blot analysis (Fig. 4) using appropriate primers and probes as shown, in addition to sequencing analyses. See the legend to Fig. 1 for a description of each gene.
A 1.5-kb pyrE marker cassette containing a putative promoter region was amplified from KGMA0119 genomic DNA using primers EP-F2 and E-R4, which were phosphorylated before use. A 2.9-kb fragment containing the aldC ORF together with its 5′ (1.0-kb) and 3′ (1.0-kb) flanking regions was amplified using primers aldC-F and aldC-R. The amplified fragment was then subcloned into pBR322 between the EcoRI and ClaI sites. The 5′ and 3′ flanking regions of aldC and the plasmid backbone, excluding 813 bp of the aldC ORF, were then amplified from the intermediary plasmid using primers aldC-i1 and aldC-i2. The PCR fragment obtained was then ligated with a 5′-phosphorylated pyrE cassette, and the resulting plasmid was designated pBR322-ΔaldC::pyrE (7,880 bp) (see Fig. 3B). In the case of the als disruption vector, a 3.4-kb fragment containing the als ORF together with its 5′ (1.0-kb) and 3′ (1.0-kb) flanking regions was amplified using primers als-F2 and als-R2. The amplified fragment was then subcloned into pBR322 between the ClaI and BamHI sites. The 5′ and 3′ flanking regions of als and the plasmid backbone, excluding 1,509 bp of the als ORF, were then amplified from the intermediary plasmid using primers als-i1 and als-i2. The PCR fragment obtained was then ligated with a 5′-phosphorylated pyrE cassette, and the resulting plasmid was designated pBR322-Δals::pyrE (7,425 bp) (see Fig. 3C).
Transformation of G. europaeus by electroporation.
Gene disruption of G. europaeus was carried out according to previous descriptions (10) with slight modifications. KGMA0119 and KGMA0704 (ΔpyrE) cells were cultured in 30 ml of YPD and YPDU (YPD with uracil), respectively. Both media contained 1.6% (wt/vol) ethanol and 1% (vol/vol) cellulase (Sigma-Aldrich, St. Louis, MO). When the culture reached logarithmic phase, cells were harvested, washed with distilled water once and with 10% (vol/vol) glycerol twice, concentrated to an optical density at 600 nm (OD660) of 30 with 10% glycerol, and stored at −80°C until use. Then, 45 μl of frozen cells was thawed on ice and mixed with DNA solution in an ice-cold 0.1-cm electroporation cuvette (Bio-Rad Laboratories, Hercules, CA). Using an ECM630 electroporation system (Harvard Apparatus, Holliston, MA), pulses of 2.5 kV, 200 Ω, and 25 μF were applied. To construct the strain with pyrE deleted, pUC19-ΔpyrE was introduced into KGMA0119 competent cells. After pulse conduction, 1 ml of ice-cold YPD was added immediately, and the cells were cultivated at 30°C for 16 h. An aliquot of culture was inoculated onto a minimal agar plate containing 5-FOA and uracil and incubated at 30°C for 2 to 3 days. To confirm the deletion of pyrE, genomic DNA was extracted from positive clones that showed 5-FOA resistance and uracil auxotrophy. The genotypes of these clones were analyzed by PCR using primers EP-F3 and E-R4 (see Fig. 3A), by sequencing of the DNA fragments obtained, and/or by Southern hybridization (see below). The strain with pyrE deleted was designated KGMA0704 and was used for the subsequent targeted gene disruption experiments.
pBR322-ΔaldC::pyrE or pBR322-Δals::pyrE was introduced into KGMA0704 competent cells by electroporation, and then the cells were allowed to recover in 1 ml of YPD for 3 h. Next, cells were harvested, washed with saline (0.85% [wt/vol] NaCl), transferred to 5 ml of minimal medium lacking uracil, and cultured for more than 48 h to concentrate the uracil prototrophs. An aliquot of the culture was diluted, spread onto a minimal agar plate lacking uracil, and incubated at 30°C for 2 to 3 days. The genotypes of the positive clones were analyzed by PCR using the primers aldC-F2 and aldC-R2 for the aldC locus (see Fig. 3B) or als-F2 and ldh-i2 for the als locus (see Fig. 3C), by sequencing of the DNA fragments obtained, and/or by Southern hybridization (see below). The strain with aldC disrupted was designated KGMA4004 (ΔpyrE ΔaldC::pyrE), and the strain with als disrupted was designated KGMA5315 (ΔpyrE Δals::pyrE), and they were used for further studies.
Southern blot analysis.
The DIG-high prime DNA labeling and detection starter kit I (Roche, Indianapolis, IN) was used for Southern blot analyses. Five micrograms of genomic DNA of KGMA0119 or its derivatives was digested with appropriate restriction enzymes (PvuII for the pyrE locus; SmaI for the aldC and als loci). DNA fragments were separated by electrophoresis on a 0.7% (wt/vol) agarose gel and blotted onto a positively charged nylon membrane (Hybond-N+; GE, Fairfield, CT). The pyrE and aldC probes were PCR amplified from KGMA0119 genomic DNA using primer pairs pyrE-probe-F1–pyrE-probe-R1 and aldC-probe-F1–aldC-probe-R1, respectively (see Fig. 3). The fragments obtained were labeled with digoxigenin (DIG)-dUTP. DNA hybridizations were performed at 50°C for 16 h. The hybridized probes were detected according to the manufacturer's instructions.
Measurement of volatile compounds by gas chromatography.
KGMA0119, KGMA4004, and KGMA5315 were cultivated in 30 ml of YPDL medium (YPD containing 0.3% [wt/vol] sodium l-lactate). Aliquots of the culture were sampled sequentially, and cell-free supernatant was prepared. Levels of ethanol, acetic acid, acetoin, and diacetyl in the supernatant were measured by gas chromatography (GC-14A; Shimadzu, Kyoto, Japan) with a packed column (polyethylene glycol 20 M [PEG20M], 10%; ShinCarbon A, 60/80; 2.1 m by 3.2 mm; Shinwa Chemical, Kyoto, Japan). The temperature was maintained at 60°C for 3 min and was then programmed to rise at a rate of 10°C per minute up to 200°C.
Nucleotide sequence accession numbers.
The nucleotide sequences of pyrE, pyrF, aldC, als, and the 16S rRNA gene have been deposited in the DDBJ, EMBL, and GenBank databases under accession numbers AB818355, AB818356, AB818357, AB828456, and AB818453, respectively.
RESULTS
De novo pyrimidine-biosynthetic pathway of G. europaeus.
We first attempted to predict the de novo pyrimidine-biosynthetic pathway of G. europaeus. Homology searches (TBLASTN) were conducted using amino acid sequences of the closely related species G. xylinus NBRC 3288 (27) against draft genome sequences of G. europaeus LMG 18890T (8). These searches revealed eight ORFs encoding the six enzymes that are essential for pyrimidine biosynthesis. The genes are not contiguous on the chromosome, with the exception of pyrB and pyrC, which encode aspartate carbamoyltransferase and dihydroorotase, respectively. pyrE, which encodes OPRTase, is located on contig 26 (CADP01000026.1) and is clustered with the glutamate racemase gene (Fig. 1A). pyrF, which encodes OMPdecase, is located on contig 8 (CADP01000008.1) and is tandemly arranged with the gene of a hypothetical protein and trpF, which encodes phosphoribosyl anthranilate isomerase. G. europaeus OPRTase consists of 253 amino acids, and its primary structure shows homology to orthologs from G. xylinus NBRC 3288 (98% identity; YP_004867468.1) and E. coli K-12 (33% identity; NP_418099.1). Likewise, G. europaeus OMPdecase consists of 235 amino acids, and its primary structure shows homology to orthologs from G. xylinus NBRC 3288 (82% identity; YP_004868701.1) and E. coli K-12 (43% identity; NP_415797.1). It has been reported that disruption of pyrE or pyrF results in 5-FOA resistance and uracil auxotrophy in some microorganisms (12–15). To investigate whether this is also the case in G. europaeus, we examined the effect of 5-FOA on the growth of the wild-type strain, KGMA0119. When 0.2% 5-FOA was added to minimal liquid medium, the growth of KGMA0119 cells was significantly inhibited (Fig. 2A). To examine whether the pyrE or pyrF mutant shows 5-FOA resistance and uracil auxotrophy, spontaneous 5-FOA-resistant mutants were selected on minimal agar plates containing 0.2% 5-FOA and 60 μg/ml uracil. When cells at mid-logarithmic phase were inoculated onto the selection plate, spontaneous mutants that showed both 5-FOA resistance and uracil auxotrophy appeared at a frequency of 2.9 × 10−6. We subsequently performed genetic characterization of 10 of these mutants. The pyrE and pyrF loci of these mutants were amplified by PCR, and the nucleotide sequences were determined. All strains had point mutations that led to amino acid substitution or nonsense mutations in either the pyrE or the pyrF ORF. These results suggest that 5-FOA-based gene disruption is applicable to G. europaeus. Therefore, we proceeded to construct a strain with pyrE deleted as follows.
Fig 2.
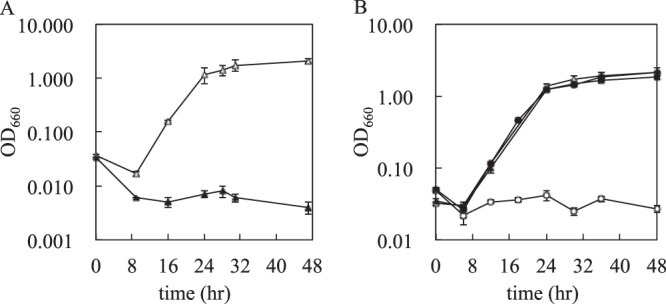
Growth profiles of KGMA0119 (wild type) and KGMA0704 (ΔpyrE). (A) Effect of 5-FOA on the growth of KGMA0119. Cells were cultivated in minimal medium either containing (solid triangles) or lacking (open triangles) 0.2% (wt/vol) 5-FOA. (B) Uracil auxotrophy of KGMA0704. KGMA0119 and KGMA0704 were cultivated in minimal medium either containing (solid symbols) or lacking (open symbols) uracil. Triangles, KGMA0119; circles, KGMA0704. Experiments were conducted in triplicate, and the error bars indicate standard deviations.
Construction of the mutant with pyrE deleted.
The pyrE disruption vector, named pUC19-ΔpyrE, was constructed as described in Materials and Methods. The plasmid was introduced into KGMA0119 cells by electroporation, and 5-FOA-resistant and uracil-auxotrophic mutants with pyrE genes that had been specifically and almost completely deleted by double-crossover homologous recombination were isolated (Fig. 3A). Several candidates were obtained, and one of the isolates, designated KGMA0704, was confirmed to exhibit uracil auxotrophy when cultivated in minimal liquid medium in the absence of uracil (Fig. 2B). In contrast to KGMA0119, which grew regardless of the absence of uracil, KGMA0704 strictly required uracil for growth. The genotype of KGMA0704 was determined by PCR using the primers EP-F3 and E-R4 and by Southern blotting using a pyrE probe that hybridized with the upstream region of the pyrE ORF (Fig. 3A). Both analyses demonstrated that the target locus of KGMA0704 was shorter than the expected length for KGMA0119 (Fig. 4A). Sequencing analysis of the target region revealed that the pyrE region was deleted, as expected. Since inactivation of the pyrF gene also results in 5-FOA resistance and uracil auxotrophy, we used PCR and sequencing analysis to confirm that the pyrF gene remained intact. These results indicated that the uracil auxotrophy of KGMA0704 was caused solely by the deletion of pyrE. Therefore, KGMA0704 was used as a host cell for the subsequent targeted gene disruption experiments. A pyrE disruptant was also obtained by introducing linear DNA (data not shown). Subsequent gene disruption experiments were carried out with circular plasmid DNA.
Fig 4.

Genotyping of the pyrE, aldC, and als loci of KGMA0119 (wild type) and its derivatives. (A to C, upper blots) Southern blot analyses. Genomic DNAs of the four strains were digested with PuvII (A) or SmaI (B and C) and hybridized with the pyrE probe (A) or the aldC probe (B and C). (A to C, lower blots) PCR analyses. The pyrE (A), aldC (B), and als (C) loci of the four strains were amplified from genomic DNA using primer pairs EP-F3–E-R4 (A), aldC-F2–aldC-R2 (B), and als-F2–ldh-i2 (C). In the case of als disruption, the PCR products were digested with EcoRI to confirm the targeted gene disruption. Figure 3 shows the restriction map and the position of each primer.
Construction of the mutant with aldC or als disrupted.
We then predicted the acetoin pathway of G. europaeus. BLAST searches revealed that the aldC gene, encoding α-acetolactate decarboxylase, is located on contig 6 (CADP01000006.1). It clusters with the als gene, which encodes α-acetolactate synthase; the ldh gene, which encodes lactate dehydrogenase; and the pdc gene, which encodes pyruvate decarboxylase (Fig. 1B). These findings suggested that G. europaeus converts lactic acid to acetoin, similarly to other AAB (24) (Fig. 1C). In addition, BLAST searches suggested that G. europaeus also has a putative diacetyl reductase gene, located on contig 48 (CADP01000048.1: range, 13474 to 14214). This gene shows relatively high identity (34%) to that of Lactococcus lactis (YP_001031692.1) (Fig. 1C). G. europaeus AldC consists of 296 amino acids, and its primary structure is homologous with that of orthologs from G. xylinus (95% identity; YP_004868152.1) and B. subtilis (40% identity; CAB07786.1). Furthermore, G. europaeus Als shows high identity (50%) with B. subtilis Als (AlsS; CAB07802.1). The acetoin pathway of G. europaeus (Fig. 1C) is considered to be similar to those of both B. subtilis and LAB. Thus, it was expected that aldC or als disruption would result in a reduction of acetoin levels.
The aldC disruption vector (pBR322-ΔaldC::pyrE) or the als disruption vector (pBR322-Δals::pyrE), which contained the pyrE marker cassette together with the putative promoter region between the 5′ upstream and 3′ downstream regions of each targeted gene, was introduced into KGMA0704. Then, uracil-prototrophic strains, in which the targeted gene had been replaced by the pyrE cassette by double-crossover homologous recombination, were isolated (Fig. 3B and C). The aldC and als disruptants were designated KGMA4004 and KGMA5315, respectively, and their genotypes were confirmed by PCR (primer pairs aldC-F2–aldC-R2 for the aldC locus and als-F2–ldh-i2 for the als locus). Southern blot analyses were also performed using an aldC probe, which hybridizes with the upstream region of the aldC ORF (Fig. 3B and C). In the case of aldC disruption, both analyses demonstrated that the aldC locus in KGMA4004 was longer than those of both the parental strain, KGMA0704, and the wild-type strain, KGMA0119, as expected (Fig. 4B). In the case of als disruption, it was expected that SmaI and EcoRI recognition sites in the als gene would disappear upon homologous recombination with the pyrE cassette. As expected, the fragment of KGMA5315, which had lost the SmaI and EcoRI sites, was larger than that of the parental strain, as detected by Southern blotting (Fig. 4C, top). The sizes of the PCR products amplified from all strains using primers als-F2 and ldh-i2 were almost identical. Therefore, als gene disruption in KGMA5315 was confirmed by EcoRI digestion of the PCR product. The PCR product derived from KGMA5315 was not digested, whereas those of both KGMA0119 and KGMA0704 were digested (Fig. 4C, lower blots). Finally, sequencing confirmation of the target region was also consistent with the ΔaldC::pyrE or Δals::pyrE genotype, both of which had been generated by double-crossover homologous recombination. Thus, aldC of KGMA4004 and als of KGMA5315 were disrupted by replacement with the pyrE cassette. No mutation was introduced in the pyrE and pyrF regions of strains KGMA4004 and KGMA5315. Next, the acetoin productivities of these strains were investigated.
Growth and volatile-compound profiles of KGMA0119 and its derivatives.
To estimate acetoin productivity, KGMA0119, KGMA4004, and KGMA5315 were cultivated in YPDL medium, and the amounts of acetoin, diacetyl, ethanol, and acetic acid were measured. Growth profiles were also monitored and are shown in Fig. 5. The amounts of ethanol and acetic acid were not very different in the three strains (Fig. 5D and E). However, the amount of acetoin in KGMA4004 culture was significantly smaller (0.009%) than that in the culture of the wild-type strain (0.042%) (Fig. 5B and 6B and C). Although KGMA4004 accumulated a trace amount of diacetyl during the late logarithmic to the early stationary growth phase, it disappeared and was not detectable at the stationary growth phase (Fig. 5C). (None of the other strains accumulated any diacetyl.) To investigate acetoin productivity under the typical conditions for rice vinegar production, KGMA0119 and KGMA4004 were cultivated in Japanese sake containing 0.3% sodium l-lactate in a jar fermentor. The amount of acetoin in KGMA4004 vinegar significantly decreased (0.025%) compared with that in KGMA0119 vinegar (0.074%). No significant difference between the two strains was observed in other profiles, such as growth, ethanol oxidization, and acetic acid production. Also, diacetyl was not detected in either vinegar (data not shown). These data actually show the strains obtained in this study produce less acetoin under the conditions for rice vinegar production. The genotypic and phenotypic stability of KGMA4004 was also confirmed: KGMA4004 was passaged for 50 generations under nonselective conditions (YPD medium), and the genotype and phenotype (growth and volatile-compound profiles) of passaged cells were investigated by PCR and cultivation in YPDL medium, respectively. These results showed that both characteristics were the same as in the parental strain, KGMA4004 (data not shown). Contrary to our expectation, the als disruptant KGMA5315 produced almost the same amount of acetoin as wild-type KMGA0119 (Fig. 5B and 6B and D).
Fig 5.

Growth and volatile-compound profiles of KGMA0119 (wild type), KGMA4004 (ΔpyrE ΔaldC::pyrE), and KGMA5315 (ΔpyrE Δals::pyrE). The three strains were cultivated in YPDL medium, and the amounts of acetoin, diacetyl, ethanol, and acetic acid in the medium were measured by gas chromatography. (A) Growth (OD660). (B) Acetoin (percent [wt/vol]). (C) Diacetyl (percent [wt/vol]). (D) Ethanol (percent [wt/vol]). (E) Acetic acid (percent [wt/vol]). Triangles, KGMA0119; diamonds, KGMA4004; circles, KGMA5315. Experiments were conducted in triplicate, and the error bars indicate standard deviations.
Fig 6.
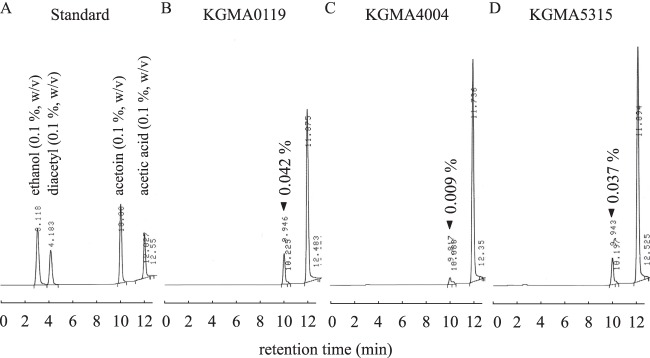
Gas chromatograms of volatile compounds that accumulated in YPDL medium after 34 h of cultivation. (A) Standards (0.1% [wt/vol]) of the four volatile compounds. (B) KGMA0119 (wild type). (C) KGMA4004 (ΔpyrE ΔaldC::pyrE). (D) KGMA5315 (ΔpyrE Δals::pyrE).
DISCUSSION
The flavor of fermented foods and beverages is characterized by many low-molecular-weight flavor compounds (e.g., short-chain alcohols, aldehydes, organic acids, and esters) (28, 29). These compounds are produced by several microorganisms that are involved in fermentation, and they have a strong impact on product quality. The subtle balance of these compounds in fermented foods and beverages is often used as a “fingerprint” for unique brands (30). Thus, the addition or removal of a specific flavor compound is expected to result in a remarkable change in the total aroma. In the construction of valuable microorganisms that produce foods, beverages, or any beneficial materials for human beings, the classical approach has long been random mutagenesis followed by selection. However, this procedure is often laborious. A large number of undesirable mutations can accumulate in the chromosome, resulting in the loss of native robustness (31). Genetic-modification techniques are more efficient than random mutagenesis for specific targeted gene disruption. 5-FOA is often used for positive selection of the uracil-auxotrophic phenotype of various microorganisms (12–15). G. europaeus KGMA0119 (wild type) is sensitive to 5-FOA. In this study, we successfully established a 5-FOA/pyrE-based gene disruption system in G. europaeus. Using this gene disruption technique, we constructed strains with aldC and als disrupted. One of these strains, KGMA4004, showed significantly lower acetoin production than the parental wild-type strain. Diacetyl has an odor similar to that of acetoin, and its sensory threshold is lower than that of acetoin. Therefore, control of the diacetyl concentration is also important for rice vinegar quality. KGMA4004 temporarily accumulated a trace amount of diacetyl, which could have been caused by spontaneous decarboxylation after accumulation of an excess of α-acetolactate. However, diacetyl in KGMA4004 culture disappeared during the stationary growth phase. This result suggests that diacetyl might be converted to acetoin by diacetyl reductase, as shown in Fig. 1C. The putative ortholog of diacetyl reductase was identified in contig 48 (CADP01000048.1; range, 13474 to 14214) of the G. europaeus draft genome sequence. Although a minute amount of acetoin was still detected in KGMA4004 culture, it is noteworthy that, in the end, diacetyl was not detectable in the cultures of all strains (Fig. 5C and 6). Temporary and slight accumulation of diacetyl should not affect rice vinegar flavor. On the other hand, aldC disruption did result in the reduction of the overall strong butter/cheese-like odor, and the use of KGMA4004 could be useful in the production of rice vinegar with a “light” flavor, which is expected to be preferable for many people. If a plasmid was available for KGMA4004 to perform a trans-complementation experiment, we could more clearly show that aldC is involved in acetoin production. We have yet to obtain plasmids for trans-complementation of mutations. In the present study, als disruption had no effect on acetoin production (Fig. 5B and 6D). As described in the introduction, many microorganisms, including G. europaeus, have two types of enzymes that catalyze the condensation of two molecules of pyruvate to yield α-acetolactate: one is Als (20), and the other is AHAS (21). AHAS is used during biosynthesis of BCAAs. These two enzymes have different physiological functions, and AHAS has a secondary function in addressing pyruvate overflow (32). Disruption of als alone might be insufficient for the reduction of acetoin in G. europaeus because of the alternative α-acetolactate synthesis by AHAS.
The technique described here could contribute to changing vinegar flavor through the use of a single-gene knockout in G. europaeus. However, multiple-gene knockout could be expected to enhance desirable phenotypes further via additional modification. In Thermococcus kodakaraensis, multiple genetic manipulations have been achieved by integration of the disruption vector into the chromosome, followed by pop-out excision of the marker gene, together with the plasmid backbone (33). A similar approach could allow multiple-gene disruption in G. europaeus. Another approach with plasmid DNA is also potent to change the characteristics of G. europaeus. In related species, such as G. xylinus (27), several plasmids have been identified. Some cryptic plasmids replicable in G. europaeus might be found, and they could expand genetic modification of AAB.
Footnotes
Published ahead of print 20 September 2013
REFERENCES
- 1.Cleenwerck I, De Wachter M, González Á, De Vuyst L, De Vos P. 2009. Differentiation of species of the family Acetobacteraceae by AFLP DNA fingerprinting: Gluconacetobacter kombuchae is a later heterotypic synonym of Gluconacetobacter hansenii. Int. J. Syst. Evol. Microbiol. 59:1771–1786 [DOI] [PubMed] [Google Scholar]
- 2.Nakai T, Sugano Y, Shoda M, Sakakibara H, Oiwa K, Tuzi S, Imai T, Sugiyama J, Takeuchi M, Yamauchi D, Mineyuki Y. 2013. Formation of highly twisted ribbons in a carboxymethylcellulase gene-disrupted strain of a cellulose-producing bacterium. J. Bacteriol. 195:958–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yadav V, Paniliatis BJ, Shi H, Lee K, Cebe P, Kaplan DL. 2010. Novel in vivo-degradable cellulose-chitin copolymer from metabolically engineered Gluconacetobacter xylinus. Appl. Environ. Microbiol. 76:6257–6265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crotti E, Rizzi A, Chouaia B, Ricci I, Favia G, Alma A, Sacchi L, Bourtzis K, Mandrioli M, Cherif A, Bandi C, Daffonchio D. 2010. Acetic acid bacteria, newly emerging symbionts of insects. Appl. Environ. Microbiol. 76:6963–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gómez-Manzo S, Chavez-Pacheco JL, Contreras-Zentella M, Sosa-Torres ME, Arreguín-Espinosa R, Pérez de la Mora M, Membrillo-Hernández J, Escamilla JE. 2010. Molecular and catalytic properties of the aldehyde dehydrogenase of Gluconacetobacter diazotrophicus, a quinoheme protein containing pyrroloquinoline quinone, cytochrome b and cytochrome c. J. Bacteriol. 192:5718–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenberg DE, Porcella SF, Zelazny AM, Virtaneva K, Sturdevant DE, Kupko JJ, III, Barbian KD, Babar A, Dorward DW, Holland SM. 2007. Genome sequence analysis of the emerging human pathogenic acetic acid bacterium Granulibacter bethesdensis. J. Bacteriol. 189:8727–8736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trček J, Jernejc K, Matsushita K. 2007. The highly tolerant acetic acid bacterium Gluconacetobacter europaeus adapts to the presence of acetic acid by changes in lipid composition, morphological properties and PQQ-dependent ADH expression. Extremophiles 11:627–635 [DOI] [PubMed] [Google Scholar]
- 8.Andrés-Barrao C, Falquet L, Calderon-Copete SP, Descombes P, Ortega Pérez R, Barja F. 2011. Genome sequences of the high-acetic acid-resistant bacteria Gluconacetobacter europaeus LMG 18890T and G. europaeus LMG 18494 (reference strains), G. europaeus 5P3, and Gluconacetobacter oboediens 174Bp2 (isolated from vinegar). J. Bacteriol. 193:2670–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okumura H, Uozumi T, Beppu T. 1985. Construction of plasmid vectors and a genetic transformation system for Acetobacter aceti. Agric. Biol. Chem. 49:1011–1017 [Google Scholar]
- 10.Shigematsu T, Takamine K, Kitazato M, Morita T, Naritomi T, Morimura S, Kida K. 2005. Cellulose production from glucose using a glucose dehydrogenase gene (gdh)-deficient mutant of Gluconacetobacter xylinus and its use for bioconversion of sweet potato pulp. J. Biosci. Bioeng. 99:415–422 [DOI] [PubMed] [Google Scholar]
- 11.Shinjoh M, Tomiyama N, Asakura A, Hoshino T. 1995. Cloning and nucleotide sequencing of the membrane-bound L-sorbosone dehydrogenase gene of Acetobacter liquefaciens IFO 12258 and its expression in Gluconobacter oxydans. Appl. Environ. Microbiol. 61:413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boeke JD, Trueheart J, Natsoulis G, Fink GR. 1987. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154:164–175 [DOI] [PubMed] [Google Scholar]
- 13.Galvão TC, de Lorenzo V. 2005. Adaptation of the yeast URA3 selection system to gram-negative bacteria and generation of a ΔbetCDE Pseudomonas putida strain. Appl. Environ. Microbiol. 71:883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yano T, Sanders C, Catalano J, Daldal F. 2005. sacB-5-Fluoroorotic acid-pyrE-based bidirectional selection for integration of unmarked alleles into the chromosome of Rhodobacter capsulatus. Appl. Environ. Microbiol. 71:3014–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato T, Fukui T, Atomi H, Imanaka T. 2003. Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 185:210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao L, Danno A, Fujii S, Fukuda W, Imanaka T, Fujiwara S. 2012. Indole-3-glycerol-phosphate synthase is recognized by a cold-inducible group II chaperonin in Thermococcus kodakarensis. Appl. Environ. Microbiol. 78:3806–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagaoka E, Hidese R, Imanaka T, Fujiwara S. 31 May 2013. Importance and determinants of induction of cold-induced DEAD RNA helicase in the hyperthermophilic archaeon Thermococcus kodakarensis. J. Bacteriol. 10.1128/JB.00332-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goupil N, Corthier G, Ehrlich SD, Renault P. 1996. Imbalance of leucine flux in Lactococcus lactis and its use for the isolation of diacetyl-overproducing strains. Appl. Environ. Microbiol. 62:2636–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang M, Oppermann-Sanio FB, Steinbüchel A. 1999. Biochemical and molecular characterization of the Bacillus subtilis acetoin catabolic pathway. J. Bacteriol. 181:3837–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frädrich C, March A, Fiege K, Hartmann A, Jahn D, Härtig E. 2012. The transcription factor AlsR binds and regulates the promoter of the alsSD operon responsible for acetoin formation in Bacillus subtilis. J. Bacteriol. 194:1100–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kisumi M, Komatsubara S, Chibata I. 1971. Valine accumulation by alpha-aminobutyric acid-resistant mutants of Serratia marcescens. J. Bacteriol. 106:493–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.el-Mansi EM, Holms WH. 1989. Control of carbon flux to acetate excretion during growth of Escherichia coli in batch and continuous cultures. J. Gen. Microbiol. 135:2875–2883 [DOI] [PubMed] [Google Scholar]
- 23.Renna MC, Najimudin N, Winik LR, Zahler SA. 1993. Regulation of the Bacillus subtilis alsS, alsD, and alsR genes involved in post-exponential-phase production of acetoin. J. Bacteriol. 175:3863–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Ley J. 1959. On the formation of acetoin by Acetobacter. J. Gen. Microbiol. 21:352–365 [DOI] [PubMed] [Google Scholar]
- 25.Akasaka N, Sakoda H, Fujiwara S. 2012. Branched-chain amino acid production by an acetic acid bacterium Gluconacetobacter europaeus. Seibutsu Kogaku 90:374–380 [Google Scholar]
- 26.Green MR, Sambrook JF. 2012. Molecular cloning: a laboratory manual, 4th ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27.Ogino H, Azuma Y, Hosoyama A, Nakazawa H, Matsutani M, Hasegawa A, Otsuyama K, Matsushita K, Fujita N, Shirai M. 2011. Complete genome sequence of NBRC 3288, a unique cellulose-nonproducing strain of Gluconacetobacter xylinus isolated from vinegar. J. Bacteriol. 193:6997–6998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romagnoli G, Luttik MA, Kötter P, Pronk JT, Daran JM. 2012. Substrate specificity of thiamine pyrophosphate-dependent 2-oxo-acid decarboxylases in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 78:7538–7548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swiegers JH, Pretorius IS. 2005. Yeast modulation of wine flavor. Adv. Appl. Microbiol. 57:131–175 [DOI] [PubMed] [Google Scholar]
- 30.Hazelwood LA, Daran JM, van Maris AJ, Pronk JT, Dickinson JR. 2008. The Ehrlich pathway for fusel alcohol production: a century of research on Saccharomyces cerevisiae metabolism. Appl. Environ. Microbiol. 74:2259–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikeda M, Mitsuhashi S, Tanaka K, Hayashi M. 2009. Reengineering of a Corynebacterium glutamicum L-arginine and L-citrulline producer. Appl. Environ. Microbiol. 75:1635–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shivers RP, Dineen SS, Sonenshein AL. 2006. Positive regulation of Bacillus subtilis ackA by CodY and CcpA: establishing a potential hierarchy in carbon flow. Mol. Microbiol. 62:811–822 [DOI] [PubMed] [Google Scholar]
- 33.Sato T, Fukui T, Atomi H, Imanaka T. 2005. Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl. Environ. Microbiol. 71:3889–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]