

Abstract
A Western blot assay using a recombinant protein, recombinant Baylisascaris procyonis RAG1 protein (rBpRAG1), was developed for the diagnosis of human baylisascariasis concurrently by the Centers for Disease Control and Prevention (CDC) in Atlanta, Georgia, and the National Reference Centre for Parasitology (NRCP) in Montreal, Canada. Assay performance was assessed by testing 275 specimens at the CDC and 405 specimens at the NRCP. Twenty specimens from 16 cases of baylisascariasis were evaluated. Eighteen were positive, with the assay correctly identifying 14 of 16 patients. The rBpRAG1 Western blot assay showed no cross-reactivity with Toxocara-positive serum and had an overall sensitivity of 88% and a specificity of 98%.
INTRODUCTION
Human baylisascariasis, caused by the raccoon roundworm Baylisascaris procyonis, results from infection of a human host with B. procyonis L3 larvae. Subsequent neural larva migrans (NLM), visceral larva migrans (VLM), and ocular larva migrans (OLM) syndromes may occur. The severity of disease is dependent on the number of infective eggs ingested and the tissues affected by larval migration (1). B. procyonis has emerged in recent years as one of the most serious causes of larva migrans in humans (2). The pathogenesis is similar to that of toxocariasis, but usually the disease is more severe and the migration of B. procyonis larvae through tissues is more aggressive than that of Toxocara spp. (3). Unlike Toxocara spp., B. procyonis larvae continue to grow while migrating, which increases the likelihood of severe neurological disease. Infected patients present with symptoms ranging from mild to severe neurological dysfunction, which may progress rapidly to marked postural and locomotor problems, blindness, seizures, coma, and death, especially if larvae invade the brain in large numbers (2, 4). Due to the limited time between the onset of symptoms and severe neurological disease, early detection is imperative for patient outcomes and survival. Currently, definitive diagnosis is dependent on the identification of larvae in biopsy or autopsy specimens (4). Although computed tomography (CT) and magnetic resonance imaging (MRI) scans are an adjunct for diagnosis, they cannot definitively differentiate between Baylisascaris infections and infections with other eosinophilic parasites that cause meningitis. In most cases, changes visualized by neuroimaging lag behind severe clinical manifestations (3, 4). The most efficient and least invasive method for diagnosis is serological analysis. Diagnostic assays using native excretory-secretory (ES) antigen from infective B. procyonis larvae were useful but cross-reactivity, particularly of Toxocara-specific antibodies, remained a problem in enzyme-linked immunosorbent assays (ELISAs) (5). Better results were obtained using Western blot tests, which could differentiate Baylisascaris from Toxocara infections, although both tests had to be performed (6). The differential diagnosis of baylisascariasis must rule out toxocariasis because the two types of parasites can cause similar clinical problems and have overlapping geographical distributions (5).
Recently, serological assays for baylisascariasis have improved greatly with the use of a recombinant protein (5, 7). An ELISA based on the recombinant Baylisascaris procyonis RAG1 protein (rBpRAG1) was developed, and early testing showed no cross-reactivity with Toxocara infections (7). However, in a more recent study testing a total of 384 serum specimens from human patients, including specimens from 20 patients with clinical Baylisascaris larva migrans, 137 patients with other parasitic infections (including 8 helminthic and 4 protozoan infections), and 227 patients with unknown/suspected parasitic infections, the rBpRAG1 ELISA showed sensitivity of 85% and specificity of 87%, with 25% cross-reactivity with Toxocara infections (5). Previously, diagnostic tests for baylisascariasis were available only at Purdue University; recently, however, the rBpRAG1 reagents were transferred to two separate national centers for assay development and independent optimization. In the present study, a sensitive and specific Western blot test for the diagnosis of B. procyonis infections in humans that was based on the recombinant protein rBpRAG1 was developed and optimized for use in both the Centers for Disease Control and Prevention (CDC) Parasitic Diseases Reference Diagnostic Laboratory in Atlanta, Georgia, and the National Reference Centre for Parasitology (NRCP) in Montreal, Canada.
MATERIALS AND METHODS
Serum samples for assay optimization.
Both institutions used a positive-control anti-B. procyonis serum from a baboon that developed severe NLM after experimental infection with embryonated B. procyonis eggs. A normal human serum pool was constructed from 5 serum samples from B. procyonis-negative, presumably healthy, U.S. residents with no history of international travel. At the CDC, a Toxocara-positive serum pool was prepared by combining 10 enzyme immunoassay (EIA)-positive serum samples.
Serum panels for determinations of diagnostic sensitivity and specificity.
To determine the diagnostic sensitivity and specificity of the assay at the CDC, a total of 275 human serum specimens were tested, including 15 serum samples and one cerebrospinal fluid (CSF) specimen from patients diagnosed with B. procyonis, 109 serum samples from patients with other diseases, and 150 serum samples from North American residents with no history of international travel. At the NRCP, a total of 405 samples were used for an independent evaluation of sensitivity and specificity, including nine serum samples and two CSF specimens from seven patients with baylisascariasis, 264 serum samples from patients with other diseases, and 130 serum samples from B. procyonis-negative, presumably healthy individuals. All serum samples were used in compliance with protocols approved by the ethical review boards of the participating institutions.
Expression and purification of rBpRAG1 and development of Western blot assays.
The pRSET C/RAG1 plasmid was obtained by the CDC and NRCP from Purdue University in 2012. Expression, purification, and Western blot optimization were performed independently at each center, based on the methods described by Dangoudoubiyam et al. (7).
Expression and purification of rBpRAG1 at the CDC.
At the CDC, the pRSET C/RAG1 plasmid was transformed into Bl21(DE3)pLysS, and the recombinant colonies were selected from LB agar plates containing 34 μg/ml chloramphenicol and 100 μg/ml ampicillin. Colonies were grown overnight at 37°C in LB broth containing 34 μg/ml chloramphenicol and 100 μg/ml ampicillin. Expression of rBpRAG1 was achieved 3 h after addition of 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to the overnight culture. Samples of the induced and noninduced cultures were collected and separated using SDS-PAGE under nonreducing conditions. The gels were visualized and analyzed using Western blotting, following previously described methods (8), with anti-His and anti-Xpress antibodies (Life Technologies), B. procyonis-infected baboon serum, and a normal human serum pool. All primary antibodies were diluted in phosphate-buffered saline (PBS) with 0.3% Tween 20 (PBS-Tw)-5% milk, with the anti-His and anti-Xpress antibodies at 1:5,000 dilutions and the baboon and normal human serum pools at 1:100 dilutions. Human serum and CSF specimens were tested at 1:100 and 1:10 dilutions, respectively, in PBS-Tw–5% milk. The secondary antibody used for the anti-His and anti-Xpress antibodies was horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG at a 1:8,000 dilution in PBS-Tw and that used for the baboon and normal human serum pools was HRP-conjugated goat anti-human IgG at a 1:8,000 dilution in PBS-Tw. The membranes were incubated with a 3,3′-diaminobenzidine (DAB) and H2O2 solution for band detection.
Purification of the polyhistidine-tagged rBpRAG1 fusion protein was performed using 6 M urea as the denaturing agent and Talon metal affinity resins (Clontech, Inc., Mountain View, CA) (7). Eluted fractions were collected and analyzed by SDS-PAGE and Western blotting with anti-His antibodies. Protein concentrations were measured using the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL).
For determination of antigen purity and optimal concentration, four concentrations of the rBpRAG1 protein, i.e., 18.3, 37.5, 75, and 150 ng/well, were run on NuPage 4 to 12% 2-bis(2-hydroxyethyl)amino-2-(hydroxymethyl)-1,3-propanediol (Bis-Tris) gradient gels (Life Technologies), blotted, and probed with anti-B. procyonis-positive baboon serum, an anti-Toxocara-positive serum pool, and a normal human serum pool, using previously described methods. For high-throughput testing of the CDC panel of sera, 2.75 μg of rBpRAG1 antigen was loaded onto a Criterion XT 4 to 12% Bis-Tris gel with an 11-cm immobilized pH gradient (IPG) well (Bio-Rad Laboratories, Hercules, CA), electrophoresed, and transferred to a nitrocellulose membrane, and the membrane was cut into 2.5-mm strips.
Expression and purification of rBpRAG1 at the NRCP.
At the NRCP, expression and purification of the rBpRAG1 protein were performed by a procedure modified from the protocol described by Dangoudoubiyam et al. (7). One liter of super optimal broth (SOB) medium with final concentrations of 34 μg/ml chloramphenicol and 100 μg/ml ampicillin was inoculated with an overnight culture of BL21(DE3)pLysS competent Escherichia coli transformed with the pRSET C/RAG1 expression plasmid. The cultures were incubated at 37°C with shaking at 225 rpm until the optical density at 600 nm (OD600) reached 1.0 to 1.2. IPTG was added to a final concentration of 0.5 mM, and the cultures were further incubated for 3.5 h at 37°C with shaking. The cells were harvested by centrifugation at 3,000 × g for 10 min at room temperature (RT) and were stored at −80°C. Cell pellets were lysed under denaturing conditions with purification buffer (20 mM Tris-HCl [pH 8.5], 8 M urea, 500 mM NaCl, 10 mM β-mercaptoethanol, 20 mM imidazole, 5 μg/ml DNase I) and were incubated on a rotary shaker for 30 min at RT. After centrifugation at 24,000 × g for 20 min at RT, the recombinant 6×His-tagged RAG1 protein was purified using SuperFlow Ni-nitrilotriacetic acid (Ni-NTA) resin (Qiagen, Venlo, Netherlands). Five milliliters of 50% saturated resin was equilibrated with 10 column volumes of purification buffer. The supernatant containing the rBpRAG1 protein was added to the equilibrated resin and allowed to bind for 1 h at RT on a rotary shaker. The resin-lysate mixture was transferred to a polypropylene column and packed under gravity flow. Six wash buffers with decreasing concentrations of urea (8, 7, 6, 5, 4, and 3 M) in 20 mM Tris-HCl (pH 8.5), 500 mM NaCl, 10 mM β-mercaptoethanol, and 20 mM imidazole were applied to the column and drained by gravity flow. The bound protein was eluted in 8 column volumes (20 ml) of elution buffer (20 mM Tris-HCl [pH 8.5], 500 mM NaCl, 3 M urea, 10 mM β-mercaptoethanol, 150 mM imidazole). The eluted protein was dialyzed against TE buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA) using Zeba spin desalting columns (Pierce, Rockford, IL) and protein concentrations were measured with the BCA assay (Pierce), according to the manufacturer's protocols.
Expression and purification of the rBpRAG1 antigen were verified by SDS-PAGE and immunoblotting. Nontransformed BL21(DE3)pLysS competent E. coli cells were used as controls to assess rBpRAG1 expression. Aliquots of both transformed and nontransformed, induced and noninduced cultures were collected at various time points, and proteins were separated on Mini-Protean 4 to 15% TGX gradient gels (Bio-Rad Laboratories) under reducing conditions. Separated proteins were transferred to nitrocellulose membranes with the iBlot dry transfer system (Life Technologies), using the default 7-min protocol. Nonspecific sites were blocked for 1 h at RT with 5% skim milk in PBS with 0.05% Tween 20. Membranes were incubated overnight at 4°C with mouse monoclonal antipolyhistidine serum at 1:6,000 (Sigma, St. Louis, MO), followed by incubation with HRP-conjugated anti-mouse IgG at a 1:50,000 dilution (GE Healthcare, Little Chalfont, United Kingdom). The purified and desalted rBpRAG1 protein was assessed for reactivity with human serum. The nitrocellulose membranes were probed with either a pool of human serum specimens positive for Baylisascaris procyonis (n = 3), at a dilution of 1:100, or a pool of healthy human serum specimens (n = 3), at 1:100, and were incubated with HRP-conjugated anti-human IgG at a 1:100,000 dilution (Perkin-Elmer, Waltham, MA). All membranes were incubated with the chemiluminescent substrate in SuperSignal West Pico (Pierce) and were exposed to autoradiographic film.
For determination of the optimal antigen concentration, four amounts of the rBpRAG1 protein (128, 256, 512, and 1,024 ng/well) were run on NuPage 4 to 12% Bis-Tris gradient gels (Life Technologies) under reducing conditions and proteins were transferred to nitrocellulose membranes, as described above. The membranes were probed with the same pools of B. procyonis-positive and healthy human serum specimens as used above, at dilutions of 1:100, but were probed with HRP-conjugated anti-human IgG at a dilution of 1:25,000. The membranes were incubated with DAB-H2O2 solution.
For high-throughput testing of sera, 6 μg of rBpRAG1 protein was separated on NuPage one-dimensional-well 4 to 12% Bis-Tris gradient gels (Life Technologies) and transferred to nitrocellulose membranes for Western blot analysis by using a Mini-Protean II multiscreen apparatus (Bio-Rad Laboratories). Human serum and CSF specimens were tested at dilutions of 1:100 and were incubated with HRP-conjugated anti-human IgG at 1:5,000. The membranes were incubated with DAB-H2O2 solution.
RESULTS
The expression of rBpRAG1 was monitored by the presence of a 37-kDa band that was detected by the anti-His antibody only in cells transformed with the expression clone. At the NRCP, recombinant RAG1 was expressed at basal levels at all times but reached optimal expression levels after 3.5 h of induction (data not shown). To improve the diagnostic potential of the rBpRAG1 antigen, a few changes in the composition of the purification and wash buffers were made by adding 10 mM β-mercaptoethanol and 20 mM imidazole. The goal was to eliminate all of the bands detected in the immunoblot probed with healthy human sera but to keep a strong signal for the 37-kDa rBpRAG1 antigen when B. procyonis-positive samples were tested. The purified and desalted rBpRAG1 antigen (37 kDa) showed a strong reaction to the human B. procyonis-positive antiserum pool (Fig. 1). No band was detected at 37 kDa when the blots were probed with individual or pooled healthy human sera, confirming the specificity of the rBpRAG1 antigen for B. procyonis. However, a single unidentified band slightly larger than 75 kDa was observed (Fig. 1). The presence of this 75-kDa band suggested that an optimal test would still require the separation of proteins, thus eliminating ELISA as a potential assay format. To further simplify the assay, the detection method was switched from enhanced chemiluminescence (ECL) to DAB detection, keeping in mind the facility and rapidity of use of the latter method. The next step was to determine the optimal rBpRAG1 antigen amount to use for detection of the 37-kDa rBpRAG1 antigen in B. procyonis-positive individuals (but not B. procyonis-negative patients) and to eliminate reactivity with the 75-kDa band in serum samples from B. procyonis-negative patients. We found that any amount of rBpRAG1 antigen between 128 and 256 ng/well was sufficient to achieve that outcome (Fig. 2A).
Fig 1.

Testing of purified and desalted rBpRAG1 antigen produced at the NRCP for reactivity against human serum pools, i.e., B. procyonis-positive sera (+) or healthy control sera (−). Detection was by ECL. A 37-kDa band corresponding to rBpRAG1 was detected only when membranes were probed with sera from individuals with baylisascariasis. A single unidentified band running slightly above 75 kDa was detected with both the positive and negative serum pools. Numbers on the left indicate molecular mass markers (in kDa).
Fig 2.
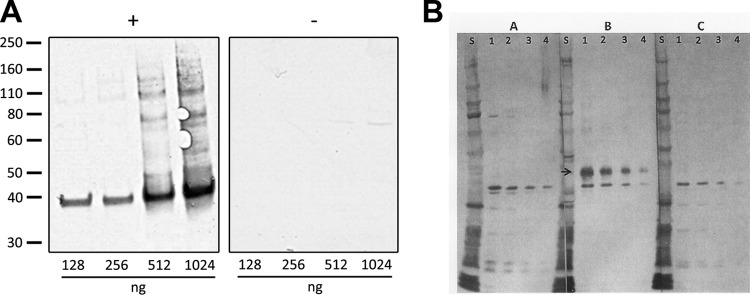
Determination of the optimal rBpRAG1 concentration. (A) At the NRCP, titration of the purified rBpRAG1 antigen was performed with 128, 256, 512, and 1,024 ng/well, and the blots were probed with the B. procyonis-positive human serum pool (+) or the healthy human serum pool (−). Numbers on the left indicate molecular mass markers (in kDa). (B) At the CDC, titration of the purified rBpRAG1 antigen was performed with concentrations of 150 ng/well (lanes 1), 75 ng/well (lanes 2), 37.5 ng/well (lanes 3), and 18.3 ng/well (lanes 4). Western blots were probed with the normal human serum pool (A lanes), anti-B. procyonis-positive baboon serum (B lanes), or the Toxocara-positive serum pool (C lanes). S, molecular mass standard. The arrow identifies rBpRAG1.
Experiments also were conducted at the CDC to determine antigen purity and optimal concentration; four concentrations of the rBpRAG1 protein, i.e., 18.3, 37.5, 75, and 150 ng/well, were evaluated using Western blotting (Fig. 2B). Baylisascaris-specific antibodies reacted with the rBpRAG1 protein with an approximate molecular mass of 37 kDa. No cross-reactivity was seen with the normal human serum pool or the Toxocara-positive serum pool. Based on protein resolution and band intensity, the rBpRAG1 concentration of 75 ng/well was chosen for further evaluation. Experiments were also performed to determine if a preblocking step was necessary. Preblocking of the membrane with PBS-Tw–5% milk for 1 h prior to the addition of serum or CSF improved assay performance and therefore was incorporated into the final protocol.
Assay performance was evaluated independently at the CDC and the NRCP (Tables 1 and 2). Sensitivity was assessed using specimens from 16 patients with baylisascariasis. A total of 15 serum samples and one CSF specimen were tested at the CDC, and nine serum samples and two CSF specimens from patients were tested at the NRCP. Of the 16 specimens from patients with baylisascariasis who were tested, 14 were positive using the rBpRAG1 Western blot assay, resulting in a sensitivity of 88%. Serum specimens from seven patients with baylisascariasis were tested at both laboratories, and all test results were positive. It was not possible to test all specimens at both institutions due to insufficient quantities of some serum and CSF specimens. A panel of 373 serum samples from patients with various diseases, including 63 Toxocara-positive samples, was tested. No serum samples from patients with toxocariasis were positive (Fig. 3A); three serum samples from patients with malaria and single serum samples from patients with giardiasis, filariasis, and strongyloidiasis reacted with rBpRAG1 (Fig. 3B). Eight serum samples from presumed healthy individuals also were reactive. Overall, 14 serum samples reacted with rBpRAG1, resulting in a specificity of 98%.
Table 1.
Reactivity of sera from patients with baylisascariasis in the rBpRAG1 assays performed at the CDC and the NRCP
| Patient no. | Collection date (mo/day/yr)a | Result from: |
|
|---|---|---|---|
| CDC | NRCP | ||
| 1 | 10/17/1990 | Positive | Positive |
| 2 | 10/23/1993 | Positive | Positive |
| 3 | 1/24/1997 | Positive | Positive |
| 4 | 8/7/1997 | Negative | NTb |
| 5 | 10/20/1997 | Positive | NT |
| 6 | 9/1/1998 | Positive | Positive |
| 7 | 1/27/2000 | Positive | Positive |
| 1/31/2000 | NT | Positive | |
| 8 | 7/17/2000 | Positive | Positive |
| 7/24/2000 (CSF) | NT | Positive | |
| 7/26/2000 | NT | Positive | |
| 9 | 5/28/2002 | Positive | Positive |
| 7/17/2002 (CSF) | NT | Positive | |
| 10 | 4/12/2004 | Negative | NT |
| 11 | 9/24/2004 | Positive | NT |
| 12 | 8/13/2005 | Positive | NT |
| 13 | 8/15/2007 | Positive | NT |
| 14 | 1/17/2008 | Positive | NT |
| 15 | 9/16/2008 | Positive | NT |
| 16 | 10/23/2008 (CSF) | Positive | NT |
All specimens were serum/plasma samples unless indicated. CSF, cerebrospinal fluid.
NT, not tested.
Table 2.
Analysis of the specificities of the rBpRAG1 assays performed at the CDC and the NRCP
| Disease | CDC |
NRCP |
||
|---|---|---|---|---|
| No. of samples | No. positive | No. of samples | No. positive | |
| None | 150 | 6 | 130 | 2 |
| Amebiasis | NTa | NT | 15 | 0 |
| Ancylostomiasis | 1 | 0 | NT | NT |
| Ascariasis | 18 | 0 | 11 | 0 |
| Cancer | 2 | 0 | NT | NT |
| Chagas disease | NT | NT | 25 | 0 |
| Cysticercosis | 2 | 0 | 8 | 0 |
| Echinococcosis | 3 | 0 | 24 | 0 |
| Eosinophilic myalgia | 2 | 0 | NT | NT |
| Fascioliasis | 1 | 0 | 35 | 0 |
| Filariasis | NT | NT | 7 | 1 |
| Giardiasis | NT | NT | 17 | 1 |
| Gnathostomiasis | 4 | 0 | NT | NT |
| Hookworm | 11 | 0 | NT | NT |
| Hymenolepiasis | 1 | 0 | NT | NT |
| Malaria | NT | NT | 10 | 3 |
| Schistosomiasis | 4 | 0 | 21 | 0 |
| Strongyloidiasis | 8 | 1 | 25 | 0 |
| Toxocariasis | 40 | 0 | 23 | 0 |
| Toxoplasmosis | 4 | 0 | 12 | 0 |
| Trichinellosis | 4 | 0 | 22 | 0 |
| Trichuriasis | 4 | 0 | 9 | 0 |
| Total | 259 | 7 | 394 | 7 |
| Specificity (%) | 97 | 98 | ||
NT, not tested.
Fig 3.

Reactivity of serum specimens. (A) At the CDC, the reactivity of serum specimens from patients with B. procyonis (strips A1 to A9) or Toxocara (strips B1 to B10) was tested using rBpRAG1 Western blot strips. Strip A1, sample from a patient diagnosed with baylisascariasis; strip A3, sample from the same patient 3 years postinfection. Patient samples that were positive for Toxocara by EIA (CDC) were tested using the rBpRAG1 strips (strips B1 to B10). The positive and negative controls were an anti-B. procyonis-positive baboon serum (strip B11) and a normal human serum sample (strip B12), respectively. The arrow identifies rBpRAG1. (B) At the NRCP, the rBpRAG1 reactivity of serum specimens from patients with Chagas disease (lanes 1 to 3), trichuriasis (lanes 4 and 5), strongyloidiasis (lanes 6 to 8), toxocariasis (lanes 9 to 11), echinococcosis (lanes 12 to 14), or filariasis (lanes 15 and 16) was tested. Western blot controls included a healthy human serum pool (−) and B. procyonis-infected baboon serum (+). Positive reactions are shown in lane 15. Numbers on the left indicate molecular mass markers (in kDa).
DISCUSSION
The performance of the rBpRAG1 Western blot assay greatly improves laboratory diagnosis of baylisascariasis. Assays based on native ES antigen showed cross-reactivity with antibodies from patients with toxocariasis, which necessitated additional testing with Toxocara antigens to determine whether Toxocara-specific antibodies were present; if they were, then a diagnosis of baylisascariasis could not be definitively made. The lack of cross-reactivity in the rBpRAG1 assay eliminates the need for additional testing to rule out toxocariasis and increases the confidence in a baylisascariasis diagnosis. The interlaboratory evaluation of the assays showed that the results from the two centers were concordant, demonstrating the robustness of the assay. Our results indicated sensitivity and specificity of the assay of 88% and 98%, respectively. However, they may actually be higher than this, since we do not know the full infection history of the test sera, e.g., if positive cross-reactors with other parasites or conditions were also infected with Baylisascaris in covert dual infections. Sera from two patients gave false-negative results in our assay. This may be related to infection levels or the kinetics of the immune response to the parasite. Future studies to examine the kinetics of the immune response and the appearance of detectable antibody could provide insight into the false-negative results seen with our assay.
The availability of a more sensitive and specific assay will also facilitate serosurveys to better examine the epidemiology of baylisascariasis. Covert infections with B. procyonis, showing mild or no symptoms, may be expected based on the widespread distribution of raccoons in North America, the prevalence of B. procyonis in raccoons (68 to 90%), and the level of human exposure to B. procyonis eggs (2, 4, 5). Covert infections have been shown in previous reports, e.g., one study showed 8% seroprevalence in children in the Chicago, Illinois, area (9), and the parents of a child with severe NLM symptoms tested positive for B. procyonis-specific antibodies without showing signs or symptoms of infection (10). Although an assay with an ELISA platform would be more efficient for service diagnostic laboratories and for serosurveys, several bands are seen in Western blots that seem to be reactive when any human serum sample, including normal human serum and Toxocara-positive serum, is tested (Fig. 2). The presence of these bands suggests that there would be lower performance for an assay in the ELISA format, and it could explain the slightly lower specificity seen with the BpRAG1 ELISA, which shows cross-reactivity with toxocariasis of 25% (5); therefore, further modifications must be performed with the rBpRAG1 antigen before an ELISA can be developed.
Currently, the rBpRAG1 diagnostic Western blot assay is being used for testing at the CDC and the NRCP. B. procyonis infections should be considered based on the presence of NLM, VLM, or OLM symptoms with supporting epidemiological evidence such as a history of pica or geophagia and exposure to raccoons or raccoon latrines. With high specificity and no cross-reactivity in Toxocara infections, a positive rBpRAG1 test result should be considered indicative of infection with B. procyonis for patients with consistent clinical presentations and risk factors.
ACKNOWLEDGMENTS
We thank Mark Eberhard of the CDC for providing the serum from a baboon experimentally infected with B. procyonis.
Work performed at the NRCP was supported by the Public Health Agency of Canada/National Microbiology Laboratory (grant HT070-010033).
Footnotes
Published ahead of print 18 September 2013
REFERENCES
- 1.Kazacos KR. 2001. Baylisascaris procyonis and related species, p 301–341 In Samuel WM, Pybus MJ, Kocan AA. (ed), Parasitic diseases of wild mammals, 2nd ed. Iowa State University Press, Ames, IA [Google Scholar]
- 2.Murray WJ, Kazacos KR. 2004. Raccoon roundworm encephalitis. Clin. Infect. Dis. 39:1484–1492 [DOI] [PubMed] [Google Scholar]
- 3.Watts CS, Liang JL, Schantz PM. 2006. Baylisascariasis: an emerging and potentially fatal form of larval migrans, p 228–238 In Holland CV, Smith HV. (ed), Toxocara: the enigmatic parasite. CAB International, Wallingford, United Kingdom [Google Scholar]
- 4.Gavin PJ, Kazacos KR, Shulman ST. 2005. Baylisascariasis. Clin. Microbiol. Rev. 18:703–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dangoudoubiyam S, Vemulapalli R, Ndao M, Kazacos KR. 2011. Recombinant antigen-based enzyme-linked immunosorbent assay for diagnosis of Baylisascaris procyonis larva migrans. Clin. Vaccine Immunol. 18:1650–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dangoudoubiyam S, Kazacos KR. 2009. Differentiation of larva migrans caused by Baylisascaris procyonis and Toxocara species by Western blotting. Clin. Vaccine Immunol. 16:1563–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dangoudoubiyam S, Vemulapalli R, Hancock K, Kazacos KR. 2010. Molecular cloning of an immunogenic protein of Baylisascaris procyonis and expression in Escherichia coli for use in developing improved serodiagnostic assays. Clin. Vaccine Immunol. 17:1933–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsang VC, Peralta JM, Simons AR. 1983. Enzyme-linked immunoelectrotransfer blot techniques (EITB) for studying the specificities of antigens and antibodies separated by gel electrophoresis. Methods Enzymol. 92:377–391 [DOI] [PubMed] [Google Scholar]
- 9.Brinkman WB, Kazacos KR, Gavin PJ, Binns HJ, Robichaud JD, O'Gorman M, Shulman ST. 2003. Seroprevalence of Baylisascaris procyonis (raccoon roundworm) in Chicago area children, abstr 1872 Abstr. Pediatr. Acad. Soc. Annu. Meet. Pediatric Academic Societies, Seattle, WA [Google Scholar]
- 10.Cunningham CK, Kazacos KR, McMillan JA, Lucas JA, McAuley JB, Wozniak EJ, Weiner LB. 1994. Diagnosis and management of Baylisascaris procyonis infection in an infant with nonfatal meningoencephalitis. Clin. Infect. Dis. 18:868–872 [DOI] [PubMed] [Google Scholar]