

Abstract
Tropical forests are being rapidly altered by logging and cleared for agriculture. Understanding the effects of these land use changes on soil bacteria, which constitute a large proportion of total biodiversity and perform important ecosystem functions, is a major conservation frontier. Here we studied the effects of logging history and forest conversion to oil palm plantations in Sabah, Borneo, on the soil bacterial community. We used paired-end Illumina sequencing of the 16S rRNA gene, V3 region, to compare the bacterial communities in primary, once-logged, and twice-logged forest and land converted to oil palm plantations. Bacteria were grouped into operational taxonomic units (OTUs) at the 97% similarity level, and OTU richness and local-scale α-diversity showed no difference between the various forest types and oil palm plantations. Focusing on the turnover of bacteria across space, true β-diversity was higher in oil palm plantation soil than in forest soil, whereas community dissimilarity-based metrics of β-diversity were only marginally different between habitats, suggesting that at large scales, oil palm plantation soil could have higher overall γ-diversity than forest soil, driven by a slightly more heterogeneous community across space. Clearance of primary and logged forest for oil palm plantations did, however, significantly impact the composition of soil bacterial communities, reflecting in part the loss of some forest bacteria, whereas primary and logged forests did not differ in composition. Overall, our results suggest that the soil bacteria of tropical forest are to some extent resilient or resistant to logging but that the impacts of forest conversion to oil palm plantations are more severe.
INTRODUCTION
The world's tropical forests are a major reservoir of biodiversity and play important roles in global climate regulation and biogeochemical cycling (1). However, in the past several decades, tropical forests have undergone rapid clearance for agriculture, with 80 million hectares of forest cleared in the 1980s and 1990s alone (2). Further, much of the forest that remains has been logged with various degrees of intensity (2, 3).
Bacteria not only make up a large proportion of the biological diversity of the rainforest environment (4) but also are a fundamental component of nutrient cycling and productivity (5, 6). Logging and forest conversion for agriculture drive changes to the soil chemistry; for instance, they can alter the pH, lead to the loss of soil carbon, and modify the C/N ratio and the content of phosphorus and calcium (7, 8). Bacterial soil communities are in many ways a product of their chemical and physical environment (9, 10, 11) and therefore can be affected by such physicochemical changes to the soil.
Moreover, the distribution of microbes not only is related to environmental factors but also can vary in relation to spatial distance (e.g., see references 12, 13, and 14). This consideration is important because logging and cultivation may have different effects on local scale richness (α-diversity) and on the turnover of species between sites (β-diversity). For example, studies on plants show that while α-diversity increases or remains the same in disturbed habitats, there is a decrease in β-diversity (15, 16), which suggests that there is a homogenization of communities across space and the loss of species from the broader-scale community (γ-diversity).
The impact of logging and forest clearance on soil bacteria is still poorly understood. In larger organisms, such as birds and invertebrates, the effects of logging cycles on diversity are not as intense as might have been expected, with only minor changes in both α- and β-diversity being detectable after 2 decades compared to old-growth forest (17, 18, 19). In contrast, wholesale forest clearance for plantation agriculture and grazing results in major decreases in biological diversity of birds and other larger organisms (18, 20, 21). Despite the fact that soil bacteria play a crucial role in ecosystem functioning, it is still unclear whether logging and forest clearance have the same impact on the α- and β-diversity of soil bacteria, as is the case for larger organisms.
Various studies have suggested that deforestation for agriculture decreases microbial biomass and alters the microbial composition of soil in tropical regions (e.g., see references 22, 23, and 24). However, most of these studies were based on techniques that offer little detail on microbial community composition at a fine taxonomic level. Nevertheless, a recent study, which used high-throughput sequencing to analyze soil bacterial communities, showed that the conversion of primary (unlogged) Amazonian forest to pasture increases bacterial α-diversity but decreases β-diversity (25), resulting in the homogenization of communities across space. Similarly, agricultural land had higher α-diversity than forests in Malaysia, with alterations of bacterial community composition being mainly driven by soil pH (11). Rodrigues et al. (25) did not include logged forests in their study, and Tripathi et al. (11) did not distinguish between logged and primary forest in their analysis, nor did they study β-diversity. Thus, there is a major knowledge gap in understanding the impacts of logging cycles on bacterial communities and of the conversion of logged forest to agriculture, since logged forests now dominate the tropics (26) and are much more likely to be converted to agriculture than true primary forests (27).
In this study, we focused on the rainforests in the global biodiversity hot spot of Sundaland, Southeast Asia. Here, the primary forest has been subject to differing degrees of logging intensity, with one or two logging cycles (28). Also, forests are being converted rapidly for agriculture, particularly to oil palm plantations (28). This provides an opportunity to study the effect of different intensities of logging on the soil bacterial community and also to evaluate if land conversion to a crop monoculture has a stronger impact on soil bacteria than logging, as is the case for larger organisms.
Our main hypotheses and predictions in this study were as follows. (i) Given the complex and subtle relationship of soil bacteria to their biological, chemical, and physical environment, we expect that a history of logging—which opens the canopy, removes certain tree species, and disturbs the soil (29, 30)—will alter the bacterial community of the forest soil with significant negative effects on α- and β-diversity, relative to undisturbed primary forest. (ii) We expect that logged-forest conversion to oil palm plantations will significantly alter bacterial community. Particularly, we predict a higher α-diversity in oil palm plantations than in forests due to the effect of artificial fertilizers on oil palm plantations (e.g., increased root growth favoring microbial activity) (31), and to competitive microbial interactions promoted by the effect of soil structure and water regime involved in cultivation (32). On the other hand, we expect a decrease in β-diversity in oil palm plantations compared to logged forest, as the former has lower diversity of larger organisms than the latter (21), potentially providing a more homogenous range of possible food substrates, mutualisms, or hosts.
MATERIALS AND METHODS
Study area.
The study was based around the 1-million-hectare Yayasan Sabah (YS) logging concession in Sabah, Malaysian Borneo (4°58.0′N, 117°48.3′E). These forests are dominated numerically by large tree species in the family Dipterocarpaceae (33), which are valuable for timber. Within the YS concession is the 238,000-ha Ulu Segama-Malua Forest Reserve (US-MFR), which was selectively logged between 1987 and 1991 with commercial stems of >0.6-m diameter at breast height (DBH) being harvested, yielding ∼113 m3 of timber per hectare (34). Between 2001 and 2007, 60% (141,000 ha) of the US-MFR was relogged, with the minimum harvested tree diameter being reduced to >0.4 m DBH for commercial species and yielding an additional 31 m3 ha−1 of timber (34). Selectively logged forest in the US-MFR is contiguous with 45,200 ha of unlogged (primary) forest in the Danum Valley Conservation Area (DVCA) and Palum Tambun Watershed Reserve. To the north and south of the US-MFR are oil palm plantations, where sampled sites had mature palms (20 to 30 years old) at a density of 100 trees per ha (21).
Field sampling.
Fieldwork was conducted from September to October 2012. Sixteen widely spaced sites (1 to 90 km apart) were established within the unlogged, once-logged, and twice-logged forests and in oil palm plantations, comprising four sites >2 km apart within each habitat type. Each site had two line transects 200 m in length and spaced 500 to 800 m apart (17). At 50-m intervals and 3 m to the left-hand side of each transect, approximately 50 g of soil was taken using a trowel from the top 5 cm of soil and excluding the leaf litter layer. Five samples were collected on each transect, combined and thoroughly mixed in the same bag, frozen within several hours of collection, and kept frozen until the time of DNA extraction (see below).
DNA extraction, PCR amplification, and sequencing.
Total soil DNA was extracted from each of the collected soil samples using the Power Soil DNA extraction kit (MoBio Laboratories, Carlsbad, CA) as directed by the manufacturer's instructions, with 0.30 g of soil. PCR amplification used primers 338F (5′-XXXXXXXXGTACTCCTACGGGAGGCAGCAG-3′) and 533R (5′TTACCGCGGCTGCTGGCAC-3′), targeting the V3 region of the bacterial 16S rRNA gene (the X sequence denotes a barcode sequence) (35). The paired-end sequencing was performed at Beijing Genome Institute (BGI), Hong Kong, China, using a paired 100-bp HiSeq 2000 sequencing system (Illumina) according to the manufacturer's instructions. Library preparation, sequencing, and initial quality filtering were performed as described in reference 36.
Sequence processing.
Illumina sequencing data were pair assembled using PANDAseq (37) with an assembly quality score of 0.9, which is the most stringent option to reduce errors. We then used the mothur pipeline (38) to align, filter, trim, and remove chimeras. DNA sequences were aligned against the EzTaxon-aligned reference sequences (39), and each operational taxonomic unit (OTU), defined at a ≥97% cutoff of sequence similarity, was classified based on the RDP database (40). As read length for Illumina is more limited than that for pyrosequencing, OTU identity can most accurately be seen as indicating genus equivalents (41). We performed all statistical analyses on a subsample of 1,592 reads per sample. Richness and diversity indices and dissimilarity matrices (Jaccard, Bray-Curtis, and unweighted-UniFrac) were estimated using mothur.
Soil analyses.
Total nitrogen, total carbon, and pH were measured at the National Instrumentation Center for Environmental Management (NICEM, South Korea) based on the standard protocol of the SSSA (Soil Science Society of America).
Statistical procedures.
To test for differences in relation to land use on the richness and diversity indices at 97% similarity, we used a linear model for normal data or generalized linear model for nonnormal data; land use was entered as a factor in each model. We used the same procedure to test whether relative abundance of the most abundant phyla differed among land uses. We also assessed the effect of land use on relative abundance at the order level within phyla that showed significant differences due to land use (see Results) and the effect of land use on the Proteobacteria-to-Acidobacteria ratio, estimated by dividing the total number of Proteobacterial sequences by the total acidobacterial sequences in each sample. Post hoc Tukey tests were used for pairwise comparisons. When neither a linear nor a generalized linear model fitted the data, we used a Kruskal-Wallis test to assess the effect of land use, with the Bonferroni correction to assess pairwise comparisons. The same procedure was used to assess differences in soil parameters among land uses. We tested whether our results for richness and diversity indices may have been influenced by spatial autocorrelation by means of Moran's I test using the model residuals.
We performed a nonmetrical multidimensional scaling (NMDS) plot using the Jaccard, Bray-Curtis, and unweighted-UniFrac dissimilarities to assess whether bacterial community composition clustered according to land use. Additionally, we used a permutational multivariate analysis of variance using dissimilarity matrices (PERMANOVA) (42) with 999 permutations to test if community composition at 97% similarity differs according to land use. We repeated these analyses removing singletons to ensure that our patterns were not driven by rarely encountered genera.
To estimate β-diversity among land uses, we used two methods. The first is a community dissimilarity-based metric, which defines β-diversity as the variation in community structure without defining a particular gradient or direction (following reference 43). Thus, community dissimilarity β-diversity was measured as the average distance from each site to the group centroid (43). To test whether community dissimilarity β-diversity differed in relation to land use, and thus whether certain communities are more homogeneous across space, we used the betadisper function in R, which assesses if the dispersion among land uses is significantly different (44). The significance of this test was evaluated using 999 permutations. The second estimated “true” β-diversity, which scales directly between α- and γ-diversity, for each land use following the equation of Whittaker (from 1960; see reference 45). True β-diversity (i.e., S/ᾱ) for each pair of samples within each of the four land uses was estimated by the following equation: S/ᾱ = (a + b + c)/[(2a + b + c)/2], where S is the total number of OTUs in two samples, ᾱ is the average number of OTUs for both samples, a is shared OTUs between both samples, b is OTUs found only in sample 1, and c is OTUs found only in sample 2. To compare true β-diversity among land uses, we used a linear model with land use as a factor and sample as a random factor to control for pseudoreplication, as each sample is used in more than one comparison within each land use. Post hoc Tukey tests were used for pairwise comparisons among land uses.
Finally, we performed a redundancy analysis (RDA) and variation partitioning (46) to assess the influence of soil parameters, land use, and spatial distance on bacterial community composition. We used the Hellinger-transformed OTU abundance as our response variable and four explanatory sets, which consisted of one set of soil parameters, a spatial trend, principal components of neighbor matrices (PCNM) functions (47, 48), and land use recoded as a dummy variable. Test of significance for each explanatory set was done by 999 permutations (see the text in the supplemental material for a detailed description of this analysis). All statistical analyses were performed using different packages in R version 2.14.1 (49).
RESULTS
From the 32 soil samples, we obtained 10,471 OTUs at ≥97% similarity from 50,942 good-quality sequences. The average number of OTUs per sample was 693 ± 91 (standard deviation [SD]), ranging from 512 to 864 OTUs. Of all OTUs, 71.7% were singletons, which accounted for 14.7% of all sequences. A high percentage of singletons is not uncommon in metagenomic studies using an Illumina sequencer (for example, see references 50 and 51). The most abundant phyla were Acidobacteria, with 22.6% of the sequences, followed by Proteobacteria (21.5%), Firmicutes (20.4%), and, to a lesser degree, Actinobacteria (7.1%), Verrucomicrobia (5.6%), and Bacteroidetes (3.2%); 16% of the sequences were unclassified (see Fig. S1 in the supplemental material).
Bacterial genus-level richness (i.e., number of OTUs) did not differ among land uses (F3,28 = 2.43, P = 0.09). Similarly, none of the α-diversity indices differed between forests in relation to logging or between forests and oil palm plantations (inverse Simpson, F3,28 = 1.69, P = 0.19; Shannon F3,28 = 1.27, P = 0.30; and nonparametric Shannon F3,28 = 1.77, P = 0.17). Moran's I tests showed no spatial autocorrelation for any of the richness and diversity indices measured (all P values were >0.10). The total number of OTUs that we recorded in each habitat type (later used as γ-diversity in the calculation of true β-diversity or Whittaker's W) was lower in primary forest (γunlogged = 3,178) than in logged forest (γonce-logged = 3,743, γtwice-logged = 3,676) or oil palm plantations (γoil palm = 4,011).
OTU community composition did, however, differ among land uses for the Jaccard (pseudo-F3,28 = 1.16, P = 0.002), Bray-Curtis (pseudo-F3,28 = 1.69, P = 0.02), and unweighted-UniFrac (pseudo-F3,28 = 1.16, P = 0.002) dissimilarities. NMDS plots using the Jaccard, Bray-Curtis, and unweighted-UniFrac indices revealed that the dissimilarities in community composition were driven by forest conversion to oil palm plantations (Fig. 1; also, see Fig. S2 in the supplemental material), with oil palm plantation soil samples being separate from forest samples (Fig. 1; also, see Fig. S2 and S3 in the supplemental material), which themselves were not distinguishable. Removing singleton OTUs did not change the pattern of our results (PERMANOVA, Jaccard, pseudo-F3,28 = 1.29, P = 0.001; Bray-Curtis, pseudo-F3,28 = 1.88, P = 0.01; unweighted UniFrac, pseudo-F3,28 = 1.23, P = 0.005) (see Fig. S4 in the supplemental material).
Fig 1.
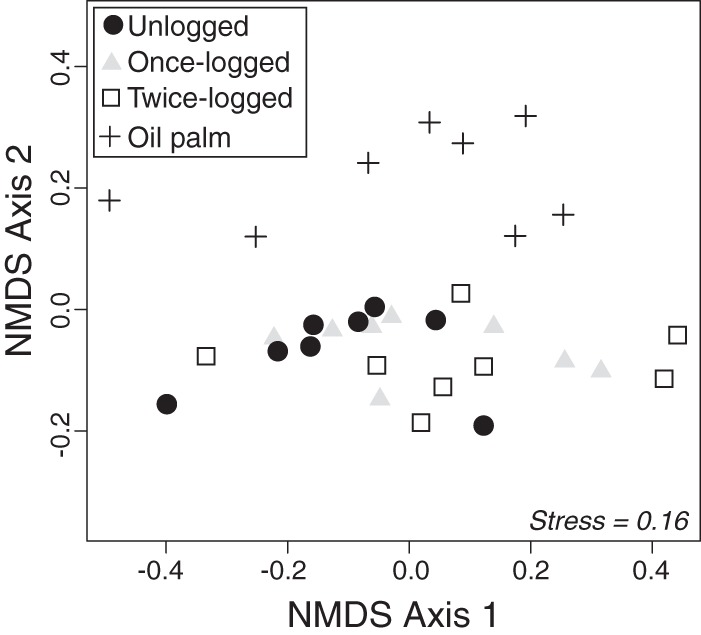
NMDS of bacterial community composition obtained using the Jaccard index in relation to land use.
Of the most abundant phyla, we found significant differences in relative abundance among different land uses for Acidobacteria (F3,28 = 3.75, P = 0.02) (Fig. 2) and Actinobacteria (F3,28 = 6.78, P = 0.001) (Fig. 2) and marginally for Verrucomicrobia (F3,28 = 2.72, P = 0.06). A comparison of Acidobacteria at the order level revealed that the relative abundance of group 2 was higher for unlogged forest than for twice-logged forest (Table 1; also, see Fig. S5 in the supplemental material). No other difference was found between primary and logged forests. Focusing on forest conversion to oil palm plantations, the relative abundance of group 5 was lower in oil palm plantations than in logged forests, and that of group 1-unclassified, group 2, and group 3 was lower in oil palm plantations than in primary forest (Table 1; also, see Fig. S5 in the supplemental material). Within Actinobacteria, Actinomycetales had higher relative abundance in oil palm plantations than in forests (Table 1; also, see Fig. S6 in the supplemental material); also, Solirubrobacterales showed a higher relative abundance in oil palm than in logged forests (Table 1; also, see Fig. S6 in the supplemental material). Among Proteobacteria, Sphingomonadales were more abundant in oil palm only than in unlogged forests (Table 1), not in logged ones. Finally, the Proteobacteria-to-Acidobacteria ratio differed in relation to land use (F3,28 = 4.08, P = 0.02) (Fig. 3), particularly between oil palm and unlogged forests (P = 0.01) and between oil palm and once-logged forests (P = 0.05).
Fig 2.
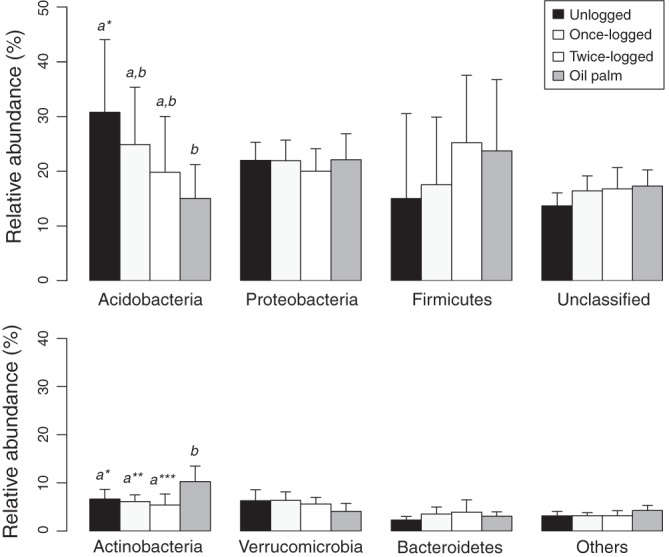
Relative abundance (means ± SD) of the most abundant phyla in different land uses. Tukey pairwise comparisons are shown; different letters denote significant differences between groups. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Table 1.
Comparison of relative abundance of the dominant orders within the phyla Acidobacteria, Actinobacteria, and Proteobacteria among land usesa
| Organism | F or χ2b | df | P | Pairwise comparisonc |
|---|---|---|---|---|
| Acidobacteriad | ||||
| Group 1-unclassified | 2.88 | 3, 28 | 0.05 | Unlogged > oil palm |
| Group 2 | 5.23 | 3, 28 | 0.005 | Unlogged > twice-logged/oil palm |
| Group 3 | 3.28 | 3, 28 | 0.04 | Unlogged > oil palm |
| Group 5 | 4.87 | 3, 28 | 0.01 | Once-logged/twice-logged > oil palm |
| Actinobacteriae | ||||
| Actinomycetales | 8.24 | 3, 28 | 0.0004 | Once-logged/twice-logged/unlogged < oil palm |
| Solirubrobacterales | 13.8* | 3 | 0.003 | Once-logged/twice-logged < oil palm |
| Proteobacteriaf | ||||
| Sphingomonadales | 10.4* | 3 | 0.01 | Unlogged < oil palm |
Only orders for which significant differences were found are shown.
Effect of land use on relative abundance evaluated by linear or generalized linear model or by the Kruskal-Wallis test (*).
Pairwise comparisons by post hoc Tukey test for linear/generalized linear models or P values Bonferroni-corrected for Kruskal-Wallis. Differences were considered significant at a P value of ≤0.05.
The 10 most abundant orders comprised 94.4% of the sequences in this phylum.
Actinomycetales, Solirubrobacterales, and unclassified orders comprised 99.3% of the sequences within Actinobacteria.
Burkholderiales, Myxococcales, Rhodospirillales, Rhizobiales, Sphingomonadales, unclassified orders within the Alpha-, Beta-, Gamma- and Deltaproteobacteria classes, and unclassified orders comprised 90.6% of sequences within Proteobacteria.
Fig 3.
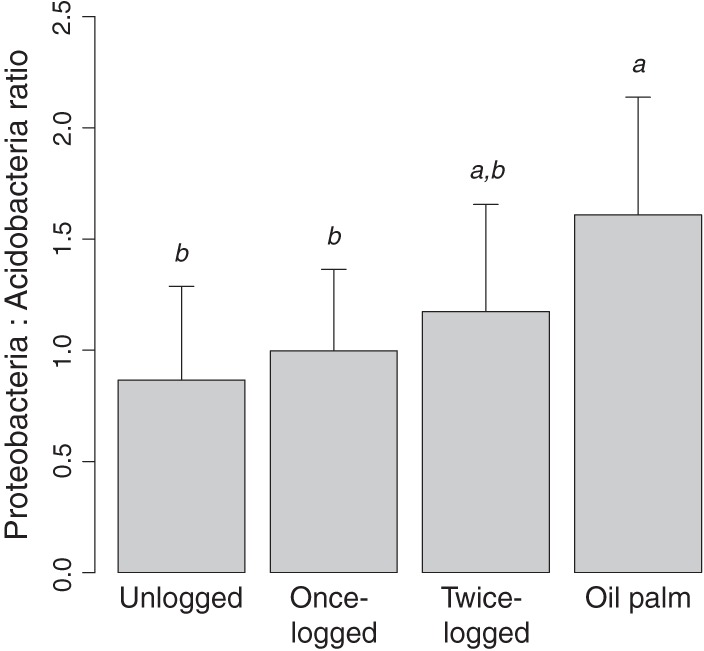
Proteobacteria-to-Acidobacteria ratio (means ± SD) among land uses. Different letters denote significant differences at a P value of ≤0.05.
Changes in community composition can to some extent be explained by changes in the rate of variation of species between habitats. Community dissimilarity-based metrics of β-diversity, measured as the average distance of all samples to the centroid within each land use, differed among land uses when the Jaccard dissimilarity was used (F3,28 = 2.88, P = 0.05) (Fig. 4A), with a marginally significant difference between primary forest and oil palm plantation (P = 0.07). Using the Bray-Curtis index did not show significant differences in β-diversity among land uses (Bray-Curtis: F3,28 = 0.54, P = 0.66). Repeating the analyses with all singleton OTUs removed, we did not find a significant difference among groups with Jaccard (F3,28 = 1.89, P = 0.15) or Bray-Curtis (F3,28 = 0.32, P = 0.82) dissimilarities.
Fig 4.
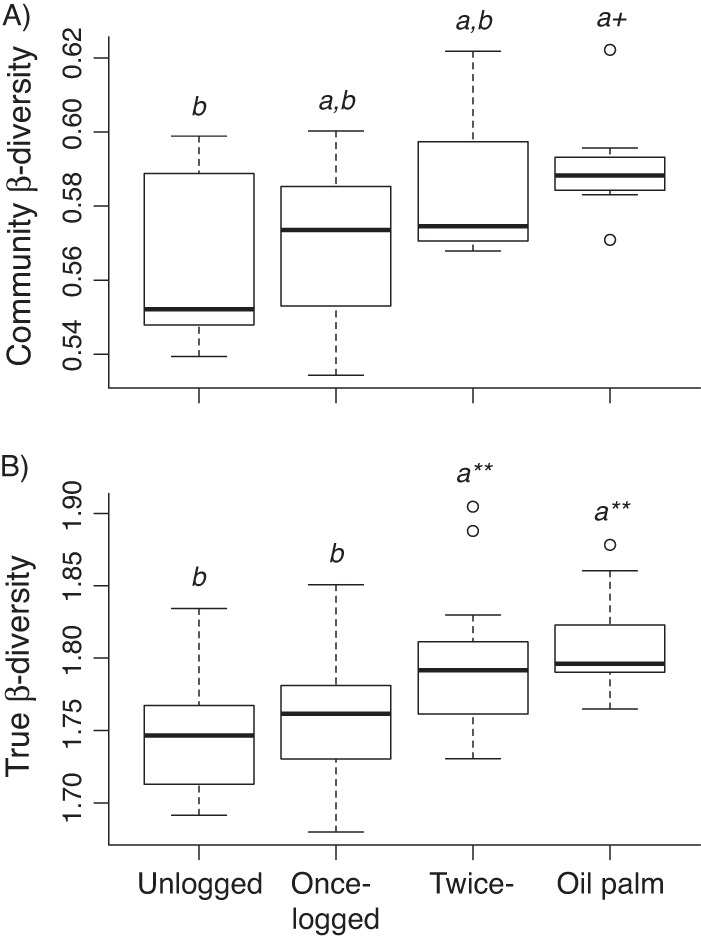
(A) Community β-diversity (average distance to group centroid) of bacterial communities in four land uses based on Jaccard dissimilarity. (B) True β-diversity (i.e., S/ᾱ) among the four land uses. Boxes show the lower quartile, the median, and the upper quartile. Tukey pairwise comparisons are shown; different letters denote significant differences between groups at P values of <0.10 (+) and <0.005 (**).
Focusing on true β-diversity (i.e., S/ᾱ), there was significant variation among land uses (LRT3 [likelihood ratio test, 3 degrees of freedom] = 27.27, P < 0.0001), with unlogged and once-logged forests having significantly lower true β-diversity than twice-logged forests and oil palm plantations (all P values were <0.005) (Fig. 4B). When the analyses were repeated with all singleton OTUs removed, results remained the same, with unlogged and once-logged forests having lower true β-diversity than twice-logged forest and oil palm plantations (LRT3 = 20.49, P = 0.0001; in Tukey pairwise comparisons, all P values were <0.05).
The variation partitioning analysis explained 52.4% of the variation in the OTU abundance; however, 85% of this was unexplained variation (see Fig. S7 in the supplemental material). Soil parameters (i.e., pH and available phosphorus) explained 13.4% of the variation (P = 0.001), of which 44.8% (i.e., 6.0% of the total variation; P = 0.005) was neither spatially structured nor related to land use (see Fig. S7). The unique components of land use and the spatial variables were not significant in explaining any of the variation observed (all P values were >0.15), but the combined effect of land use and the spatial variables explained 9.1% of the variation (P = 0.02).
DISCUSSION
We predicted that logging would significantly alter the bacterial community composition of soil compared to primary forest, altering diversity at the α- and β-levels. Our results showed that forest logging cycles had no effect on α-diversity and a small positive effect on true β-diversity. Thus, our first hypothesis is only partially supported. The fact that we did not observe clear differences in overall community composition between logged and unlogged forest suggests that soil bacteria can recover, at least on a time scale of decades after logging, or that they are resilient to such disturbance. It is possible, however, that if we had analyzed the bacterial community structure at a higher taxonomic resolution, for example, by 454 pyrosequencing, we would have found differences between logging treatments.
Bacterial community composition was significantly altered by conversion of either logged or unlogged forest to oil palm plantations, in support of our second hypothesis. However, contrary to our expectation, α-diversity was similar between forests and oil palm plantations. In contrast, true β-diversity was higher in oil palm plantations than in once-logged and primary forests, whereas community dissimilarity-based metrics of β-diversity using the Jaccard index was only marginally higher in oil palm plantations than in primary forest. While the Illumina sequencing method we used does not have as high a level of taxonomic definition as 454 pyrosequencing, due to shorter read lengths, it is unlikely that this could produce false positives in terms of differences in β-diversity. Moreover, it has been observed that paired-end Illumina reads are as accurate at the genus level and that taxon abundance is consistent with that obtained by pyrosequencing, with both methods giving similar results in overall microbial community composition (51, 52, 53). If anything, lower taxonomic resolution may be expected to minimize the apparent differences in β-diversity, giving false negatives.
Bacterial community.
Logging did not significantly alter bacterial community composition of primary forest soil at the phylum or order level. The only difference we found was for Acidobacteria group 2, which was more abundant in primary forest than in twice-logged forest. In contrast, significant differences were observed between forest and oil palm plantation using the Jaccard, Bray-Curtis, and unweighted-UniFrac dissimilarities (Fig. 1; also, see Fig. S1 and S2 in the supplemental material). Oil palm plantation communities remained different from forest communities even after singletons were removed (see Fig. S4 in the supplemental material), suggesting that shifts in soil bacterial composition between forest and oil palm plantations are related not only to the presence of certain taxonomic groups but also to differences in the relative abundance of shared OTUs.
Soils from oil palm plantations had significantly lower abundances of Acidobacteria than primary forest, whereas Actinobacteria was more abundant in oil palm plantations than in all forests. Similar results have been found for these phyla in studies of soils in the Amazon and forested land in the United States (25, 54). Acidobacteria are oligotrophic organisms abundant in carbon-poor soils (55). Carbon and nitrogen content often decreases in cultivated soils (8, 56), and in this respect our study was no exception, with lower concentrations of total carbon and total nitrogen and a lower C/N ratio in soil from oil palm plantations than from forests (all P values were <0.05). Thus, we might have expected a higher abundance of Acidobacteria in the carbon-reduced oil palm plantation soils than in forests. However, Acidobacteria are also strongly influenced by pH, favoring soils with pHs of <4 (57, 58). Primary forest soils had lower pH (4.23 ± 0.37) than oil palm plantation soils (4.75 ± 0.59), probably promoting Acidobacterial abundance. In fact, groups 1-unclassified, 2, and 3, which were more abundant in unlogged forest, were negatively related to pH (Spearman correlation, all ρ < −0.80; P < 0.001) (59). The only difference between oil palm plantation and logged forests in Acidobacteria was for group 5, whose relative abundance was correlated with available phosphorus (ρ = 0.37, P = 0.04) and C/N ratio (r = 0.45, P = 0.01) but not with pH, suggesting a less significant effect of pH for this group.
Actinobacteria showed an increased abundance in oil palm plantations compared to logged forests, mainly due to the higher abundance of Actinomycetales and Solirubrobacterales in oil palm plantations. Actinomycetales are dominant in cultivated land compared to forest (54, 60), suggesting its potential use as a biomarker of land use effects (54). Solirubrobacterales are less known, but they may behave similarly to Actinomycetales in relation to disturbance (54). In the Proteobacteria, only Sphingomonadales had higher relative abundances in oil palm plantations, and only compared to primary forest, probably because this group favors cultivation (61, 62) and the low C/N ratio (62) of oil palm plantations.
The Proteobacteria-to-Acidobacteria ratio was higher in oil palm plantation soils than in primary and once-logged forest soils. This ratio tends to be lower in oligotrophic soils and higher in soils with organic input, indicating the nutritional status of soil (32). Clearly, the difference in this ratio among land uses in our study was due to the difference in relative abundance of Acidobacteria, as Proteobacteria were equally abundant among land uses. Notably, total carbon and C/N ratio were lower in oil palm plantation soils than in forests (all P < 0.05), suggesting that in this case, other factors that impact Acidobacteria, such as pH, had a stronger role than any effect the carbon concentration and C/N ratio might have had on the copiotrophic Proteobacteria (63).
Alpha and beta diversity.
OTU richness and diversity indices in our samples did not differ among land uses, indicating that α-diversity was similar in forests and oil palm plantations. In contrast, community dissimilarity β-diversity was marginally lower in primary forests than oil palm plantations when the Jaccard index was used, which takes into account only community membership. Also, true β-diversity, which compares the total OTU richness (γ-diversity) in relation to the average OTU richness (α-diversity) for each land use, was higher for twice-logged forests and oil palm plantations than for primary and once-logged forests, even after singletons were removed. Combined, these results suggest that oil palm plantation soil has a higher turnover of bacterial diversity at large spatial scales and is more heterogeneous, whereas bacterial composition in forest soils tends to be more homogeneous. However, our results also suggest that the marginal increase in community dissimilarity β-diversity in oil palm plantation soil is due to the presence of rare OTUs, because differences disappeared when singleton OTUs were removed. There could thus be a long-term degradation of soil bacterial diversity in oil palm plantations if small populations suffer stochastic extinction.
Land conversion for agriculture tends to homogenize biodiversity of larger organisms (64), and a recent study in Amazonia, in which bacterial β-diversity (measured as species turnover and variation in community dissimilarity) in pasture was lower than in primary forest (25), similarly suggests that this is also the case for bacteria. However, we found higher β-diversity in oil palm plantations than in forest, especially for true β-diversity. One possible explanation is that the relatively dissimilar bacterial communities among our oil palm plantation samples reflect dissimilarities that were already present in soil bacterial communities due to underlying variations in soil chemistry or hydrology before the conversion of the original forest cover to oil palm plantations, and perhaps homogenization will occur in the longer term. We do not know exactly how long ago this site was converted to oil palm plantations (due to lack of records for this area), but it can be no less than 30 years, as mature oil palms here are at least that age (21). The effects of past land use can persist for many decades and even centuries (64, 65); thus, we cannot discard the possibility that the higher β-diversity observed in oil palm plantation soils is more related to past legacies from the original forest soils than to factors related to oil palm plantation.
Other studies have found that cultivated soils have a more diverse bacterial community than undisturbed ones (54, 66), suggesting that some of the activities performed for cultivation, such as tillage and residues left by crops, can either promote or ease competition resulting in a more diverse community (32, 54). Although these studies reveal differences in α-diversity, it is possible that factors that promote a higher α-diversity in cultivated land also do so for β-diversity. This might suggest that while some factors tend to make bacterial composition more distinct between oil palm plantations and forests, there are others that promote considerable variation in bacterial composition in oil palm plantation soils (Fig. 1; also, see Fig. S2 in the supplemental material; both show considerable spread among oil samples on axis 1). This is partially supported by our variation partitioning results, in which soil parameters (i.e., pH and available phosphorus) explained the largest proportion of variation in OTU abundance (13.4%), with 45% of this variation being neither spatially structured nor related to land use.
Several studies have shown that bacterial communities, including those in soil, are sensitive to disturbance and not resilient, at least in the short term (see review 67). Our results suggest that diversity of tropical forest soil bacteria is partly either resilient or resistant to land use change. Past logging cycles appear to have no long-term effects, which is promising for the recovery of logged forests soils and for the continued provisioning of ecosystem functions. This reinforces the general picture obtained by studying larger organisms. Even clearance of forest for oil palm plantation does not depress the overall diversity of bacteria. However, differences in certain taxonomic groups (Table 1) and changes in community composition between forests and oil palm plantations indicate that oil palm cultivation does have some negative impacts. Our results are only a snapshot in time, and only long-term studies will be able to elucidate if the changes in soil bacterial community composition observed here between forests and oil palm plantations are an intermediate disturbance effect or a more permanent one. Future studies should also focus on functional changes associated with changes in the microbial composition, something not addressed in this study. If the bacterial community retains its function in spite of changes in its composition due to cultivation, then ecosystem processes might be little affected. This could represent good possibilities of long-term restoration of rainforest, both in cases where oil palm plantation lands are purchased to reconnect forest fragments (e.g., Kinabatangan River, Sabah) or more generally should the economics of oil palm change such that these lands are abandoned.
Supplementary Material
ACKNOWLEDGMENTS
We thank Glen Reynolds, the Royal Society's SEARRP, and the Borneo Rainforest Lodge for logistical support and Suzan Benedick for collaborative support. We also thank Yayasan Sabah, Danum Valley Management Committee, the State Secretary, Sabah Chief Minister's Departments, and the Economic Planning Unit of the Prime Minister's Department for permission to conduct research. We also thank two anonymous reviewers for their comments on a previous version of the manuscript.
This project was partially funded by a National Research Foundation grant (NRF-2013-031400), Ministry of Education, Science and Technology, South Korea.
Footnotes
Published ahead of print 20 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02541-13.
REFERENCES
- 1.Orians GH, Dirzo R, Cushman JH. 1996. Biodiversity and ecosystem processes in tropical forests. Springer-Verlag, New York, NY [Google Scholar]
- 2.Gibbs HK, Ruesch AS, Achard F, Clayton MK, Holmgren P, Ramankutty N, Foley JA. 2010. Tropical forests were the primary sources of new agricultural land in the 1980s and 1990s. Proc. Natl. Acad. Sci. U. S. A. 107:16732–16737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geist HJ, Lambin EF. 2002. Proximate causes and underlying driving forces of tropical deforestation. Bioscience 52:143–150 [Google Scholar]
- 4.Whitman WB, Coleman DC, Wiebe WJ. 1998. Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U. S. A. 95:6578–6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang JC, Insam H. 1991. Microbial biomass and relative contributions of bacteria and fungi in soil beneath tropical rain forest, Hainan Island, China. J. Trop. Ecol. 7:385–393 [Google Scholar]
- 6.van der Heijden MGA, Bardgett RD, Van Straalen NM. 2008. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 11:296–310 [DOI] [PubMed] [Google Scholar]
- 7.McGrath DA, Smith CK, Gholz HL, Oliveira FDA. 2001. Effects of land-use change on soil nutrient dynamics in Amazônia. Ecosystems 4:625–645 [Google Scholar]
- 8.Murty D, Kirschbaum MUF, McMurtrie RE, McGilvray H. 2002. Does conversion of forest to agricultural land change soil carbon and nitrogen? A review of the literature. Glob. Change Biol. 8:105–123 [Google Scholar]
- 9.Cornejo FH, Varela A, SJW 1994. Tropical forest litter decomposition under seasonal drought: nutrient release, fungi and bacteria. Oikos 70:183–190 [Google Scholar]
- 10.Lauber CL, Strickland MS, Bradford MA, Fierer N. 2008. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 40:2407–2415 [Google Scholar]
- 11.Tripathi B, Kim M, Singh D, Lee-Cruz L, Lai-Hoe A, Ainuddin AN, Go R, Rahim RA, Husni MHA, Chun J, Adams JM. 2012. Tropical soil bacterial communities in Malaysia: pH dominates in the Equatorial tropics too. Microb. Ecol. 64:474–484 [DOI] [PubMed] [Google Scholar]
- 12.Ramette A, Tiedje JM. 2007. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc. Natl. Acad. Sci. U. S. A. 104:2761–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL. 2008. Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc. Natl. Acad. Sci. U. S. A. 105:11505–11511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martiny JBH, Eisen JA, Penn K, Allison SD, Horner-Devine C. 2011. Drivers of bacterial β-diversity depend on spatial scale. Proc. Natl. Acad. Sci. U. S. A. 108:7850–7854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cousins SAO, Eriksson O. 2002. The influence of management history and habitat on plant species richness in a rural hemiboreal landscape, Sweden. Landscape Ecol. 17:517–529 [Google Scholar]
- 16.Smart SM, Thompson K, Marrs RH, Le Duc MG, Maskell LC, Firbank LG. 2006. Biotic homogenization and changes in species diversity across human-modified ecosystems. Proc. R. Soc. Lond. B Biol. Sci. 273:2659–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edwards DP, Larsen TH, Docherty TDS, Ansell FA, Hsu WW, Derhé MA, Hamer KC, Wilcove SC. 2011. Degraded lands worth protecting: the biological importance of Southeast Asia's repeatedly logged forests. Proc. R. Soc. Lond. B Biol. Sci. 278:82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson L, Lee TM, Koh LP, Brook BW, Gardner TA, Barlow J, Peres CA, Bradshaw CJA, Laurance WF, Lovejoy TE, Sodhi NS. 2011. Primary forests are irreplaceable for sustaining tropical biodiversity. Nature 478:378–381 [DOI] [PubMed] [Google Scholar]
- 19.Woodcock P, Edwards DP, Fayle TM, Newton RJ, Chey VK, Bottrell SH, Hamer KC. 2011. The conservation value of Southeast Asia's highly degraded forests: evidence from leaf-litter ants. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366:3256–3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fitzherbert EB, Struebig MJ, Morel A, Danielsen F, Brühl CA, Donald PF, Phalan B. 2008. How will oil palm expansion affect biodiversity? Trends Ecol. Evol. 23:538–545 [DOI] [PubMed] [Google Scholar]
- 21.Edwards DP, Hodgson J, Hamer KC, Mitchell SL, Ahmad AH, Cornell S, Wilcove DS. 2010. Wildlife-friendly oil palm plantations fail to protect biodiversity effectively. Conserv. Lett. 3:236–242 [Google Scholar]
- 22.Borneman J, Triplett EW. 1997. Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl. Environ. Microbiol. 63:2647–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carney KM, Matson PA, Bohannan BJM. 2004. Diversity and composition of tropical soil nitrifiers across a plant diversity gradient and among land-use types. Ecol. Lett. 7:684–694 [Google Scholar]
- 24.Bossio DA, Girvan MS, Verchot L, Bullimore J, Borelli T, Albrecht A, Scow KM, Ball AS, Pretty JN, Osborn AM. 2005. Soil microbial community response to land use change in an agricultural landscape of western Kenya. Microb. Ecol. 49:50–62 [DOI] [PubMed] [Google Scholar]
- 25.Rodrigues JLM, Pellizari VH, Mueller R, Baek K, Ederson DCJ, Paula FS, Mirza B, Hamaoui GS, Jr, Tsai SM, Feigl B, Tiedje JM, Bohannan BJM, Nüsslein K. 2013. Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities. Proc. Natl. Acad. Sci. U. S. A. 110:988–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blaser J, Sarre A, Poore D, Johnson S. 2011. Status of tropical forest management. ITTO technical series 38. International Tropical Timber Organization, Yokohama, Japan [Google Scholar]
- 27.Asner GP, Knapp DE, Broadbent EN, Oliveira PJC, Keller M, Silva JN. 2005. Selective logging in the Brazilian Amazon. Science 310:480–482 [DOI] [PubMed] [Google Scholar]
- 28.Wilcove DS, Giam X, Edwards DP, Fisher B, Koh P. 2013. Navjot's nightmare revisited: logging, agriculture, and biodiversity in Southeast Asia. Trends Ecol. Evol. 28:531–540 [DOI] [PubMed] [Google Scholar]
- 29.Cannon CH, Peart DR, Leighton M. 1998. Tree species diversity in commercially logged Bornean rainforest. Science 281:1366–1368 [DOI] [PubMed] [Google Scholar]
- 30.Chazdon RL. 2003. Tropical forest recovery: legacies of human impact and natural disturbances. Perspect. Plant Ecol. Evol. Syst. 6:51–71 [Google Scholar]
- 31.Dick RP. 1992. A review: long-term effects of agricultural systems on soil biochemical and microbial parameters. Agric. Ecosyst. Environ. 40:25–36 [Google Scholar]
- 32.Torsvik V, Øvreås L. 2002. Microbial diversity and function in soil: from genes to ecosystems. Curr. Opin. Microbiol. 5:240–245 [DOI] [PubMed] [Google Scholar]
- 33.Fisher B, Edwards DP, Giam X, Wilcove DS. 2011. The high costs of conserving Southeast Asia's lowland rainforest. Front. Ecol. Environ. 9:329–334 [Google Scholar]
- 34.Fisher B, Edwards DP, Larsen TH, Ansell FA, Hsu WW, Roberts C, Wilcove S. 2011. Cost-effective conservation: calculating biodiversity and logging tradeoffs in Southeast Asia. Conserv Lett. 4:443–450 [Google Scholar]
- 35.Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable Tag sequencing. PLoS Genet. 4:e1000255. 10.1371/journal.pgen.1000255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou H-W, Li D-F, Tam NF-Y, Jiang X-T, Zhang H, Sheng H-F, Qin J, Liu X, Zou F. 2011. BIPES, a cost-effective high-throughput method for assessing microbial diversity. ISME J. 5:741–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masella A, Bartram A, Truszkowski J, Brown D, Neufeld J. 2012. PANDAseq: paired-end assembler for Illumina sequences. BMC Bioinformatics 13:31. 10.1186/1471-2105-13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schloss PD, Westcott SI, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chun J, Lee J-H, Jung Y, Kim M, Kim S, Kim BK, Lim YW. 2007. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 57:2259–2261 [DOI] [PubMed] [Google Scholar]
- 40.Maidak BL, Cole JR, Lilburn TG, Parker CT, Jr, Saxman PR, Farris RJ, Garrity GM, Olsen GJ, Schmidt TM, Tiedje JM. 2001. The RDP-II (Ribosomal Database Project). Nucleic Acids Res. 29:173–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soergel DAW, Dey N, Knight R, Brenner SE. 2012. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 6:1440–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 26:32–46 [Google Scholar]
- 43.Anderson MJ, Crist TO, Chase JM, Vellend M, Inouye BD, Freestone AL, Sanders NJ, Cornell HV, Comita LS, Davies KF, Harrison SP, Kraft NJB, Stegen JC, Swenson NG. 2011. Navigating the multiple meanings of β diversity: a roadmap for the practicing ecologist. Ecol. Lett. 14:19–28 [DOI] [PubMed] [Google Scholar]
- 44.Anderson MJ. 2006. Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62:245–253 [DOI] [PubMed] [Google Scholar]
- 45.Koleff P, Gaston KJ, Lennon JJ. 2003. Measuring beta diversity for presence–absence data. J. Anim. Ecol. 72:367–382 [Google Scholar]
- 46.Peres-Neto PR, Legendre P, Dray S, Borcard D. 2006. Variation partitioning of species data matrices: estimation and comparison of fractions. Ecology 87:2614–2625 [DOI] [PubMed] [Google Scholar]
- 47.Borcard D, Legendre P. 2002. All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol. Model. 153:51–68 [Google Scholar]
- 48.Borcard D, Legendre P, Avois-Jacquet C, Tuomisto H. 2004. Dissecting the spatial structure of ecological data at multiple scales. Ecology 85:1826–1832 [Google Scholar]
- 49.R Development Core Team 2008. R: a language, environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 50.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U. S. A. 108:4516–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bartram AK, Lynch MDJ, Stearns JC, Moreno-Hagelsieb G, Neufeld JD. 2011. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 77:3846–3852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Claesson MJ, Wang Q, O'Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O'Toole PW. 2010. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 38:e200. 10.1093/nar/gkq873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT. 2012. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One 7:e30087. 10.1371/journal.pone.0030087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shange RS, Ankumah RO, Ibekwe AM, Zabawa R, Dowd SE. 2012. Distinct soil bacterial communities revealed under a diversely managed agroecosystem. PLoS One 7:e40338. 10.1371/journal.pone.0040338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nemergut DR, Cleveland CC, Wieder WR, Washenberger CL, Townsend AR. 2010. Plot-scale manipulations of organic matter inputs to soils correlate with shifts in microbial community composition in a lowland tropical rain forest. Soil Biol. Biochem. 42:2153–2160 [Google Scholar]
- 56.Waldrop MP, Balser TC, Firestone MK. 2000. Linking microbial community composition to function in a tropical soil. Soil Biol. Biochem. 32:1837–1846 [Google Scholar]
- 57.Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N. 2009. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J. 3:442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lauber CL, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as predictor of soil bacteria community structure at the continental scale. Appl. Environ. Microbiol. 75:5111–5120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin C, Jones KL, Peterson DE, Garrett KA, Hulbert SH, Paulitz TC. 2010. Members of soil bacterial communities sensitive to tillage and crop rotation. Soil Biol. Biochem. 42:2111–2118 [Google Scholar]
- 60.Hill P, Krištùfek V, Dijkhuizen L, Boddy C, Kroetsch D, van Elsas JD. 2011. Land use intensity controls actinobacterial community structure. Microb. Ecol. 61:286–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ceja-Navarro JA, Rivera-Orduña FN, Patiño-Zúñiga L, Vila-Sanjurjo A, Crossa J, Govaerts B, Dendooven L. 2010. Phylogenetic and multivariate analyses to determine the effects of different tillage and residue management practices on soil bacterial communities. Appl. Environ. Microbiol. 76:3685–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuramae EE, Yergeau E, Wong LC, Pijl AS, van Veen JA, Kowalchuk GA. 2012. Soil characteristics more strongly influence soil bacterial communities than land-use type. FEMS Microbiol. Ecol. 79:12–24 [DOI] [PubMed] [Google Scholar]
- 63.Fierer N, Bradford MA, Jackson RB. 2007. Toward an ecological classification of soil bacteria. Ecology 88:1354–1364 [DOI] [PubMed] [Google Scholar]
- 64.Foster D, Swanson F, Aber J, Burke I, Brokaw N, Tilman D, Knapp A. 2003. The importance of land-use legacies to ecology and conservation. Bioscience 53:77–88 [Google Scholar]
- 65.Fraterrigo JM, Balser TC, Turner MG. 2006. Microbial community variation and its relationship with nitrogen mineralization in historically altered forests. Ecology 87:570–579 [DOI] [PubMed] [Google Scholar]
- 66.Acosta-Martínez V, Dowd S, Sun Y, Allen V. 2008. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 40:2762–2770 [Google Scholar]
- 67.Allison SD, Martiny JBH. 2008. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. U. S. A. 105:11512–11519 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.