

Abstract
Listeria monocytogenes can cause the serious infection listeriosis, which despite antibiotic treatment has a high mortality. Understanding the response of L. monocytogenes to antibiotic exposure is therefore important to ensure treatment success. Some bacteria survive antibiotic treatment by formation of persisters, which are a dormant antibiotic-tolerant subpopulation. The purpose of this study was to determine whether L. monocytogenes can form persisters and how bacterial physiology affects the number of persisters in the population. A stationary-phase culture of L. monocytogenes was adjusted to 108 CFU ml−1, and 103 to 104 CFU ml−1 survived 72-h treatment with 100 μg of norfloxacin ml−1, indicating a persister subpopulation. This survival was not caused by antibiotic resistance as regrown persisters were as sensitive to norfloxacin as the parental strain. Higher numbers of persisters (105 to 106) were surviving when older stationary phase or surface-associated cells were treated with 100 μg of norfloxacin ml−1. The number of persisters was similar when a ΔsigB mutant and the wild type were treated with norfloxacin, but the killing rate was higher in the ΔsigB mutant. Dormant norfloxacin persisters could be activated by the addition of fermentable carbohydrates and subsequently killed by gentamicin; however, a stable surviving subpopulation of 103 CFU ml−1 remained. Nitrofurantoin that has a growth-independent mode of action was effective against both growing and dormant cells, suggesting that eradication of persisters is possible. Our study adds L. monocytogenes to the list of bacterial species capable of surviving bactericidal antibiotics in a dormant stage, and this persister phenomenon should be borne in mind when developing treatment regimens.
INTRODUCTION
One of the greatest achievements in medicine was the discovery of antibiotics that have enabled treatment of a wide range of microbial diseases. However, the extensive use of antibiotics is also a known driver for the development of resistance, rendering even simple infections difficult to treat (1). The presence of bacterial biofilms is another cause of treatment failure, since biofilm cells tend to be more tolerant to antibiotics. This tolerance is often caused by the presence of dormant antibiotic-tolerant bacterial persisters (1–3). The first description of bacterial persisters was published only a few years after the discovery of penicillin, when Joseph Bigger observed that a small subpopulation of Staphylococcus aureus survived treatment with penicillin. Bigger concluded that penicillin was incapable of sterilizing an infection as persisting cells, temporarily in a nondividing phase, were tolerant to penicillin (4). Persisters have been described in other pathogens such as Escherichia coli, Pseudomonas aeruginosa, Mycobacterium tuberculosis, and Candida albicans (5–9). Recent studies have shown a direct link between relapse of chronic infections such as tuberculosis, cystic fibrosis pneumonia, and candidacies and the presence of persister (6, 10, 11). Persisters are a subpopulation of the total population that are in (or develop into) a dormant, nondividing state that enables them to survive treatment with otherwise bactericidal antibiotic (3, 12). This dormant state can be observed by a typical biphasic antibiotic killing pattern with an initial rapid killing of bulk population and plateau where only the persister subpopulation remains alive or are slowly killed. This biphasic pattern is observed both with increasing concentrations of antibiotics or with increasing treatment time (3, 12).
The entry of bacterial cells into dormancy can be regarded as an insurance policy wherein a small subpopulation allows the survival of the organism during stress exposure (13). The best-studied example of dormancy is spore formation; however, nonsporulating species can also enter dormancy such as the persister state (14). Persister formation is a stochastic event caused by phenotypic switching (15). Recent studies have suggested that the exit of dormancy, i.e., the revival postantibiotic treatment is also a stochastic event (14, 16). Although being a stochastic event, the formation of persisters can be induced by environmental factors (type I persisters), but they are also continuously generated during growth (type II persisters) (15). The level of persisters, i.e., the number of persisters surviving antibiotic treatment compared to the initial total population, is influenced by cell physiology and environmental conditions such as the length of stationary phase (9, 17), biofilm growth (8, 18), sublethal antibiotic concentration (19), indole (20), and oxidative stress (21). Shah et al. (22) compared the transcriptome of E. coli persisters with stationary-phase cells and showed that specific genes such as toxin-antitoxin (TA) systems are induced in persisters. Persisters were therefore described as a distinct physiological state, but the regulatory and genetic mechanisms involved in persister formation are not fully understood. TA systems contribute to bacteriostasis and can facilitate survival during stress such as antibiotic exposure (23), since fluctuations of toxin levels above and below a certain threshold cause the phenotypic switch and thereby the coexistence of actively growing and dormant cells (24).
The food-borne pathogen Listeria monocytogenes can cause a noninvasive gastroenteritis in people with normal immune defense or an invasive infection in the immunocompromised, the elderly, or the unborn fetus. The mortality in patients with listeriosis is high and can reach 20 to 30% (25). The clinical symptoms of invasive listeriosis can appear within days but may not be evident for up to 2 months (26, 27). Relapse of listeriosis is rare, but cases where unique strains have caused a second infection have been reported (28, 29). However, in most of the reported relapse cases, there has been an underlying illness causing the host to be immunocompromised (28, 29). Although L. monocytogenes is susceptible to a wide range of antibiotics, treatment of listeriosis is complex. Most antibiotics have a bacteriostatic effect on L. monocytogenes, and only a few have a bactericidal effect (30, 31). On the other hand, the bacterium is only intrinsically resistant to a few antibiotics, i.e., nalidixic acid, fosfomycin, and broad-spectrum cephalosporins (31), and acquired resistance to other antibiotics is only slowly increasing (32). L. monocytogenes is an intracellular organism and replicate in the cytosol of the host cell, where only a limited number of antibiotics are effective (33). For years the standard treatment has been a combination of gentamicin and ampicillin (31). A third obstacle to antibiotic treatment success could be formation of persisters but this phenomenon has not been described in L. monocytogenes. However, in a previous study we observed a biphasic killing pattern when L. monocytogenes was exposed to increasing concentrations of norfloxacin and gentamicin (34). A similar biphasic killing patterns can be found in older literature when the bacterium was treated with bactericidal antibiotics (35), although some reports also demonstrate a complete killing of L. monocytogenes by bactericidal antibiotics (36, 37).
Based on biphasic killing pattern of L. monocytogenes observed in previous studies (34, 35), we hypothesized that L. monocytogenes could form persisters, as observed in other pathogens, and the aim of the present study was to investigate whether L. monocytogenes can form persisters and, if so, how physiological factors affect the number of persisters in the population and finally whether dormant L. monocytogenes persisters can be killed by different strategies.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
L. monocytogenes strains used in the present study were shown in Table 1. The panel included well-studied laboratory strains (EGD and EGDe) and lineage I and II strains, i.e., a human clinical isolate L. monocytogenes 4446 (38), a persistent strain isolated from the fish smokehouse L. monocytogenes N53-1 (39), and an outbreak strain caused by Mexican-style soft cheese L. monocytogenes F2365 (40). Bacterial stock cultures were stored at −80°C and grown on brain heart infusion (BHI; Oxoid, catalog no. CM 1135) agar at 37°C. One colony was inoculated in 5 ml of BHI and incubated overnight aerobically at 37°C with shaking (250 rpm). To obtain a standardized stationary-phase culture, the overnight culture was diluted 1,000-fold, and 10 μl of this diluted culture was inoculated in 10 ml of fresh BHI in 50-ml Falcon tubes (corresponding to a 106 dilution) and incubated at 37°C for 16 h at 250 rpm.
Table 1.
L. monocytogenes strains used in this study
Determining the presence and level of persisters.
To allow comparison between different treatments and cells in different growth phases, we used an experimental setup with a constant stoichiometric ratio between biomass and antibiotics consistent with other studies (5, 9). All cultures, i.e., exponential- and stationary-phase cultures, as well as surface-associated cells, were adjusted to optical density at 600 nm (OD600) of 0.2 in either fresh BHI or peptone saline (0.9% saline with 0.1% peptone) before addition of antibiotics. The treatments included bactericidal antibiotics: 4 to 200 μg of norfloxacin ml−1 (dissolved in 0.99 ml of sterile MilliQ water with 10 μl of glacial acetic acid; Fluka, catalog no. N9890), 0.5 to 40 μg of gentamicin ml−1 (dissolved in sterile MilliQ water; Sigma, catalog no. G3632), a combination of gentamicin and ampicillin (dissolved in sterile MilliQ water; Sigma, catalog no. A9518) in 0.2 to 20 μg ml−1 and 1 to 100 μg ml−1, respectively, or 1 to 500 μg of nitrofurantoin ml−1 (dissolved in dimethylformamide; Sigma, catalog no. N7878). Cultures were incubated at 37°C at 250 rpm during antibiotic treatment. Bacterial counts were determined just before treatment and at subsequent time points by plate counting on BHI agar. All experiments were performed with two or three independent biological replicates, and the number of replicates in each experiment has been indicated in the figure legends.
Effect of physiological state of L. monocytogenes on the persister level.
The effect of growth phase on persister level in the population was investigated by comparing stationary phase cells to exponentially growing cells. The latter were obtained by diluting an overnight culture 109 or 1010 times in fresh BHI in a 50-ml Falcon tube and incubating at 37°C at 250 rpm. This resulted in a balanced exponentially growing culture (OD600 = 0.2 to 0.6) after 16 h. The effect of static/aerated conditions on persister level in the population was investigated by diluting an overnight culture 106 times in BHI and incubating it under static conditions at 37°C for 16 h. The shaken and static cultures were also sampled after 3 and 6 days of incubation. The effect of surface-associated growth on persister level was performed as described by Singh et al. (18). An overnight culture was diluted 1,000-fold, and 10 μl was spotted onto 0.2-μm-pore-size GE polycarbonate filter (GE Water & Process Technologies, catalog no. K02CP04700) that was placed on a BHI agar and incubated at 37°C. The persister level was determined at 16 h, 3 days, and 6 days by submerging a filter with bacterial growth in 10 ml of BHI and vortexing the sample vigorously for 1 min before adjusting the OD600 to an 0.2 and exposing the sample to antibiotics. The effect of indole was investigated as described by Vega et al. (20). L. monocytogenes was grown in BHI with or without 15 μM indole before treatment with antibiotics. The effect of short-term exposure to 500 μM indole was investigated by adjusting an exponentially growing culture to an OD600 of 0.2, adding 500 μM indole, and incubating the sample for 1 h. The OD600 was adjusted to 0.2 after 1 h, and the persister level was determined as described. All experiments were performed with three independent biological replicates except the indole experiments that were performed twice.
Killing of persisters by carbohydrate and antibiotic addition.
The addition of fermentable carbohydrate allows killing of E. coli persisters by gentamicin (41). In the present study, norfloxacin persisters were obtained as described above by using 100 or 200 μg of norfloxacin ml−1. The persister population was isolated after 4 h treatment by centrifugation at 8,000 × g for 5 min, washed with improved minimal medium (IMM [42]), centrifuged, and finally resuspended in IMM without any carbon source. The persister population was then treated with 10 μg of gentamicin ml−1 with or without addition of carbohydrates: 10 or 20 mM glucose, 10 mM fructose, 10 nM mannitol, or 10 mM glycerol. Controls with no gentamicin or glucose were included. Bacterial counts were done as described previously. The experiment was performed with at two or three independent biological replicates.
Statistical analysis.
Bacterial counts from each biological replicate were log10 transformed prior to statistical analysis using Microsoft Excel. A Student t test with a significance level of P < 0.05 was used to determine whether the number of surviving persisters was significantly different upon the different conditions.
RESULTS
L. monocytogenes persisters are formed when treated with bactericidal antibiotics.
We previously observed a surviving subpopulation when L. monocytogenes was treated with bactericidal antibiotics (34), and to investigate this possible persister phenomenon, we exposed stationary-phase L. monocytogenes EGDe to increasing norfloxacin concentrations and found a similar biphasic killing pattern (Fig. 1A). The bulk population (108 CFU ml−1) was killed rapidly reaching a plateau of surviving cells of 104 to 105 CFU ml−1 consistent with our previous study (34). A similar biphasic killing pattern was observed in treatments with increasing concentrations of gentamicin for 4 h (Fig. 1B) and in treatments with a combination of gentamicin and ampicillin (Fig. 1C). Biphasic killing patterns such as these are characteristic of the presence of persisters (3, 12). Similar results were obtained when testing an EGD strain instead of EGDe (see Fig. S1A to C in the supplemental material).
Fig 1.

Concentration-dependent killing kinetics of L. monocytogenes treated with bactericidal antibiotic. (A) Treatment of L. monocytogenes EGDe (■) with increasing concentrations of norfloxacin for 4 h. The experiment was performed with two biological replicates, and error bars indicate the standard deviations. (B) Treatment of L. monocytogenes EGDe (■) with increasing concentrations of gentamicin for 4 h. The experiment was performed with two biological replicates, and error bars indicate the standard deviations. (C) Treatment of L. monocytogenes EGDe (■) with increasing concentrations of a mixture of gentamicin and ampicillin. The ratio of gentamicin (G) to ampicillin (A) was 1:5. The experiment was performed with three biological replicates, and error bars indicate the standard deviations. (D) Treatment of L. monocytogenes wild types—EGD (◆), 4446 (▲), N53-1 (×), and F2365 (●)—with increasing concentrations of norfloxacin for 4 h. The experiment was performed with two biological replicates, and error bars indicate the standard deviations.
Persisters are defined as the surviving population when a microbial culture is exposed either to increasing concentration of bactericidal antibiotics or to a fixed concentration over longer time (12). A biphasic killing pattern was also seen, when L. monocytogenes was treated with 100 μg of norfloxacin ml−1 over a 72-h period (Fig. 2A). The bulk of the population was rapidly killed; however, from 24 h and onward a low, but stable, subpopulation of ∼103 CFU of L. monocytogenes ml−1 survived. To ensure that the surviving bacteria were in fact persisters and not resistant bacteria, we isolated surviving persister cells from 24 h treatment with 100 μg of norfloxacin ml−1. These persister isolates were inoculated in fresh medium and grown to stationary-phase cultures and treated with 100 μg of norfloxacin ml−1. They were as sensitive to norfloxacin treatment as the parental strain EGDe and similar biphasic killing pattern was observed (Fig. 2B). Collectively, this indicates that the surviving subpopulation was persisters since they had not acquired resistance allowing survival upon norfloxacin treatment.
Fig 2.
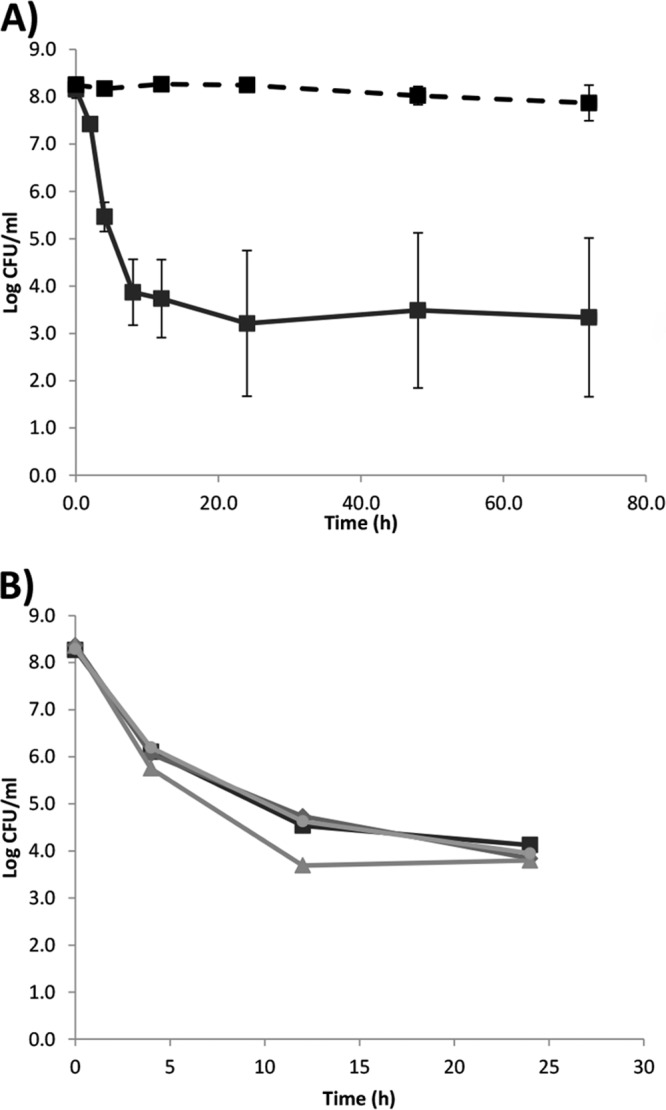
Time-dependent killing of L. monocytogenes treated with 100 μg of norfloxacin ml−1. (A) L. monocytogenes EGDe (■) in BHI (full line) or peptone saline (broken line). Experiments were performed with three and two biological replicates, respectively, and error bars indicate the standard deviations. (B) Killing of surviving persisters from panel A. Two persister isolates, B1 (◆) and B2 (▲), which survived 24 h of treatment with 100 μg of norfloxacin ml−1 in BHI, were exposed to a second 24-h treatment with 100 μg of norfloxacin ml−1 in BHI. One persister isolate, B3 (●), which survived the second cycle (B1) of 24 h of treatment with 100 μg of norfloxacin ml−1 in BHI, was exposed to a third 24-h treatment with 100 μg of norfloxacin ml−1. As a control, a nonpersister EGDe (■) was included. The experiment was performed once.
To verify that the persister phenomenon was not unique to the laboratory strain EGDe wild type, we treated a small panel of wild-type strains with norfloxacin. The killing kinetics of all strains were similar (Fig. 1D), although the strains differed slightly in the level of surviving cells. For all strains, more cells survived when treated for 4 h with 200 as opposed to 100 μg of norfloxacin ml−1. This paradoxical phenomenon of reduced killing by increasing concentration of antibiotics is known as the Eagle effect (43, 44). Although the EGDe strain showed Eagle effect at 4 h, the persister level after 24 h norfloxacin treatment was similar at 100 and 200 μg ml−1 (data not shown), indicating that increased incubation time eliminated the Eagle effect for this strain.
The L. monocytogenes persister level is dependent on the growth phase and conditions.
Persisters are assumed to generated during growth, i.e., preexist in the population before antibiotic treatment (15, 45, 46); however, the number of persisters in the total population is dependent on the growth condition and/or growth phase (8, 9). Compared to the stationary-phase culture (16 h), an exponentially growing culture had significantly fewer surviving bacteria (P = 0.01) when treated for 4 h with 100 μg of norfloxacin ml−1 (Fig. 3A), but the persister level was similar in an exponentially growing and stationary phase when treated for 24 h (P = 0.95, Fig. 3A). A significantly (P = 0.01 and 0.01) higher level of persisters were seen when 3- and 6-day-old stationary-phase cultures were treated with 100 μg of norfloxacin ml−1 compared to a 16-h stationary-phase culture (Fig. 3A), indicating the age of the stationary-phase population influenced the level of persisters. Killing kinetic of the different cultures also indicated a relationship between the age of the L. monocytogenes culture and the rapidness of the killing (Fig. 3A).
Fig 3.
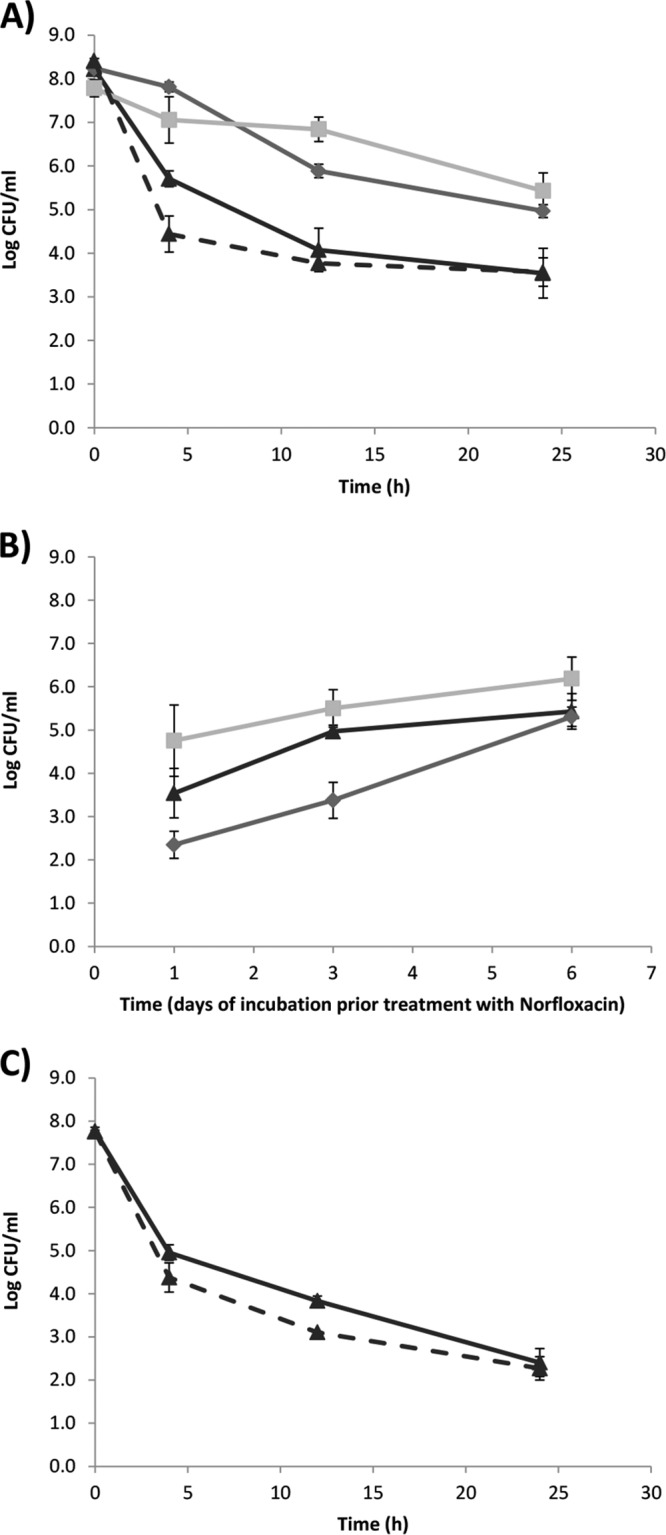
Effect of physiological factors on the level of L. monocytogenes persisters. (A) Time-dependent killing of L. monocytogenes EGDe with 100 μg of norfloxacin ml−1. The results for exponentially growing cells at 16 h (▲, broken line), stationary-phase cells at 16 h (▲, full line), stationary-phase cells at day 3 (◆), and stationary-phase cells at day 6 (■) are shown. The experiment was performed with three biological replicates, and error bars indicate the standard deviations. (B) Effect of different growth conditions and lengths of incubation on the persister level of L. monocytogenes EGDe when treated with 100 μg of norfloxacin ml−1 for 24 h. The results for stationary-phase planktonic cells grown aerated (▲), stationary-phase planktonic cells grown statically (◆), and surface-associated cells (■) are shown. The experiment was performed with three biological replicates, and error bars indicate the standard deviations. (C) Time-dependent killing of L. monocytogenes EGD and ΔsigB mutant with 100 μg of norfloxacin ml−1. The results for stationary-phase cells of EGD at 16 h (▲, full line) and stationary-phase cells of the ΔsigB mutant at 16 h (▲, broken line) are shown. The experiment was performed with two biological replicates, and error bars indicate the standard deviations.
Surface-associated growth or biofilm growth generate higher levels of persisters in P. aeruginosa and S. aureus compared to planktonic growth (8, 18). There is also a tendency toward increased levels of L. monocytogenes persister during surface-associated growth compared to planktonic culture at all investigated time points, i.e., 16 h, day 3, and day 6; however, the increase is not significant (P = 0.10, P = 0.11, and P = 0.11, respectively; Fig. 3B). With increased age, the difference in the persister level between surface-associated and planktonic shaken cultures were reduced (Fig. 3B), possibly due to increased heterogeneity in planktonic aged cultures since the biofilm cells are known to have high heterogeneity (47).
Further, we analyzed whether the level of oxygen affected the L. monocytogenes persister level. The persister level was lower in static cultures compared to shaken cultures at 16 h (P = 0.03) and day 3 (P = 0.003) but not at day 6 (P = 0.67; Fig. 3B). This difference could in part be caused by lower growth rate during static growth and the growth phases, i.e., the ages of static and shaken cultures may not be comparable.
Since the persister level was dependent on the length of the stationary phase and surface-associated growth, this could indicate a relationship with the general stress response, and we compared the persister level in a wild-type EGD and isogenic ΔsigB mutant. In an early-stationary-phase culture, the number of surviving persisters was lower in the ΔsigB mutant compared to the wild-type at 4 h (P = 0.06) and 12 h (P = 0.01) of norfloxacin treatment (Fig. 3C), but at 24 h there was no difference between the wild type and the ΔsigB mutant (P = 0.69). When 3- or 6-day stationary-phase cultures were treated, there was no difference in the persister levels of the wild type and the ΔsigB mutant (data not shown).
In E. coli, the stationary-phase signal, indole, increases the level of persisters (20); however, we found that growth in BHI with 15 μM indole had no effect on the L. monocytogenes persister level compared to BHI control (data not shown). Similarly, a short time exposure to 500 μM indole did not affect persister level (data not shown). This suggests that indole does not affect the persister level in L. monocytogenes and might not act as a stationary-phase signal in L. monocytogenes.
Addition of fermentable carbohydrates to gentamicin reduced the L. monocytogenes persisters.
Several studies have been dedicated to approaches that resensitize persisters to antibiotics (5, 41, 48). We added fermentable carbohydrates and gentamicin to norfloxacin persisters of L. monocytogenes EGDe and found that addition of 10 mM glucose to 10 μg of gentamicin ml−1 killed two log units of the L. monocytogenes persisters compared L. monocytogenes persisters to which no carbohydrate was added during treatment with 10 μg of gentamicin ml−1 (Fig. 4A). In IMM without carbohydrates, no growth was observed but L. monocytogenes persisters would regain growth capacity by addition of 10 mM glucose to IMM without antibiotic. Addition of 20 mM glucose instead of 10 mM had no effect further on the killing of L. monocytogenes persister. Similar to glucose, 10 mM fructose induced rapid gentamicin killing of the persisters, whereas 10 mM glycerol induced a slow killing of persisters (Fig. 4A). In contrast, 10 mM mannitol, which cannot be metabolized by L. monocytogenes, was not capable of inducing gentamicin killing of L. monocytogenes persisters (Fig. 4A). Similar results were obtained when testing EGD persisters isolated after treatment with 100 or 200 μg of norfloxacin ml−1 (see Fig. S2A and B in the supplemental material).
Fig 4.
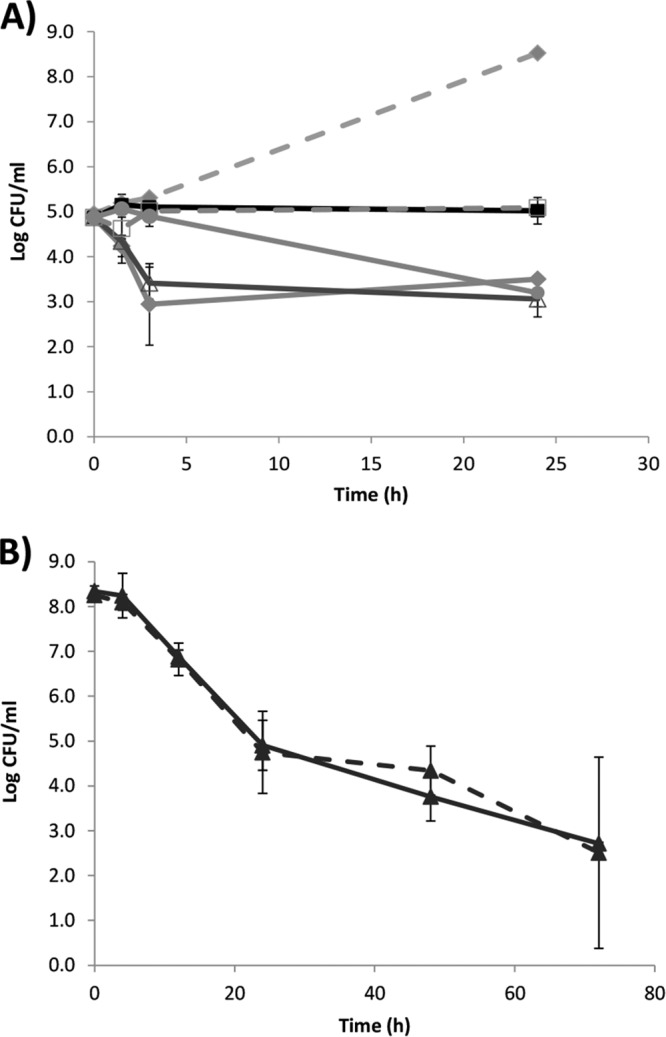
Killing kinetics of L. monocytogenes persisters treated with gentamicin and carbohydrates or by nitrofurantoin. (A) Gentamicin killing of persisters with different fermentable carbohydrates. The results obtained for gentamicin in IMM (■, full line), gentamicin plus 10 mM glucose in IMM (◆, full line), gentamicin plus 10 mM fructose in IMM (Δ, full line), gentamicin plus 10 mM mannitol in IMM (□, broken line), gentamicin plus 10 mM glycerol in IMM (●, full line), and the growth control of 10 mM glucose in IMM (◆, broken line) are shown. The experiment was performed with two biological replicates, and error bars indicate the standard deviations. (B) Time-dependent killing of L. monocytogenes EGDe (▲) with 400 μg of nitrofurantoin ml−1 in either BHI (full line) or peptone saline (broken line). The experiment was performed with three biological replicates, and error bars indicate the standard deviations. At 48- and 72-h time points, only two biological replicates were performed.
Although the addition of fermentable carbohydrates allowed killing L. monocytogenes persisters with gentamicin, this was not a complete eradication since ∼103 CFU ml−1 survived for 24 h of treatment, a finding consistent with the persisting population after norfloxacin treatment.
Active metabolism is required for killing of L. monocytogenes by norfloxacin but not by nitrofurantoin.
Many antibiotics, including gentamicin (Fig. 4A), have a mechanism of action that is dependent on active bacterial metabolism. We investigated norfloxacin killing when a stationary-phase L. monocytogenes culture was diluted in peptone saline instead of BHI. In peptone saline, less than 1 order of magnitude of killing was observed over the 72 h tested (Fig. 2A), indicating that the killing of L. monocytogenes by norfloxacin was dependent on an active metabolism. However, other antibiotics do not require growth or active metabolism to exert their effect and one such antibiotic is nitrofurantoin (49). Indeed, nitrofurantoin caused the same killing pattern of L. monocytogenes cells when diluted in either BHI or peptone saline, indicating a killing not dependent on active metabolism (Fig. 4B). The nitrofurantoin killing of L. monocytogenes continued, albeit at a slower pace, after 24 h, suggesting that nitrofurantoin is capable of reducing the otherwise stable L. monocytogenes persister population.
DISCUSSION
We here demonstrate that a subpopulation of a L. monocytogenes culture can survive treatment with bactericidal antibiotics such as norfloxacin and gentamicin. This dormant, antibiotic-tolerant state has been described in several pathogens, including E. coli, S. aureus, M. tuberculosis, P. aeruginosa, and C. albicans (2, 4, 6–8), and we can now add L. monocytogenes to this list. Persister formation is a significant challenge in treatment of chronic diseases (3, 10), and bacterial populations forming high levels of persisters have been isolated in cystic fibrosis lungs (11) and from cancer patients with oral candidiasis (6). Although there are only few reports on treatment failure or relapse of listeriosis (28, 29), our data indicate that the persister phenomenon should be borne in mind, especially since persisters were formed when L. monocytogenes was exposed to a combination of gentamicin and ampicillin, which is the standard clinical treatment.
The persister phenomenon was not restricted to laboratory strains, i.e., EGD and EGDe, but was also found in clinical and outbreak wild types such as 4446 and F2365. Increasing concentrations of norfloxacin did not increase or stabilize the killing but rather reduced the killing of several L. monocytogenes strains. This so-called Eagle effect (44) was first described over 60 years ago; however, the mechanism is still not fully understood (43). Recently, Tanouchi et al. (50) observed that altruistic death could generate an Eagle effect as death and lysis of some cells in the population caused the release of enzymes, which increased the surviving chances for the rest of the population. Other mechanisms, such as reduced antibiotic activity or the induction of resistance mechanisms, have been suggested as cause of the Eagle effect (referred to in reference 50). In E. coli, the Eagle effect appears to be strain specific (51), and we similarly observed strain differences in L. monocytogenes. Therefore, the Eagle effect is more likely caused by altruistic death or differences in resistance and/or tolerance than reduced antibiotic activity since this likely would not have caused differences between strains. The Eagle effect was, however, time dependent, and this could have serious implications for short but high doses of antibiotic treatment compared to lower doses of antibiotics used for longer treatments.
Low levels of persisters are found in exponentially growing E. coli and P. aeruginosa, whereas the persister level increase in stationary phase (8, 9). In agreement with these studies, we found that an extended incubation time of the culture, i.e., the length of the stationary phase, increased the persister level, especially after 6 days of incubation. A difference in killing between exponentially growing cells and early-stationary-phase cultures was seen after 4 h treatment with norfloxacin but not after 24 h, indicating that the killing of exponentially growing cells was faster than of an early stationary-phase culture. The level of persisters in E. coli increase with 4 logs during the first 18 h of stationary phase (9), whereas the L. monocytogenes persister level increased only ∼2 logs over 6 days. This slower and lower increase in levels of persisters could be caused by the slower growth rate of L. monocytogenes compared to E. coli. However, other factors could affect this differential increase of persisters in stationary phase. One such factor could be the lack the sucB gene encoding the 2-oxoglutarate dehydrogenase (52), causing a noncyclic tricarboxylic acid pathway in L. monocytogenes. An E. coli sucB mutant has a very low level of persisters, and no increase in persister level was observed upon entry to stationary phase (9). However, the L. monocytogenes persister level in exponentially growing cultures and the early stationary phase are higher than those observed E. coli and P. aeruginosa (8, 9). This initial high level of persisters could be due to the lack of hydroxyl radical production upon antibiotic exposure, which appears to be absent in L. monocytogenes (34). Persisters produce fewer hydroxyl radicals after antibiotic treatment than normally growing cells, and this could be one of the mechanisms by which they escape the antibiotic activity and killing (53).
We had expected SigB to play a role in persister formation in L. monocytogenes, since the only TA systems in L. monocytogenes lmo0887-lmo0888, which is a homolog to the S. aureus MazEF system, is located immediately upstream of the sigB operon (54). In S. aureus, mazEF is cotranscribed with sigB and induces bacteriostasis during exposure to the antibiotics and other stressors (55). However, SigB did not appear to be involved in L. monocytogenes persister formation, and the role of its putative TA system is unknown.
Eradication of persisters is important due to their clinical impact (5, 41, 48, 49), and studies have targeted specific characteristics of persisters, such as activating metabolism by the addition of fermentable carbohydrate (41) or inducing hydroxyl production with the antibiotic clofazimine (5). The addition of fermentable carbohydrate to L. monocytogenes persisters could activate the dormant cells, as seen for E. coli and S. aureus (41). However, even after activation and gentamicin killing, a stable surviving subpopulation was still present, and we speculate that the addition of fermentable carbohydrate do not kill the truly dormant L. monocytogenes population. In contrast, nitrofurantoin seems to be a promising candidate for killing L. monocytogenes persisters in vitro, a finding consistent with reports in Mycobacterium bovis (49).
Our discovery of L. monocytogenes persisters could potentially, in part, explain the behavior of this organism in both food production and during infection. L. monocytogenes has a saprophytic lifestyle with a niche in soil and decaying plant material (56) and is a common contaminant of raw materials and subsequently food products. Complete eradication of the bacterium from food processing is almost impossible, and particular molecular subtypes of L. monocytogenes can persist in production facilities for up to 5 to 10 years (39, 57); one could speculate that a persister response to the cleaning and sanitation treatment is a factor enabling persistence. During infection, clinical symptoms of listeriosis often appear after a long incubation time (26, 27), and the bacterium survives the host immune system for extended periods. Using a bio-luciferase-tagged L. monocytogenes, Hardy et al. (58, 59) found the bacteria were undetectable in mice for up to 15 days prior spreading to gallbladder, bone marrow, etc., and they suggested that the persisting bacteria were dormant during this period (59). L. monocytogenes enters the host by crossing the gastrointestinal barrier, and it quickly translocate to the spleen and liver, where it grows intracellularly in macrophages (25). At this stage, L. monocytogenes is capable of modulating the innate immune system (25); however, while this hampering can explain the immediate evasion of the immune system, it does explain the long-term survival of L. monocytogenes in the host. One could speculate that the persister phenomenon could be a survival strategy of the organism when exposed to the host immune defense factors. However, further studies on mechanisms and regulation are needed to disclose the full implications of L. monocytogenes persisters.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Gram lab and Christian Munck from Morten Sommer's lab for suggestions and good discussions.
This study was supported by Danish Council for Independent Research, Technology, and Production Sciences grant 09-066098 (274-08-0531).
Footnotes
Published ahead of print 20 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02184-13.
REFERENCES
- 1.Rogers GB, Carroll MP, Bruce KD. 2012. Enhancing the utility of existing antibiotics by targeting bacterial behaviour? Br. J. Pharmacol. 165:845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fauvart M, De GV, Michiels J. 2011. Role of persister cells in chronic infections: clinical relevance and perspectives on anti-persister therapies. J. Med. Microbiol. 60:699–709 [DOI] [PubMed] [Google Scholar]
- 3.Lewis K. 2007. Persister cells, dormancy, and infectious disease. Nat. Rev. Microbiol. 5:48–56 [DOI] [PubMed] [Google Scholar]
- 4.Bigger J. 1944. Treatment of Staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244:497–500 [Google Scholar]
- 5.Grant SS, Kaufmann BB, Chand NS, Haseley N, Hung DT. 2012. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc. Natl. Acad. Sci. U. S. A. 109:12147–12152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LaFleur MD, Qi Q, Lewis K. 2010. Patients with long-term oral carriage harbor high-persister mutants of Candida albicans. Antimicrob. Agents Chemother. 54:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moyed HS, Bertrand KP. 1983. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 155:768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spoering AL, Lewis K. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183:6746–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luidalepp H, Joers A, Kaldalu N, Tenson T. 2011. Age of inoculum strongly influences persister frequency and can mask effects of mutations implicated in altered persistence. J. Bacteriol. 193:3598–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Yew WW, Barer MR. 2012. Targeting persisters for tuberculosis control. Antimicrob. Agents Chemother. 56:2223–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulcahy LR, Burns JL, Lory S, Lewis K. 2010. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 192:6191–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kint CI, Verstraeten N, Fauvart M, Michiels J. 2012. New-found fundamentals of bacterial persistence. Trends Microbiol. 20:577–585 [DOI] [PubMed] [Google Scholar]
- 13.Kussell E, Kishony R, Balaban NQ, Leibler S. 2005. Bacterial persistence: a model of survival in changing environments. Genetics 169:1807–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buerger S, Spoering A, Gavrish E, Leslin C, Ling L, Epstein SS. 2012. Microbial scout hypothesis, stochastic exit from dormancy, and the nature of slow growers. Appl. Environ. Microbiol. 78:3221–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622–1625 [DOI] [PubMed] [Google Scholar]
- 16.Epstein SS. 2009. Microbial awakenings. Nature 457:1083. [DOI] [PubMed] [Google Scholar]
- 17.Klapper I, Gilbert P, Ayati BP, Dockery J, Stewart PS. 2007. Senescence can explain microbial persistence. Microbiology 153:3623–3630 [DOI] [PubMed] [Google Scholar]
- 18.Singh R, Ray P, Das A, Sharma M. 2009. Role of persisters and small-colony variants in antibiotic resistance of planktonic and biofilm-associated Staphylococcus aureus: an in vitro study. J. Med. Microbiol. 58:1067–1073 [DOI] [PubMed] [Google Scholar]
- 19.Dörr T, Vulic M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8:e1000317. 10.1371/journal.pbio.1000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vega NM, Allison KR, Khalil AS, Collins JJ. 2012. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 8:431–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dörr T, Lewis K, Vulic M. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5:e1000760. 10.1371/journal.pgen.1000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah D, Zhang Z, Khodursky A, Kaldalu N, Kurg K, Lewis K. 2006. Persisters: a distinct physiological state of Escherichia coli. BMC Microbiol. 6:53. 10.1186/1471-2180-6-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerdes K, Maisonneuve E. 2012. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 66:103–123 [DOI] [PubMed] [Google Scholar]
- 24.Rotem E, Loinger A, Ronin I, Levin-Reisman I, Gabay C, Shoresh N, Biham O, Balaban NQ. 2010. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. U. S. A. 107:12541–12546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cossart P. 2011. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc. Natl. Acad. Sci. U. S. A. 108:19484–19491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goulet V, King LA, Vaillant V, de Valk H. 2013. What is the incubation period for listeriosis? BMC Infect. Dis. 13:11. 10.1186/1471-2334-13-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.CDC 2011. Multistate outbreak of listeriosis associated with Jensen Farms cantaloupe—United States, August-September 2011. MMWR Morb. Mortal. Wkly. Rep. 60:1357–1358 [PubMed] [Google Scholar]
- 28.Sauders BD, Wiedmann M, Desjardins M, Fenlon C, Davenport N, Hibbs JR, Morse DL. 2001. Recurrent Listeria monocytogenes infection: relapse or reinfection with a unique strain confirmed by molecular subtyping. Clin. Infect. Dis. 33:257–259 [DOI] [PubMed] [Google Scholar]
- 29.McLauchlin J, Audurier A, Taylor AG. 1991. Treatment failure and recurrent human listeriosis. J. Antimicrob. Chemother. 27:851–857 [DOI] [PubMed] [Google Scholar]
- 30.Espaze EP, Reynaud AE. 1988. Antibiotic susceptibilities of Listeria: in vitro studies. Infection 16(Suppl 2):S160–S164 [DOI] [PubMed] [Google Scholar]
- 31.Hof H, Nichterlein T, Kretschmar M. 1997. Management of listeriosis. Clin. Microbiol. Rev. 10:345–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morvan A, Moubareck C, Leclercq A, Herve-Bazin M, Bremont S, Lecuit M, Courvalin P, Le MA. 2010. Antimicrobial resistance of Listeria monocytogenes strains isolated from humans in France. Antimicrob. Agents Chemother. 54:2728–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hof H, Waldenmeier G. 1988. Therapy of experimental listeriosis—an evaluation of different antibiotics. Infection 16(Suppl 2):S171–S174 [DOI] [PubMed] [Google Scholar]
- 34.Feld L, Knudsen GM, Gram L. 2012. Bactericidal antibiotics do not appear to cause oxidative stress in Listeria monocytogenes. Appl. Environ. Microbiol. 78:4353–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boisivon A, Guiomar C, Carbon C. 1990. In vitro bactericidal activity of amoxicillin, gentamicin, rifampicin, ciprofloxacin, and trimethoprim-sulfamethoxazole alone or in combination against Listeria monocytogenes. Eur. J. Clin. Microbiol. Infect. Dis. 9:206–209 [DOI] [PubMed] [Google Scholar]
- 36.Gordon RC, Wofford RE, Hsu T, Fechner LL. 1980. Ampicillin-gentamicin synergism against Listeria monocytogenes in vitro. Infection 8:221–222 [Google Scholar]
- 37.Winslow DL, Damme J, Dieckman E. 1983. Delayed bactericidal activity of beta-lactam antibiotics against Listeria monocytogenes: antagonism of chloramphenicol and rifampin. Antimicrob. Agents Chemother. 23:555–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen CN, Nørrung B, Sommer HM, Jakobsen M. 2002. In vitro and in vivo invasiveness of different pulsed-field gel electrophoresis types of Listeria monocytogenes. Appl. Environ. Microbiol. 68:5698–5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wulff G, Gram L, Ahrens P, Vogel BF. 2006. One group of genetically similar Listeria monocytogenes strains frequently dominates and persists in several fish slaughter- and smokehouses. Appl. Environ. Microbiol. 72:4313–4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linnan MJ, Mascola L, Lou XD, Goulet V, May S, Salminen C, Hird DW, Yonekura L, Hayes P, Weaver R, Audurier A, Plikaytis BD, Fanning SL, Kleks A, Broome CV. 1988. Epidemic listeriosis associated with Mexican-style cheese. N. Engl. J. Med. 319:823–828 [DOI] [PubMed] [Google Scholar]
- 41.Allison KR, Brynildsen MP, Collins JJ. 2011. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 473:216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phan-Thanh L, Gormon T. 1997. A chemically defined minimal medium for the optimal culture of Listeria. Int. J. Food Microbiol. 35:91–95 [DOI] [PubMed] [Google Scholar]
- 43.Yeh PJ, Hegreness MJ, Aiden AP, Kishony R. 2009. Drug interactions and the evolution of antibiotic resistance. Nat. Rev. Microbiol. 7:460–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eagle H, Musselman AD. 1948. The rate of bactericidal action of penicillin in vitro as a function of its concentration, and its paradoxically reduced activity at high concentrations against certain organisms. J. Exp. Med. 88:99–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230:13–18 [DOI] [PubMed] [Google Scholar]
- 46.Balaban NQ, Gerdes K, Lewis K, McKinney JD. 2013. A problem of persistence: still more questions than answers? Nat. Rev. Microbiol. 11:587–591 [DOI] [PubMed] [Google Scholar]
- 47.Stewart PS, Franklin MJ. 2008. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6:199–210 [DOI] [PubMed] [Google Scholar]
- 48.Kim JS, Heo P, Yang TJ, Lee KS, Cho DH, Kim BT, Suh JH, Lim HJ, Shin D, Kim SK, Kweon DH. 2011. Selective killing of bacterial persisters by a single chemical compound without affecting normal antibiotic-sensitive cells. Antimicrob. Agents Chemother. 55:5380–5383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murugasu-Oei B, Dick T. 2000. Bactericidal activity of nitrofurans against growing and dormant Mycobacterium bovis BCG. J. Antimicrob. Chemother. 46:917–919 [DOI] [PubMed] [Google Scholar]
- 50.Tanouchi Y, Pai A, Buchler NE, You L. 2012. Programming stress-induced altruistic death in engineered bacteria. Mol. Syst. Biol. 8:626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldstein K, Rosdahl VT. 1981. High concentration of ampicillin and the Eagle effect among gram-negative rods. Chemotherapy 27:313–317 [DOI] [PubMed] [Google Scholar]
- 52.Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couve E, de Daruvar A, Dehoux P, Domann E, Dominguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, Garcia-del Portillo F, Garrido P, Gautier L, Goebel W, Gomez-Lopez N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, de Pablos B, Perez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, Simoes N, Tierrez A, Vazquez-Boland JA, Voss H, Wehland J, Cossart P. 2001. Comparative genomics of Listeria species. Science 294:849–852 [DOI] [PubMed] [Google Scholar]
- 53.Kim JS, Heo P, Yang TJ, Lee KS, Jin YS, Kim SK, Shin D, Kweon DH. 2011. Bacterial persisters tolerate antibiotics by not producing hydroxyl radicals. Biochem. Biophys. Res. Commun. 413:105–110 [DOI] [PubMed] [Google Scholar]
- 54.Pandey DP, Gerdes K. 2005. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33:966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu Z, Donegan NP, Memmi G, Cheung AL. 2007. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189:8871–8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freitag NE, Port GC, Miner MD. 2009. Listeria monocytogenes: from saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 7:623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carpentier B, Cerf O. 2011. Review: persistence of Listeria monocytogenes in food industry equipment and premises. Int. J. Food Microbiol. 145:1–8 [DOI] [PubMed] [Google Scholar]
- 58.Hardy J, Francis KP, DeBoer M, Chu P, Gibbs K, Contag CH. 2004. Extracellular replication of Listeria monocytogenes in the murine gallbladder. Science 303:851–853 [DOI] [PubMed] [Google Scholar]
- 59.Hardy J, Chu P, Contag CH. 2009. Foci of Listeria monocytogenes persist in the bone marrow. Dis. Model. Mech. 2:39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brøndsted L, Kallipolitis BH, Ingmer H, Knöchel S. 2003. kdpE and a putative RsbQ homologue contribute to growth of Listeria monocytogenes at high osmolarity and low temperature. FEMS Microbiol. Lett. 219:233–239 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.