

Abstract
The genus Aspergillus is a burden to public health due to its ubiquitous presence in the environment, its production of allergens, and wide demographic susceptibility among cystic fibrosis, asthmatic, and immunosuppressed patients. Current methods of detection of Aspergillus colonization and infection rely on lengthy morphological characterization or nonstandardized serological assays that are restricted to identifying a fungal etiology. Collagen-like genes have been shown to exhibit species-specific conservation across the noncollagenous regions as well as strain-specific polymorphism in the collagen-like regions. Here we assess the conserved region of the Aspergillus collagen-like (acl) genes and explore the application of PCR amplicon size-based discrimination among the five most common etiologic species of the Aspergillus genus, including Aspergillus fumigatus, A. flavus, A. nidulans, A. niger, and A. terreus. Genetic polymorphism and phylogenetic analysis of the aclF1 gene were additionally examined among the available strains. Furthermore, the applicability of the PCR-based assay to identification of these five species in cultures derived from sputum and bronchoalveolar fluid from 19 clinical samples was explored. Application of capillary electrophoresis on nanogels was additionally demonstrated to improve the discrimination between Aspergillus species. Overall, this study demonstrated that Aspergillus acl genes could be used as PCR targets to discriminate between clinically relevant Aspergillus species. Future studies aim to utilize the detection of Aspergillus acl genes in PCR and microfluidic applications to determine the sensitivity and specificity for the identification of Aspergillus colonization and invasive aspergillosis in immunocompromised subjects.
INTRODUCTION
Members of the fungal genus Aspergillus are ubiquitous saprophytic environmental fungi (1) that have a variety of industrial applications, including the production of citric acid and amylases, fermentation of soybeans by A. oryzae, and production of lovastatin, a cholesterol-lowering medication, by Aspergillus terreus (2, 3). Other members of the genus, such as A. flavus and A. parasiticus, are common crop contaminants and producers of aflatoxin, a potent human carcinogen and immunosuppressant (4). Aspergillus spp. are also important opportunistic pathogens. Among these, A. fumigatus is the most common etiologic agent of invasive aspergillosis (IA), a disease associated with high rates of mortality (17% to 60%) in severely immunocompromised patients (5–8). As a noninvasive pathogen, A. fumigatus is also associated with allergic sensitization for 6% to 24% of the general population, with A. fumigatus-specific allergies noted in 45% of pediatric asthmatics and 70% in adult asthmatics (9). Aspergillus spp. are also potent inducers of complex hypersensitivities such as allergic bronchopulmonary aspergillosis in up to 15% of cystic fibrosis patients (10, 11), and this group has also been known to cause life-threatening allergic episodes in 12% to 40% of asthmatic patients (10, 12). The ubiquitous nature of Aspergillus conidia may further exacerbate respiratory morbidity, with an average inhalation rate of A. fumigatus spores alone being as high as 104 conidia/m3/day in certain environments (1). The incidence of any of the Aspergillus-related infections in the United States in 2003 was 36 per million, with a mean total hospital charge of $96,731 which ranged as high as $442,233 for the HIV patient subgroup (13). Ironically, A. fumigatus was also identified in December 2012 in a national contamination of triamcinolone, a corticosteroid that has historically been used for management of allergic aspergillosis (14, 15).
A. fumigatus, A. flavus, A. nidulans, A. niger, and A. terreus are among the most common agents of Aspergillus-opportunistic disease in humans (8). However, the identification of these organisms remains challenging. Commonly, laboratories have to rely on morphological analysis of colonies based on viable growth on selective media and the morphology of reproductive structures of the isolated organism. This remains an area of intense research focus, as early and rapid diagnosis is crucial for improved patient survival (16). Recently, PCR-based assays have been shown to have promising sensitivity for Aspergillus infections relative to other biochemical diagnostic methods such as galactomannan (GM) and (1, 3)-β-d-glucan assays (17, 18). The lack of specificity of serologically based assays detecting common fungal cell wall components has additionally remained a significant limitation for the diagnosis of opportunistic Aspergillus infections (19). To date, PCR-based assays for diagnosis of Aspergillus infections have been generally excluded from diagnostic protocols as widely nonstandardized (20); however, PCR sensitivity has been shown to be dependent on the methods of DNA extraction from bronchoalveolar lavage (BAL) and whole-blood samples; extraction methods from specimens have been additionally demonstrated (21).
To date, there are few PCR-based assays that can exercise specificity based on amplicon size. In this study, collagen-like (CL) genes, which commonly harbor conserved, species-specific noncollagenous domains and variable CL regions, have been demonstrated to be practical biomarkers for detection and fingerprinting, respectively, of prokaryotic organisms such as Bacillus spp. through band size discrimination by slab gel and capillary electrophoresis, mass spectrometry, or microchannel fluidics (22–24). The purpose of this study was to evaluate the utility of size-based amplicon discrimination for the species-specific detection of the most common etiologic Aspergillus species, A. fumigatus, A. flavus, A. nidulans, A. niger, and A. terreus, through targeting the Aspergillus collagen-like (acl) genes. Further, an examination of the potential use of the CL region of the aclF1 gene for the purpose of strain fingerprinting was undertaken in addition to explore the utility of nanogel-based capillary electrophoresis for increased sensitivity to amplicon size compared to conventional slab-gel electrophoresis.
MATERIALS AND METHODS
Fungal collections.
Two fungal collections were used in this study. The first collection was obtained from the National Institute of Occupational Safety and Health (NIOSH), Centers for Disease Control and Prevention, Morgantown, WV, and was used to extract genomic DNA representing a broad spectrum of Aspergillus species. This collection included the following strains of species harboring known acl genes: 12 A. fumigatus strains, 4 A. terreus strains, 2 A. flavus strains, 1 A. nidulans strain, and 1 A. niger strain, as well as species lacking evidence of known acl genes, such as A. candidus, A. chevalieri, A. clavatus, A. parasiticus, A. penicillioides, A. sydowii, A. ustus, and two A. versicolor strains (Table 1).
Table 1.
Aspergillus species strain collection
| Fungal species | Abbreviation | Accession no. in other collections | Origina |
|---|---|---|---|
| A. fumigatus | Af293 | AF293, FGSC A1100 | IPA patient |
| Af163b | ATCC 1022, ATCC 4813, BCRC 30502, CBS 133.61, IMI 16152, NCTC 982, NRRL 163, WB 163 | Chicken lung, CT, United States | |
| Af164b | CBS 113.26, IMI 360453, NRRL 164, WB 164 | Soil, Germany | |
| Af165b | NRRL 165 | Pharmaceutical contaminant, France | |
| Af166b | NRRL 166 | Soil, Adirondack Mountains, NY | |
| Af174b | CBS 110.46, IMI 16153, NCTC 5911, NRRL 174/1940, WB 174 | Melanin-deficient mutant from dust, Birmingham, United Kingdom | |
| Af5109b | ATCC 16903, CBS 487.65, IMI 172286, NRRL 5109/A-12321, WB 5109 | Emphysema patient, Chicago, IL | |
| Af5517b | ATCC 22268, NRRL 5517, CBS 158.71 | Soil, USSR | |
| Af5587b | ATCC 36962, CBS 457.75, NRRL 5587, WB 5452 | Soil, Mohanlalganj, India | |
| Af6113b | ATCC 26606, CBS 542.75, NRRL/A-20355 | Sinusitis patient, UCSF Hospital, CA | |
| Af37 | NIOSH 17-30-37 | Dust, Shanghai, China | |
| Af01 | ATCC 13073, NIOSH 35-11-01 | Human pulmonary lesion, MA, United States | |
| A. candidus | CAN | NIOSH 17-28-24 | Dust, Shanghai, China |
| A. chevalieri | CHV | ATCC 16443, CBS 522.65, IMI 211382, NRRL 78 | Beans |
| A. clavatus | CLAVA | NIOSH 06-22-78 | |
| A. flavus | FL07 | NIOSH 15-41-07 | |
| FL86 | PRC-86a | Oats, Courtland, AL, United States | |
| A. nidulans | NID | NIOSH 15-22-08 | Cooperstown, NY, United States |
| A. niger | NIG | ATCC 9029, BCRC 32720, CBS 120-49, FGSC A1143, IMI 041876, NRRL 3/566, WB 3/566 | United States |
| A. parasiticus | PAR | ATCC 26691, BCRC 30164/31490, CBS 100309/921.70, IMI 091019/283883, NRRL 2999 | |
| A. penicilloides | PEN | ATCC 16910, BCRC 33421CBS 540.65, IMI 211342211392, NRRL 4548, WB 4548 | Skin of patient with lobomycosis, Brazil |
| A. sydowii | SYD | ATCC 9507 | |
| A. terreus | T04 | ATCC 1012/10071, BCRC 32068, CBS 601.65/601.956011.65, IMI 017294017294iiNBRC 33026, NCTC 981, NRRL 225/255/543, WB 255 | Soil, CT, United States |
| T05 | ATCC 16794, CBS 594.65, IMI 135817, NRRL 680, WB 680 | Soil | |
| T06 | SRC 2174 | ||
| T31 | NIOSH 17-30-31 | Dust, Shanghai, China | |
| A. ustus | UST | ATCC 104116818, BCRC 30198, CBS 261.67, IMI 211805, NRRL 275/1734/A-969, QM 7477, WB 275 | Contaminant from A. Sydowii culture, United States |
| A. versicolor | VSC3 | ATCC 44408 | Cheese |
| A. versicolor | VSC6 | NIOSH 32-46-03 | New Orleans, LA, United States |
IPA, invasive pulmonary aspergillosis, UCSF, University of California, San Francisco; USSR, Union of Soviet Socialist Republics.
Reference isolates were obtained from the USDA ARS Culture Collection (NRRL).
Aspergillus strains isolated from clinical specimens and banked by the clinical laboratory at West Virginia University Healthcare (WVUH) between October 2011 and November 2012 were also utilized (and are referred to here as “clinical isolates”). These isolates were deidentified of all patient information by the clinical laboratory prior to analysis, rendering the study “Not Human Subjects Research” as determined by the Institutional Review Board. The 19 isolates were obtained predominately from respiratory sources (cystic fibrosis sputa, 11; other sputa, 3; bronchoalveolar lavage, 4), and one was obtained from a skin biopsy specimen. Four isolates were referred to the clinical laboratory by outside facilities for purpose of identification, while the remaining 15 were obtained from local patients. Each isolate was recovered on Sabouraud dextrose agar incubated at 26°C to 30°C. Routine identification to the species level was achieved using standard phenotypic characteristics, including (i) colony morphology, (ii) absence of growth on media containing cycloheximide, (iii) phialide configuration and distribution along the vesicle, conidium size and morphology, and presence of cleistothecia or Hülle cells discerned by the use of cellophane tape mounts, and (iv) growth at 45°C. All isolates were prospectively stocked at −80°C per laboratory protocol using M17 broth (Oxoid, Basingstoke, England) supplemented with 20% glycerol. For isolates with discordant phenotypic identification and acl PCR assay results, organisms were propagated from stocks, subcultured to M17 broth (Oxoid, Basingstoke, England), and incubated in ambient air at 37°C for 14 to 21 days with daily shaking to disrupt aerial mycelia along the liquid-air interface. DNA was extracted by the phenol-chloroform method for downstream analyses.
Bioinformatic analyses.
Searches for Aspergillus collagen-like proteins (CLPs) (Acl) were performed among the sequences of the collagen family (PF013910) of the Sanger Institute's Pfam Protein Families Database (Table 2) (25). Subsequent sequence searches were carried out using PSI-BLAST (26) and the NCBI nonredundant database. The presence of a signal peptide for Acl proteins was predicted with the SignalP 4.1 webserver provided by the Center for Biological Sequence Analysis at the Technical University of Denmark (27), and glycosylphosphatidylinositol (GPI)-anchor prediction was performed using the Institute of Molecular Pathology's GPI Prediction Server (version 3.0) (28). The DNA and protein sequence data were analyzed with the Lasergene v.10 software suite (DNASTAR, Inc., Madison, WI).
Table 2.
Basic characteristics of the putative Acl proteins
| Species | Pfam designation | Acl proteinb |
|||||
|---|---|---|---|---|---|---|---|
| Name | Length (aa) | SSa | GPI | CL region |
|||
| GXY type | Location (aa) | ||||||
| A. fumigatus | |||||||
| 293 | Q4WBU6_ASPFU | AclF1 | 359 | 1–15 | GPy | 168–244 | |
| Q4WW98_ASPFU | AclF2 | 143 | GQy | 11–106 | |||
| CBS 144.89 | B0Y9H1_ASPFC | AclF1 | 343 | 1–15 | GPy | 167–257 | |
| B0Y8Y8_ASPFC | AclF2 | 125 | GQy | 11–88 | |||
| A. terreus | |||||||
| NIH 2624 | Q0CXL1_ASPTN | AclT1 | 358 | 336 | GAP | 179–262 | |
| NIH 2624 | Q0CUF0_ASPTN | AclT2 | 265 | 1–17 | GHy | 68–196 | |
| A. flavus | |||||||
| ATCC 200026 | B8NLK1_ASPFN | AclFL1 | 554 | 1–23 | GTP | 277–366 | |
| A. nidulans | |||||||
| NCBI 162425 | Q5B8U8_EMENI | AclN1 | 432 | 1–17 | 411 | GQP.GQS | 258–315 |
| A. oryzae | |||||||
| NCBI 5062 | Q2TZV0_ASPOR | AclO1 | 114 | GLy | 25–110 | ||
| A. kawachii | |||||||
| NBRC 4308 | G7XZR3_ASPKW | AclK1 | 239 | 1–19 | GNS.GNR | 179–220 | |
| A. niger | |||||||
| ATCC 1015 | G3YG25_ASPNA | AclNi1 | 556 | GAT.GKK | 484–528 | ||
SS, signal sequence.
aa, amino acids. GPI, residue number of predicted cleavage site.
Molecular phylogenetic analyses.
Maximum-parsimony (MP) and Bayesian analyses were performed with A. fumigatus aclF1 sequences derived from clinical and environmental samples. The aclF1 nucleotide sequences were aligned using MUSCLE (29) and visually verified. The MP analysis was performed in PAUP 4.0 (30) with 1,000 nonparametric bootstrap replicates to measure lineage support. MP heuristic search settings included creating starting trees by stepwise additions, subsequent branch swapping with the tree-bisection-reconnection (TBR) algorithm, and 200 Max trees. The MP analysis was performed twice, with gaps treated both as “missing data” and as a “fifth character state,” and no differences were observed in the resulting phylogenies.
Bayesian analysis was performed using MrBayes 3.2 (31), and posterior probabilities (PP) were calculated as a measure of lineage support. The evolutionary model implemented was the F81 model (32) and allowed for a proportion of invariable sites (F81+I), which was determined to be the best fit model by the Akaike Information Criterion in MrModelTest version 2.3 (33). The Markov chain Monte Carlo parameters were set to 1,000,000 generations and 6 simultaneous chains. Burn-in and stabilization of model parameters occurred at 800,000 generations, and every 100th tree after burn-in was used to generate a 50% majority rule consensus phylogeny. All trees were constructed in FigTree v 1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
DNA methods. (i) Extraction of genomic DNA.
DNA was isolated using phenol-chloroform extraction and an ethanol precipitation method, as previously described (34). To extract DNA, 0.1 g of mycelium was harvested with a 70-μm-pore-size nylon mesh filter (BD Biosciences, Bedford, MA) and placed into a 2-ml BioSpec bead-beater tube containing 0.5-mm-diameter soda lime glass beads (BioSpec Products Inc., Bartlesville, OK). Next, 0.2 ml DNA extraction buffer (0.2 M Tris-HCl [pH 7.5], 0.5 M NaCl, 0.01 M EDTA, 1% sodium dodecyl sulfate solution) and 0.2 ml phenol-chloroform-isoamyl alcohol (25:24:1) solution were added to the tube and the mix was bead-beaten for 1 min. An additional 0.3 ml of DNA extraction buffer and 0.3 ml of phenol-chloroform-isoamyl alcohol were added to the tube and vortexed. The phases were separated by centrifugation at 17,000 × g for 1 min. The aqueous phase was transferred to a new 1.5-ml microcentrifuge tube and extracted again with an equal volume of the phenol-chloroform-isoamyl alcohol. The entire sample was transferred to a 2.0-ml Phase Lock Gel tube (5PRIME, Gaithersburg, MD), mixed, and centrifuged at 15,000 × g for 5 min. After centrifugation, the gel-separated aqueous phase was transferred to a new microcentrifuge tube and DNA was precipitated with ethanol overnight at −20°C. Following centrifugation at 17,000 × g for 5 min, the ethanol was discarded, and the DNA pellet was washed with 70% ethanol followed by centrifugation as described above. The DNA pellet was air-dried and dissolved in 200 μl of double-distilled water (ddH2O); DNA concentrations were measured spectrophotometrically using a Spectramax 190 (Molecular Devices, Sunnyvale, CA), and DNA concentrations were diluted to ∼30 ng/μl.
Crude DNA extraction was performed by boiling mycelia harvested from Sabouraud dextrose agar cultures of 19 clinical specimens (with identification numbers assigned in a blind manner) in 200 μl of PrepMan Ultra sample preparation reagent (Applied Biosystems, Foster City, CA) for 10 min. The samples were pelleted at 20,000 × g for 5 min, and the supernatant was transferred to a fresh microcentrifuge tube and stored at 4°C until further analysis. Similarly, the DNA concentration was measured spectrophotometrically as described above and standardized to a concentration of ∼30 ng/μl.
(ii) Primer design for acl gene detection of Aspergillus spp.
Primers were designed based on the genomic acl gene sequences from the strains of sequenced Aspergillus species deposited in the NCBI GenBank database (Table 3). A. fumigatus-specific detection primers targeted both the 5′ and 3′ hypothetically conserved regions of the aclF1 allele present in the complete genomic sequence of A. fumigatus clinical isolate Af293 (culture identification no. [ID] ATC MYA-4609). Similarly, primer pairs were designed targeting the aclT2-5′ region of A. terreus NIH 2624, the aclFL-3′ region of A. flavus ATCC 200026, the aclN1-3′ region of A. nidulans NCBI 162425, and the aclNi1-5′ region of A. niger ATCC 1015. The predicted sizes of the amplicons ranged between 227 and 489 bp. Additional primers flanking the collagen-like region of aclF1 (aclF1-CL) were designed to assess the CL-region length polymorphism, and the aclF1 gene primers amplified the entire gene for sequencing purposes.
Table 3.
Primers used for amplification of the acl genes
| Species | Amplicon | Length (bp) | Primer name | Sequence (5′–3′) |
|---|---|---|---|---|
| A. fumigatus | aclF1-5′ | 489 | AclF1_ATGF2a,b | ATGCTCCTCCTACCACTCCTTGCC |
| AclF1_NtermR2 | GTTTACACATCCTTGGTAGTGC | |||
| aclF1-3′ | 338 | AclF1_CtermF2b | GCAATCATGGGTGCAATAAACC | |
| AclF1_CtermR1 | CTGCCCCGAGGGATTCTT | |||
| aclF1-CL | 327 | AclF1_VarF3 | AGTGGCTACCCGGTCCACG | |
| AclF1_VarR5b | TTCGTCGGTGTACTTCGGACCAT | |||
| aclF1a | 1,080 | AclF1_ATGF2a | ATGCTCCTCCTACCACTCCTTGCC | |
| AclF1_TGAR2a | TCAAGCAACCCCAATCCCCTGCC | |||
| AclF1_Nterm_R1b | GGATCGTGTTATGCGGGTAG | |||
| A. flavus | aclFL-3′ | 403 | AclFL1_CF2 | CCCTCCGATTGGTATTACCACC |
| AclFL1_CR1 | CATTCCTCATCTTCGTCCTGATC | |||
| A. terreus | aclT2-5′ | 262 | AclT2_NtermF1 | ATGAAGATCCCCGTCGTCGTTG |
| AclT2_NtermR1 | CCTTCATGCTCTTTACCGGGG | |||
| A. nidulans | aclN1-3′ | 227 | AclN1_CtermF1 | CTCCTAGCGTTCCTACTGCTCCT |
| AclN1_CtermR1 | CTAGAGGAGAGCCATGATTGTC | |||
| A. niger | aclNi-5′ | 297 | AclNi1_IntrINTF1 | ACCATCCTCCAAAGCACCC |
| AclNi1_IntrINTR1 | GCCAGCGCAGCCGTATCC |
Primer used for amplification of the whole aclF1 gene.
Primer used for sequencing.
(iii) PCR amplification.
The amplification conditions for all detection primer pairs were tested by the use of a temperature gradient of 55°C to 65°C; a uniform annealing temperature was established at 61°C. The PCR mix contained a 0.2 mM concentration of each primer, 0.2 mM deoxynucleoside triphosphate (dNTPs) in a 1× PCR buffer (10 mM Tris-HCl, 1.5 mM MgCl2, 50 mM KCl; pH 8.3). For the two-amplicon detection of A. fumigatus involving two-plex PCR of the aclF1-5′ and aclF1-3′ regions, the primer pairs were used at 0.23 mM and 0.20 mM concentrations, respectively, using Q5 High-Fidelity DNA polymerase (New England BioLabs Inc., Boston, MA). PCR protocols for amplification of the whole aclF1 gene, as well as the collagen-like region, required optimization by temperature gradient and the addition of 1.0 M betaine (Sigma-Aldrich, St. Louis, MO).
The amplification was performed in a Bio-Rad S1000 Thermal Cycler as follows: initial denaturation at 94°C for 3 min, followed by 32 cycles of denaturation at 94°C for 30 s, annealing at 61°C for 30 s, and elongation at 72°C for 30 s, with a final elongation step at 72°C for 3 min. For two-plex PCR amplification, the initial denaturation step was at 98°C for 30 s, followed by 32 cycles of 10 s of denaturation at 98°C, 20 s of annealing at 61°C, and 10 s of elongation at 72°C, ending with a final 2-min elongation step. The PCR products were analyzed on a 2% agarose gel. Electrophoresis was performed in 1× Tris-acetate-EDTA buffer at 120 mV. The DNA standard used was a TrackIt 1-kb Plus DNA ladder (Invitrogen, Carlsbad, CA). Images were captured using Eagle Eye II (Stratagene, La Jolla, CA) and the FOTO/Analyst Investigator/Eclipse gel documentation workstation (Fotodyne, Hartland, WI) and processed with Adobe Design Standard Creative Suite 6 (Adobe Systems Inc. San Jose, CA).
(iv) Capillary electrophoresis.
The following reagents were used for capillary electrophoresis: phospholipids dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) (Avanti Polar Lipids, Alabaster, AL), 3-(N-morpholino)-propanesulfonic acid (MOPS) (Alfa Aesar, Ward Hill, MA), and SYBR green 1 (Life Technologies, Grand Island, NY). Deionized water was obtained using an Elga PureLab Ultra water filtration system (Lowell, MA). An aqueous solution of 100 mM MOPS buffered to pH 7 was used for the phospholipid preparation and was the solution used in the anodic and cathodic reservoirs. The phospholipid additive was prepared as described previously (35) to obtain molar ratios of [DMPC]/[DHPC] = 0.5 at 5% (wt/vol) phospholipid (95% weight phospholipid/volume of aqueous MOPS buffer) as well as [DMPC]/[DHPC] = 2.5 at 10%. A preparation of intercalating dye was made from a 50-μl volume of 1× SYBR green 1 diluted in nanogel. The 50-bp DNA ladder (New England BioLabs, Ipswich, MA) was used as a size marker.
Separations were performed on a Beckman Coulter P/ACE MDQ system with a laser induced-fluorescence detection module equipped with a 3 mW air-cooled argon ion laser (λex = 488 nm and λem = 520 nm). The fused silica capillary (Polymicro Technologies, Phoenix, AZ) had an outer diameter of 360 μm and an inner diameter of 25 μm. Prior to separation, the capillary was prepared as previously reported (36). Briefly, the capillary was rinsed with 1 N NaOH (140 kPa for 30 min), deionized water (140 kPa for 15 min), methanol (140 kPa for 15 min), and deionized water (140 kPa for 15 min). Following this rinse protocol, the inner surface of the capillary was passivated in order to suppress electro-osmotic flow, using a previously characterized method based on phospholipids (37). This was accomplished by a coating procedure performed twice at 140 kPa with a semipermanent coating consisting of 5% phospholipid of [DMPC]/[DHPC] = 0.5 containing 1.25 mM CaCl2 for 20 min followed by a 2-min flush with 100 mM MOPS buffered to pH 7. The capillary was then filled at an ambient temperature of 19°C with a 10% solution of [DMPC]/[DHPC] = 2.5. Once the capillary was filled with the phospholipid nanogel, the capillary temperature was increased to 30°C. After each separation procedure, the capillary was flushed at 140 kPa for 3 min with 5% [DMPC]/[DHPC] = 0.5, for 2 min with MOPS (pH 7), and for 3 min with [DMPC]/[DHPC] = 2.5. The sample was injected as described previously (38) by first introducing a preplug of run buffer (6.9 kPa for 7 s), performing a reverse-polarity electrokinetic injection of the DNA, and finally providing a postplug of run buffer (3.4 kPa for 5 s). Separations were achieved with reverse polarity. Data collection and analysis were performed using 32 Karat Software version 5.0 (Beckman Coulter).
(v) DNA sequencing.
For aclF1 gene sequencing, a primer pair was designed that encompassed the entire gene (Table 3). Hypothetically, the primer pair should produce an amplicon of 1,080 bp in A. fumigatus strain Af293. Amplification conditions were set at an initial denaturation step at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C, annealing at 61°C for 1 min, and extension at 72°C for 1 min 30 s, followed by a final extension step at 72°C for 10 min. In addition, the PCR cocktail contained a 0.20 mM concentration solution for each primer, a 0.20 mM dNTP solution, and a 0.83 M betaine solution in a 1× PCR buffer. DNA templates were used from all 12 available A. fumigatus strains, and amplification products were purified using a Millipore Amicon Ultra 50K cartridge before sequencing; sequences were assembled using the Lasergene Genomics Suite (DNA Star, Madison, WI) and analyzed to assess the natural aclF1 gene polymorphism among A. fumigatus strains.
Sequencing with universal primers of an internal transcribed spacer region with ITS-1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS-2 (5′-GCTGCGTTCTTCATCGATGC-3′) and/or of a partial 28S rRNA gene with NL-1 (5′-GCATATCAATAAGCGGAGGAAAAG-3′) and NL-4 (5′-GGTCCGTGTTTCAAGACGG-3′) was performed using standard protocols (39–41). Sequencing results were queried through the NCBI BLAST to establish best-fit identities.
Nucleotide sequence accession numbers.
The aclF1 sequences of A. fumigatus (Af) reported here have been deposited in GenBank under the following accession numbers: Af6113, KF704160; Af5587, KF704161; Af5517, KF704162; Af5109, KF704163; Af174, KF704164; Af166, KF704165; Af165, KF704166; Af01, KF704167; Af164, KF704168; Af163, KF704169; Af37, KF704170; Af (clinical) sample no. 3, KF704171; Af sample no. 5, KF704172; Af sample no. 8, KF704173; Af sample no. 9, KF704174; Af sample no. 10, KF704175; Af sample no. 11, KF704176; Af sample no. 12, KF704177; Af sample no. 13, KF704178; Af sample no. 15, KF704179; Af sample no. 17, KF704180; Af sample no. 18, KF704181; Af sample no. 19, KF704182. The acl/Acl gene/protein sequences previously deposited by third parties that were used here can be retrieved using the information reported in Table 2 or directly via hyperlinks included in Fig. S1 in the supplemental material.
RESULTS
Identification and characterization of Aspergillus collagen-like proteins.
Recent studies demonstrated that collagen-like genes can be effectively used for the detection and fingerprinting of Bacillus cereus group organisms (22–24). Therefore, the Pfam collagen database (http://pfam.sanger.ac.uk/family?PF01391) was queried in order to assess the distribution of the collagen-like proteins (CLPs) among Aspergillus species. A total of 62 sequences were annotated among 44 fungi (see Fig. S1 in the supplemental material). Nine Aspergillus collagen-like proteins, designated Acl, were identified that were harbored by 7 species within the mitosporic Trichocomaceae subfamily (Fig. 1); only one additional CLP was annotated within the order Eurotiales, outside Trichocomaceae, found in Talaromyces stipitatus. Phylogenetic analyses of all 62 fungal collagen-like proteins were performed (data not shown). However, the amino acid sequences were too divergent for MP analysis to determine relatedness within these samples. Additionally, Bayesian analysis, performed using 3,000,000 generations, did not reach a stationary posterior probability distribution (split frequencies P = 0.2), indicating that a model-based phylogeny could not be resolved. The large variation among collagen-like proteins provides evidence that sequences of the Aspergillus Acl proteins are unique to this subfamily. These included two proteins (AclF1 and AclF2) of A. fumigatus, two proteins (AclT1 and AclT2) of A. terreus, and one protein each in species of A. flavus (AclFL1), A. nidulans (AclN1), A. niger (AclNi1), A. oryzae (AclO1), and A. kawachii (AclK1). Although the number of deposited sequences of each Acl protein was limited to one or two strains, these results positively identified the presence of the potential acl gene targets in all major Aspergillus species that are responsible for human infections, especially A. fumigatus.
Fig 1.
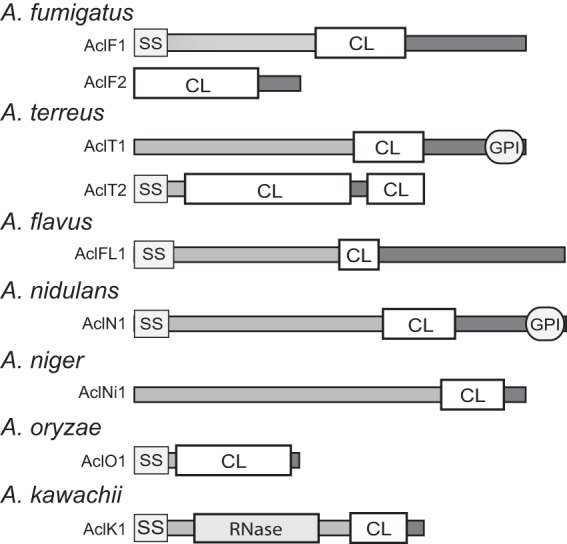
Identification and characterization of the Acl proteins among members of the Trichocomaceae family. The general architecture of nine Acl proteins (not to scale) found in seven different Aspergillus species is shown. Main Acl-protein regions are designated as follows: SS, signal sequence; CL, collagen-like; GPI, cell wall anchor motif; RNase, RNase domain.
The basic features of the Acl proteins were rendered from deposited sequences and indicated their variable characteristics (Fig. 1 and Table 2). Thus, in addition to the presence of a common collagen-like region in all nine Acls, candidates from six species had predicted signal peptides (AclF1, AclT2, AclFL1, AclN1, and AclK1), suggesting possible extracellular localization. Preliminary Western blot experiments using rabbit polyclonal antibodies raised against a synthetic peptide located within a C-terminal region of the AclF1 protein of A. fumigatus identified an immunoreactive band of the predicted size in the hyphal protein fraction but not in conidial extracts or the culture supernatant fraction (data not shown). Two proteins (AclT1 and AclN1) also contained the GPI anchor, which suggested that they were surface attached. As expected, the CL regions of the Acl proteins differed in length, depending on the number of GXY-triplet repeats containing between 13× GXY in AclFL1 and 42× GXY in AclT2 protein. The type of triplet repeats used also differed between different Acl-CL regions, although they consisted of relatively few distinct triplets. For example, AclT1-CL consisted solely of GAP triplets and AclFL1-CL of GTP repeats. In two proteins, AclF1 and AclT2, two-residue interruptions, GV and GH, respectively, were embedded within their CL sequences. Only one protein, AclK1, was predicted to harbor a known RNase noncollagenous domain. Altogether, the presumed Acl proteins share common general architecture but their acl-encoding sequences differ significantly, thus allowing for specific targeting by PCR.
acl-based detection of A. fumigatus.
Since A. fumigatus species are by far the most frequent causes of opportunistic Aspergillus infections, the initial focus was on designing PCR primers that would specifically target and amplify DNA fragments of the aclF1 and aclF2 genes. Several primer pairs were designed based on gene sequences found in the sequenced strain Af293 (FGSC A1100) and were subsequently tested using the collection of 11 more A. fumigatus strains, as well as 17 non-A. fumigatus controls (Fig. 2A). The aclF2 gene target failed to produce consistent and reliable results and was not considered for further analysis (data not shown). In contrast, two aclF1-based primer pairs demonstrated consistent amplicons from all 12 A. fumigatus templates but not from non-A. fumigatus controls (Fig. 2A). The observed sizes of the amplicons were the same in all strains based upon separation in a 2% agarose gel. Amplicon sizes of 489 bp (aclF1-5′) and 338 bp (aclF1-3′) were observed for all Af strains (Fig. 2A), which was in agreement with the expected sequence lengths based on the Af293 strain. These amplicons were also produced in a single two-plex reaction (Fig. 2B; see also Fig. S2 in the supplemental material) and separated by standard gel electrophoresis and by capillary electrophoresis (Fig. 2B and C). These results suggested that conserved regions of the aclF1 gene had been targeted, allowing for specific and rapid detection of the A. fumigatus DNA.
Fig 2.

PCR detection of A. fumigatus. (A) PCR amplicons of the aclF1-5′ and aclF1-3′ conserved regions were analyzed by 2% agarose gel electrophoresis. Total genomic DNA templates were used from 12 A. fumigatus strains and 17 non-A. fumigatus controls; strain abbreviations are shown in Table 1. M, 1-kb Plus TrackIt DNA ladder. (B) Two-plex PCR of the aclF1-5′ and -3′ regions. Results of amplification with the A. fumigatus Af293 strain are shown in the first lane. In addition, control genomic DNA from the non-A. fumigatus species was used to demonstrate specificity of two-plex PCR. Genomic DNA templates were standardized to approximately 30 ng/μl. PCR amplification was resolved by 2% agarose gel electrophoresis. M, 1-kb Plus TrackIt DNA ladder. (C) Nanogel electrophoretic separations of two-plex PCR. The DNA base ladder (top trace) and PCR-amplified markers of A. fumigatus (bottom trace) detected with the intercalating dye SYBR green 1 are shown. The separation is accomplished using a 25-μm-inner-diameter capillary with an effective length of 40.2 cm, E = 100 V/cm, 30°C, and a 10% nanogel with [DMPC]/[DHPC] = 2.5 at 6 kV for a 2-s injection.
aclF1 sequence polymorphism.
Since initial sequence data were limited to two strains of A. fumigatus, Af293 and Af1163 (Table 2), the aclF1 gene was sequenced in the remaining additional 11 strains available in our collection that represented both clinical and environmental samples collected within and outside the United States (Table 1). First, length polymorphism of the aclF1-CL region among 12 A. fumigatus strains was assessed by PCR using AclF1_VarF3 and VarR5 primers (Fig. 3A). Only strain Af6113 yielded an obviously smaller DNA fragment, while the remaining 11 products, including Af293, seemed to produce the same size amplicons. Subsequently, the entire aclF1 gene was sequenced for those strains to deduce the natural DNA and protein sequence polymorphisms.
Fig 3.
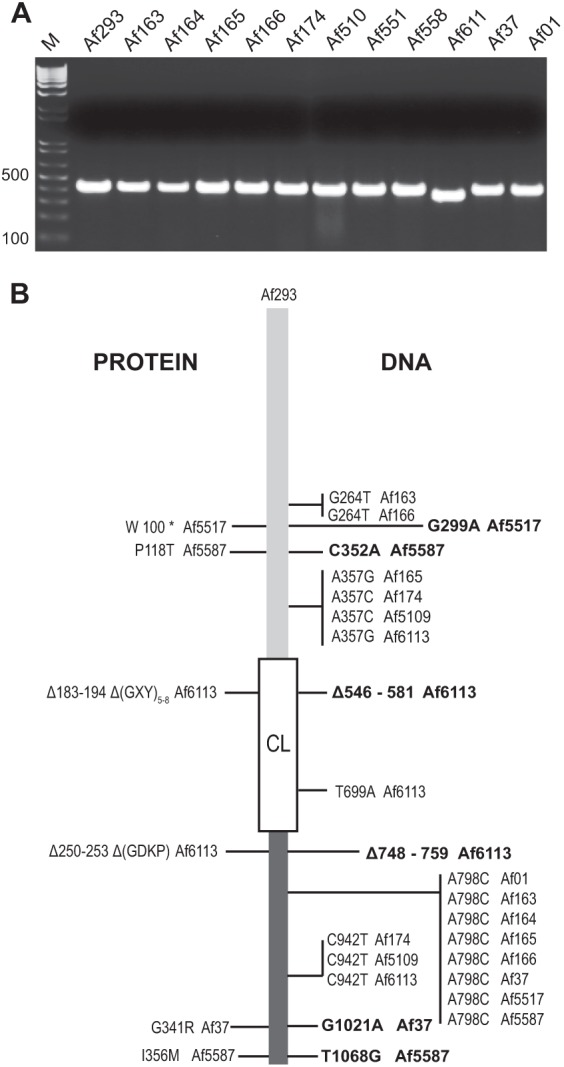
Allelic polymorphism in aclF1 gene and AclF1 protein. (A) Length polymorphism of the aclF1-collagen-like region. PCR amplicons were generated with the aclF1-CL primers (Table 1) flanking the CL region of the aclF1 gene in 12 A. fumigatus laboratory strains and resolved in a 2% agarose gel. M, 1-kb Plus TrackIt DNA ladder. (B) aclF1/AclF1 gene/protein sequence polymorphism among A. fumigatus strains compared to aclF1 in the sequenced Af293 strain. A summary of nucleotide and amino acid polymorphisms is shown; nonsynonymous polymorphisms are indicated in bold. The asterisk (*) indicates a stop codon.
The sequencing data revealed 11 polymorphic sites resulting in 9 different aclF1 alleles, including the allele represented by strain Af293 (Fig. 3B). In addition to 9 single nucleotide polymorphism (SNP) changes, polymorphisms included two deletions: three GXY repeats in the CL region and four amino acid residues (GDKP) in a downstream region. Both deletions were present in one strain, Af6113, thereby verifying the basis for the smaller amplicon previously detected by PCR. Strain Af6113 exhibited the greatest sequence polymorphism among the tested strains, with three SNPs and two deletions. Of the 11 polymorphisms found, 6 were nonsynonymous, resulting in 5 protein variants: the AclF1 variant in strains Af293, Af6113 (Δ183–194, Δ250–253), Af5587 (P118T, I356M), and Af37 (G341R) and one null mutation in strain Af5517 (W100*). These sequence data identified the conserved regions of aclF1 gene and validated the location of our detection primers.
Detection of acl genes in other Aspergillus species.
Inasmuch as A. fumigatus is the predominant cause of IA among immunocompromised patients, other Aspergillus species are also etiological agents of IA and may additionally elicit allergic sensitization (16, 42). Therefore, acl-based primers were designed to specifically target the aclFL1 gene of A. flavus, aclN1 of A. nidulans, aclNi1 of A. niger, and aclT2 of A. terreus (Fig. 4A, top to bottom). The selected primer pairs all produced amplicons of the predicted sizes for their corresponding species, suggesting that conserved sequences or at least regions without clear length polymorphism were targeted. The observed sizes of the amplicons were approximately 403 bp for the aclFL1-3′, 227 bp for the aclN1-3′, 297 bp for the aclNi1-5′, and 262 bp for the aclT2-5′ amplicons. All PCRs were performed under the same amplification conditions as those described above for A. fumigatus (Fig. 2). None of the primer pairs produced detectable amplicons with any A. fumigatus template, and they detected the acl genes only in targeted species, with the sole exception of A. parasiticus, which consistently yielded a positive PCR band with aclFL1-3′ primers designed for A. flavus. Partial sequencing of PCR products confirmed the presence of an acl allele encoding an Acl variant in A. parasiticus, which was similar to AclFL1 of A. flavus. In aggregate, four additional acl targets for the specific detection of four additional species of clinical importance were successfully developed.
Fig 4.

acl-based detection of non-A. fumigatus species. (A) PCR amplification and gel electrophoresis of various acl genes among non-A. fumigatus species. The species (from top to bottom) and assays are shown as follows: A. flavus (amplification of the 403-bp aclFl1-3′ region; A. flavus-specific amplicons are marked with arrowheads, and an amplicon detected in A. parasiticus is marked with an asterisk); A. nidulans (amplification of the 227-bp aclN1-3′ region); A. niger (amplification of the 297-bp aclNi1-5′ region); and A. terreus (amplification of the 262-bp aclT2-5′ conserved region). PCR amplification was resolved by 2% agarose gel electrophoresis. M, 1-kb Plus TrackIt DNA ladder. (B) Nanogel electrophoretic separation. PCR amplicons specific for A. fumigatus, A. flavus, A. nidulans, A. niger, and A. terreus were simultaneously resolved and detected with the intercalating dye SYBR green 1. The separation is accomplished using a 25-μm-inner-diameter capillary with an effective length of 40.2 cm, E = 100 V/cm, 30°C, and a 10% nanogel with [DMPC]/[DHPC] = 2.5 at 6 kV for a 2-s injection. RFU, relative fluorescence units.
Analysis by capillary electrophoresis using phospholipid nanogels.
Capillary nanogel electrophoresis was used to estimate the size of acl-based PCR products. Figure 2C demonstrates the separation of the acF1-5′ and aclF1-3′ two-plex amplicons from A. fumigatus, whereas the electropherogram shown in Fig. 4B demonstrates a separation of multiple amplicons, including the aclFL1-3′ amplicon from A. flavus, the aclN1-3′ amplicon from A. nidulans, the aclNi1-5′ amplicon from A. niger, and the aclT2-5′ amplicon from A. terreus in addition to the two-plex aclF1-5′ and -3′ amplicons of A. fumigatus. Using internal size standards of 150, 250, and 350 bp, the sizes of the amplicons were estimated to be 226 ± 1 bp (aclN1-3′), 259 ± 2 bp (aclT2-5′), 296 ± 1 bp (aclNi1-5′), 336 ± 2 bp (aclF1-3′), 396 ± 3 bp (aclFL1-3′), and 482 ± 6 bp (acF1-5′) for n = 5. Hence, nanogel separation can provide an accurate size evaluation of all acl targets with great certainty.
Detection of clinical isolates: proof of principle.
The acl-based PCR assays were tested by screening 19 clinical isolates with confirmed Aspergillus morphology. Crude DNA preparations obtained in the clinical microbiology laboratory directly from mycelia on Sabouraud dextrose agar were arbitrarily numbered 1 to 19 and assayed in separate PCRs for each acl target, including the two-plex PCR for A. fumigatus, along with corresponding positive controls. PCRs were performed under single amplification conditions and analyzed in 2% agarose gel (Fig. 5).
Fig 5.

acl-based detection of Aspergillus species in clinical samples. PCR-based detection of (from top to bottom) A. fumigatus, A. flavus, A. nidulans, A. niger, and A. terreus is shown, representing bronchoalveolar lavage cultures from 19 different clinical samples. Control PCR results for each corresponding species are shown to the right of lanes M. Top labels, species identified based on positive amplification with species-specific amplicons; bottom labels, sample numbers (1 to 19) provided for the specimens in a blind manner. PCR amplification was resolved by 2% agarose gel electrophoresis. M, 1-kb Plus DNA ladder.
Samples 3, 5, 8, 9, 10, 11, 12, 13, 15, 17, 18, and 19 yielded dual amplicons (aclF1-5′ and aclF1-3′), signifying identification of A. fumigatus. Sample 6 was positive for aclN1-3′ of A. nidulans. Samples 4 and 14 yielded strong amplification bands of equal sizes with primers targeting the aclNi1-5′ region of A. niger; interestingly, sample 3 also yielded weak amplification with the A. niger-specific primers, suggesting a mixed culture with A. fumigatus. Samples 1, 2, and 7 yielded the same amplicons with A. terreus-specific primers targeting the aclT2-5′ region, though samples 1 and 2 were weaker. Surprisingly, none of the samples were amplified for the aclFL1-5′ target of A. flavus, which is known to be responsible for a significant number of infections (43). Finally, sample 16 was PCR negative with all primer pairs.
To confirm the identity of amplicons of all non-A. fumigatus samples, organisms were cultured from stocks for DNA isolation. Samples were then amplified using the 18S-internal transcribed spacer (ITS) region and the partial nuclear large (NL) subunit 28S rRNA gene primers, as described elsewhere (39, 41). Sequencing of all acl-positive amplicons from these samples was also performed. All sequence data were analyzed using NCBI BLAST. In general, results agreed well with clinical identifications of the specimens. Sample 3 was originally identified as A. fumigatus, and yet growth on subculture was contaminated with A. niger. Both organisms were accurately detected by acl PCR, and each was subsequently reisolated in pure culture and confirmed by the corresponding acl-based PCR. Interestingly, sample 16 was negative for all acl targets and was identified in the clinical laboratory as A. fumigatus. Repeat culture from stock, with DNA purification by the phenol-chloroform extraction method and subsequent retesting with all acl primer pairs, again failed to yield any acl target. Ultimately, sample 16 was identified based on ITS and NL data as Neosartorya pseudofischeri (the telomorph of Aspergillus thermomutatus).
aclF1 sequence polymorphism in clinical isolates of A. fumigatus.
First, we performed the PCR using AclF1_VarF3 and VarR5 primers in order to assess aclF1-CL length polymorphism among A. fumigatus clinical strains (Fig. 6A) and to compare it with the polymorphism previously found in laboratory strains (Fig. 3A). Compared with Af293 aclF1-CL, samples 3, 5, 8, 9, 10, 11, 17, and 18 produced shorter amplicons, potentially similar to the amplicon found in Af6113. Subsequently, the entire aclF1 gene was sequenced in all clinical strains to deduce the natural DNA and protein sequence polymorphisms.
Fig 6.
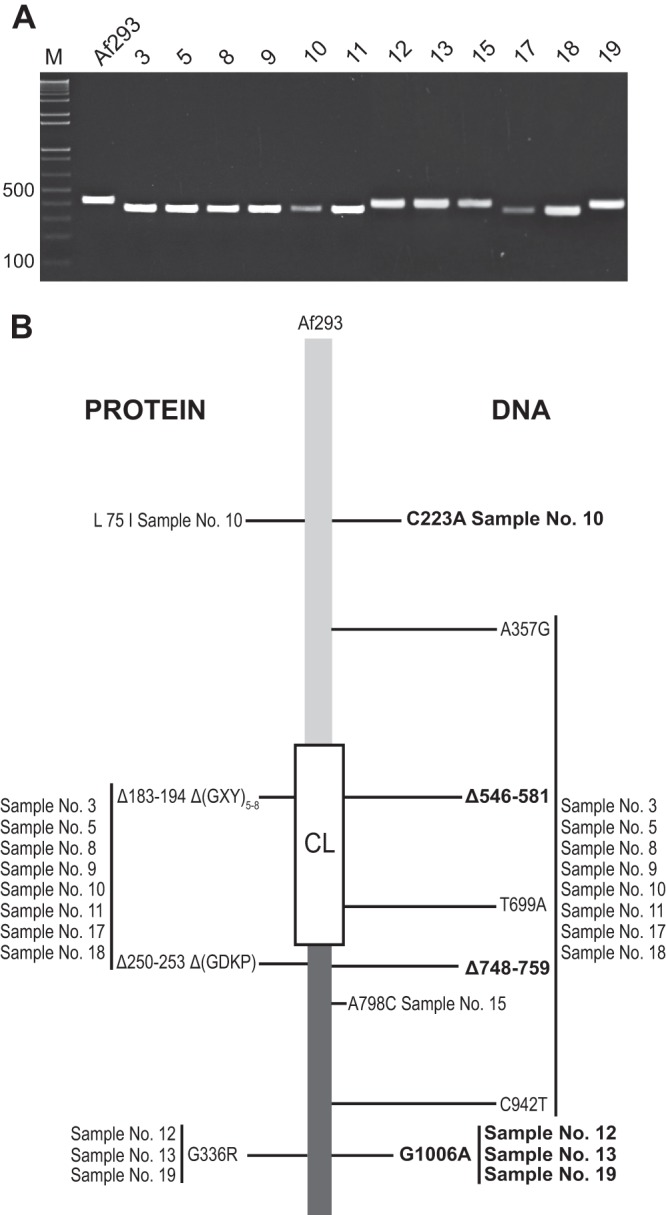
Allelic polymorphism in aclF1 gene and AclF1 protein among clinical isolates of A. fumigatus. (A) Length polymorphism of the aclF1-collagen-like region. PCR amplicons were generated with the aclF1-CL primers (Table 1) flanking the CL region of the aclF1 gene in 12 clinical A. fumigatus strains and resolved in a 2% agarose gel. M, 1-kb Plus TrackIt DNA ladder. (B) aclF1/AclF1 gene/protein sequence polymorphism among clinical A. fumigatus strains compared to aclF1 in the sequenced strain Af293. A summary of nucleotide and amino acid polymorphisms is shown; nonsynonymous polymorphisms are indicated in bold.
DNA sequencing revealed eight polymorphic sites that identified four aclF1 alleles, resulting in three protein variants (Fig. 6B). The most common aclF1 allele was found in samples 3, 5, 8, 9, 11, 17, and 18 and was identical to that previously identified in strain Af6113 (Fig. 3B); in addition, sample 10 harbored one extra SNP (C223A). These sequencing results verified smaller aclF1-CL amplicons yielded by these samples (Fig. 6A). The second-most-common SNP (G1006A) was present in samples 12, 13, and 19. Finally, a single SNP (A798C) was found in sample 15, resulting in an allele previously identified in Af164 and Af01. At the protein level, only two SNPs resulted in new AclF1 variants: sample 10 (L75I) and samples 12, 13, and 19 (G336R).
Molecular phylogeny of A. fumigatus aclF1.
The diversity among A. fumigatus strains was assessed using molecular phylogenetic analyses of the sequenced aclF1 gene (Fig. 7). MP and Bayesian methods produced identical topologies. While there was high sequence conservation, the SNPs and deletions within the aclF1 alleles provided enough information to separate the isolates into three clades, although the results were not statistically supported. A combination of 4 SNPs and two deletions (36 and 12 bp) distinguished the two major branches. The majority of isolates grouped with the annotated A. fumigatus 293 reference strain and included both clinical and environmental samples from a broad range of geographical locations (Table 1). The clade harboring A. fumigatus strain 6113 did not contain any environmental isolates, suggesting either that the two deletions within the aclF1 allele, characteristic of this clade, may provide an advantage during infection or that environmental members have not yet been identified. Additionally, A. fumigatus strains 174 and 5109 formed their own sister clade, as they share SNPs with the two other clades and also encode a unique SNP (A357C). The aclF1 gene can therefore be utilized for A. fumigatus species identification as well as to further understand strain diversification.
Fig 7.

Molecular phylogenetic analysis of aclF1 nucleotide sequences from A. fumigatus environmental and clinical isolates. An unrooted Bayesian analysis tree, created from 1,080 aligned nucleotides, is shown. Maximum-parsimony (MP) analysis yielded the same topology (data not shown). Percent Bayesian posterior probability values and MP bootstrap values > 50% are indicated on branches (−, <50% bootstrap value). The scale bar signifies the number of substitutions per site. Boxed letters indicate clinical (C) and environmental (E) sources.
DISCUSSION
Collagens comprise a large family of proteins that are found in many diverse members of the animal kingdom. Structurally, all collagens adopt a triple-helical conformation in the tertiary structure of the protein composed of three left-handed polyproline II-type chains that intertwine around a central axis, forming a right-handed superhelix (44). There have been 19 different types of vertebrate collagens classified that are essential building elements of connective tissues, but other unique collagens were found in invertebrates, including mussels, worms, and sponges (45, 46). More recently, prokaryotic collagens were also described in bacteria and phages (47) that form collagen-like triple helices (48).
In the fungal kingdom, collagens have been less well characterized since the discovery of the fimbriae containing collagen-like protein in the haploid cells of the smut fungus, Microbotryum violaceum (49, 50). More recently, the Metarhizium anisopliae Mcl1 collagen-like protein has been identified and characterized as a component of protective coating that mediated immune evasion in a Manduca sexta insect model (51). Interestingly, Mcl1 expression was induced upon transfer into a M. sexta hemolymph and impeded phagocytosis. In the aggregate, several known domains were predicted in fungal collagen-like proteins that were also found in the transcription initiation factor RNase, calapain family cysteine protease, or mucin-like protein; a complete list of all fungal CLPs and their domain organization is shown in Fig. S1 in the supplemental material. Here, we report nine Aspergillus collagen-like proteins, designated Acl, that were annotated in the collagen Pfam database among members of the environmental molds belonging to the fungal order Eurotiales (family Trichocomaceae). In addition to the common collagen-like (CL) region, six Acl proteins contain predicted amino-terminal signal peptides and two had a predicted carboxyl-terminal cell-anchor GPI signature; one protein, AclN1 of A. nidulans, had both, strongly suggesting that it is cell associated. In preliminary experiments, we detected the AclF1 protein in association with hyphal extracts of A. fumigatus 293 by Western blotting (data not shown); however, expression of the AclF1 protein in different morphological units among A. fumigatus strains is a subject of future studies.
The Acl proteins differ by size and primary sequence, but their presumed function(s) cannot be assumed since only the AclK1 protein of A. kawachii contains a known predicted RNase domain. Based on available data, the CL regions of the Acls contain 13 to 42 triplet repeats that are often composed of a single triplet or a few distinct triplets. Interestingly, two proteins, AclF1 and AclT2, contain two-amino-acid interruptions within their CL regions. Similar interruptions that form kinks are found in the triple-helical domains of several mammalian proteins, such as complement factor C1q and mammalian lectins (52–54). These proteins possess globular carbohydrate recognition domains (CRD) and are arranged in higher-ordered cross-shaped or bouquet-shaped quaternary structures for increased binding potential of the CRDs (53, 55). Kinks in the collagen-like domains serve to accommodate the packaging of the globular domains. In higher eukaryotes, posttranslational hydroxylation of proline residues at position Y is considered essential for the stability of the triple helix (56–58). Sequencing databases suggest that the putative enzyme prolyl 4-hydroxylase is present in several Aspergillus species and thereby could facilitate the stabilization of the Acl-CL triple helix through proline hydroxylation; however, proline residues at position Y are found only in the CL regions of AclF1, AclT1, AclFL1, and AclN1, whereas they are missing in 5 remaining Acls. Further studies are needed to formally demonstrate that Acl proteins are trimeric and that they form collagen-like triple helices.
In this study, we identified the acl genes among several species belonging to the genus Aspergillus. Our initial screening of the available 12 A. fumigatus strains, as well as positive detection of the 12 clinical isolates, provides promising data about the applicability of acl-based detection of Aspergillus using PCR or other molecular-based platforms. This finding is significant, as this organism is responsible for more than 90% of IA cases (43). Unfortunately, we had limited numbers of A. flavus, A. nidulans, A. niger, and A. terreus strains that have not yet been sequenced, and yet the acl-based detection still produced amplicons of the predicted size for their respective organisms. These results suggest the potential use of the noncollagenous acl regions as species-specific detection targets.
Molecular epidemiology involves strain fingerprinting, which often employs length polymorphism associated with the presence of sequence repeats. Microsatellite repeats and genes with internal repeats were identified and characterized in Aspergillus and Candida, respectively (59, 60). Length polymorphism associated with triplet repeats of the collagen-like region has also been used for bacterial strain differentiation (22–24). Here, we assessed length variation of the aclF1 gene by PCR with primers flanking the CL region. Unexpectedly, only 1 of 12 A. fumigatus strains showed length polymorphism as detected by gel electrophoresis. Sequence analysis confirmed the deletion of three GXY repeats as well as an additional deletion of four amino acids outside the CL region. These data suggest either greater-than-expected stability of the aclF1-CL region or the need for additional data.
Two commercially available assays include the broad-pathogen-range SeptiFast assay (Roche Molecular Systems, Mannheim, Germany) and the MycAssay Aspergillus (Myconostica, Cambridge, United Kingdom); PCR detection of DNA is based on bacterial 16S-23S (SeptiFast) and fungal 18S-5.8S internal transcribed space (ITS) regions of rRNA genes (SeptiFast and MycAssay Aspergillus). The MycAssay Aspergillus, a Conformité Européenne-marked quantitative PCR (qPCR) assay, was comparable in performance to an existing galactomannan enzyme-linked immunosorbent assay (GM-ELISA), which has been incorporated into the European Organization for Research and Treatment of Cancer/Mycoses Study Group (EORTC/MSG) as a disease-defining criterion (61). Although the assay is capable of detecting 15 different Aspergillus spp. according to the manufacturer product insert, it does not discriminate between the species, which can be a crucial determinant for selection of appropriate antifungal treatment (62–66). A similar ITS1-based qPCR assay for detection of the four medically most important Aspergillus species, i.e., A. fumigatus, A. flavus, A. niger, and A. terreus, has been successfully developed and tested in the United States (67). For comparison, a diagnostic approach based on the current acl model may provide rationalized treatment of the five most common disease-causing agents but may not be able to detect other Aspergillus species that rarely cause diseases.
Our current preliminary assessment of the acl-based detection produced unexpected results in two cases: (i) a lack of positive identification of Neosartorya fischeri in clinical sample no. 16 and (ii) an unexpected positive amplification of A. parasiticus with A. flavus-specific primers. Neosartorya fischeri (the telomorph of A. thermomutatus) and N. pseudofischeri (the telomorph of A. thermomutatus) have great phenotypic similarity to some A. fumigatus strains (68), which may explain why the aforementioned organisms were phenotypically identified as A. fumigatus. Despite its morphological similarity to A. fumigatus, however, sample no. 16 consistently failed to amplify with any of the acl assays. Upon sample reculturing, the microscopic appearance (with absence of cleistothecia) and growth at 45°C remained consistent with A. fumigatus, although colonies on Sabaroud dextrose agar were pink rather than green. Subsequent ITS- and NL-region sequencing confirmed the identity of N. pseudofischeri (A. thermomutatus). Therefore, the lack of amplification with the acl assays indicates that this organism either lacks the acl gene or has an acl sequence with limited similarity to that of the A. fumigatus aclF1 gene reported in this study.
In contrast, A. flavus and A. parasiticus are notorious for their aflatoxin production in agriculture. As such, their relationship has been the subject of genomic and proteomic studies. Unfortunately, the entire genome for A. flavus has not been assembled, and apart from the aflatoxin pathway, there is very little genomic data on A. parasiticus. Nevertheless, the close genetic relationship between these organisms has been demonstrated through the above-mentioned pathway, as well as for additional genetic loci analyzed, including 5.8S-ITS, aflR, nor-1, and β-tubulin loci (4, 69, 70). Furthermore, upon the discovery of a sexual life cycle of several Aspergillus species, an experimental telomorph of A. flavus was demonstrated to be morphologically indistinguishable from that of A. parasiticus by Horn et al. (71). Our finding that the aclFL1-based primer pairs produced amplicons in both A. flavus and A. parasiticus is consistent with the trend of genetic homology between these species.
The use of microscale separation was assessed for the acl-based amplicons as an alternative to agarose gel electrophoresis. Separations performed in a capillary with an inner diameter ranging from 10 to 100 μm utilize nano-to-femtoliter sample volumes. Microscale separations may be performed in automated instruments or in portable microfluidic devices. In addition, the decreased channel diameter generates lower separation current, and electric field strengths ranging from 100 to 600 V/cm can be used. Higher field strengths generate more-efficient analyte peaks in shorter separation times. Chemical sieving of DNA fragments has been accomplished using capillary electrophoresis separations in cross-linked gels (72). Although separations with cross-link gels are possible, linear polymers are more frequently used for capillary sieving of DNA (73–75). Linear gels are viscous additives that provide good separation performance and do not need to be polymerized in the capillary. Issues associated with the introduction of highly viscous materials into narrow-bore capillaries have spawned the use of thermally reversible sieving materials (76–78), including phospholipid nanogels (36). These materials are introduced into the capillary at a temperature that generates low viscosity and then converted into a highly viscous separation additive in the capillary by changing the separation temperature. For phospholipid nanogels, the viscosity increases by 2 orders of magnitude when the temperature is increased from 19°C to 30°C and the shear rate is changed (79). This thermal response provides a means to easily fill or expel the sieving material at 19°C. Sieving is then accomplished at 30°C. Separations performed under conditions that do not denature DNA provided nearly single-base resolution of short tandem repeats relevant to human identification (36).
The similarity between a slab gel separation and a capillary nanogel separation is demonstrated in Fig. 2C with a size ladder and the separation of the acF1-5′ and aclF1-3′ amplicons from A. fumigatus. The ladder and amplicons are separated in two different runs, in a manner analogous to the use of separate lanes in a slab gel. Slab gel separations of double-stranded DNA (dsDNA) typically incorporate a fluorescent intercalating dye, such as SYBR green 1, in the sieving material. For the capillary nanogel separations, the intercalating dye is not included in the media. Instead, a nanoliter-volume plug of dye loaded nanogel is loaded into the capillary near the detection window. At the onset of separation, the cationic dye migrates toward the DNA fragments. Once the DNA fragments and cationic dye intersect, the resulting DNA-dye complex continues to migrate toward the detection window. In order to quantify the size of DNA fragments, accurate size measurements of DNA are achieved by coinjecting internal size standards with the PCR marker (see Fig. S3 in the supplemental material). This obviates the run-to-run variability in migration time that may occur with variability in the degree of dye loading.
The separations shown in Fig. 4 yield size determinations with an accuracy of 1 bp (aclN1-3′, aclNi1-5′), 2 bp (aclT2-5′, aclF1-3′), 3 bp (aclFL1-3′), or 6 bp (acF1-5′). The accuracy of size standardization is a function of the size of the DNA separated, the available standard size, and the applied voltage. The size range of the measurement affects accuracy, as the DNA migration is linear (i.e., in Ogston sieving) up to ∼450 bp (36). DNA fragments larger than this undergo reptation-based transport. More accurate size measurement is obtained for the size standards that are closest to the expected size of the DNA fragment; however, overlap with the other amplicons to be measured limits the standards that may be used. Lower applied voltages reduce the effect of DNA alignment in the electric field, which improves size determination. However, separation times are longer for lower field strengths. If the precision of the size determination is not critical, capillary nanogel separations can be obtained in shorter separation times (∼5 to 10 min) than those shown in Fig. 2C and 4B and yet still yield baseline resolution of the DNA fragments (36).
Based on the current results presented here, the acl genes are species specific and appear to be widely distributed among strains tested. Notably, the broad diversity of fungal collagen-like proteins suggests that the utilization of acl genes should not result in false-positive species identifications in clinical settings. The collagen-like region of aclF1 exhibits some length polymorphism, but a screening of a broader spectrum of strains is necessary to further understand the applicability of the acl-based assay for strain fingerprinting. The phylogeny created from the available strains provides evidence that this locus may be informative for further examination and identification of clinical strains. While the environmental and clinical isolates were obtained from diverse geographical areas (Table 1), two major aclF1 clades were obtained. The grouping of solely clinical isolates (i.e., of A. fumigatus strain 6113 with eight contemporary clinical samples [no. 3, 5, 8, 9, 10, 11, 17, and 18]) may indicate that the basal aclF1 allele may be undergoing changes linked to a higher propensity for clinical infections and also may be incorporated for diagnostics of disease-causing strains. Interestingly, atypical A. fumigatus strain Af6113, which was isolated from a sinusitis case of a patient at the University of California, San Francisco Medical Center, was morphologically distinct and was initially designated A. phialiseptus (80). The acl-based assay was able to correctly identify 18 of 19 clinically isolated plate cultures of Aspergillus spp. to the species level, missing only one species (N. pseudofischeri/A. thermomutatus) for which primers were not targeted. This is due to the fundamental difference of the acl-based assay from existing commercial PCR-based assays, namely, its focusing on species differentiation among the most common etiologic agents of aspergillosis rather than on pan-Aspergillus sensitivity. As the concept of the acl assay is amplicon size-based discrimination, capillary electrophoresis provides a more accurate tool for multiplex separation of several products. Finally, given that the AclF1 protein of A. fumigatus harbors a signal peptide and is associated with production and display by hyphae (the predominant fungal form in invasive disease), the potential for serologic detection of these molecules in diagnostic applications would appear promising. In summary, here we demonstrate the potential of the acl genes as biomarkers for detection of several Aspergillus species.
Supplementary Material
ACKNOWLEDGMENTS
We thank William R. Rittenour and Yael Tarlovsky for their contribution in the project.
This work was supported in part by National Science Foundation Cooperative Agreement EPS-1003907 (to S.L.). L.A.H. acknowledges support for capillary nanogel separations by the National Science Foundation under grant no. CHE1212537. R.V.M.R. is supported by NSF-IOS 1025274. K.T. was supported by NIH grant 5P20RR016477 to the West Virginia IDeA Network for Biomedical Research Excellence. B.C.D. was supported by the NSF-EPSCoR Graduate Fellowship Program (under Cooperative Agreement 1003907).
The findings and conclusions in this report are ours and do not necessarily represent the views of the National Institute of Occupational Safety and Health.
Footnotes
Published ahead of print 11 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02835-13.
REFERENCES
- 1.Eduard W. 2009. Fungal spores: a critical review of the toxicological and epidemiological evidence as a basis for occupational exposure limit setting. Crit. Rev. Toxicol. 39:799–864 [DOI] [PubMed] [Google Scholar]
- 2.Machida M, Yamada O, Gomi K. 2008. Genomics of Aspergillus oryzae: learning from the history of Koji mold and exploration of its future. DNA Res. 15:173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jahromi MF, Liang JB, Ho YW, Mohamad R, Goh YM, Shokryazdan P. 2012. Lovastatin production by Aspergillus terreus using agro-biomass as substrate in solid state fermentation. J. Biomed. Biotechnol. 2012:196264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu J, Chang PK, Cary JW, Wright M, Bhatnagar D, Cleveland TE, Payne GA, Linz JE. 1995. Comparative mapping of aflatoxin pathway gene clusters in Aspergillus parasiticus and Aspergillus flavus. Appl. Environ. Microbiol. 61:2365–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zmeili OS, Soubani AO. 2007. Pulmonary aspergillosis: a clinical update. QJM 100:317–334 [DOI] [PubMed] [Google Scholar]
- 6.Latgé JP. 1999. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 12:310–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groll AH, Kurz M, Schneider W, Witt V, Schmidt H, Schneider M, Schwabe D. 1999. Five-year-survey of invasive aspergillosis in a paediatric cancer centre. Epidemiology, management and long-term survival. Mycoses 42:431–442 [DOI] [PubMed] [Google Scholar]
- 8.Dimopoulos G, Frantzeskaki F, Poulakou G, Armaganidis A. 2012. Invasive aspergillosis in the intensive care unit. Ann. N. Y. Acad. Sci. 1272:31–39 [DOI] [PubMed] [Google Scholar]
- 9.Hsieh KH, Shen JJ. 1988. Prevalence of childhood asthma in Taipei, Taiwan, and other Asian Pacific countries. J. Asthma 25:73–82 [DOI] [PubMed] [Google Scholar]
- 10.Chaudhary N, Marr KA. 2011. Impact of Aspergillus fumigatus in allergic airway diseases. Clin. Transl. Allergy 1:4. 10.1186/2045-7022-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patterson K, Strek ME. 2010. Allergic bronchopulmonary aspergillosis. Proc. Am. Thorac. Soc. 7:237–244 [DOI] [PubMed] [Google Scholar]
- 12.Simon-Nobbe B, Denk U, Poll V, Rid R, Breitenbach M. 2008. The spectrum of fungal allergy. Int. Arch. Allergy Immunol. 145:58–86 [DOI] [PubMed] [Google Scholar]
- 13.Tong KB, Lau CJ, Murtagh K, Layton AJ, Seifeldin R. 2009. The economic impact of aspergillosis: analysis of hospital expenditures across patient subgroups. Int. J. Infect. Dis. 13:24–36 [DOI] [PubMed] [Google Scholar]
- 14.Pingleton WW, Hiller FC, Bone RC, Kerby GR, Ruth WE. 1977. Treatment of allergic aspergillosis with triamcinolone acetonide aerosol. Chest 71:782–784 [DOI] [PubMed] [Google Scholar]
- 15.FDA 2013. Multistate outbreak of fungal meningitis and other infections. U.S. Food and Drug Administration, Silver Spring, MD: http://www.fda.gov/Drugs/DrugSafety/FungalMeningitis/default.htm [Google Scholar]
- 16.Denning DW. 2000. Aspergillus species, p 2306–2310 In Mandell GL, Bennett JE, Dolin R. (ed), Principles and practices of infectious diseases, part III—infectious diseases and their etiologic agents. Churchill Livingstone, Philadelphia, PA [Google Scholar]
- 17.Hachem RY, Kontoyiannis DP, Chemaly RF, Jiang Y, Reitzel R, Raad I. 2009. Utility of galactomannan enzyme immunoassay and (1,3) β-D-glucan in diagnosis of invasive fungal infections: low sensitivity for Aspergillus fumigatus infection in hematologic malignancy patients. J. Clin. Microbiol. 47:129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kourkoumpetis TK, Fuchs BB, Coleman JJ, Desalermos A, Mylonakis E. 2012. Polymerase chain reaction-based assays for the diagnosis of invasive fungal infections. Clin. Infect. Dis. 54:1322–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hope WW, Walsh TJ, Denning DW. 2005. Laboratory diagnosis of invasive aspergillosis. Lancet Infect. Dis. 5:609–622 [DOI] [PubMed] [Google Scholar]
- 20.Loeffler J, Barnes R, Donnelly JP. 2012. Standardization of Aspergillus PCR diagnosis. Bone Marrow Transplant. 47:299–300 [DOI] [PubMed] [Google Scholar]
- 21.White PL, Bretagne S, Klingspor L, Melchers WJ, McCulloch E, Schulz B, Finnstrom N, Mengoli C, Barnes RA, Donnelly JP, Loeffler J. 2010. Aspergillus PCR: one step closer to standardization. J. Clin. Microbiol. 48:1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leski TA, Caswell CC, Pawlowski M, Klinke DJ, Bujnicki JM, Hart SJ, Lukomski S. 2009. Identification and classification of bcl genes and proteins of Bacillus cereus group organisms and their application in Bacillus anthracis detection and fingerprinting. Appl. Environ. Microbiol. 75:7163–7172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castanha ER, Swiger RR, Senior B, Fox A, Waller LN, Fox KF. 2006. Strain discrimination among B. anthracis and related organisms by characterization of bclA polymorphisms using PCR coupled with agarose gel or microchannel fluidics electrophoresis. J. Microbiol. Methods 64:27–45 [DOI] [PubMed] [Google Scholar]
- 24.Sylvestre P, Couture-Tosi E, Mock M. 2002. A collagen-like surface glycoprotein is a structural component of the Bacillus anthracis exosporium. Mol. Microbiol. 45:169–178 [DOI] [PubMed] [Google Scholar]
- 25.Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. 2000. The Pfam protein families database. Nucleic Acids Res. 28:263–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786 [DOI] [PubMed] [Google Scholar]
- 28.Eisenhaber B, Bork P, Eisenhaber F. 1999. Prediction of potential GPI-modification sites in proprotein sequences. J. Mol. Biol. 292:741–758 [DOI] [PubMed] [Google Scholar]
- 29.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swofford DL. 2002. PAUP 4.0—phylogenetic analysis using parsimony, version 4. Sinauer Associates, Sunderland, MA [Google Scholar]
- 31.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Felsenstein J. 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17:368–376 [DOI] [PubMed] [Google Scholar]
- 33.Nylander JAA. 2004. MrModeltest v2. Evolutionary Biology Centre, Uppsala University, Uppsala, Sweden [Google Scholar]
- 34.Chow TY-K, Käfer E. 1993. A rapid method for isolation of total nucleic acids from Aspergillus nidulans. Fungal Genet. Newslett. 40:25–27 [Google Scholar]
- 35.Archer-Hartmann SA, Sargent LM, Lowry DT, Holland LA. 2011. Microscale exoglycosidase processing and lectin capture of glycans with phospholipid assisted capillary electrophoresis separations. Anal. Chem. 83:2740–2747 [DOI] [PubMed] [Google Scholar]
- 36.Durney BC, Lounsbury JA, Poe BL, Landers JP, Holland LA. 2013. A thermally responsive phospholipid pseudogel: tunable DNA sieving with capillary electrophoresis. Anal. Chem. 85:6617–6625 [DOI] [PubMed] [Google Scholar]
- 37.White CM, Luo R, Archer-Hartmann SA, Holland LA. 2007. Electrophoretic screening of ligands under suppressed EOF with an inert phospholipid coating. Electrophoresis 28:3049–3055 [DOI] [PubMed] [Google Scholar]
- 38.Luo R, Archer-Hartmann SA, Holland LA. 2010. Transformable capillary electrophoresis for oligosaccharide separations using phospholipid additives. Anal. Chem. 82:1228–1233 [DOI] [PubMed] [Google Scholar]
- 39.Henry T, Iwen PC, Hinrichs SH. 2000. Identification of Aspergillus species using internal transcribed spacer regions 1 and 2. J. Clin. Microbiol. 38:1510–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White TB, Lee TJS, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. Academic Press, Inc., New York, NY [Google Scholar]
- 41.Kurtzman CP, Robnett CJ. 1997. Identification of clinically important ascomycetous yeasts based on nucleotide divergence in the 5′ end of the large-subunit (26S) ribosomal DNA gene. J. Clin. Microbiol. 35:1216–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogaboam CM, Carpenter KJ, Schuh JM, Buckland KF. 2005. Aspergillus and asthma—any link? Med. Mycol. 43(Suppl 1):S197–S1202 [DOI] [PubMed] [Google Scholar]
- 43.Torres HA, Rivero GA, Lewis RE, Hachem R, Raad II, Kontoyiannis DP. 2003. Aspergillosis caused by non-fumigatus Aspergillus species: risk factors and in vitro susceptibility compared with Aspergillus fumigatus. Diagn. Microbiol. Infect. Dis. 46:25–28 [DOI] [PubMed] [Google Scholar]
- 44.Berisio R, Vitagliano L, Mazzarella L, Zagari A. 2002. Crystal structure of the collagen triple helix model [(Pro-Pro-Gly)10]3. Protein Sci. 11:262–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hulmes DJ. 1992. The collagen superfamily—diverse structures and assemblies. Essays Biochem. 27:49–67 [PubMed] [Google Scholar]
- 46.Engel J. 1997. Versatile collagens in invertebrates. Science 277:1785–1786 [DOI] [PubMed] [Google Scholar]
- 47.Rasmussen M, Jacobsson M, Bjorck L. 2003. Genome-based identification and analysis of collagen-related structural motifs in bacterial and viral proteins. J. Biol. Chem. 278:32313–32316 [DOI] [PubMed] [Google Scholar]
- 48.Munk R, Ghosh P, Ghosh MC, Saito T, Xu M, Carter A, Indig F, Taub DD, Longo DL. 2011. Involvement of mTOR in CXCL12 mediated T cell signaling and migration. PLoS One 6:e24667. 10.1371/journal.pone.0024667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Celerin M, Ray JM, Schisler NJ, Day AW, Stetler-Stevenson WG, Laudenbach DE. 1996. Fungal fimbriae are composed of collagen. EMBO J. 15:4445–4453 [PMC free article] [PubMed] [Google Scholar]
- 50.Celerin M, Day AW, Smith RJ, Laudenbach DE. 1997. Immunolocalization of fimbrial epitopes in thin sections of Microbotryum violaceum. Can. J. Microbiol. 43:136–142 [Google Scholar]
- 51.Wang C, St Leger RJ. 2006. A collagenous protective coat enables Metarhizium anisopliae to evade insect immune responses. Proc. Natl. Acad. Sci. U. S. A. 103:6647–6652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Håkansson K, Reid KB. 2000. Collectin structure: a review. Protein Sci. 9:1607–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van de Wetering JK, van Golde LM, Batenburg JJ. 2004. Collectins: players of the innate immune system. Eur. J. Biochem. 271:1229–1249 [DOI] [PubMed] [Google Scholar]
- 54.Kilchherr E, Hofmann H, Steigemann W, Engel J. 1985. Structural model of the collagen-like region of C1q comprising the kink region and the fibre-like packing of the six triple helices. J. Mol. Biol. 186:403–415 [DOI] [PubMed] [Google Scholar]
- 55.Haagsman HP. 1994. Surfactant proteins A and D. Biochem. Soc. Trans. 22:100–106 [DOI] [PubMed] [Google Scholar]
- 56.Persikov AV, Ramshaw JA, Kirkpatrick A, Brodsky B. 2003. Triple-helix propensity of hydroxyproline and fluoroproline: comparison of host-guest and repeating tripeptide collagen models. J. Am. Chem. Soc. 125:11500–11501 [DOI] [PubMed] [Google Scholar]
- 57.Vitagliano L, Berisio R, Mazzarella L, Zagari A. 2001. Structural bases of collagen stabilization induced by proline hydroxylation. Biopolymers 58:459–464 [DOI] [PubMed] [Google Scholar]
- 58.Holmgren SK, Taylor KM, Bretscher LE, Raines RT. 1998. Code for collagen's stability deciphered. Nature 392:666–667 [DOI] [PubMed] [Google Scholar]
- 59.Levdansky E, Romano J, Shadkchan Y, Sharon H, Verstrepen KJ, Fink GR, Osherov N. 2007. Coding tandem repeats generate diversity in Aspergillus fumigatus genes. Eukaryot. Cell 6:1380–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sabino R, Sampaio P, Rosado L, Stevens DA, Clemons KV, Pais C. 2010. New polymorphic microsatellite markers able to distinguish among Candida parapsilosis sensu stricto isolates. J. Clin. Microbiol. 48:1677–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torelli R, Sanguinetti M, Moody A, Pagano L, Caira M, De Carolis E, Fuso L, De Pascale G, Bello G, Antonelli M, Fadda G, Posteraro B. 2011. Diagnosis of invasive aspergillosis by a commercial real-time PCR assay for Aspergillus DNA in bronchoalveolar lavage fluid samples from high-risk patients compared to a galactomannan enzyme immunoassay. J. Clin. Microbiol. 49:4273–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lass-Flörl C, Follett SA, Moody A, Denning DW. 2011. Detection of Aspergillus in lung and other tissue samples using the MycAssay Aspergillus real-time PCR kit. Can. J. Microbiol. 57:765–768 [DOI] [PubMed] [Google Scholar]
- 63.Arendrup MC, Jensen RH, Grif K, Skov M, Pressler T, Johansen HK, Lass-Florl C. 2012. In vivo emergence of Aspergillus terreus with reduced azole susceptibility and a Cyp51a M217I alteration. J. Infect. Dis. 206:981–985 [DOI] [PubMed] [Google Scholar]
- 64.Blum G, Hortnagl C, Jukic E, Erbeznik T, Pumpel T, Dietrich H, Nagl M, Speth C, Rambach G, Lass-Florl C. 2013. New insight into amphotericin B resistance in Aspergillus terreus. Antimicrob. Agents Chemother. 57:1583–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Georgiadou SP, Kontoyiannis DP. 2012. The impact of azole resistance on aspergillosis guidelines. Ann. N. Y. Acad. Sci. 1272:15–22 [DOI] [PubMed] [Google Scholar]
- 66.Hadrich I, Makni F, Neji S, Cheikhrouhou F, Bellaaj H, Elloumi M, Ayadi A, Ranque S. 2012. Amphotericin B in vitro resistance is associated with fatal Aspergillus flavus infection. Med. Mycol. 50:829–834 [DOI] [PubMed] [Google Scholar]
- 67.Walsh TJ, Wissel MC, Grantham KJ, Petraitiene R, Petraitis V, Kasai M, Francesconi A, Cotton MP, Hughes JE, Greene L, Bacher JD, Manna P, Salomoni M, Kleiboeker SB, Reddy SK. 2011. Molecular detection and species-specific identification of medically important Aspergillus species by real-time PCR in experimental invasive pulmonary aspergillosis. J. Clin. Microbiol. 49:4150–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balajee SA, Gribskov J, Brandt M, Ito J, Fothergill A, Marr KA. 2005. Mistaken identity: Neosartorya pseudofischeri and its anamorph masquerading as Aspergillus fumigatus. J. Clin. Microbiol. 43:5996–5999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klich MA, Cary JW, Beltz SB, Bennett CA. 2003. Phylogenetic and morphological analysis of Aspergillus ochraceoroseus. Mycologia 95:1252–1260 [DOI] [PubMed] [Google Scholar]
- 70.El Khoury A, Atoui A, Rizk T, Lteif R, Kallassy M, Lebrihi A. 2011. Differentiation between Aspergillus flavus and Aspergillus parasiticus from pure culture and aflatoxin-contaminated grapes using PCR-RFLP analysis of aflR-aflJ intergenic spacer. J. Food. Sci. 76:M247–M253 [DOI] [PubMed] [Google Scholar]
- 71.Horn BW, Moore GG, Carbone I. 2009. Sexual reproduction in Aspergillus flavus. Mycologia 101:423–429 [DOI] [PubMed] [Google Scholar]
- 72.Guttman A. 1993. Separation of DNA by capillary electrophoresis, p 129–143 In Landers J. (ed), Handbook of capillary and microchip electrophoresis and associated microtechniques, 3 ed. Taylor & Francis, Boca Raton, FL [Google Scholar]
- 73.Tian H, Landers JP. 2002. Hydroxyethylcellulose as an effective polymer network for DNA analysis in uncoated glass microchips: optimization and application to mutation detection via heteroduplex analysis. Anal. Biochem. 309:212–223 [DOI] [PubMed] [Google Scholar]
- 74.Barron AE, Soane DS, Blanch HW. 1993. Capillary electrophoresis of DNA in uncross-linked polymer solutions. J. Chromatogr. A 652:3–16 [DOI] [PubMed] [Google Scholar]
- 75.Ruiz-Martinez MC, Berka J, Belenkii A, Foret F, Miller AW, Karger BL. 1993. DNA sequencing by capillary electrophoresis with replaceable linear polyacrylamide and laser-induced fluorescence detection. Anal. Chem. 65:2851–2858 [DOI] [PubMed] [Google Scholar]
- 76.Buchholz BA, Shi W, Barron AE. 2002. Microchannel DNA sequencing matrices with switchable viscosities. Electrophoresis 23:1398–1409 [DOI] [PubMed] [Google Scholar]
- 77.Case WS, Glinert KD, LaBarge S, McGown LB. 2007. Guanosine gel for sequence-dependent separation of polymorphic ssDNA. Electrophoresis 28:3008–3016 [DOI] [PubMed] [Google Scholar]
- 78.Yu Y, Nakamura D, DeBoyace K, Neisius AW, McGown LB. 2008. Tunable thermoassociation of binary guanosine gels. J. Phys. Chem. B 112:1130–1134 [DOI] [PubMed] [Google Scholar]
- 79.Wu X, Langan TJ, Durney BC, Holland LA. 2012. Thermally responsive phospholipid preparations for fluid steering and separation in microfluidics. Electrophoresis 33:2674–2681 [DOI] [PubMed] [Google Scholar]
- 80.Kwon-Chung KJ. 1975. A new pathogenic species of Aspergillus in the Aspergillus fumigatus series. Mycologia 67:770–779 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.