

Abstract
Mycobacterial infections in fish are commonly referred to as piscine mycobacteriosis, irrespectively of the specific identity of the causal organism. They usually cause a chronic disease and sometimes may result in high mortalities and severe economic losses. Nearly 20 species of Mycobacterium have been reported to infect fish. Among them, Mycobacterium marinum, M. fortuitum, and M. chelonae are generally considered the major agents responsible for fish mycobacteriosis. As no quick and inexpensive diagnostic test exists, we tested the potential of high-resolution melting analysis (HRMA) to rapidly identify and differentiate several Mycobacterium species involved in fish infections. By analyzing both the melting temperature and melting profile of the 16S-23S rRNA internal transcribed spacer (ITS), we were able to discriminate 12 different species simultaneously. Sensitivity tests conducted on purified M. marinum and M. fortuitum DNA revealed a limit of detection of 10 genome equivalents per reaction. The primers used in this procedure did not lead to any amplification signal with 16 control non-Mycobacterium species, thereby demonstrating their specificity for the genus Mycobacterium.
INTRODUCTION
The nontuberculous mycobacteria (NTM) are Gram-positive, acid-fast, aerobic bacteria that belong to the order Actinomycetales. They are widespread in the aquatic environment, in both fresh and marine waters, where they can survive under hostile conditions by forming biofilms (1). Some of them are the causative agents of fish mycobacteriosis, which occurs predominantly as a chronic disease and occasionally in an acute form (2, 3). The primary pathological lesions associated with the disease are grayish white nodules (granulomas) in the internal organs, such as the liver, spleen, and kidney, which may further lead to high fish mortality and severe losses in the aquaculture industry (4–6). Fish mycobacteriosis has been reported to affect nearly 200 freshwater and saltwater species (7). Mycobacterium marinum, M. fortuitum, and M. chelonae are considered the main causative agents of fish mycobacteriosis (2). However, many other Mycobacterium species have been found to be associated with granulomas in aquarium, cultured, and wild fish, among which are Mycobacterium abscessus, M. gastri, M. smegmatis, M. bohemicum, M. gordonae, etc. (3, 6). NTM causing infection in fish are divided between rapid growers (which develop visible colonies on solid media within 7 days) and slow growers (which require longer incubation times) (3, 8). Although comprehensive surveys are rare, the frequency of NTM infection in cultured fish seems to be increasing (9). As an illustration, 135 out of 312 ornamental fish collected during an 18-month survey were positive for NTM, with 55% being positive by Ziehl-Neelsen staining (10). Another study reported the isolation of Mycobacterium spp. from 29.9% of 127 ornamental fish batches imported into Italy (11). It was also shown that a low-dose infection of M. marinum results in the development of a latent disease (12). However, there are no validated treatments for mycobacteriosis in fish; complete depopulation of asymptomatic carriers and disinfection are the primary methods for controlling the disease (13). Some NTM species are also responsible for human infections (9, 14), with an increasing incidence.
The rapid development of fish farming and of the ornamental fish trade has led to a worldwide increase in the number of reports of mycobacterial infections in fish, with two major consequences: (i) a substantial financial loss in the two above-mentioned industries and (ii) an increased risk of contamination for people who handle fish (15–18). Therefore, early surveillance systems based on a rapid identification of fish pathogens are critical for effective disease control in aquaculture and for improved epidemiological surveys. Furthermore, some authors have discussed specific recommendations for the policy on the importation of ornamental fish (18), which should include the evaluation of biosecurity procedures and disease monitoring.
In recent years, a great number of molecular methods, mostly based on nucleic acid amplification, have been developed for the diagnosis of fish mycobacteriosis (reviewed in reference 6). Recently, a commercial kit, the GenoType Mycobacterium CM (common mycobacteria) kit was introduced for identification of mycobacterial cultures (mainly of clinical origin). This kit is able to identify 25 different species based on 16S rRNA gene hybridization. Combined with another version, the AS (additional species) kit, the test can discriminate among 44 species in total, with a success rate of ∼96% over 219 tested isolates (19). However, the use of this kit remains both time-consuming and costly, since in most cases it requires the prior growth of mycobacterial isolates, and the cost for one reaction remains far more expensive than sequencing. Another commercial test exclusively targeting fish pathogens, the INNO-LipA Mycobacteria v2 assay, was developed (20). This kit is also based on the hybridization between the mycobacterial 16S-23S rRNA internal transcribed spacer and the corresponding oligonucleotide probes immobilized on membrane strips. Although this kit is able to distinguish 16 different Mycobacterium species with a rather good success rate (21), it still relies on the isolate cultivation, which does not fulfill the speed requirements for large-scale prevalence studies and epidemiological surveys.
Therefore, there is an urgent need for a fast, accurate, sensitive, and cost-effective method adapted to veterinary needs. Over the last few years, real-time PCR methods have been developed and widely evaluated in studies for detection of Mycobacterium spp. (22, 23). High-resolution melting analysis (HRMA), further developed from real-time PCR, is an emerging technique in medical microbiology that may allow simultaneous detection and diagnosis of pathogens at the species and subspecies levels (24–26). This technique, first reported in 2002, is based on the difference in melting behaviors of DNA molecules, according to their sequence, lengths, and GC content (25, 27). HRMA requires only nanogram amounts of DNA and has the potential to discriminate closely related microorganisms with high accuracy, speed, and sensitivity.
The present study specifically aimed at developing an HRMA-based identification test of the major Mycobacterium species affecting fishes. Targeting the 16S-23S rRNA internal transcribed spacer (ITS), this assay relies on the measurement of differences in both the melting temperature and melting profile.
MATERIALS AND METHODS
Bacterial strains.
Twelve NTM isolates were used as reference species in the present study. Among them, 5 were purchased from Pasteur Institute (Paris, France) as pure isolates, 2 were obtained from the Laboratoire Départemental Vétérinaire (LDV, Montpellier, France) and consisted of strains isolated from fish tissues, and 5 were isolated from human patients in Arnaud de Villeneuve Hospital (Montpellier, France). A list of these strains is presented in Table 1. All of these strains, which had previously been identified by biochemical tests and/or sequencing, were cultured on Lowenstein-Jensen (LJ) slants and grown at 37°C for several days to several weeks. In addition, 16 nonmycobacterial field isolates, including bacteria that are pathogenic and opportunistically pathogenic for fish, were also used for evaluating the specificity of the assay (Table 1). These bacteria, which comprised 6 Gram-positive and 10 Gram-negative species, were grown in their specific culture medium. All isolates were manipulated in a biosafety level 2 containment laboratory.
Table 1.
List of Mycobacterium and non-Mycobacterium species used in this study
| Strain | Gram positivity | Source |
|---|---|---|
| Mycobacterium spp. | ||
| M. phlei (CIP 105389T) | + | Pasteur Institute |
| M. bohemicum (CIP 105811T) | + | Pasteur Institute |
| M. gastri (CIP 104530T) | + | Pasteur Institute |
| M. pseudoshottsii (CIP 109775T) | + | Pasteur Institute |
| M. smegmatis (CIP 104444T) | + | Pasteur Institute |
| M. fortuitum subsp. fortuitum | + | Arnaud de Villeneuve Hospital |
| M. marinum | + | Arnaud de Villeneuve Hospital |
| M. chelonae | + | Arnaud de Villeneuve Hospital |
| M. abscessus | + | Arnaud de Villeneuve Hospital |
| M. gordonae | + | Arnaud de Villeneuve Hospital |
| M. avium | + | LDV Montpellier |
| M. haemophilum | + | LDV Montpellier |
| Other species | ||
| Flavobacterium psychrophilum | − | LDV Montpellier |
| Pseudomonas fluorescens | − | LDV Montpellier |
| Aeromonas sobria | − | LDV Montpellier |
| Aeromonas hydrophila | − | LDV Montpellier |
| Vibrio vulnificus | − | LDV Montpellier |
| Citrobacter braaki | − | LDV Montpellier |
| Shewanella putrefaciens | − | LDV Montpellier |
| Photobacterium damselae | − | LDV Montpellier |
| Chryseobacterium indologenes | − | LDV Montpellier |
| Lactococcus garvieae | + | LDV Montpellier |
| Carnobacterium piscicola | + | LDV Montpellier |
| Streptococcus parauberis | + | LDV Montpellier |
| Carnobacterium maltaromaticum | + | LDV Montpellier |
| Enterococcus faecalis | + | LDV Montpellier |
| Citrobacter freundii | − | LDV Montpellier |
| Nocardia sp. | + | CVI Wageningena |
Central Veterinary Institute.
Fish tissue samples.
As one of the French approved laboratories, the Laboratoire Départemental Vétérinaire (LDV34) of Montpellier (France) is requested by local fish farmers or owners to diagnose their fish when the latter are suspected of carrying infections. As a result, LDV34 holds a small collection of fish samples that were either diagnosed for mycobacterial infections or collected from infected farms. From this collection, 30 fish samples were used for validating the PCR-HRM test described in this study, together with 3 DNA samples extracted from mycobacterial cultures isolated from some of these fish. The list of these samples and their origins are presented in Table 2.
Table 2.
Fish tissue samples
| Original sample (fish species) | DNA concna | NTM species identification |
|---|---|---|
| Tissue samples | ||
| 080109000133-01 (Anableps anableps) | 7.41E+05 | M. phlei |
| 070213001086-01 (Dicentrarchus labrax) | 6.17E+06 | M. marinum |
| 061107005301-01 (Hemigrammus bleheri) | 1.61E+07 | Mix of M. phlei and M. marinum? |
| 070821004737-01 (undetermined cichlid) | 1.67E+06 | M. phlei |
| 070821004737-01 (undetermined cichlid) | 1.04E+07 | M. phlei |
| 090805003940-01(Sparus aurata) | 1.45E+06 | M. malmoense |
| 090805003940-01 (Sparus aurata) | 3.89E+06 | M. phlei |
| 090805003940-01 (Sparus aurata) | 2.07E+06 | M. phlei |
| 120124000342-01 (Scophthalmus maximus) | 5.39E+06 | M. marinum |
| 071108006385-02 (Sciaenops ocellatus) | 2.49E+07 | M. marinum |
| DNA samples | ||
| 120417001884-01 (Danio rerio) | M. abscessus | |
| 120124000342-01 (Scophthalmus maximus) | M. marinum | |
| 120127000443-01 (Danio rerio) | M. marinum |
Expressed as genome equivalents per g of tissue.
Genomic DNA isolation.
DNA from all samples (bacterial strains and fish tissues) was purified with the Wizard Genomic DNA purification kit (Promega), following the appropriate protocol provided in the kit. For Gram-positive cultures, a slightly modified protocol was used. Bacterial colonies were resuspended in 480 μl of EDTA solution (50 mM, pH 8). After addition of 120 μl of lysozyme (10 mg/ml), bacterial cells were incubated for 1 h at 37°C, centrifuged for 5 min at 13,000 × g, resuspended in 600 μl of nuclear lysis solution, incubated again for 10 min at 100°C, and cooled to room temperature. This lysate was then supplemented with 20 μl of proteinase K (20 mg/ml) and incubated for another 3 h at 55°C, with gentle shaking. The rest of the procedure was performed according to the manufacturer's instructions. DNA from Gram-negative isolates and from fish tissues was extracted following the Gram-negative and animal tissue protocols provided in the kit, respectively. In all cases, DNA was eluted in 70 μl of the provided Tris-EDTA solution, and its concentration was measured by UV spectrometry (NanoDrop ND-1000 spectrophotometer; NanoDrop Technologies Inc.). Bacterial genomic DNA was adjusted to approximately 0.5 × 104 or 0.5 × 106 genome equivalents μl−1, based on an average genome size of 6.6 Mb, whereas fish DNA was diluted to 10 ng μl−1.
Assay design.
Since the assay aimed at being as simple as possible, it relied on the use of a double-strand intercalating—and thus non-sequence-specific—fluorophore for measuring differences in melting profiles of amplification products. Under such conditions, the targeted genomic region (i) had to be unique for each of the investigated species and (ii) had to harbor conserved sequences at its extremities, enabling genus-specific amplification. Multiple alignments of different genomic regions, including the 16S-23S ribosomal operon, the β subunit of RNA polymerase (rpoB), the 65-kDa heat shock protein (hsp65), and the B subunit of DNA gyrase (gyrB) genes, were realized with ClustalX v2 (28) on sequences imported from the NCBI collection (www.ncbi.nlm.nih.gov). They revealed that the internal transcribed spacer (ITS) region could fulfill these requirements. From these alignments, a single primer pair was subsequently designed to amplify a fragment of ∼220 to ∼320 bp in all the targeted mycobacterial species (forward, GCTGGATCACCTCCTTTCTA; reverse, AGATGCTCGCAACCACTAT). The primers were verified for the absence of secondary structures with GeneRunner v3.01 (Hasting Software, Inc.) and purchased from Eurofins-MWG-Operon.
The amplification and melting steps were achieved using the LightCycler480 high-resolution melting master kit (Roche). The reaction mixture was composed of 2× master mix, MgCl2, forward and reverse primers, genomic DNA, and PCR-grade water, in a final volume of 10 μl. The amplification procedure consisted of an initial denaturation followed by 45 cycles of denaturation, annealing, and elongation. After amplification, the melting program was set up by heating to 95°C for 1 min, cooling to 40°C for 1 min, and applying a temperature ramp from 65 to 95°C with a transition rate of 0.2°C s−1 and a continuous fluorescence monitoring. Each reaction was run in triplicate in 96-well plates, with the LightCycler 480 system (Roche). Each PCR-HRMA run included one negative control where the DNA template was replaced by water.
Sensitivity and specificity.
For determination of the assay sensitivity, serial 10-fold dilutions of known genomic DNA concentrations of M. fortuitum and M. marinum were prepared in (i) sterilized distilled water and (ii) 100 ng of genomic DNA extracted from Pangasianodon hypophthalmus fish liver. The number of genome equivalents was estimated from the measured DNA concentrations and the size of the fully sequenced M. marinum genome (6.66 Mb). Serial dilutions of M. fortuitum and M. marinum DNA covered the range of 106 to 1 genome equivalents, and standard curves were drawn from these measurements performed under the conditions described above. Because mycobacterial genomes may carry 1 or 2 rRNA operons (29, 30), results were always expressed as genome copies or genome equivalents, and not as 16S-23S (ITS) copies.
The specificity of the assay was evaluated on 16 nonmycobacterial isolates, including 7 Gram-positive and 9 Gram-negative species (Table 1). The amount of nonmycobacterial DNA in each reaction mixture was adjusted to approximately 106 genome equivalents (based on an average genome size of 6 Mb). Positive controls consisted of 2 mycobacterial species (M. marinum and M. fortuitum), and their DNA amount was set to ∼104 genome equivalents only. To ensure the integrity of these 18 genomic DNAs, they were subsequently amplified with a pair of 16S universal primers (31) in a 2720 thermal cycler (Applied Biosystems). PCR mixtures contained 5 μl of 2× master mix (Fast-Start PCR kit; Roche), 0.8 μM forward (5′-GCACAAGCGGTGGAGCATGTGG-3′) and reverse (5′-GCCCGGGAACGTATTCACCG-3′) primers and 2 μl of template DNA, in a final volume of 10 μl. Amplification consisted of 30 cycles of denaturation (95°C, 30 s), annealing (60°C, 30 s), and elongation (72°C, 30 s), and PCR products were observed by 1% agarose gel electrophoresis containing SYBR Safe DNA gel stain (Invitrogen).
High-resolution melting analysis.
The LightCycler480 software package (version 1.5.0.39) was utilized for both PCR and HRM analyses. After each run, cycles of quantification (Cq) were calculated in order to ensure that each DNA template had been successfully amplified. All amplifications that resulted in Cq values of >30 were arbitrarily considered negative and were therefore excluded from subsequent analyses. Melting profiles were analyzed with the gene scanning function, in a standardized way. First, melting curves were normalized in the premelting and postmelting regions; the normalization temperature ranges were 81 to 81.5°C and 92 to 92.5°C, respectively. Then, a temperature shift was applied on normalized curves with a threshold set at 5% of normalized fluorescence. Finally, melting curves were classified into groups with a default sensitivity of 0.3 and using the autogroup function. This way, curves showing nearly identical shapes were grouped together and were depicted in the same color for easy visualization. However, since the software enables a classification into a maximum of 6 groups only, a Tm (melting temperature) calling was also applied in order to record the melting temperature of each product. Species were then discriminated according to both their melting profile and their melting temperature.
Validation of the assay with blind samples.
To evaluate the ability of this PCR-HRM assay to identify unknown specimens, 30 tissues originating from either infected fish or fish collected from infected ponds, as well as 3 DNA samples extracted from mycobacterial cultures isolated from some of these fish, were investigated (Table 2). DNAs from these samples were blindly subjected to PCR-HRMA, together with the 12 Mycobacterium reference species (M. marinum, M. fortuitum, M. chelonae, M. gordonae, M. smegmatis, M. phlei, M. bohemicum, M. pseudoshottsii, M. abscessus, M. haemophilum, M. avium, and M. gastri) and one negative control (no DNA). Amplification yield was first verified, and samples that displayed a Cq value of <30 were tentatively assigned a species name through the successive analysis of the melting profile and Tm. For confirmation, samples that displayed one single amplification product were sent for sequencing to Genoscreen (Lille, France).
RESULTS
Specificity of the primers.
Tested on genomic DNA extracted from pure bacterial cultures, the primers specifically designed for this assay successfully amplified all of the 12 assessed Mycobacterium species, including M. marinum, M. fortuitum, and M. chelonae (3). The amplification resulted in products of the expected size, between approximately 220 and 320 bp. For 7 of the analyzed strains, a single product was amplified, as revealed by melting peaks obtained from the first derivative of fluorescence over temperature. However, for M. abscessus, M. gastri, and M. haemophilum strains, a small secondary melting peak was always observed, whereas the peak shape of M. chelonae and M. fortuitum contained a minor “shoulder” (data not shown). Probably because the secondary melting peaks were small, the LightCycler 480 software detected two Tm values solely for M. gastri. In this case, only the Tm corresponding to the main peak was taken into consideration. The occurrence of secondary melting peaks, mostly in fast-growing species, may be due to the existence of 2 rRNA operons (29, 32). However, the melting curves for these species were highly reproducible between experiments, as they were identical in all subsequent HRMA runs.
When tested on a range of nonmycobacterial species (see Table 1), the primers always yielded Cq values above 30, i.e., below the detection threshold, with no detectable band on agarose gel electrophoresis (Fig. 1). It is noteworthy that the amount of tested genomic DNA for these 16 nonmycobacterial strains was elevated, as it approximated ∼106 genome equivalents. In comparison, the two major pathogens M. marinum and M. fortuitum, tested at a 100-fold-lower DNA concentration (about 104 genome copies), resulted in Cq values around 19 to 20. When universal primers for 16S rRNA genes were tested on the same DNAs, all the strains yielded an amplification product of nearly the same intensity, demonstrating that neither PCR inhibition nor DNA degradation had occurred (Fig. 1C).
Fig 1.

Specificity of the PCR-HRM assay. Amplification curves of the 16 nonmycobacterial species (∼106 genome equivalents) are presented together with those of M. marinum and M. fortuitum at a 100-fold-lower concentration (∼104 genome equivalents) (A) and corresponding gel electrophoresis with the Mycobacterium-specific primers (B) and 16S universal primers (C). Lane 1, molecular weight marker; lanes 2 to 19, Mycobacterium marinum, Mycobacterium fortuitum, Carnobacterium piscicola, Streptococcus parauberis, Carnobacterium maltaromaticum, Enterococcus faecalis, Citrobacter freundii, Lactococcus garvieae, Nocardia sp., Flavobacterium psychrophilum, Pseudomonas fluorescens, Aeromonas sobria, Aeromonas hydrophila, Vibrio vulnificus, Citrobacter braakii, Shewanella putrefaciens, Photobacterium damselae, and Chryseobacterium indologenes.
Sensitivity of the PCR-HRM assay.
Sensitivity tests were conducted on M. marinum and M. fortuitum DNAs, assuming that results would be comparable for the other strains, since the use of equivalent DNA concentrations yielded nearly similar Cq values for all the tested strains. As described previously, the detection threshold was set at 30 cycles, because Cq values of negative controls were always between 30 and 35, probably because of a slight primer-dimer formation that was undetectable by agarose gel electrophoresis and by melting curve analysis. Using this threshold, the assay was able to accurately and reproducibly detect as few as 10 copies of M. marinum and M. fortuitum genomes in two experimental backgrounds, i.e., water and fish DNA. Figure 2 presents the amplification results obtained for M. marinum and M. fortuitum diluted in 100 ng of fish DNA. The faint band observed on gel electrophoresis for 1 genome equivalent of M. marinum (Fig. 2b) was not considered significant. Furthermore, the relationship between fluorescence intensity and DNA quantity was linear over 6 logs for the dilutions in fish DNA (R2 = 0.999 and 0.998 for M. marinum and M. fortuitum, respectively) as well as for the dilutions in water (R2 = 0.997 and 0.998 for M. marinum and M. fortuitum, respectively) (Fig. 3). Therefore, the presence of fish DNA, which reflects the actual nature of biological samples, did not alter the sensitivity of Mycobacterium detection.
Fig 2.

Sensitivity of the PCR-HRM assay. Serial dilutions of bacterial DNA were prepared in 100 ng of fish genomic DNA and subsequently used as the template for PCR amplification. (A) Amplification curves; (B) corresponding gel electrophoresis.
Fig 3.
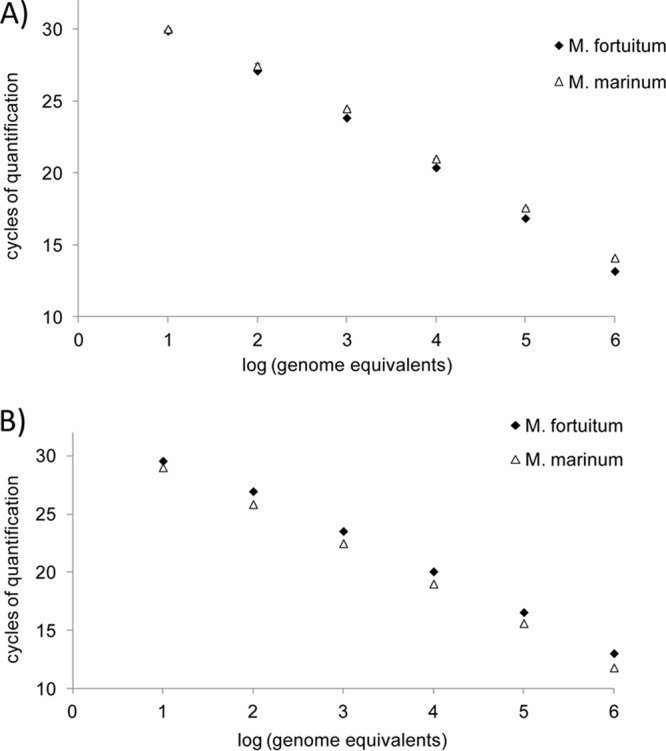
Standard curves obtained from serial dilutions of bacterial DNA in H2O (A) and in 100 ng of fish DNA resuspended in Tris-EDTA (B).
Analysis of melting profiles.
Twelve strains were tested for their ability to be discriminated according to their melting profile and melting temperature. Analysis of the melting curves showed the presence of 9 distinct melting profiles, as illustrated by the difference plots in Fig. 4. However, the strains that shared identical melting profiles could be differentiated by distinct Tms (Table 3), thereby enabling a total discrimination of the 12 species. Reliability of such strain classification was evaluated through different means. First, the intrarun repeatability of difference plots was verified by (i) running 6 replicates of each strain and (ii) running 6 serial dilutions of M. marinum and M. fortuitum, each in triplicate. As shown by Fig. 5, the replicate curves could be superimposed. More importantly, the difference plots, and hence the grouping ability, was not affected by the template DNA concentration, at least between 10 and 106 genome copies, in the reaction mixture. Second, measurement of the melting temperature of all 12 mycobacterial strains from three separate runs revealed a high reproducibility, as shown in Table 3. All but 3 standard deviations (SD) were ≤0.04°C, and the maximum SD was 0.12°C (for M. fortuitum). Finally, when repeated 3 times from 3 independent runs, the HRM analysis showed consistent grouping patterns. It is noteworthy that the 3 most frequently detected Mycobacterium fish pathogens—M. marinum, M. fortuitum, and M. chelonae—display somewhat different Tms. Therefore, their identification could be simply done according to their Tms (Table 3).
Fig 4.

Strain grouping by HRMA. Difference plots were obtained after a temperature shift on the normalized melting curves, using the curve for M. pseudoshottsii as a base curve. For clarity, the strain differentiation is displayed in 2 plots, one showing data for 5 groups (A) and one for 4 groups (B). This experiment was repeated 3 times independently and yielded exactly the same grouping results. When 2 species grouped together, they were differentiated by their Tms. This was the case for group 1 (M. marinum and M. gordonae), group 2 (M. phlei and M. pseudoshottsii), and group 3 (M. fortuitum and M. haemophilum). Refer to Table 3 for Tm values.
Table 3.
Reproducibility of the melting temperature measurementa
| Species |
Tm (°C) |
|||
|---|---|---|---|---|
| 1 | 2 | 3 | Mean ± SD | |
| M. abscessus | 86.12 | 86.07 | 86.09 | 86.10 ± 0.03 |
| M. marinum | 86.71 | 86.68 | 86.67 | 86.68 ± 0.02 |
| M. chelonae | 87.31 | 87.23 | 87.26 | 87.27 ± 0.04 |
| M. haemophilum | 87.20 | 87.14 | 87.16 | 87.16 ± 0.03 |
| M. gordonae | 88.21 | 88.20 | 88.17 | 88.19 ± 0.02 |
| M. fortuitum subsp. fortuitum | 89.13 | 89.34 | 89.33 | 89.27 ± 0.12 |
| M. gastri | 89.46 | 89.39 | 89.46 | 89.44 ± 0.04 |
| M. avium | 89.05 | 88.87 | 88.88 | 88.94 ± 0.10 |
| M. phlei | 90.92 | 90.92 | 90.82 | 90.89 ± 0.06 |
| M. smegmatis | 90.12 | 90.11 | 90.10 | 90.11 ± 0.01 |
| M. pseudoshottsii | 89.97 | 89.97 | 89.97 | 89.97 ± 0.01 |
| M. bohemicum | 89.98 | 89.96 | 89.93 | 89.96 ± 0.03 |
Each measurement is the average for 3 replicates.
Fig 5.

Reproducibility of difference plots. (A) Each mycobacterial DNA was replicated 6 times, and difference plots were drawn for the 6 replicates. For clarity, data for only 6 species are presented. (B) Difference plots were drawn from serial dilutions of M. marinum and M. fortuitum (106 to 10 genomes/reaction), using 3 replicates per dilution.
Mycobacterium detection and species assignment in blind samples.
Among the 30 fish tissue samples that were tested, only 10 could be successfully amplified with the HRMA primers, since the 20 others yielded Cq values higher than 30. The concentrations in these 10 samples varied between 20 and 1,200 genome equivalents per reaction, or between ∼1 and ∼60 genome equivalents per ng of total genomic DNA, since 20 ng of total DNA were used in each reaction. Considering the mass of tissue used (20 mg) and the DNA yield (ng of total DNA per g of tissue) for each extraction, such concentrations corresponded to approximately 7.5 × 105 to 2.5 × 107 genome copies per g tissue. The HRM analysis allowed us to assign 3 of these samples as M. marinum and 6 as M. phlei (Fig. 6). Sequencing of the 9 corresponding amplification products confirmed the species identification for 8 of them, while the remaining one turned out to belong to Mycobacterium malmoense, which is not included in the present assay. The last positive sample presented a melting curve with double inflection, with Tm values comparable to those of M. marinum (86.7°C) and M. phlei (90.9°C), suggesting the presence of a mixed infection with those 2 species (Fig. 6C). Regarding the 3 unknown DNAs, 2 could be unambiguously assigned to M. marinum, while the third one was associated with M. abscessus (data not shown). Sequencing of the corresponding amplification products confirmed this species identification for the 3 samples.
Fig 6.

Identification of unknown samples. Difference plots of 4 reference strains (one each of M. marinum, M. fortuitum, M. chelonae, and M. phlei) are displayed alone (A) and together with 9 unknown samples identified as M. marinum or M. phlei (B), using the M. fortuitum curve as a base curve. (C) Melting curve of an unknown sample, showing two inflections with Tms comparable to those of M. marinum and M. phlei.
DISCUSSION
To our knowledge, this is the first report of an HRM-based assay that enables rapid detection and identification of several nontuberculous mycobacterial species without relying on costly probes such as molecular beacons or TaqMan probes. When genomic DNA extracted from pure cultures was used, the assay was able to confidently detect approximately 10 mycobacterial genomes per reaction. This low detection limit was comparable to that obtained by Zerihun et al. (33), who used a TaqMan probe, and slightly better than that obtained by Salati et al. (34) with a nested-PCR test. It is also comparable to the detection levels described by Pakarinen et al. (35), whose results showed that the use of a hybridization probe did not significantly improve the assay sensitivity. Moreover, the ability of species identification was not affected by the initial DNA template amount, as melting curves obtained from 10-fold dilutions of genomic DNA (corresponding to 106 to 10 genome equivalents) could be superimposed (Fig. 5B).
The mycobacterial concentrations of the unknown fish samples were estimated to range between 20 and 1,200 genome equivalents per reaction, which corresponded to approximately 7.5 × 105 and 2.5 × 107 genome equivalents per g of tissue. The lowest concentration was close to the detection limit of the present assay, indicating that lower bacterial loads would hardly be detected under the conditions used here. It is difficult to compare these values with those obtained by other groups (33, 34), as the procedures used to estimate the minimal detectable bacterial load from fish tissues were different. Technical culture may be considered the gold standard with a limit of detection close to 10 CFU per g of tissue (36). However, in some situations, culture-based methods may underestimate the amount of mycobacterial cells, because of (i) the presence of viable but noncultivable mycobacteria (37) and (ii) nonoptimal culture conditions. The detection limit obtained in our study (∼10 bacterial genomes per reaction) is comparable to that reported by others (33) on DNA extracted from pure cultures (∼6.5 CFU per reaction). Based on their calculations, our lowest estimated concentration would thus correspond to ∼150 CFU g−1 tissue. Such a detection limit would therefore allow to reveal the presence of mycobacterium in most infected fish, as the scarce information reporting mycobacterial loads in tissues of infected fish indicate values between 102 to 109 CFU per g tissue (38, 39). Optimization of the DNA extraction protocol from fish tissue would possibly improve this sensitivity level. Moreover, our present results confirm previous findings obtained by LDV34 (not published), i.e., the 4 samples found positive for M. marinum correspond to organs that showed granulomas and were taken from sick fishes that had been diagnosed with M. marinum infection; the 26 remaining ones corresponded to fish that did not show any lesions but were in contact with other infected fishes. However, the fact that M. phlei was identified in 6 of them is rather surprising and requires further investigation, as M. phlei is not known as a common NTM species in fish.
High-resolution melting analysis has already been successfully used for discriminating related species of bacteria (26, 40). In addition, by measuring the Tm of a small amplicon of the hsp65 gene, it has been found to be possible to differentiate Mycobacterium abscessus and M. chelonae, 2 very closely related, rapidly growing species that cannot be discriminated biochemically (41). The authors hypothesized that with a Tm SD of less than 0.1°C, it should be possible to differentiate many different species. In our case, M. abscessus and M. chelonae had pretty different melting profiles and could be discriminated by both their melting profile and melting temperature, which differed by approximately 1.2°C. However, even though most Tm SDs were lower than 0.1°C, some species could not be identified solely according to their Tm (e.g., M. chelonae and M. haemophilum or M. pseudoshottsii and M. bohemicum). Thus, the combination of both Tm and melting profile analysis increased the discriminative power of the assay.
Selection of the target sequence was made bearing in mind that some mycobacterial species carry 2 rRNA operons and that strains belonging to the same species may display sequence differences. Therefore, the relatively long length of the target amplicon (∼220 to 320 bp, depending on the species) was expected to confer a greater tolerance of the melting profile to sequence changes. Among the unknown samples that were identified as M. marinum (Table 2), the sequence of two of them differed from the strain used as a reference by 2 mismatches located near the 5′ extremity of the amplification product. However, the presence of 2 mismatches did not significantly modify the Tm and did not prevent us from classifying these 2 strains as M. marinum. The extent to which sequence variations affect melting profiles and temperatures has not been precisely quantified. However, it is known that melting differences decrease as the amplicon size increases (42), and the use of such a long amplicon in the present study is likely to represent a limitation for the number of species that can be simultaneously discriminated.
In any case, the results presented here show that this assay confidently discriminates 12 mycobacterial species, i.e., M. phlei, M. smegmatis, M. gastri, M. bohemicum, M. marinum, M. fortuitum, M. chelonae, M. gordonae, M. pseudoshottsii, M. abscessus, M. haemophilum, and M. avium. Though it includes the most frequently reported fish pathogens, we cannot exclude the possibility of a wrong species determination in the case of fish carrying uncommon mycobacterial species not accounted for here. An alternative solution to increase the number of species that can be simultaneously differentiated and reduce the probability of wrong species assignment has recently been described (43). Although it looks very attractive, the use of a combination of labeled probes dramatically increases the cost of the experiment. In addition, it also seems to decrease the assay sensitivity, since the lowest bacterial concentration that was tested (100 genome copies) yielded relatively high Cq values (around 35). This drop in sensitivity is probably the result of a higher level of constraints induced by the simultaneous use of 4 different probes that require consensus experimental conditions for optimal behavior.
Finally, compared to the 2 existing commercial kits (19, 21), the assay described here presents many advantages. First, the real-time PCR format makes it possible to analyze many samples at the same time. Indeed, using the 384-well plate format and running both reference and target samples in triplicate, it is possible to simultaneously analyze more than 100 samples. Second, since this PCR-HRM assay does not require prior mycobacterial culture, it takes only a couple of hours to obtain results from fish tissue samples. Third, the cost of such a test will be much lower than that of the existing kits, since it is a one-step assay and it does not rely on any costly labeled probe. A first estimation would place the cost of one reaction, from sample to result, below 5 €. To conclude, the present PCR-HRM assay is accessible, quick, and inexpensive. It enables the detection of the presence of any mycobacterial genome, since it uses genus-specific primers, as well as the identification of 12 mycobacterial species directly from fish samples, without prior bacterial cultivation. Its format allows the analysis of more than 100 unknown samples simultaneously, thus making it possible to investigate the prevalence of these pathogens on a large scale and at a reasonable cost.
ACKNOWLEDGMENTS
This work was made possible thanks to an EU postdoctoral MAHEVA fellowship.
We thank Stéphanie Laurence, Hélène Boulet, Céline Fourré, and Laura Boschiroli for their technical help in strain cultivation. We are also grateful to Marc Engelsma and Jean-François Bernardet for providing Gram-positive bacteria.
D.C. and J.-C.A. declare competing financial interests, as they intend to make commercial use of the method described in this paper. All other authors declare no conflict of interest.
Footnotes
Published ahead of print 11 October 2013
This is ISEM publication IRD-DIVA-ISEM 2013-104.
REFERENCES
- 1.Hall-Stoodley L, Brun OS, Polshyna G, Barker LP. 2006. Mycobacterium marinum biofilm formation reveals cording morphology. FEMS Microbiol. Lett. 257:43–49 [DOI] [PubMed] [Google Scholar]
- 2.Decostere A, Hermans K, Haesebrouck F. 2004. Piscine mycobacteriosis: a literature review covering the agent and the disease it causes in fish and humans. Vet. Microbiol. 99:159–166 [DOI] [PubMed] [Google Scholar]
- 3.Gauthier DT, Rhodes MW. 2009. Mycobacteriosis in fishes: a review. Vet. J. 180:33–47 [DOI] [PubMed] [Google Scholar]
- 4.Beran V, Matlova L, Dvorska L, Svastova P, Pavlik I. 2006. Distribution of mycobacteria in clinically healthy ornamental fish and their aquarium environment. J. Fish Dis. 29:383–393 [DOI] [PubMed] [Google Scholar]
- 5.Bruno DW, Griffiths J, Mitchell CG, Wood BP, Fletcher ZJ, Drobniewski FA, Hastings TS. 1998. Pathology attributed to Mycobacterium chelonae infection among farmed and laboratory-infected Atlantic salmon Salmo salar. Dis. Aquat. Org. 33:101–109 [DOI] [PubMed] [Google Scholar]
- 6.Jacobs JM, Stine CB, Baya AM, Kent ML. 2009. A review of mycobacteriosis in marine fish. J. Fish Dis. 32:119–130 [DOI] [PubMed] [Google Scholar]
- 7.Toranzo AE, Magarinos B, Romalde JL. 2005. A review of the main bacterial fish diseases in mariculture systems. Aquaculture 246:37–61 [Google Scholar]
- 8.Stahl DA, Urbance JW. 1990. The division between fast- and slow-growing species corresponds to natural relationships among the mycobacteria. J. Bacteriol. 172:116–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falkinham JO. 1996. Epidemiology of infection by nontuberculous mycobacteria. Clin. Microbiol. Rev. 9:177–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prearo M, Zanoni RG, Dall'Orto BC, Pavoletti E, Florio D, Penati V, Ghittino C. 2004. Mycobacterioses: emerging pathologies in aquarium fish. Vet. Res. Commun. 28:315–317 [DOI] [PubMed] [Google Scholar]
- 11.Zanoni RG, Florio D, Fioravanti ML, Rossi M, Prearo M. 2008. Occurrence of Mycobacterium spp. in ornamental fish in Italy. J. Fish Dis. 31:433–441 [DOI] [PubMed] [Google Scholar]
- 12.Parikka M, Hammaren MM, Harjula S-KE, Halfpenny NJA, Oksanen KE, Lahtinen MJ, Pajula ET, Iivanainen A, Pesu M, Ramet M. 2012. Mycobacterium marinum causes a latent infection that can be reactivated by gamma irradiation in adult zebrafish. PLoS Pathog. 8:e1002944. 10.1371/journal.ppat.1002944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noga EJ. 1996. Fish disease: diagnosis and treatment. Iowa State University Press, Ames, IA [Google Scholar]
- 14.Wagner D, Young LS. 2004. Nontuberculous mycobacterial infections: a clinical review. Infection 32:257–270 [DOI] [PubMed] [Google Scholar]
- 15.Durborow RM. 1999. Health and safety concerns in fisheries and aquaculture. Occup. Med. 14:373–406 [PubMed] [Google Scholar]
- 16.Finkelstein R, Oren I. 2011. Soft tissue infections caused by marine bacterial pathogens: epidemiology, diagnosis, and management. Curr. Infect. Dis. Rep. 13:470–477 [DOI] [PubMed] [Google Scholar]
- 17.Lehane L, Rawlin GT. 2000. Topically acquired bacterial zoonoses from fish: a review. Med. J. Aust. 173:256–259 [DOI] [PubMed] [Google Scholar]
- 18.Passantino A, Macri D, Coluccio P, Foti F, Marino F. 2008. Importation of mycobacteriosis with ornamental fish: medico-legal implications. Travel Med. Infect. Dis. 6:240–244 [DOI] [PubMed] [Google Scholar]
- 19.Makinen J, Marjamaki M, Marttila H, Soini H. 2006. Evaluation of a novel strip test, GenoType Mycobacterium CM/AS, for species identification of mycobacterial cultures. Clin. Microbiol. Infect. 12:481–483 [DOI] [PubMed] [Google Scholar]
- 20.Tortoli E, Mariottini A, Mazzarelli G. 2003. Evaluation of INNO-LiPA MYCOBACTERIA v2: improved reverse hybridization multiple DNA probe assay for mycobacterial identification. J. Clin. Microbiol. 41:4418–4420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pourahmad F, Thompson KD, Taggart JB, Adams A, Richards RH. 2008. Evaluation of the INNO-LiPA mycobacteria v2 assay for identification of aquatic mycobacteria. J. Fish Dis. 31:931–940 [DOI] [PubMed] [Google Scholar]
- 22.O'Mahony J, Hill C. 2002. A real time PCR assay for the detection and quantitation of Mycobacterium avium subsp paratuberculosis using SYBR Green and the Light Cycler. J. Microbiol. Methods 51:283–293 [DOI] [PubMed] [Google Scholar]
- 23.Parashar D, Chauhan DS, Sharma VD, Katoch VM. 2006. Applications of real-time PCR technology to mycobacterial research. Indian J. Med. Res. 124:385–398 [PubMed] [Google Scholar]
- 24.Castellanos E, Aranaz A, De Buck J. 2010. PCR amplification and high-resolution melting curve analysis as a rapid diagnostic method for genotyping members of the Mycobacterium avium-intracellulare complex. Clin. Microbiol. Infect. 16:1659–1662 [DOI] [PubMed] [Google Scholar]
- 25.Ruskova L, Raclavsky V. 2011. The potential of high resolution melting analysis (HRMA) to streamline, facilitate and enrich routine diagnostics in medical microbiology. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 155:239–252 [DOI] [PubMed] [Google Scholar]
- 26.Šimenc J, Potočnik U. 2011. Rapid differentiation of bacterial species by high resolution melting curve analysis. Prikl. Biokhim. Mikrobiol. 47:283–290 [PubMed] [Google Scholar]
- 27.Reed GH, Kent JO, Wittwer CT. 2007. High-resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics 8:597–608 [DOI] [PubMed] [Google Scholar]
- 28.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 29.Bercovier H, Kafri O, Sela S. 1986. Mycobacteria possess a surprisingly small number of ribosomal-RNA genes in relation to the size of their genome. Biochem. Biophys. Res. Commun. 136:1136–1141 [DOI] [PubMed] [Google Scholar]
- 30.Klappenbach JA, Saxman PR, Cole JR, Schmidt TM. 2001. rrndb: the Ribosomal RNA Operon Copy Number Database. Nucleic Acids Res. 29:181–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwamoto T, Tani K, Nakamura K, Suzuki Y, Kitagawa M, Eguchi M, Nasu M. 2000. Monitoring impact of in situ biostimulation treatment on groundwater bacterial community by DGGE. FEMS Microbiol. Ecol. 32:129–141 [DOI] [PubMed] [Google Scholar]
- 32.Menendez MC, Garcia MJ, Navarro MC, Gonzalez-y-Merchand JA, Rivera-Gutierrez S, Garcia-Sanchez L, Cox RA. 2002. Characterization of an rRNA operon (rrnB) of Mycobacterium fortuitum and other mycobacterial species: implications for the classification of mycobacteria. J. Bacteriol. 184:1078–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zerihun MA, Hjortaas MJ, Falk K, Colquhoun DJ. 2011. Immunohistochemical and Taqman real-time PCR detection of mycobacterial infections in fish. J. Fish Dis. 34:235–246 [DOI] [PubMed] [Google Scholar]
- 34.Salati F, Meloni M, Fenza A, Angelucci G, Colorni A, Orru G. 2010. A sensitive FRET probe assay for the selective detection of Mycobacterium marinum in fish. J. Fish Dis. 33:47–56 [DOI] [PubMed] [Google Scholar]
- 35.Pakarinen J, Nieminen T, Tirkkonen T, Tsitko I, Ali-Vehmas T, Neubauer P, Salkinoja-Salonen MS. 2007. Proliferation of mycobacteria in a piggery environment revealed by mycobacterium-specific real-time quantitative PCR and 16S rRNA sandwich hybridization. Vet. Microbiol. 120:105–112 [DOI] [PubMed] [Google Scholar]
- 36.Kaattari IM, Rhodes MW, Kator H, Kaattari SL. 2005. Comparative analysis of mycobacterial infections in wild striped bass Morone saxatilis from Chesapeake Bay. Dis. Aquat. Org. 67:125–132 [DOI] [PubMed] [Google Scholar]
- 37.Lescenko P, Matlova L, Dvorska L, Bartos M, Vavra O, Navratil S, Novotny L, Pavlik I. 2003. Mycobacterial infection in aquarium fish. Vet. Med. Czech 48:71–78 [Google Scholar]
- 38.Gauthier DT, Rhodes MW, Vogelbein WK, Kator H, Ottinger CA. 2003. Experimental mycobacteriosis in striped bass Morone saxatilis. Dis. Aquat. Org. 54:105–117 [DOI] [PubMed] [Google Scholar]
- 39.Yanong RPE, Pouder DB, Falkinham JO. 2010. Association of mycobacteria in recirculating aquaculture systems and mycobacterial disease in fish. J. Aquat. Anim. Health 22:219–223 [DOI] [PubMed] [Google Scholar]
- 40.Cheng JC, Huang CL, Lin CC, Chen CC, Chang YC, Chang SS, Tseng CP. 2006. Rapid detection and identification of clinically important bacteria by high-resolution melting analysis after broad-range ribosomal RNA real-time PCR. Clin. Chem. 52:1997–2004 [DOI] [PubMed] [Google Scholar]
- 41.Odell ID, Cloud JL, Seipp M, Wittwer CT. 2005. Rapid species identification within the Mycobacterium chelonae-abscessus group by high-resolution melting analysis of hsp65 PCR products. Am. J. Clin. Pathol. 123:96–101 [DOI] [PubMed] [Google Scholar]
- 42.Gundry CN, Vandersteen JG, Reed GH, Pryor RJ, Chen J, Wittwer CT. 2003. Amplicon melting analysis with labeled primers: a closed-tube method for differentiating homozygotes and heterozygotes. Clin. Chem. 49:396–406 [DOI] [PubMed] [Google Scholar]
- 43.El-Hajj HH, Marras SAE, Tyagi S, Shashkina E, Kamboj M, Kiehn TE, Glickman MS, Kramer FR, Alland D. 2009. Use of sloppy molecular beacon probes for identification of mycobacterial species. J. Clin. Microbiol. 47:1190–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]