

Abstract
Dimethyl adenosine transferase (KsgA) performs diverse roles in bacteria, including ribosomal maturation and DNA mismatch repair, and synthesis of KsgA is responsive to antibiotics and cold temperature. We previously showed that a ksgA mutation in Salmonella enterica serovar Enteritidis results in impaired invasiveness in human and avian epithelial cells. In this study, we tested the virulence of a ksgA mutant (the ksgA::Tn5 mutant) of S. Enteritidis in orally challenged 1-day-old chickens. The ksgA::Tn5 mutant showed significantly reduced intestinal colonization and organ invasiveness in chickens compared to those of the wild-type (WT) parent. Phenotype microarray (PM) was employed to compare the ksgA::Tn5 mutant and its isogenic wild-type strain for 920 phenotypes at 28°C, 37°C, and 42°C. At chicken body temperature (42°C), the ksgA::Tn5 mutant showed significantly reduced respiratory activity with respect to a number of carbon, nitrogen, phosphate, sulfur, and peptide nitrogen nutrients. The greatest differences were observed in the osmolyte panel at concentrations of ≥6% NaCl at 37°C and 42°C. In contrast, no major differences were observed at 28°C. In independent growth assays, the ksgA::Tn5 mutant displayed a severe growth defect in high-osmolarity (6.5% NaCl) conditions in nutrient-rich (LB) and nutrient-limiting (M9 minimum salts) media at 42°C. Moreover, the ksgA::Tn5 mutant showed significantly reduced tolerance to oxidative stress, but its survival within macrophages was not impaired. Unlike Escherichia coli, the ksgA::Tn5 mutant did not display a cold-sensitivity phenotype; however, it showed resistance to kasugamycin and increased susceptibility to chloramphenicol. To the best of our knowledge, this is the first report showing the role of ksgA in S. Enteritidis virulence in chickens, tolerance to high osmolarity, and altered susceptibility to kasugamycin and chloramphenicol.
INTRODUCTION
In bacteria, the ksgA gene encodes a dimethyl adenosine transferase (KsgA) protein that belongs to the KsgA/Dim1 family of universally conserved methyltransferases. According to Harris et al. (1), the KsgA/Dim1 family is one of the 50 factors conserved in all kingdoms of life and probably the only one of its kind that was part of the genetic core of the last universal ancestor. Despite being highly conserved, KsgA mediates diverse functions in bacteria. For example, in Escherichia coli, KsgA acts as a 16S rRNA adenine methyltransferase by adding two methyl groups to the two highly conserved adenine residues located at positions 1518 and 1519 (numbered in the E. coli system) within the universally conserved helix 45 at the 3′ end of the translationally inactive form of the 16S rRNA subunit (2). These methyl groups are donated by S-adenosylmethionine, which is also highly conserved among bacteria, to produce N6,N6-dimethyladenosine bases (3). Methylation of 16S rRNA is important in ribosomal biogenesis and impacts ribosome functions during translation initiation and elongation phases (4). Deficiency of KsgA in E. coli results in altered ribosome profiles characterized by accumulation of free immature small ribosomal subunits (SSU) that are unable to enter the translation cycle. Current models indicate that the KsgA-mediated 30S rRNA methylation is a conserved maturation signal that enables release of KsgA from mature SSUs, resulting in conformational changes that permit SSUs to join the large subunit and IF3 to initiate translation (5).
KsgA also possesses a DNA glycosylase/AP lyase activity that prevents chromosomal mutations by repairing mismatched DNA strands. More specifically, KsgA excises mismatched cytosine bases opposing oxidatively damaged thymine bases by a β-excision mechanism in E. coli (6). Lack of RNA methylase activity caused by mutations within the ksgA locus in E. coli and Neisseria gonorrhoeae results in resistance to the aminoglycoside antibiotic kasugamycin (KSG) (3, 7). KSG inhibits translation initiation in bacteria by blocking tRNA binding to the 30S ribosomal subunit, mimicking the mRNA molecule and occupying its place in the peptidyl (P) and exit (E) sites of the ribosome, which eventually disturbs the mRNA-tRNA-ribosome spatial interaction (8). Exogenous supplementation of wild-type KsgA can rescue KSG sensitivity in KSG-resistant strains of E. coli (3). In addition, E. coli strains lacking KsgA also show a 4-fold reduction in the MIC of gentamicin (9). In contrast, a ksgA mutant of Staphylococcus aureus was more sensitive to kanamycin and paromomycin, probably due to the conformational changes distal to the aminoglycoside binding site in the SSU, which are further propagated from the KsgA methylation site (10). Recently, disruption of ksgA in a clarithromycin-resistant Mycobacterium tuberculosis strain resulted in abolishment of resistance (11), suggesting that KsgA-mediated drug resistance is likely to be strain and species dependent.
Depending on the bacterial system, lack of methylation of the 16S rRNA subunit due to KsgA deficiency also leads to a temperature-sensitive phenotype. Connolly et al. (5) showed that E. coli mutant strains lacking KsgA display growth defects at suboptimal temperatures (25°C and 20°C). This phenotype was characterized by less efficient ribosome biogenesis as fewer mature and translationally active ribosomes were available at low temperature and immature ribosomal subunits accumulate in these cells (5). In contrast, a Bacillus subtilis KsgA-deficient mutant showed a significant growth disadvantage at 37°C when grown in competition assays against E. coli, Streptomyces coelicolor, and Mycobacterium smegmatis (12). Unlike E. coli, S. aureus displays a mild cold-sensitive phenotype that is not characterized by differential accumulation of free immature 30S ribosomal subunits, suggesting that KsgA may not be critical for ribosome biogenesis in this organism (10). Interestingly, overexpression of wild-type KsgA at low temperatures (25°C) can rescue the cold-sensitive phenotype in E. coli; however, its overexpression at 37°C exerts a negative impact on growth in both the wild type and the KsgA-deficient mutant strain (5). In concordance, overexpression of the chlamydial KsgA ortholog, in a ksgA E. coli deletion mutant, inhibited the growth of E. coli at 37°C (13). While this phenotype has not been observed in S. aureus at either low or high temperatures, overexpression of catalytically inactive KsgA in S. aureus at 37°C had a negative effect on growth (10). Similarly, overexpression of a catalytically inactive form of KsgA resulted in significant growth defects in KsgA-deficient and wild-type E. coli strains at both 37°C and 25°C. This catalytically inactive form, produced by an alanine substitution at position 66 of KsgA, does not methylate its adenine targets, although it remains attached to them, thereby blocking binding sites for other ribosomal factors and preventing ribosomal maturation from entering the translation cycle, resulting in accumulation of free SSUs (2). In Saccharomyces cerevisiae, growth assays comparing catalytically inactive Dim1 (KsgA) mutants versus their wild-type counterparts showed no difference in growth rate of yeast at 18, 25, 30, or 37°C (14). In general, the above-mentioned studies show that KsgA deficiency may confer temperature sensitivity; however, the dependence on KsgA may not be similar among different microorganisms.
The role of KsgA in bacterial virulence has been recently recognized in at least one bacterial model. A KsgA-deficient mutant of Yersinia pseudotuberculosis was attenuated after oral infection in BALB/c mice. This mutant was significantly impaired in its survival in the intestine, in its invasiveness in internal organs, such as liver and spleen, and in cultured HeLa cells (15, 16). We recently reported that KsgA deficiency in Salmonella enterica serovar Enteritidis, one of the most important food-borne pathogens, results in multiple phenotypes, including (i) KSG resistance, (ii) reduced invasiveness in cultured human intestinal epithelial cells (Caco-2) and chicken liver cells (LMH), and (iii) reduced survival in egg albumen (17). In this study, we hypothesized that KsgA plays a role in virulence of S. Enteritidis in chickens. Consequently, we tested the virulence of a ksgA mutant of S. Enteritidis (ksgA::Tn5) in a model of orally infected day-old chickens and demonstrated that the mutant was attenuated. We also found that, unlike E. coli, the KsgA-deficient mutant of S. Enteritidis does not display temperature sensitivity; however, it does confer resistance to kasugamycin and, in striking contrast, increased susceptibility to chloramphenicol. Finally, we also demonstrate that KsgA deficiency in S. Enteritidis confers susceptibility to oxidative stress and high osmolarity. To the best of our knowledge, this is the first report showing the role of ksgA in S. Enteritidis virulence in chickens, tolerance to high osmolarity, and increased susceptibility to chloramphenicol.
MATERIALS AND METHODS
Bacterial strains.
The S. Enteritidis G1 Nalr (phage type 4) strain, which is invasive in human intestinal epithelial cells (Caco-2) and virulent in orally infected mice and chickens (17, 18), was used as a wild-type (WT) parental strain. A Caco-2 cell invasion-attenuated S. Enteritidis mutant (ksgA::Tn5), kanamycin and nalidixic acid resistant, was identified previously (17). A complemented mutant (ksgA::Tn5-pACYC184-ksgA) was generated by cloning the full-length ksgA gene into the tetracycline resistance site of a low-copy-number plasmid, pACYC184 (New England BioLabs, USA), bearing a chloramphenicol resistance cassette, as described previously (17). Unless otherwise stated, strains were cultured in Luria-Bertani (LB) broth, on LB agar plates containing 30 μg/ml of nalidixic acid (WT and ksgA::Tn5), or in 30 μg/ml of chloramphenicol (ksgA::Tn5-pACYC184-ksgA).
Chicken virulence assay.
Specific-pathogen-free (SPF) fertile eggs were obtained from Sunrise Farms (Catskill, NY) and hatched in isolation at an animal facility at Washington State University. One-day-old birds were distributed in three groups of 9 birds each (experiment 1) or three groups of 15 birds each (experiment 2). Cloacal swabs were taken before placement in environmentally controlled isolation cages to screen for Salmonella by enrichment in tetrathionate broth (TTB) and plating onto xylose-lysine deoxycholate (XLD; Difco) agar. Antibiotic-free flock raiser diet (Purina, St. Louis, MO) and water were provided ad libitum throughout the experimental period. Bacterial inoculum was prepared from an overnight culture grown at 37°C and diluted in maximum recovery diluent (MRD; 1 g/liter peptone, 8.5 g/liter NaCl, pH 7.0) to obtain the desired concentration. Chicks were orally infected with 200 μl of LB broth containing ∼108 (experiment 1) or ∼109 (experiment 2) CFU of the ksgA::Tn5 mutant or the G1 Nalr WT parent strain. Negative-control groups in both experiments were mock inoculated with 200 μl of LB broth. Three birds per group were sacrificed at 4 days, 8 days, and 12 days postinfection (p.i.; experiment 1) or 24 h, 48 h, 4 days, 8 days, and 12 days p.i. (experiment 2). Small intestine, cecum, liver, and spleen were aseptically collected and homogenized in sterile phosphate-buffered saline (PBS), and serial dilutions were plated onto XLD agar (Difco) to obtain the number of viable colonies per gram of each tissue. Animal experiments were performed according to protocols approved by the WSU Institutional Animal Care and Use Committee. Data were analyzed by two-way analysis of variance (ANOVA) and a Tukey-Kramer test (NCSS 2007).
Growth assays.
Single colonies of the WT S. Enteritidis G1 Nalr strain, the ksgA::Tn5 mutant, and a ksgA::Tn5-pACYC184-ksgA complemented mutant were inoculated separately in 5 ml of LB broth with the appropriate antibiotics and incubated overnight (16 h) at 37°C with shaking at 200 rpm. Approximately 100 CFU of each strain was inoculated in 5 ml of LB broth with the appropriate antibiotics and incubated at 42°C, 37°C, 25°C, and 20°C for temperature effect experiments. Ten-fold dilutions of each strain were prepared in MRD at 24, 48, 72, and 96 h postincubation and spotted in triplicate on LB agar plates to determine CFU at each time point. Additionally, turbidity was measured at each time point (optical density at 600 nm [OD600]) by using a BioTek EL808 spectrophotometer (BioTek Instruments, USA). Each strain was tested in duplicates in three independent experiments. Results were transformed to log10 units, and independent replicates were analyzed by ANOVA followed by a Tukey-Kramer test using NCSS 2007 statistical software (NCSS, Kaysville, UT).
Phenotype microarray.
A total of 10 96-well phenotype microarray (PM) plates constituting eight metabolic panels (PM1 to PM8) and 2 sensitivity panels (PM9 and PM10) were used according to published protocols (19). To assess the altered phenotypes of the ksgA::Tn5 mutant, the respiratory activity (RA) units of the mutant were compared with those of its WT parent at 42°C, 37°C, and 28°C. Cell respiration is measured by reduction of tetrazolium violet dye, which turns purple upon reduction caused by respiration processes along the electron transport chain and accumulates irreversibly within the cells, allowing for colorimetric detection (20). A mean RA threshold of >50 was considered a significant difference (21). The data were further confirmed by a Student t test. Selected results of the phenotype microarray were confirmed by culture in LB and M9 media (3.4 mM Na2HPO4, 2.2 mM KH2PO4, 0.85 mM NaCl, 0.93 mM NH4Cl, 1 mM MgSO4, 0.3 mM CaCl2, 25 mM sodium pyruvate). To assess effects of osmolarity, 100 CFU of each strain was inoculated into 5 ml of LB or M9 medium supplemented with 6.5% NaCl and incubated at 42°C. Ten-fold dilutions of cultures were plated on LB agar plates at 24 h, 48 h, 72 h, 96 h, and 6 days postincubation. Growth was also monitored by measuring OD600. Each strain was tested in two independent experiments. An unpaired Student t test was used to assess differences (P < 0.05) in growth between strains by NCSS 2007 software.
Antibiotic resistance assay.
The emergence of chloramphenicol-resistant spontaneous mutants was assessed using an agar dilution method. Briefly, an average of approximately 3.8 × 102 CFU of each strain (WT and ksgA::Tn5 mutant) was inoculated in duplicate on LB agar plates containing 10 μg/ml of chloramphenicol and incubated for 48 h at 37°C. Colonies were counted in three independent experiments to obtain an average number of CFU/ml. The frequency of resistant colonies was calculated by dividing the recovered colony number by the initial inoculum and multiplying by 100. Data were analyzed by conducting a Z test between two independent proportions (NCSS 2007).
Oxidative stress responses in KsgA-deficient S. Enteritidis.
A single colony of each strain was inoculated in 5 ml of LB broth with appropriate antibiotics at 37°C and incubated overnight (16 h) with shaking at 200 rpm. An aliquot of overnight culture was diluted to determine initial CFU followed by centrifugation at 5,000 rpm for 10 min at 25°C. Oxidative stress was tested by resuspending bacterial pellets in 5 ml normal saline (0.9% NaCl, pH 7.2) preheated to 42°C followed by the addition of H2O2 to a final concentration of 15 mM and incubation at 42°C for 30 min with constant agitation (200 rpm). At the end of exposure, suspensions were diluted 10-fold in MRD and plated on LB agar. Each strain was tested in duplicate in three independent experiments. Percent survival was calculated as follows: (CFU at 30 min/initial CFU) × 100. Data were analyzed by conducting a Z test between two independent proportions (NCSS 2007).
Infection assays in chicken macrophages.
The uptake and survival within chicken macrophages (HD-11 cells) was tested using a gentamicin protection assay as described previously with minor modifications (17). Briefly, HD-11 cells were cultured in Iscove's modified Dulbecco's medium (IMDM) in two 12-well plates at a density of 1 × 106 cells per well and incubated for 2 days at 37°C with 5% CO2. Bacterial inoculum was prepared by culturing strains overnight at 37°C in LB broth with the appropriate antibiotics. Cells were inoculated with the bacterial preparations at a multiplicity of infection (MOI) of approximately 20, centrifuged for 3 min at 1,000 rpm, and incubated at 42°C with 5% CO2 for 30 min to allow bacterial uptake by the cells. Next, plates were washed three times in PBS (pH 7.4) to remove extracellular bacteria followed by treatment with gentamicin (200 μg/ml for 30 min) in IMDM with 10% fetal bovine serum (FBS) to kill any remaining extracellular bacteria. Subsequently, gentamicin was removed by washing cells three times in PBS. At this point, cells in one plate were lysed with the addition of 0.5% (vol/vol) Triton X-100 for 10 min at 42°C, and dilutions of cell lysates were plated on LB agar to determine bacterial uptake (30 min). The second plate was incubated for 8 h and treated as described above to determine intramacrophage survival. Bacterial uptake was calculated using the following formula: (intracellular CFU at 30 min/inoculum CFU) × 100. The intramacrophage survival of the ksgA::Tn5, ksgA::Tn5-pACYC184-ksgA, and G1 Nalr strains was calculated using the following formula: (CFU at 24 h/CFU at 30 min) × 100. Data were analyzed by conducting a Z test for difference of proportions (NCSS 2007).
RESULTS AND DISCUSSION
KsgA-deficient S. Enteritidis is virulence attenuated in chickens.
We recently reported that KsgA deficiency significantly reduced invasiveness of S. Enteritidis in cultured human intestinal epithelial cells (Caco-2), impaired growth of S. Enteritidis in egg albumen at 25 ± 2°C, and resulted in moderate reduction in the invasiveness in chicken liver (LMH) cells compared to that of the WT parent (17). These finding led us to our hypothesis that ksgA plays a role in virulence of S. Enteritidis in the target host chicken. Consequently, we conducted two experiments to test this hypothesis. In experiment 1, we inoculated 1-day-old chickens with the ksgA mutant and WT strain at an initial dose of 108 CFU per bird and monitored kinetics of Salmonella infection in the small intestine, cecum, liver, and spleen by determining viable colonies at 4 days, 8 days, and 12 days p.i. Significant differences in the CFU of the ksgA mutant and WT were found throughout the experimental period (Fig. 1). The ksgA mutant showed a 3- to 4-log reduction in CFU in the small intestine and cecum at any given time compared with the WT parent, indicating that the ksgA mutant was significantly attenuated in its ability to colonize the chicken gut. Unlike the WT, the mutant strain was not recovered from internal organs, such as liver and spleen, from any of the infected birds at any time points (Fig. 1), suggesting that the mutant was significantly attenuated in its organ invasiveness.
Fig 1.

KsgA deficiency decreases the ability of S. Enteritidis to survive within chicken small intestine (SI) (a), cecum (b), liver (c), and spleen (d). One-day-old chicks were orally infected with 108 or 109 CFU of the ksgA::Tn5 mutant or the WT strain. Mean log10 CFU values ± standard error (SE) per gram of tissue were determined on XLD agar at different time points. *, significant difference, P < 0.05; **, significant difference between all groups at a specific time point, P < 0.05.
In the second experiment, we increased the infection dose to 109 CFU per bird, included a group of chickens challenged with a strain expressing KsgA in trans (pACYC184-ksgA), and sacrificed infected birds early during the infection process (i.e., 24 h and 48 h). With these modifications, the number of viable colonies of the WT parent and mutant strain were similar at 24 and 48 h p.i. (P > 0.05, ANOVA) in the small intestine. After this period, the numbers of CFU of the mutant strain were consistently lower than those of the WT parent strain (P < 0.02), reaching a maximum difference of 2.4 log at 12 days p.i. (Fig. 1a). A similar trend was observed in the ceca of birds infected with the high dose (109 CFU); however, these differences were statistically significant only at 12 days p.i. (P = 0.000014) (Fig. 1b). While these results demonstrate an infectious dose response, the number of mutant bacteria recovered tapered off later during infection, consistent with attenuation. In liver, the ksgA mutant showed significantly lower numbers of CFU at 48 h (2.11 ± 0.21), 8 days (3.35 ± 0.04), and 12 days (1.96 ± 0.03) p.i. when a 109 CFU dose was used (P < 0.05), yielding a maximum average difference of 5.10 log at 48 h (P = <0.05), although this difference was not significant (P = 0.054) at 4 days p.i. (Fig. 1c), which also coincided with increased numbers of CFU in the small intestine and ceca (Fig. 1a and b). At a high dose, the CFU counts in spleen also showed the same decreasing trends, both for the WT parent and the mutant strain (Fig. 1d). The mutant strain showed significantly lower CFU counts than the WT parent at 48 h, 8 days, and 12 days (P < 0.05). The most remarkable difference between the WT and the mutant was observed at 48 h p.i., reaching up to 5.5 log (P = 0.003). In trans complementation of KsgA on a low-copy-number plasmid, pACYC184, carrying a chloramphenicol resistance cassette was unable to rescue the virulence phenotype in the KsgA-deficient strain (Fig. 1). The numbers of CFU of the complemented mutant were consistently lower than those of the wild type and the KsgA-deficient mutant throughout the experimental period in all organs tested (P < 0.05).
The role for ksgA in virulence has been previously examined only for Y. pseudotuberculosis in a murine model. Oral inoculation of BALB/c mice with 5 × 108 CFU of a KsgA-deficient mutant of Y. pseudotuberculosis resulted in a bacterial burden significantly lower than that of its wild-type parent in small intestine, Peyer's patches, spleen, and liver until 10 days p.i., which also translated in higher survival rates (15, 22). More importantly, this mutant was able to confer protection against the WT Y. pseudotuberculosis challenge (16). Similar to that in Y. pseudotuberculosis, KsgA deficiency in S. Enteritidis impacts the bacterial burden in intestinal and extraintestinal tissues, rendering possible attenuation in this bacterium. The molecular mechanism underlying this attenuation is currently unknown. Given the role of KsgA in ribosomal maturation, translational initiation, and protein synthesis, it is possible that KsgA deficiency may have pleotropic effects in S. Enteritidis. While dissecting molecular mechanism of virulence attenuation is beyond the scope of this study, we performed a comprehensive phenotypic characterization to identify other altered phenotypes in the ksgA mutant strain of S. Enteritidis.
KsgA deficiency does not confer growth defects in S. Enteritidis.
Growth defects caused by ksgA deletion, particularly at low temperatures, have been reported in E. coli and other bacterial systems (5); the extent of this effect, however, remains unclear due to differential dependence on KsgA (10). Consequently, we tested whether lack of KsgA activity in a ksgA mutant might confer growth defects in S. Enteritidis at a wide range of temperatures. In contrast to the aforementioned reports, we did not observe a significant difference (P > 0.05, Tukey's test) in bacterial growth between the ksgA::Tn5 mutant, a WT strain, and a complemented mutant (data not shown) cultured in LB broth at 20°C, 25°C, 37°C, or 42°C (Fig. 2). These data suggest that unlike in E. coli, KsgA deficiency does not significantly alter the growth of S. Enteritidis at either optimal or suboptimal temperature for growth in vitro, nor does it display a cold-sensitive phenotype observed in other bacterial systems.
Fig 2.
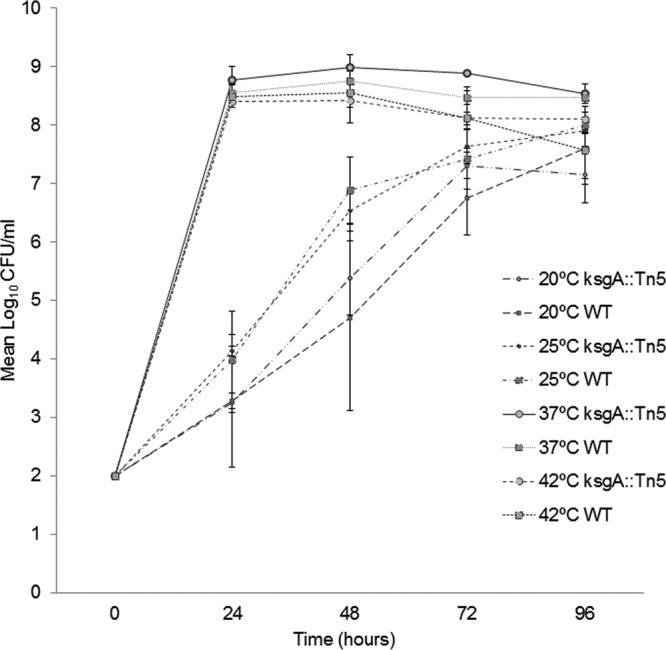
KsgA deficiency does not impair growth in S. Enteritidis in vitro. Growth of the S. Enteritidis ksgA::Tn5, ksgA::Tn5-pACYC184-ksgA (not shown), and WT parent strains was assessed in LB broth with 30 μg/ml of nalidixic acid for up to 96 h at 20°C, 25°C, 37°C, and 42°C. Mean log10 CFU values ± standard deviation (SD) per ml were determined on XLD agar at different experimental time points for three independent replicates.
Deficiency of KsgA significantly alters respiratory activity of S. Enteritidis.
Phenotype microarray (PM) technology was used to assess respiratory activity (RA) of a ksgA mutant and its WT parent at 42°C, 37°C, and 28°C for up to 48 h, testing a total of 920 different phenotypes arranged in 10 96-well microplates (PM1 to PM10) (Table 1). At low temperatures (28°C), the RA of the mutant was not significantly different from that of the WT parent strain with the exception of three (0.33%) out of the 920 phenotypes, all within the carbon panel. The ksgA mutant had an RA significantly higher than that of the wild type in the presence of l-asparagine, whereas it showed significantly lower RA in the presence of d,l-α-glycerol phosphate and d-glucuronic acid as carbon sources (Table 1). The largest numbers of differences were found at avian body temperature (42°C), comprising 15 (1.63%) out of 920 phenotypes tested. The ksgA mutant showed significantly impaired utilization of d-alanine (PM1-A09) and Tween 20 (PM1-C05) as carbon sources, glucuronamide (PM3B-E06) as the nitrogen source, and pyrophosphate (PM4A-A03) as the phosphorus source (Table 1). The RA was also impaired for five different peptide-nitrogen combinations (Table 1). The greatest differences were observed under multiple high-osmolarity panels (4 out of 15 phenotypes) composed of 6% or 6.5% NaCl with and without supplementation with a variety of osmoprotectants (Table 1). Similar results were observed at 37°C, at which at least 4 out of 8 differences were observed in osmolyte panels (Table 1), indicating that KsgA deficiency in S. Enteritidis alters susceptibility to high osmolarity. As expected, no significant differences in the RA were observed when the principle compatible solutes, such as glycine-betaine, carnitine, proline, ectoine, and trehalose, were supplemented as osmoprotectants (see Table 1), further confirming the reduced tolerance of the mutant strain to high osmolarity. When mild osmoprotectants, such as choline, trimethylamine, and trigonelline, were supplemented, the RA of the ksgA mutant was significantly lower than that of the WT parent (Table 1). This is not surprising, because choline is not directly used as an osmoprotectant. It is taken up by cells either by the common ProU transporter or by the specific BetT system and must be enzymatically processed to betaine by the BetA and BetB enzymes induced during osmotic stress (23). Similarly, trigonelline is a cationic betaine and is usually accumulated by Gram-negative bacteria, such as E. coli, but it does not function as an osmoprotectant in Salmonella (24, 25). Finally, trimethylamine is an osmoprotectant in many marine invertebrates and some vertebrates (26), where it induces refolding of thermodynamically misfolded proteins (27), while plant microorganisms obtain this compound from vegetable sources, such as alfalfa seeds (28). Based on these data, it is reasonable to suggest that transport systems for osmoprotectants in the ksgA mutant remain viable, although internal enzymatic machinery might be impaired, potentially due to impaired protein synthesis as a result of accumulation of unmethylated immature ribosomes. Alternatively, these differences might also reflect the poor osmoprotectant activity of choline, trimethylamine, and trigonelline under high-osmolarity conditions tested in this study. Nonetheless, these results indicate that RA of the ksgA mutant was significantly impaired in the presence of high-osmolarity conditions.
Table 1.
Differences in the RA between the WT and ksgA-deficient S. Enteritidis mutant grown at different temperatures as tested by phenotype microarray (Biolog, USA)a
| Temp | Plate type | PM well | Phenotype | RA ±SE |
Mean difference | P value | |
|---|---|---|---|---|---|---|---|
| WT | ksgA mutant | ||||||
| 42°C | Carbon source | PM1-B05 | d-Glucuronic acid | 258 ± 3 | 179 ± 1 | 78.5 | 0.023 |
| Carbon source | PM1-A09 | d-Alanine | 237 ± 1 | 181 ± 0.3 | 56 | 0.023 | |
| Carbon source | PM1-C05 | Tween 20 | 207 ± 10 | 125 ± 10 | 81.5 | 0.004 | |
| Nitrogen source | PM3-E06 | Glucuronamide | 199 ± 0.3 | 119 ± 1 | 79.5 | 0.020 | |
| Phosphorus and sulfur source | PM4A-A03 | Pyrophosphate | 209 ± 3 | 156 ± 1 | 53.5 | 0.042 | |
| Peptide-nitrogen source | PM6-C08 | Asn-Val | 197 ± 0.3 | 146 ± 3 | 51 | 0.050 | |
| Peptide-nitrogen source | PM6-G07 | Ile-His | 177 ± 2 | 125 ± 0.3 | 52.5 | 0.042 | |
| Peptide-nitrogen source | PM6-H05 | Leu-Arg | 165 ± 0 | 91 ± 1 | 74.5 | 0.021 | |
| Peptide-nitrogen source | PM6-H12 | Leu-Phe | 150 ± 3 | 97 ± 2 | 53 | 0.024 | |
| Peptide-nitrogen source | PM8-E09 | Val-Lys | 120 ± 2 | 63 ± 2 | 57.5 | 0.006 | |
| Osmolyte | PM9-A08 | 6.5% NaCl | 170 ± 12 | 107 ± 13 | 63 | 0.020 | |
| Osmolyte | PM9-B02 | 6% NaCl + betaine | 198 ± 8 | 177 ± 15 | 21 | 0.335 | |
| Osmolyte | PM9-B03 | 6% NaCl + N,N-dimethyl glycine | 189 ± 3 | 151 ± 4 | 38 | 0.034 | |
| Osmolyte | PM9-B07 | 6% NaCl + ectoine | 197 ± 7 | 112 ± 11 | 85.5 | 0.231 | |
| Osmolyte | PM9-B08 | 6% NaCl + choline | 189 ± 1 | 129 ± 1 | 60 | 0.011 | |
| Osmolyte | PM9-B12 | 6% NaCl + l-carnitine | 193 ± 3 | 144 ± 1 | 50 | 0.032 | |
| Osmolyte | PM9-C02 | 6% NaCl + l-proline | 192 ± 3 | 160 ± 8 | 32 | 0.156 | |
| Osmolyte | PM9-C08 | 6% NaCl + trehalose | 173 ± 4 | 125 ± 5 | 49 | 0.033 | |
| Osmolyte | PM9-C10 | 6% NaCl + trimethylamine | 189 ± 3 | 123 ± 0 | 66 | 0.048 | |
| Osmolyte | PM9-C12 | 6% NaCl + trigonelline | 188 ± 3 | 121 ± 3 | 67.5 | 0.005 | |
| Osmolyte | PM9-D04 | 6% potassium chloride | 226 ± 5 | 167 ± 6 | 59.5 | 0.027 | |
| 37°C | Carbon source | PM1-B05 | d-Glucuronic acid | 264 ± 11 | 156 ± 9 | 108 | 0.02 |
| Carbon source | PM1-H06 | l-Lyxose | 64 ± 1 | 132 ± 5 | −68 | 0.004 | |
| Osmolyte | PM9-A07 | 6% NaCl | 221 ± 1 | 164 ± 8 | 57 | 0.020 | |
| Osmolyte | PM9-B01 | 6% NaCl | 224 ± 3 | 171 ± 6 | 53 | 0.016 | |
| Osmolyte | PM9-B05 | 6% NaCl + dimethyl sulfonyl propionate | 114 ± 7 | 62 ± 4 | 52 | 0.020 | |
| Osmolyte | PM9-C05 | 6% NaCl + γ-amino-N-butyric acid | 208 ± 3 | 158 ± 8 | 51 | 0.025 | |
| Osmolyte | PM9-E03 | 3% sodium formate | 166 ± 8 | 110 ± 8 | 56 | 0.037 | |
| Osmolyte | PM9-F09 | 9% sodium lactate | 181 ± 1 | 109 ± 7 | 73 | 0.008 | |
| 28°C | Carbon source | PM1-B05 | d-Glucuronic acid | 229 ± 5 | 140 ± 9 | 89 | 0.013 |
| Carbon source | PM1-B07 | d,l-α-glycerol phosphate | 225 ± 6 | 164 ± 4 | 61 | 0.011 | |
| Carbon source | PM1-D01 | l-Asparagine | 62 ± 4 | 146 ± 3 | −85 | 0.003 | |
Phenotypes with a mean difference in RA of >50 and a P value of <0.05 were considered significantly different. RA, respiratory activity; WT, wild type.
Utilization of d-glucuronic acid (PM1-B05), a structural element of the repeating unit of the colanic acid capsule (29), was significantly impaired in the ksgA mutant at all temperatures tested. Liver is considered a rich source of d-glucuronic acid, in which it acts as a precursor for a glucuronidation reaction that is involved in metabolic conversion of endogenous and exogenous substances to more water-soluble compounds that are excreted into urine and bile (30). Several enzymes are involved in d-glucuronic acid metabolism to yield glyceraldehyde-3-phosphate and pyruvate that is used in the Embden-Meyerhof-Parnas pathway in the liver (31). Moreover, Salmonella can use glucuronic acid as a sole source of carbon (32). Therefore, it is possible that deficiency of KsgA may impair the ability of S. Enteritidis to use d-glucuronic acid as the sole carbon source in certain tissues, such as liver, thereby diminishing its ability to invade and survive within such tissues. This effect is consistent with our chicken experiment, in which we were not able to recover viable colonies in liver or spleen when 108 CFU was used for oral infection (Fig. 1c) and the bacterial burden was fairly diminished even when chickens were infected with a high dose (109 CFU).
At 42°C, the ksgA mutant showed significantly lower RA than the WT parent in the presence of leucine-arginine or leucine-phenylalanine as the peptide nitrogen source (Table 1). Leucine plays an important role in protein synthesis, in which more than 90% of intracellular leucine is incorporated for protein production, implying protein synthesis may be impaired at 42°C in the ksgA mutant strain. Deficiency of KsgA may result in production of immature unmethylated ribosomes, which may in turn lead to impaired protein synthesis during initiation and elongation steps and stimulate translation errors (33). It is possible that reduced tolerance to osmotic stress at avian physiological temperature (42°C) along with the likelihood of impaired protein synthesis could impact expression of genes necessary for intestinal invasion and colonization of extraintestinal tissues in chickens. Peptide nitrogen source utilization was impaired only at 42°C when five different peptide-nitrogen sources were used. These results indicate potential pleiotropic effects of KsgA deficiency in S. Enteritidis that are more obvious at the physiological temperature of its natural reservoir host. Interestingly, these defects did not seem to affect growth in vitro (Fig. 2), highlighting the importance of using physiological appropriate models when testing bacterial strains with genetic mutations.
KsgA deficiency confers susceptibility to high osmolarity in S. Enteritidis.
The PM analysis indicated that high osmolarity (6 to 6.5% NaCl) negatively impacted the ksgA mutant. Therefore, we compared the WT, ksgA mutant, and complemented mutant strains for their ability to grow in nutrient-rich LB broth containing 6.5% NaCl (equivalent to 1.1 M) incubated at 42°C for up to 6 days (34). The mutant strain failed to grow at any of the time points tested, whereas both the WT parent and the complemented mutant strain were detectable at 72 h postincubation (Fig. 3). The average doubling times were similar between all three strains when cultured in LB alone at 42°C, being close to an average of 34 min for the three strains. In the presence of 6.5% NaCl, the WT doubling time increased to 273 min (4.5 h), whereas the complement had an average doubling time of 500 min (8.33 h), indicating a partial ability of in trans complementation to rescue this specific phenotype. Increased doubling time, from 1 h to 2.6 h, in response to high osmolarity (7.5% NaCl) has also been observed in Listeria monocytogenes (35). While complementation of ksgA completely rescued the ksgA mutant's inability to grow in high osmolarity at 6 days postincubation, this effect was also partially observed at 72 and 96 h, suggesting that ksgA is indeed involved in tolerance of S. Enteritidis to high osmolarity. We also tested tolerance of the ksgA mutant to high osmolarity in M9 medium containing 6.5% (1.1 M) NaCl for up to 4 days (96 h) at 42°C. In this assay, sodium pyruvate was provided as the carbon source, and viable colonies were counted in LB agar at time points of 24, 48, 72, and 96 h postincubation. Similar to the growth assay in LB medium, the mutant strain did not grow in M9 medium up to 96 h; however, 5.64 ± 0.27 log10 CFU/ml of the WT strain was detected at 24 h postincubation, confirming susceptibility of the mutant to high osmolarity. Unlike with growth assay in LB, we were unable to rescue this phenotype by in trans complementation of ksgA in M9 minimal salts medium.
Fig 3.
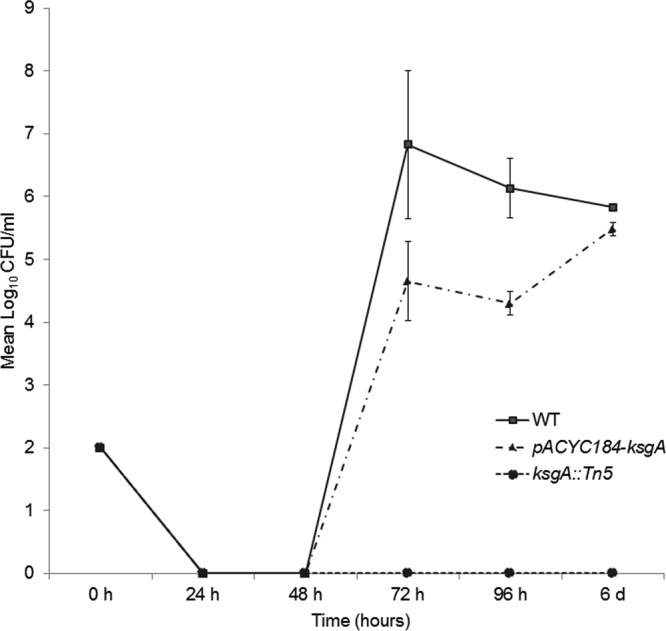
KsgA deficiency confers susceptibility to high osmolarity in S. Enteritidis. Growth of the S. Enteritidis ksgA::Tn5, ksgA::Tn5-pACYC184-ksgA, and WT parent strains was assessed in LB broth and 1.1 M NaCl with 30 μg/ml of nalidixic acid for up to 6 days at 42°C. Mean log10 CFU values ± standard deviation (SD) per ml were determined on XLD agar at different time points for two independent replicates.
To our knowledge, this is the first report showing that ksgA is required for survival under high osmolarity in any bacterial model. Similar to other Gram-negative bacteria, Salmonella has evolved two major mechanisms that function biphasically to respond to this type of stress. A primary response involves an inducible high-affinity system (Kdp) and two low-affinity systems (Trk, Kup) that stimulate uptake of potassium (36). This system tolerates up to 0.5 M of NaCl before inducing a dramatic increase in cytoplasmic concentration (by uptake or synthesis) of osmoprotective compounds, such as glycine-betaine, carnitine, ectoine, proline, trehalose, and amino acids, that are part of the secondary response (37). Additionally, outer membrane porins OmpC and OmpF also play a role in bacterial response to osmotic shock by modulating pore size and number, thus regulating permeability (38). It remains unclear which mechanism is specifically affected in the ksgA mutant. The osmolarity condition (6.5% NaCl) used in this study is well above the theoretical cutoff for the potassium uptake system. It is also important to note that the WT parent and mutant strain showed similar growth under moderate-osmolarity (LB with 300 mM NaCl, data not shown) conditions encountered in the intestinal environment (39). Consequently, we surmise that uptake of potassium is less likely to be affected in the mutant strain.
Deficiency of KsgA alters sensitivity to chloramphenicol.
Deficiency of KsgA or its homologs is known to promote resistance to the aminoglycoside KSG in E. coli, Chlamydia trachomatis, and Neisseria gonorrheae (13). KsgA deficiency in the S. Enteritidis mutant strain used in this study was confirmed by demonstration of resistance to the antibiotic KSG by monitoring growth under increasing concentrations of KSG (from 50 to 1,000 μg/ml). The average MIC of KSG for the ksgA mutant was 500 μg/ml, whereas for the WT and complemented strains, the MIC was 150 μg/ml and 100 μg/ml, respectively, confirming that deficiency of KsgA indeed conferred resistance to the antibiotic KSG (Fig. 4). KSG is a potent inhibitor of translation initiation which acts by preventing binding of the initiator fMet-tRNA to the P site of the 30S ribosomal subunit (40). Resistance is caused indirectly because of unstable interactions between helix 45 (where A1519 and A1518 are located) and helix 44 (primary binding site of kasugamycin) (8). Recent data suggest a new role for KsgA in resistance of M. tuberculosis to the macrolide clarithromycin (11). While the molecular basis of ksgA-mediated macrolide resistance is unknown, macrolides bind to the 50S subunit, causing premature detachment of incomplete polypeptide chains, resulting in impaired protein synthesis (41). Chloramphenicol also binds to the 50S ribosomal subunit, thereby inhibiting protein synthesis by preventing growth of the polypeptide chain (42). Interestingly, one binding site of chloramphenicol lies at the entrance of the peptide exit tunnel (E site) overlapping partially the macrolide erythromycin binding site (43). Therefore, we examined the frequency of resistant (breakout) colonies to moderate doses of the antibiotic chloramphenicol by means of an agar dilution method. The complemented strain was excluded from this assay because the pACYC184 carrier plasmid contains a chloramphenicol resistance cassette for appropriate selection. In this assay, the S. Enteritidis mutant showed significantly increased susceptibility to chloramphenicol (10 μg/ml) at 48 h of incubation. The frequency of appearance of breakouts for the KsgA-deficient mutant was 21.6 ± 11.3 per 108 colonies, whereas the WT strain had an average of 149.3 ± 31.5 breakouts per 108 colonies (P < 0.05) (Fig. 5). These differences indicate that KsgA deficiency in S. Enteritidis confers increased susceptibility to chloramphenicol. Dimethylation caused by KsgA occurs within the loop conformation of helix 45. This loop consists of bases G1516, G1517, A1518, and A1519, which compress the conserved GNRA tetraloop where the second to fourth bases stack toward the 3′ end of the loop. Structurally, the fully methylated 30S lacks the hydrogen bonding between N2 of G1516 and N7 of A1519, so the loop is wider and allows binding of 50S and IF3 and direct contact of helix 45 and helix 44. Lack of methylation provides a tighter loop that inhibits these functions and impairs correct folding and stabilization of the mature ribosome (33). This structural defect might account for the difference in sensitivity to antibiotics that target ribosome sites near helixes 44 and 45 since such unstable interaction might spread throughout the ribosomal P and E sites. Further studies that uncover the mechanism of ksgA-mediated altered drug susceptibility in Salmonella and other bacterial models may provide clues to develop antimicrobials that can specifically target KsgA to render the bacterium more susceptible to specific therapeutic interventions.
Fig 4.
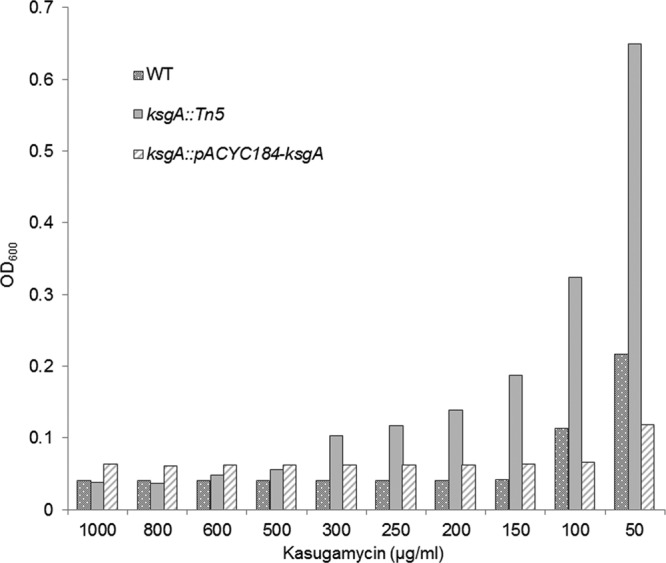
KsgA deficiency confers resistance to kasugamycin (KSG) in S. Enteritidis. Growth was monitored under increasing concentrations of KSG (50 to 1,000 μg/ml) in LB broth by measuring OD600. The average MIC of KSG was 500 μg/ml, 150 μg/ml, and 100 μg/ml for the ksgA::Tn5, ksgA::Tn5-pACYC184-ksgA, and WT strains, respectively.
Fig 5.
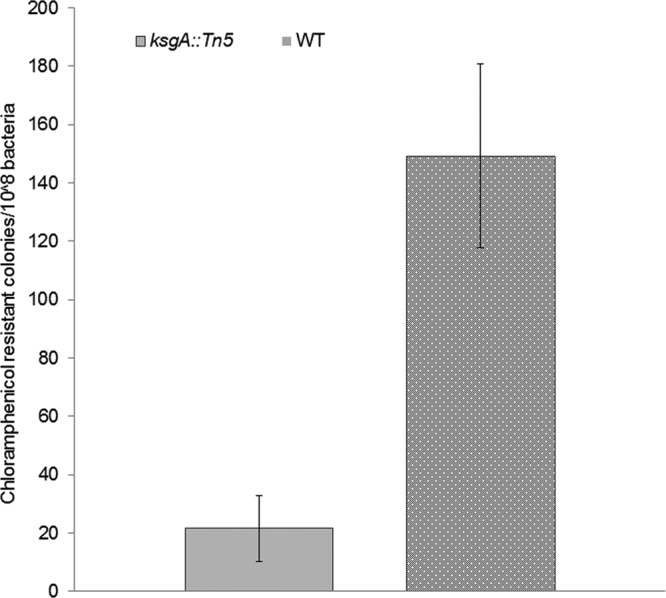
KsgA deficiency reduces tolerance to chloramphenicol in S. Enteritidis. Frequency of resistant colonies to chloramphenicol (10 μg/ml) was assessed through the agar dilution method in wild-type (WT) and KsgA-deficient mutant (ksgA::Tn5) strains. Results from three independent experiments are expressed as average numbers of resistant colonies per 108 bacteria ± SD.
KsgA deficiency reduces tolerance of S. Enteritidis to oxidative stress and impacts uptake by avian macrophages.
Hydrogen peroxide produced by chicken macrophages and heterophils can penetrate cell membranes and act on intracellular targets, thereby playing a crucial role in Salmonella killing. We tested whether the KsgA deficiency would alter the oxidative stress response of S. Enteritidis to treatment with 15 mM H2O2 for 30 min at 42°C. Interestingly, 58 ± 2.8% of the ksgA mutant survived the H2O2 treatment, whereas 71% ± 4% of the WT strain survived this treatment (P < 0.05) (Fig. 6). We were unable to rescue this phenotype by in trans complementation of ksgA, for which the proportion of survivors for the complemented mutant was 16% ± 1% (P < 0.05). Nevertheless, these results corroborate with those of Shah et al. (17), who demonstrated that stress-sensitive S. Enteritidis strains treated with 15 mM H2O2 show a survival proportion as low as 66%. In addition, uptake and intramacrophage survival assays showed that the avian macrophages were able to engulf a significantly higher proportion (14%) of the ksgA mutant than the WT parent (7%) (Fig. 7). However, the intramacrophage survival was similar for both strains, suggesting that mechanisms other than intramacrophage killing may be responsible for reduced virulence of the ksgA mutant in chicken.
Fig 6.
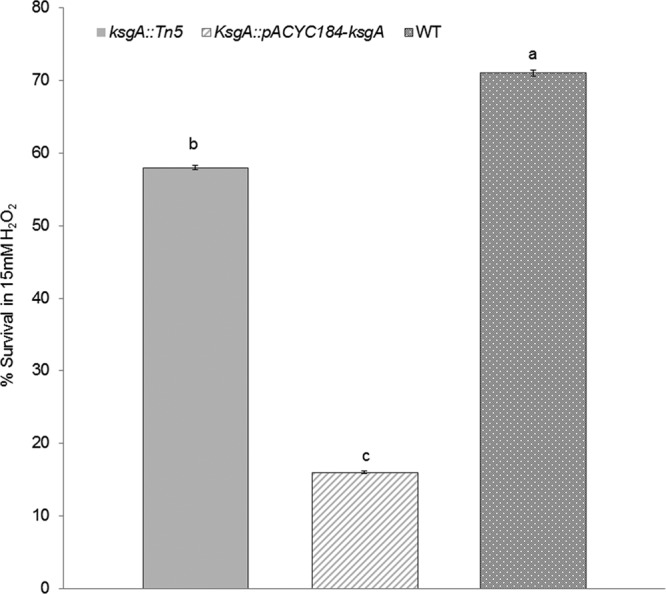
KsgA deficiency reduces tolerance to oxidative stress in S. Enteritidis. Oxidative stress response was assessed as average percentage of survival ± SD after treatment with 15 mM H2O2 for 30 min at 42°C in the S. Enteritidis ksgA::Tn5, ksgA::Tn5-pACYC184-ksgA, and wild-type (WT) parent strains. Three independent experiments were performed; different letters (a, b, and c) represent statistical significant difference (P < 0.05).
Fig 7.
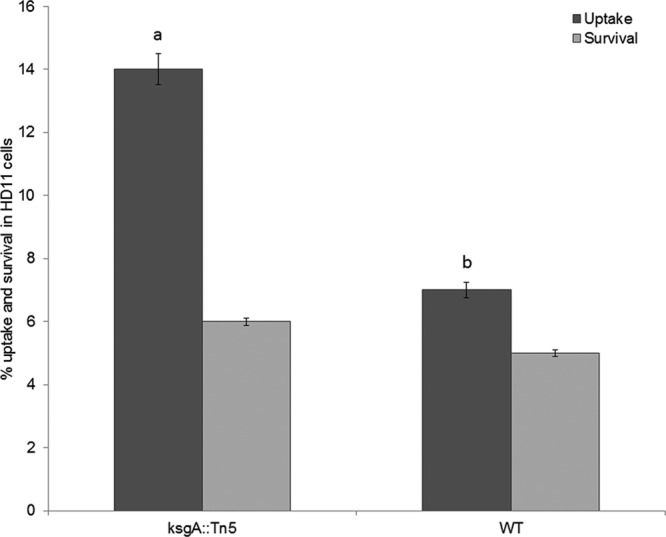
KsgA-deficient S. Enteritidis reduces the ability to survive within chicken macrophage HD11 cells. A gentamicin protection assay with an MOI of 20 was performed at 42°C. Mean log10 CFU values ± standard deviation (SD) per ml were determined at 30 min (invasion) and 8 h (survival) postinfection. The uptake of the ksgA::Tn5 mutant (a) was significantly higher than that of the wild-type parent strain (b) (P < 0.05). Three independent replicates were included in these experiments.
It has been reported that the ksgA gene has a weak promoter, lacks a recognizable ribosomal binding site (44), and displays an autogenous regulation during translation (45). Additionally, overexpression of ksgA in E. coli results in growth defects, suggesting that the controlled expression of this enzyme may be critical to overcome such defects (5). Therefore, while constructing a complementation plasmid, we incorporated a ribosomal binding site upstream of the ksgA gene to enable a heterologous gene to be controlled by the promoter of the tetracycline resistance gene (17). Apparently, this strategy, combined with the use of a low-copy-number pACYC184 plasmid vector, was suitable, as it did not impact growth in vitro at a range of temperatures and also successfully restored the sensitivity of the ksgA mutant to KSG and resistance to high osmolarity in LB medium. However, we were unable to restore other phenotypes, such as resistance to high osmolarity in M9 medium, oxidative stress, uptake by cultured macrophages, and virulence in orally challenged chickens (Fig. 1). While the pACYC plasmid has been successfully used as a low-copy-number plasmid to complement gene function in Salmonella (46), it is possible that unlike for the WT parent strain, the expression levels of ksgA in our complemented strain are not tightly regulated, which may have resulted in detrimental effects on the mutant in some cases. In addition, some reports indicate that either carriage of pACYC184 or the chloramphenicol resistance cassette may also impact invasion efficiency of S. Typhimurium in human epithelial (HeLa) and phagocytic (RAW 264.7) cells and can suppress expression of Salmonella pathogenicity island 1 genes involved in intestinal pathogenesis (47, 48). Therefore, one or more of the above-mentioned factors may have impacted the ability of the complemented strain to restore certain phenotypes. In cis complementation may circumvent some of these difficulties.
In summary, our results clearly show that ksgA contributes to intestinal colonization and organ invasiveness of S. Enteritidis in chickens. Deficiency of KsgA in S. Enteritidis confers no apparent growth defects in vitro at a wide range of temperatures under nutrient-rich conditions. KsgA deficiency does, however, confer increased susceptibility to (i) high osmolarity, (ii) chloramphenicol, and (iii) oxidative stress, suggesting potential pleotropic effects on Salmonella physiology. Given the impaired kinetics of infection of the ksgA mutant in the target host, it appears that the sum of all these defects might become evident within the host environment, where Salmonella must outgrow the local microbiota and also overcome antimicrobial defenses produced by the host (49). For instance, exposure of Salmonella to intestinal high osmolarity and bile salts serves as a cue to modulate its own gene expression (50). The effects of bile salts include DNA damage, secondary structure formation in RNA, and misfolding or denaturation of proteins. Salmonella uses multiple mismatch repair proteins, such as MutH, MutL, and MutS, to prevent DNA damage from bile activity (51). It is important to note that KsgA performs a DNA glycosylase/AP lyase activity to prevent such mutations in E. coli, and KsgA deficiency results in increased spontaneous mutations in mutM and mutY, which also contribute to DNA repair (6). Similar conditions might be encountered in chicken intestinal lumen, which may result in virulence attenuation and lower numbers of physiologically adjusted bacteria within the host.
ACKNOWLEDGMENTS
We thankfully acknowledge the technical assistance of Gail Deckert, Dubraska Diaz (WAADL), and Russell McClanahan (FDIU). We also thank Jake Elder for helpful technical discussions and Douglas Call for critical review of the manuscript.
This project was funded in part with funds from the Agricultural Animal Health Program, College of Veterinary Medicine, Washington State University. Kim Lam Chiok was supported in part by the LASPAU Academic and Professional Programs for the Americas and by the Agricultural Research Center at Washington State University.
Footnotes
Published ahead of print 11 October 2013
REFERENCES
- 1.Harris JK, Kelley ST, Spiegelman GB, Pace NR. 2003. The genetic core of the universal ancestor. Genome Res. 13:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Farrell HC, Pulicherla N, Desai PM, Rife JP. 2006. Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA 12:725–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Helser TL, Davies JE, Dahlberg JE. 1972. Mechanism of kasugamycin resistance in Escherichia coli. Nat. New Biol. 235:6–9 [DOI] [PubMed] [Google Scholar]
- 4.van Buul CPJJ, Visser W, van Knippenberg PH. 1984. Increased translational fidelity caused by the antibiotic kasugamycin and ribosomal ambiguity in mutants harbouring the ksgA gene. FEBS Lett. 177:119–124 [DOI] [PubMed] [Google Scholar]
- 5.Connolly K, Rife JP, Culver G. 2008. Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol. Microbiol. 70:1062–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang-Akiyama QM, Morinaga H, Kikuchi M, Yonekura S, Sugiyama H, Yamamoto K, Yonei S. 2009. KsgA, a 16S rRNA adenine methyltransferase, has a novel DNA glycosylase/AP lyase activity to prevent mutations in Escherichia coli. Nucleic Acids Res. 37:2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duffin PM, Seifert HS. 2009. ksgA mutations confer resistance to kasugamycin in Neisseria gonorrhoeae. Int. J. Antimicrob. Agents 33:321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schluenzen F, Takemoto C, Wilson DN, Kaminishi T, Harms JM, Hanawa-Suetsugu K, Szaflarski W, Kawazoe M, Shirouzu M, Nierhaus KH, Yokoyama S, Fucini P. 2006. The antibiotic kasugamycin mimics mRNA nucleotides to destabilize tRNA binding and inhibit canonical translation initiation. Nat. Struct. Mol. Biol. 13:871–878 [DOI] [PubMed] [Google Scholar]
- 9.Zarubica T, Baker MR, Wright HT, Rife JP. 2011. The aminoglycoside resistance methyltransferases from the ArmA/Rmt family operate late in the 30S ribosomal biogenesis pathway. RNA 17:346–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Farrell HC, Rife JP. 2012. Staphylococcus aureus and Escherichia coli have disparate dependences on KsgA for growth and ribosome biogenesis. BMC Microbiol. 12:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phunpruch S, Warit S, Suksamran R, Billamas P, Jaitrong S, Palittapongarnpim P, Prammananan T. 2013. A role for 16S rRNA dimethyltransferase (ksgA) in intrinsic clarithromycin resistance in Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 41:548–551 [DOI] [PubMed] [Google Scholar]
- 12.Ochi K, Kim JY, Tanaka Y, Wang G, Masuda K, Nanamiya H, Okamoto S, Tokuyama S, Adachi Y, Kawamura F. 2009. Inactivation of KsgA, a 16S rRNA methyltransferase, causes vigorous emergence of mutants with high-level kasugamycin resistance. Antimicrob. Agents Chemother. 53:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binet R, Maurelli AT. 2009. The chlamydial functional homolog of KsgA confers kasugamycin sensitivity to Chlamydia trachomatis and impacts bacterial fitness. BMC Microbiol. 9:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pulicherla N, Pogorzala LA, Xu Z, O'Farrell HC, Musayev FN, Scarsdale JN, Sia EA, Culver GM, Rife JP. 2009. Structural and functional divergence within the Dim1/KsgA family of rRNA methyltransferases. J. Mol. Biol. 391:884–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mecsas J, Bilis I, Falkow S. 2001. Identification of attenuated Yersinia pseudotuberculosis strains and characterization of an orogastric infection in BALB/c mice on day 5 postinfection by signature-tagged mutagenesis. Infect. Immun. 69:2779–2787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergman MA, Loomis WP, Mecsas J, Starnbach MN, Isberg RR. 2009. CD8(+) T cells restrict Yersinia pseudotuberculosis infection: bypass of anti-phagocytosis by targeting antigen-presenting cells. PLoS Pathog. 5:e1000573. 10.1371/journal.ppat.1000573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah DH, Zhou X, Kim HY, Call DR, Guard J. 2012. Transposon mutagenesis of Salmonella enterica serovar Enteritidis identifies genes that contribute to invasiveness in human and chicken cells and survival in egg albumen. Infect. Immun. 80:4203–4215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah DH, Zhou X, Addwebi T, Davis MA, Orfe L, Call DR, Guard J, Besser TE. 2011. Cell invasion of poultry-associated Salmonella enterica serovar Enteritidis isolates is associated with pathogenicity, motility and proteins secreted by the type III secretion system. Microbiology 157:1428–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morales CA, Porwollik S, Frye JG, Kinde H, McClelland M, Guard-Bouldin J. 2005. Correlation of phenotype with the genotype of egg-contaminating Salmonella enterica serovar Enteritidis. Appl. Environ. Microbiol. 71:4388–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bochner BR, Gadzinski P, Panomitros E. 2001. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 11:1246–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Biswas I. 2009. A phenotypic microarray analysis of a Streptococcus mutans liaS mutant. Microbiology 155:61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamark T, Rokenes TP, McDougall J, Strom AR. 1996. The complex bet promoters of Escherichia coli: regulation by oxygen (ArcA), choline (BetI), and osmotic stress. J. Bacteriol. 178:1655–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peddie BA, Lever M, Randall K, Chambers ST. 1999. Osmoprotective activity, urea protection, and accumulation of hydrophilic betaines in Escherichia coli and Staphylococcus aureus. Antonie Van Leeuwenhoek 75:183–189 [DOI] [PubMed] [Google Scholar]
- 25.Ly A, Henderson J, Lu A, Culham DE, Wood JM. 2004. Osmoregulatory systems of Escherichia coli: identification of betaine-carnitine-choline transporter family member BetU and distributions of betU and trkG among pathogenic and nonpathogenic isolates. J. Bacteriol. 186:296–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kempf B, Bremer E. 1998. Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch. Microbiol. 170:319–330 [DOI] [PubMed] [Google Scholar]
- 27.Baskakov I, Bolen DW. 1998. Forcing thermodynamically unfolded proteins to fold. J. Biol. Chem. 273:4831–4834 [DOI] [PubMed] [Google Scholar]
- 28.Perroud B, Le Rudulier D. 1985. Glycine betaine transport in Escherichia coli: osmotic modulation. J. Bacteriol. 161:393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stevenson G, Andrianopoulos K, Hobbs M, Reeves PR. 1996. Organization of the Escherichia coli K-12 gene cluster responsible for production of the extracellular polysaccharide colanic acid. J. Bacteriol. 178:4885–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watkins JB, Gregus Z, Thompson TN, Klaassen CD. 1982. Induction studies on the functional heterogeneity of rat liver UDP-glucuronosyltransferases. Toxicol. Appl. Pharmacol. 64:439–446 [DOI] [PubMed] [Google Scholar]
- 31.Shulami S, Gat O, Sonenshein AL, Shoham Y. 1999. The glucuronic acid utilization gene cluster from Bacillus stearothermophilus T-6. J. Bacteriol. 181:3695–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutnick D, Calvo JM, Klopotowski T, Ames BN. 1969. Compounds which serve as the sole source of carbon or nitrogen for Salmonella typhimurium LT-2. J. Bacteriol. 100:215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demirci H, Murphy F, IV, Belardinelli R, Kelley AC, Ramakrishnan V, Gregory ST, Dahlberg AE, Jogl G. 2010. Modification of 16S ribosomal RNA by the KsgA methyltransferase restructures the 30S subunit to optimize ribosome function. RNA 16:2319–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warriss PD, Knowles TG, Brown SN, Edwards JE, Kettlewell PJ, Mitchell MA, Baxter CA. 1999. Effects of lairage time on body temperature and glycogen reserves of broiler chickens held in transport modules. Vet. Rec. 145:218–222 [DOI] [PubMed] [Google Scholar]
- 35.Patchett RA, Kelly AF, Kroll RG. 1992. Effect of sodium chloride on the intracellular solute pools of Listeria monocytogenes. Appl. Environ. Microbiol. 58:3959–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvarez-Ordonez A, Begley M, Prieto M, Messens W, Lopez M, Bernardo A, Hill C. 2011. Salmonella spp. survival strategies within the host gastrointestinal tract. Microbiology 157:3268–3281 [DOI] [PubMed] [Google Scholar]
- 37.Galinski EA. 1995. Osmoadaptation in bacteria. Adv. Microb. Physiol. 37:272–328 [PubMed] [Google Scholar]
- 38.Sleator RD, Hill C. 2002. Bacterial osmoadaptation: the role of osmolytes in bacterial stress and virulence. FEMS Microbiol. Rev. 26:49–71 [DOI] [PubMed] [Google Scholar]
- 39.Tran QT, Gomez G, Khare S, Lawhon SD, Raffatellu M, Baumler AJ, Ajithdoss D, Dhavala S, Adams LG. 2010. The Salmonella enterica serotype Typhi Vi capsular antigen is expressed after the bacterium enters the ileal mucosa. Infect. Immun. 78:527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuwirth BS, Day JM, Hau CW, Janssen GR, Dahlberg AE, Cate JH, Vila-Sanjurjo A. 2006. Structural analysis of kasugamycin inhibition of translation. Nat. Struct. Mol. Biol. 13:879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giguere S. 2006. Macrolides, azalides, and ketolides, 4th ed. Blackwell Publishing Professional, Hoboken, NJ [Google Scholar]
- 42.Dowling P. 2006. Chloramphenicol, thiamphenicol, and florfenicol, 4th ed. Blackwell Publishing Professional, Hoboken, NJ [Google Scholar]
- 43.Long KS, Porse BT. 2003. A conserved chloramphenicol binding site at the entrance to the ribosomal peptide exit tunnel. Nucleic Acids Res. 31:7208–7215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Gemen B, Koets HJ, Plooy CA, Bodlaender J, Van Knippenberg PH. 1987. Characterization of the ksgA gene of Escherichia coli determining kasugamycin sensitivity. Biochimie 69:841–848 [DOI] [PubMed] [Google Scholar]
- 45.van Gemen B, Twisk J, van Knippenberg PH. 1989. Autogenous regulation of the Escherichia coli ksgA gene at the level of translation. J. Bacteriol. 171:4002–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deiwick J, Nikolaus T, Shea JE, Gleeson C, Holden DW, Hensel M. 1998. Mutations in Salmonella pathogenicity island 2 (SPI2) genes affecting transcription of SPI1 genes and resistance to antimicrobial agents. J. Bacteriol. 180:4775–4780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knodler LA, Bestor A, Ma C, Hansen-Wester I, Hensel M, Vallance BA, Steele-Mortimer O. 2005. Cloning vectors and fluorescent proteins can significantly inhibit Salmonella enterica virulence in both epithelial cells and macrophages: implications for bacterial pathogenesis studies. Infect. Immun. 73:7027–7031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clark L, Martinez-Argudo I, Humphrey TJ, Jepson MA. 2009. GFP plasmid-induced defects in Salmonella invasion depend on plasmid architecture, not protein expression. Microbiology 155:461–467 [DOI] [PubMed] [Google Scholar]
- 49.Baumler AJ, Winter SE, Thiennimitr P, Casadesus J. 2011. Intestinal and chronic infections: Salmonella lifestyles in hostile environments. Environ. Microbiol. Rep. 3:508–517 [DOI] [PubMed] [Google Scholar]
- 50.Prouty AM, Brodsky IE, Falkow S, Gunn JS. 2004. Bile-salt-mediated induction of antimicrobial and bile resistance in Salmonella typhimurium. Microbiology 150:775–783 [DOI] [PubMed] [Google Scholar]
- 51.Prieto AI, Ramos-Morales F, Casadesus J. 2006. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics 174:575–584 [DOI] [PMC free article] [PubMed] [Google Scholar]